

Abstract
Background
Human Papillomavirus (HPV) infection is the main risk factor for the development and progression of cervical cancer. HPV-16 E6 and E7 expression is essential for induction and maintenance of the transformed phenotype. These oncoproteins interfere with the function of several intracellular proteins, including those controlling the PI3K/AKT/mTOR pathway in which Phospolipase D (PLD) and Phosphatidic acid (PA) play a critical role.
Methods
PLD activity was measured in primary human keratinocytes transduced with retroviruses expressing HPV-16 E6, E7 or E7 mutants. The cytostatic effect of rapamycin, a well-known mTOR inhibitor with potential clinical applications, was evaluated in monolayer and organotypic cultures.
Results
HPV-16 E7 expression in primary human keratinocytes leads to an increase in PLD expression and activity. Moreover, this activation is dependent on the ability of HPV-16 E7 to induce retinoblastoma protein (pRb) degradation. We also show that cells expressing HPV-16 E7 or silenced for pRb acquire resistance to the antiproliferative effect of rapamycin.
Conclusion
This is the first indication that HPV oncoproteins can affect PLD activity. Since PA can interfere with the ability of rapamycin to bind mTOR, the use of combined strategies to target mTOR and PLD activity might be considered to treat HPV-related malignancies.
Electronic supplementary material
The online version of this article (10.1186/s12885-018-4392-8) contains supplementary material, which is available to authorized users.
Keywords: HPV, E7, PLD, Phospholipase, Rapamycin, Phosphatidic acid, PA, mTOR, pRb
Background
Human papillomavirus (HPV) is the most prevalent sexually transmitted infection and a necessary cause of cervical cancer, the third most common cancer in women worldwide [1]. More than 99% of cervical cancers contain DNA of HPV high-risk types [2]. In addition, HPV DNA is also found in a significant percentage of other anogenital lesions as well as oral and oropharyngeal tumors [3–5]. HPV-16 is the most prevalent type and it is found in almost 50% of all cervical cancer cases [6]. The two major HPV oncoproteins, E6 and E7, are consistently expressed in all HPV positive cancers. The expression of these proteins in primary human keratinocytes effectively induces their immortalization [7]. Furthermore, when grown under conditions that allow stratification and the formation of skin-like structures, cells immortalized with E6 and E7 from high-risk HPV types display morphological hallmarks of high-grade squamous intra-epithelial lesions, well-established precursors of cervical cancer [8]. The best characterized activity of HPV-16 E6 and E7 is their ability to bind to and induce the proteasome-mediated degradation of tumor suppressors p53 and pRb, respectively [9, 10]. Besides, both HPV-16 E6 and E7 are able to bind to and alter the biological function of several other cellular proteins [11]. Among them are many members of the phosphatidylinositol (PI)-3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) signaling pathway [12]. The PI3K/Akt/mTOR signaling axis plays a very important role in HPV-induced carcinogenesis by acting through multiple cellular and molecular events [13]. Although the regulation of mTOR through PI3K/AKT has been extensively described, another mechanism contributing to activation of mTOR has been proposed. Phosphatidic acid (PA), a product of phospholipase D, is required for mTORC1 activation by mitogens as well as amino acid signals [14–16]. More recently, PA was identified as a major product capable of displacing DEPTOR, a mTOR binding protein that normally functions to inhibit both mTORC1 and mTORC2 pathways [17]. PA species with unsaturated fatty acids chains, such as those produced by PLD, bind with high affinity to the FRB domain of mTOR in a manner that is competitive with its inhibitor rapamycin. As a consequence, elevated PLD activity has been associated to rapamycin resistance [18, 19]. In addition, phospholipase D enzymes play a fundamental role in cells: they maintain the integrity of cellular membranes and they participate in cell signaling including cytoskeletal dynamics, cell migration, intracellular protein trafficking, and cell proliferation. Consistent with this data, increased PLD activity has been reported in a large number of human cancers, including breast, colon, gastric, and kidney [20].
Our results show that upon HPV-16 E7 expression, primary human foreskin keratinocytes upregulate PLD protein levels and activity. Such effect is dependent on the integrity of the E7 LxCxE binding motif and, ultimately to the ability of HPV-16 E7 to induce pRb degradation and promote immortalization. We also show that organotypic cultures of keratinocytes expressing HPV-16 E7 become resistant to the antiproliferative effect of rapamycin.
Methods
Cell culture
Low passage-pooled neonatal foreskin keratinocytes or Primary Human Keratinocytes (cat no. 192906) (Lonza Walkersville, Inc., Walkersville, MD) were maintained in K-SFM media supplemented with 5 μg/L EGF and 50 mg/L bovine pituitary extract (BPE) (Invitrogen, Carlsbad, CA, USA).
Recombinant retroviruses and retroviral-mediated gene transfer
Retroviral vectors pLXSN-neo, pLXSN-E6, −E7, −E6E7, E7E26G and -E7CVQ68-70AAA were kindly provided by Dr. Denise Galloway (Fred Hutchinson Cancer Center, Seattle, USA). Recombinant retroviruses were produced from the amphotropic packaging cell line GP + envAm12 as described earlier [21]. Early passage PHKs were infected with different retroviruses at the same multiplicity of infection (MOI).
pRb downregulation
Specific shRNAs clones for pRB silencing were selected from the MISSION® shRNA Human Gene Family Set-Tumor Suppressors (SH0531, Sigma-Aldrich, St. Louis, MO, USA) and transfected in HEK-293 T cells (ATCC® cat. no. 3216) together with MISSION® Lentiviral Packaging Mix (Sigma-Aldrich, St. Louis, MO, USA) using FuGENE® HD Transfection Reagent (Promega, Madison, WI, USA) according to the manufacturer’s instructions. Supernatants containing lentivirus were collected after 48 and 72 h after transfection. Lentiviral particles titer was determined using an HIV-1 p24 antigen enzyme-linked immunosorbent assay (ELISA) kit (ZeptoMetrix Corporation, Buffalo, NY, USA). Sub-confluent cultures of primary human keratinocytes were infected with lentiviral particles (MOI 5) expressing specific shRNA. After 2 days cells were selected with 2.5 μg/ml of puromycin (Sigma-Aldrich, St. Louis, MO, USA) until complete death of uninfected cells was observed.
Organotypic raft cultures
Organotypic raft cultures of PHK were prepared as described elsewhere [22]. Raft cultures were allowed to differentiate for 9 days and rapamycin (Calbiochem, San Diego, CA, USA) was added to a final concentration of 100 ng/ml at day 6. Bromodeoxyuridine (BrdU) (Sigma-Aldrich, St Louis, MO, USA) was added to a final concentration of 50 μg/ml for the last 12 h. The rafts were then harvested, fixed in formalin and paraffin embedded.
Immunohistochemistry
Raft sections were deparaffinized and dehydrated in xylene and alcohol sequential baths. The endogenous peroxidase activity was quenched with 3% H2O2 after antigen retrieval in boiling citrate buffer. Primary antibodies against BrdU (1:400; Zymed, San Francisco, CA, USA), p53 DO-7 (1:400; DakoCytomation, Glostrup, Denmark) and pRb (1:500; Novocastra, Newcastle-upon-Tyne, UK) were incubated for 18 h in 1% bovine serum albumin-phosphate buffered solution. After incubation with secondary antibody, antigens were detected with streptavidin-biotin-peroxidase complex (StreptABComplex/HRP Duet Mouse/Rabbit DakoCytomation, Dako). Chromogenic detection of peroxidase was performed with diaminobenzidine (DAB) substrate (Sigma-Aldrich, St Louis, MO, USA). The percentage of BrdU-positive/total nuclei was determined by direct counting cell nuclei. At least 3000 nuclei were counted per experiment.
Cell proliferation assays
For Alamar blue based proliferation analysis cells silenced with lentiviral particles (MOI 5) expressing the shRNA described above and appropriate controls were seeded in octuplicate in 96 wells plates (2000 cells/well). After 72 h 10 μL of Alamar blue (Life Technologies, Carlsbad, CA) were added per well and cells were incubated at 37 °C for 4 to 7 h. After this period, Alamar Blue reduction was monitored in at 570 e 600 nm in an Epoch Microplate Spectrophotometer (Bio-Tek, Winooski, VT, USA). For growth curves, cells were seeded in low density in six-well plates and treated with rapamycin (100 ng/ml) in the next day. Cell proliferation was assessed by cell counting in the following 7 days.
Western-blot
Total protein extracts were obtained from monolayer cultures of infected PHKs using lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1% NP-40, 0.5% Sodium deoxycholate, 0.1% SDS) supplemented with complete mini proteases inhibitors cocktail (Roche Diagnostics GmbH, Mannheim, Germany). Whole cell lysates (60 μg) were subjected to SDS-PAGE and transferred to PVDF membranes (GE Healthcare, Buckinghamshire, UK). Membranes were blocked with 5% non-fat dry milk and incubated with PLD antibody (1:300; Upstate Biotechnology, Lake Placid, NY, USA), pRB antibody (1:500; Novocastra NCL-RB-358; Leica Biosystems, Newcastle-upon-Tyne, UK), actin antibody (1:1000; Sigma-Aldrich, St. Louis, MO, USA), anti-HPV-16 E6 antibody (1:500) and anti-HPV-16 E7 antibody (1:500) from Arbor Vita Corporation (Fremont, CA, USA). The immunoreactive bands were visualized by chemiluminescence using ECL reagents (GE Healthcare, Buckinghamshire, UK).
Measurement of phospholipase D activity
To measure PLD activity, PHK infected with different recombinant retrovirus were plated in 100 mm culture dishes and allowed to reach about 80% confluence. Cells were then pre-labeled with [3H] myristic acid (Perkin-Elmer, Waltham, MA, USA) (6 μCi, 30 Ci/mmol) in 4 ml of culture medium for 5 h. PLD activity was determined by measuring the formation of [3H] phosphatidylbutanol (PtBt), the product of PLD-mediated trans-phosphatidylation, in the presence of 0.8% 1-Butanol (Sigma-Aldrich, St. Louis, MO, USA). Lipid extraction and characterization were performed by thin layer chromatography (TLC). Briefly, lipid extracts were counted in a scintillation counter to normalize all samples. Approximately 500.000 CPM of each sample were spotted onto TLC plates (silica gel 60A, Fisher Scientific, Fair Lawn, NJ, USA). TLC were developed with the upper organic phase of a mixture containing ethyl acetate:iso-octane:glacial acetic acid:H2O (1.1:0.5:0.2:1, vol/vol) and allowed to dry before being sprayed with En3hancer Spray (Perkin Elmer, IL, USA). Phosphatidilbutanol was detected by autoradiography with Kodak BioMax MR Film (Kodak, Rochester, NY, USA).
PLD activity in control and HPV gene-expressing primary human keratinocytes after pRb downregulation was determined using the Phospholipase D Assay Kit (#700590, Cayman Chemicals, Ann Arbor, MI, USA) according to the manufacturer’s instructions.
Statistical analysis
Statistical analyses were performed by student’s unpaired t-test using Graphpad Prism Software (Graphpad Software, La Jolla, CA). All the experiments were performed at least three times. p-values were considered two-tailed and significance was defined as p < 0.05.”
Results
HPV-16 E7 expression increases PLD protein expression and activity
PLD1 and PLD2 catalyze the hydrolysis of phosphatidylcholine (PC) to phosphatidic acid (PA) and choline [23]. Due to their vital role in cell signaling and proliferation we sought to investigate if HPV-16 E7 expression could promote any alteration in PLD expression and activity. Our results show that primary human keratinocytes (PHK), expressing HPV-16 E6 and E7 or E7 alone, show an increase in both PLD1 and PLD2 isoforms when compared with keratinocytes transduced only with the empty vector (Fig. 1a). This fact was paralleled by an increase in PLD activity (Fig. 1b). E6 expression alone did not promote any alterations in PLD activation (Additional file 1: Figure S1). Interestingly, we observed that E7 ability to induce PLD expression and activation is dependent on the integrity of its LxCxE motif, as the E7E26G mutant failed to induce PLD expression or increase its activity (Fig. 1a and b). An intact LxCxE motif enables E7 protein to bind pRb protein leading to the release of E2F transcriptional factor and progression into S phase [24, 25]. On the other hand, the E7 mutant (E7CVQ68-70AAA), that keeps the ability to destabilize pRb family members [26], was able to upregulate PLD like the WT E7 protein. Altogether these data indicates that HPV-16 E7 promotes an upregulation in PLD expression and activity and this effect is dependent on the integrity of its LxCxE motif.
Fig. 1.
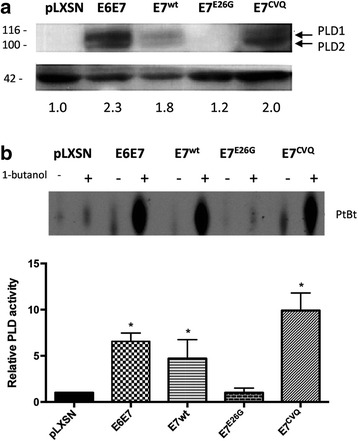
HPV-16 E7 expression increases PLD expression and activity. a PLD expression levels were analyzed by Western-blot in PHK transduced with empty vector, HPV-16 E6E7, wild-type E7 or E7 mutants E7E26G and E7CVQ68-70AAA. b PLD activity was determined by trans-phosphatidylation reaction in the presence of 1-butanol. Generation of phosphatidylbutanol (PtBt) indicates increased PLD activity. PLD activity was normalized to that in pLXSN transduced cells, which was given an arbitrary value of 1
HPV-16 E7 induces PLD activation in a pRb dependent-manner
We investigated if pRb inactivation, induced by HPV-16 E7, could be linked to PLD increased activation. To address this question, foreskin primary human keratinocytes (PHK) were transduced with two different shRNAs targeting pRb. Retinoblastoma protein was strongly downregulated in both cell lines when compared to PHK transducing scrambled shRNA (Fig. 2a). Moreover, PLD activity was strongly upregulated in cells with pRb depleted (Fig. 2b). This result indicates that PLD activation, induced by E7, is linked to its ability to induce pRb degradation.
Fig. 2.
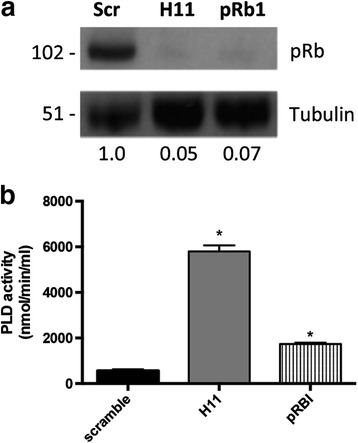
pRb downregulation leads to PLD activation in PHK. a Expression of pRb in PHK was silenced using lentiviral particles codifying specific shRNAs. Silencing efficiency was determined by western blot in 30 micrograms of total protein extracts. b The effect of pRb silencing on PLD activity was determined using Phospholipase D kit according to manufacture instructions. Error bars represent the standard deviation for three independent experiments. (*) p < 0.05
HPV-16 E7 expression confers resistance to the antiproliferative effect of rapamycin
Elevated PLD activity has been previously linked to rapamycin resistance [18]. Rapamycin is a highly specific inhibitor of mTOR and has been widely used to block cell proliferation [27]. Considering this, we sought to study the effect of rapamycin in the proliferation of human keratinocytes expressing HPV-16 main oncogenes in organotypic raft cultures of human keratinocytes. These cultures exhibit highly reduced p53 and pRb expression levels as an indication of the presence of functional E6 and E7 (Additional file 2: Figure S2a). HPV-16 E6 and E7 expression was also confirmed by Western-blot (Additional file 2: Figure S2b). In keratinocytes transduced with empty vector, from now on referred as control cultures, DNA synthesis was observed only in cells from the basal and parabasal layers of the epithelium (Fig. 3a). After being treated with rapamycin, control cultures showed 50% less BrdU incorporation than the untreated tissue (Fig. 3b). On the other hand, organotypic cultures of keratinocytes expressing E6E7 were significantly resistant to the antiproliferative effect of rapamycin (Fig. 3a and b). In organotypic cultures expressing only HPV-16 E6, BrdU incorporation rate decreased by 40% after rapamycin treatment (Fig. 3a and b). Surprisingly, when PHK were transduced only with HPV-16 E7 alone, rapamycin treatment promoted an increase in BrdU incorporation. Growth curve of monolayer cultures of keratinocytes treated with 100 ng/ml rapamycin for 7 days also indicate that PHK transduced with HPV-16 E7 alone proliferate much faster than PHK transduced with the empty vector (Fig. 3c). Interestingly, we also found that rapamycin treatment increased rather than decreased PLD activity in PHK expressing HPV-16 E7 (Fig. 3d). Our results show that HPV-16 E7 expression in primary human keratinocytes not only confers resistance to the antiproliferative effect of rapamycin, but also promotes proliferation in organotypic cultures expressing only this oncoprotein.
Fig. 3.
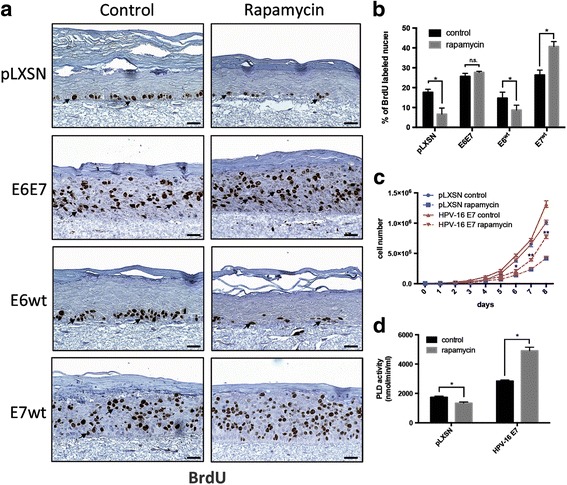
HPV-16 E7 expression confers resistance to the antiproliferative effect of rapamycin. a DNA synthesis in organotypic cultures of keratinocytes was detected by BrdU incorporation followed by immunohistochemistry. Arrows indicate BrdU-positive nuclei. b The percentage of BrdU-positive/total nuclei was determined by direct nuclei counting. The bars correspond to values obtained in at least three independent different experiments. (*) p < 0.05. c Growth curve of monolayer cultures of keratinocytes treated with 100 ng/ml rapamycin for 7 days. Results are representative of three independent experiments performed in triplicates. (*) p < 0.05; (**) p < 0.001. d Rapamycin treatment is associated with increased PLD activity in cultures of PHK expressing HPV16 E7. PLD activity was determined as described in Fig. 2b. Scale bars, 20 μm
We also investigated if the integrity of the LxCxE E7 motif could affect rapamycin resistance. Our results show that E7E26G expressing cultures were as sensitive to rapamycin as control samples (Fig. 4a and b). On the other hand, DNA synthesis was not affected by rapamycin in cultures expressing the E7CVQ68-70AAA mutant (Fig. 4a and b). We also observed that PHK expressing shRNA against pRb are much more resistant to rapamycin treatment than those transduced with scrambled shRNA (Fig. 4c and d). This result indicates that rapamycin resistance observed in these cultures is associated to the ability of E7 to induce pRb degradation.
Fig. 4.
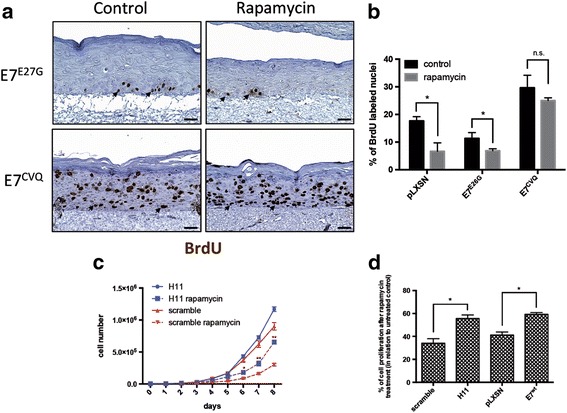
E7-mediated rapamycin resistance requires an intact LxCxE motif. a DNA synthesis in organotypic culture of keratinocytes expressing HPV-16 E7 mutants was detected by BrdU staining. Arrows indicate positive nuclei. b The percentage of BrdU-positive/total nuclei was determined by direct nuclei counting. The bars correspond to values obtained in at least three independent experiments. (*) p < 0.05. c Growth curve of monolayer cultures of keratinocytes treated with 100 ng/ml rapamycin for 7 days. Results are representative of three independent experiments performed in triplicates. (*) p < 0.05; (**) p < 0.001. d Alamar Blue cell proliferation assay was used to evaluate the effect of rapamycin on the proliferation of PHK silenced for pRb expression HPV16 E7 transduced PHKs. Gene silencing was performed as described in Fig. 2. Experiments were carried out in triplicate for each time point. Scale bars, 20 μm
Discussion
A growing body of evidences gathered during the past few years point toward the role of PA in mTOR activation [15]. More recently, PA was identified as a major product capable of displacing DEPTOR, a mTOR binding protein that normally functions to inhibit both mTORC1 and mTORC2 pathways [17]. The major cellular mechanism for generating PA is through the hydrolysis of phosphatidylcholine by PLD [20]. Here we show that HPV-16 E7, one of the major HPV-16 oncoproteins, is able to induce both PLD expression and activity. Interestingly, PLD has been considered a critical regulator of cell proliferation and abnormalities in its activity have been observed in many human cancers [23]. Additionally, PLD activity is elevated in cells transformed by a variety of oncogenes including v-Src, v-Ras, v-Fps and v-Raf [23]. We also demonstrate that PLD activation induced by E7 is dependent on the integrity of the LxCxE motif. This particular region of HPV-16 E7 is known to promote AKT activation in primary human keratinocytes grown in organotypic cultures [28]. The LxCxE motif of high-risk HPVs is also responsible for binding pRb leading to E2F release. For this reason it was previously linked to the ability of HPV-16 E7 to induce cellular transformation [25, 26]. Considering this, we sought to investigate if pRb inactivation could promote PLD activation. Our results show that PHK depleted of pRb present an increase in PLD expression and activity. Interestingly, E2F putative binding sites were identified in PLD1 and PLD2 promoters using the TRANSFAC database [29] and resistance to mTOR inhibitors could be found in cells with defective regulation of the retinoblastoma protein checkpoint [30]. In line with this observation and the fact that PLD overexpression also confers resistance to the mTOR inhibitor rapamycin, we investigated if cells expressing HPV-16 E7 could also display rapamycin resistance. Our results indicate that HPV-16 E7 is also capable to confer resistance to the antiproliferative effect of rapamycin. Supporting our previous observation, this resistance was also associated to the integrity of the LxCxE motif. This result is not without precedent once E7 is also capable of inducing resistance to other cytostatic agents such as TGF-β and TNF in an LxCxE motif dependent-manner [31–33].
One limitation of our study is related to the fact that exogenous PLD expression is associated to human keratinocyte differentiation [21] which precludes the possibility to test directly the effect of PLD overexpression in rapamycin resistance in normal PHK. Nonetheless, our results highlight the importance of HPV-16 E7 in bypassing negative growth regulatory signals [34].
Intriguingly, we found that cultures of keratinocytes expressing only HPV-16 E7 presented an increase in proliferation and an increase in PLD activity after rapamycin treatment. Although this effect was not seen in cells expressing HPV-16 E6 concomitantly, this observation could help us to understand why HPV-associated tumor xenografts generated in immuno-compromised mice grow slower when rapamycin is administered daily to the animals, but fail to lead to any long-term cure [35]. Even with the treatment prolonging survival and delaying cell proliferation, tumors cells could still grow ultimately affecting survival [35]. For this reason, several studies are suggesting the use of rapamycin in combination with other therapeutic drugs [36]. In fact, rapamycin and rapamycin derivatives are being proposed as a concurrent agent to standard-of-care cisplatin/radiation therapy to attenuate tumor lactate production and induce regression of (HPV)-related head and neck squamous cell carcinomas (HNSCC) [36]. A proposed model describing our findings is presented in Fig. 5.
Fig. 5.
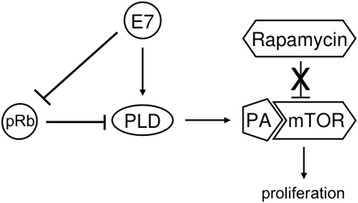
A proposed model for HPV-16 E7 induced resistance to rapamycin and PLD activation. In HPV-16 E7 expressing cells, high intracellular PA levels, generated by increased PLD activity, competes with rapamycin for mTOR binding leading to an increase in cell proliferation
Conclusions
Our work present evidences that HPV-16 E7 up-regulates PLD activity. It also shows that the increase in PLD activation is related to the ability of E7 to induce pRb degradation. Moreover, we show that cells depleted of pRb expression exhibit higher PLD activity. Supporting our findings we present data indicating that both HPV-16 E7 expression and pRb depletion lead to resistance to the antiproliferative effect of rapamycin. Considering the fact that rapamycin and rapamycin analogs are being combined with other chemotherapeutic drugs, it is possible that rapamycin may be associated with PLD inhibitors to circumvent rapamycin resistance exhibited by many types of human cancers, including those related to HPV. Further studies are warranted.
Additional files
Figure S1. HPV-16 E6 did not affect PLD activity. (PDF 127 kb)
Figure S2. HPV-16 E6 and E7 expression affect p53 and pRb expression. (PDF 116 kb)
Acknowledgments
We thank Dr. David Foster (Hunter College of the City University of New York) for sharing the protocol for PLD activity detection and Carlos Ferreira do Nascimento and Severino Ferreira from the ACCamargo Cancer Center for technical assistance with histological samples. We also thank Vanesca de Souza Lino for technical assistance with Western-blots.
Funding
This work was supported by grants from: Ludwig Institute for Cancer Research; Fundação de Amparo a pesquisa do Estado de São Paulo (FAPESP) to Tatiana Rabachini (2003/14008–9), Enrique Boccardo (2010/20002–0), Katia Regina Perez (05/59142–2), Iolanda Midea Cuccovia, Luisa Lina Villa (2008/03232–1 and 2008/57889–1); Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) to Rubiana Andrade and Conselho Nacional de Pesquisa e Desenvolvimento (CNPq) to Rubiana Andrade and Luisa Lina Villa (573799/2008–3), Enrique Boccardo (480552–2011) and Iolanda Midea Cuccovia. The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Availability of data and materials
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- BPE
Bovine pituitary extract
- Brdu
Bromodeoxyuridine
- DAB
Diaminobenzidine
- HNSCC
Head and neck squamous cell carcinomas
- HPV
Human Papillomavirus
- MOI
Multiplicity of infection
- mTOR
Mammalian target of rapamycin
- mTORC1
MTOR complex 1
- PA
Phosphatidic acid
- PC
Phosphatidylcholine
- PHK
Primary human keratinocytes
- PI3K
Phosphatidylinositol (PI)-3-kinase
- PLD
Phospholipase D
- pRb
Retinoblastoma protein
- PtBt
Phosphatidylbutanol
Authors’ contributions
TRA, EB, IMC, and LLV designed the research, supervised all experiments, performed the statistical analysis and drafted this paper. TRA, KRP and RA executed PLD assays, WB and proliferation assays. TRA, EB and SN performed PHK infections, organotypic cultures of keratinocytes and immunohistochemistry. TRA, EB and LLV discussed the results and revised the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12885-018-4392-8) contains supplementary material, which is available to authorized users.
Contributor Information
Tatiana Rabachini, Phone: +55 17 3227 5648, Email: tatianarabachini@hotmail.com.
Enrique Boccardo, Email: eboccardo@usp.br.
Rubiana Andrade, Email: biahbb@hotmail.com.
Katia Regina Perez, Email: perez.katiaregina@gmail.com.
Suely Nonogaki, Email: nonogaki@terra.com.br.
Iolanda Midea Cuccovia, Email: imcuccov@iq.usp.br.
Luisa Lina Villa, Email: l.villa@hc.fm.usp.br.
References
- 1.Pisani P, Bray F, Parkin DM. Estimates of the world-wide prevalence of cancer for 25 sites in the adult population. Int J Cancer. 2002;97(1):72–81. doi: 10.1002/ijc.1571. [DOI] [PubMed] [Google Scholar]
- 2.Walboomers JM, Jacobs MV, Manos MM, Bosch FX, Kummer JA, et al. Human papillomavirus is a necessary cause of invasive cervical cancer worldwide. J Pathol. 1999;189(1):12–19. doi: 10.1002/(SICI)1096-9896(199909)189:1<12::AID-PATH431>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 3.D'Souza G, Kreimer AR, Viscidi R, Pawlita M, Fakhry C, et al. Case-control study of human papillomavirus and oropharyngeal cancer. N Engl J Med. 2007;356(19):1944–1956. doi: 10.1056/NEJMoa065497. [DOI] [PubMed] [Google Scholar]
- 4.Gillison ML, D'Souza G, Westra W, Sugar E, Xiao W, et al. Distinct risk factor profiles for human papillomavirus type 16-positive and human papillomavirus type 16-negative head and neck cancers. J Natl Cancer Inst. 2008;100(6):407–420. doi: 10.1093/jnci/djn025. [DOI] [PubMed] [Google Scholar]
- 5.Paavonen J. Human papillomavirus infection and the development of cervical cancer and related genital neoplasias. Int J Infect Dis. 2007;11(Suppl 2):S3–S9. doi: 10.1016/S1201-9712(07)60015-0. [DOI] [PubMed] [Google Scholar]
- 6.Clifford GM, Smith JS, Plummer M, Munoz N, Franceschi S. Human papillomavirus types in invasive cervical cancer worldwide: a meta-analysis. Br J Cancer. 2003;88(1):63–73. doi: 10.1038/sj.bjc.6600688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hawley-Nelson P, Vousden KH, Hubbert NL, Lowy DR, Schiller JT. HPV16 E6 and E7 proteins cooperate to immortalize human foreskin keratinocytes. EMBO J. 1989;8(12):3905–3910. doi: 10.1002/j.1460-2075.1989.tb08570.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blanton RA, Perez-Reyes N, Merrick DT, McDougall JK. Epithelial cells immortalized by human papillomaviruses have premalignant characteristics in organotypic culture. Am J Pathol. 1991;138(3):673–685. [PMC free article] [PubMed] [Google Scholar]
- 9.Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63(6):1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 10.Boyer SN, Wazer DE, Band V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996;56(20):4620–4624. [PubMed] [Google Scholar]
- 11.Hebner CM, Laimins LA. Human papillomaviruses: basic mechanisms of pathogenesis and oncogenicity. Rev Med Virol. 2006;16(2):83–97. doi: 10.1002/rmv.488. [DOI] [PubMed] [Google Scholar]
- 12.Buchkovich NJ, Yu Y, Zampieri CA, Alwine JC. The TORrid affairs of viruses: effects of mammalian DNA viruses on the PI3K-Akt-mTOR signalling pathway. Nat Rev Microbiol. 2008;6(4):266–275. doi: 10.1038/nrmicro1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang L, Wu J, Ling MT, Zhao L, Zhao KN. The role of the PI3K/Akt/mTOR signalling pathway in human cancers induced by infection with human papillomaviruses. Mol Cancer. 2015;14:87. doi: 10.1186/s12943-015-0361-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fang Y, Vilella-Bach M, Bachmann R, Flanigan A, Chen J. Phosphatidic acid-mediated mitogenic activation of mTOR signaling. Sci. 2001;294(5548):1942–1945. doi: 10.1126/science.1066015. [DOI] [PubMed] [Google Scholar]
- 15.Sun Y, Chen J. mTOR signaling: PLD takes center stage. Cell Cycle. 2008;7(20):3118–3123. doi: 10.4161/cc.7.20.6881. [DOI] [PubMed] [Google Scholar]
- 16.Yoon MS, Du G, Backer JM, Frohman MA, Chen J. Class III PI-3-kinase activates phospholipase D in an amino acid-sensing mTORC1 pathway. J Cell Biol. 2011;195(3):435–447. doi: 10.1083/jcb.201107033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yoon MS, Rosenberger CL, Wu C, Truong N, Sweedler JV, Chen J. Rapid mitogenic regulation of the mTORC1 inhibitor, DEPTOR, by phosphatidic acid. Mol Cell. 2015;58(3):549–556. doi: 10.1016/j.molcel.2015.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chen Y, Zheng Y, Foster DA. Phospholipase D confers rapamycin resistance in human breast cancer cells. Oncogene. 2003;22(25):3937–3942. doi: 10.1038/sj.onc.1206565. [DOI] [PubMed] [Google Scholar]
- 19.Toschi A, Lee E, Xu L, Garcia A, Gadir N, Foster DA. Regulation of mTORC1 and mTORC2 complex assembly by phosphatidic acid: competition with rapamycin. Mol Cell Biol. 2009;29(6):1411–1420. doi: 10.1128/MCB.00782-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Foster DA. Regulation of mTOR by phosphatidic acid? Cancer Res. 2007;67(1):1–4. doi: 10.1158/0008-5472.CAN-06-3016. [DOI] [PubMed] [Google Scholar]
- 21.Jung EM, Betancourt-Calle S, Mann-Blakeney R, Griner RD, Bollinger Bollag W. Sustained phospholipase D activation is associated with keratinocyte differentiation. Carcinogenesis. 1999;20(4):569–576. doi: 10.1093/carcin/20.4.569. [DOI] [PubMed] [Google Scholar]
- 22.McLaughlin-Drubin ME, Münger K. The human papillomavirus E7 Oncoprotein. Virology. 2009;384(2):335–344. doi: 10.1016/j.virol.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Foster DA, Xu L. Phospholipase D in cell proliferation and cancer. Mol Cancer Res. 2003;1(11):789–800. [PubMed] [Google Scholar]
- 24.Helt AM, Galloway DA. Mechanisms by which DNA tumor virus oncoproteins target the Rb family of pocket proteins. Carcinogenesis. 2003;24(2):159–69. 10.1093/carcin/24.2.159. [DOI] [PubMed]
- 25.Demers GW, Espling E, Harry JB, Etscheid BG, Galloway DA. Abrogation of growth arrest signals by human papillomavirus type 16 E7 is mediated by sequences required for transformation. J Virol. 1996;70(10):6862–6869. doi: 10.1128/jvi.70.10.6862-6869.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Helt AM, Galloway DA. Destabilization of the retinoblastoma tumor suppressor by human papillomavirus type 16 E7 is not sufficient to overcome cell cycle arrest in human keratinocytes. J Virol. 2001;75(15):6737–6747. doi: 10.1128/JVI.75.15.6737-6747.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hidalgo M, Rowinsky EK. The rapamycin-sensitive signal transduction pathway as a target for cancer therapy. Oncogene. 2000;19(56):6680–6686. doi: 10.1038/sj.onc.1204091. [DOI] [PubMed] [Google Scholar]
- 28.Menges CW, Baglia LA, Lapoint R, McCance DJ. Human papillomavirus type 16 E7 up-regulates AKT activity through the retinoblastoma protein. Cancer Res. 2006;66(11):5555–5559. doi: 10.1158/0008-5472.CAN-06-0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wingender E, Chen X, Hehl R, Karas H, Liebich I, Matys V, Meinhardt T, Prüss M, Reuter I, Schacherer F. TRANSFAC: an integrated system for gene expression regulation. Nucleic Acids Res. 2000;28:316–319. doi: 10.1093/nar/28.1.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kurmasheva RT, Huang S, Houghton PJ. Predicted mechanisms of resistance to mTOR inhibitors. Br J Cancer. 2006;95(8):955–960. doi: 10.1038/sj.bjc.6603353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Basile JR, Zacny V, Munger K. The cytokines tumor necrosis factor-alpha (TNF-alpha ) and TNF-related apoptosis-inducing ligand differentially modulate proliferation and apoptotic pathways in human keratinocytes expressing the human papillomavirus-16 E7 oncoprotein. J Biol Chem. 2001;276(25):22522–22528. doi: 10.1074/jbc.M010505200. [DOI] [PubMed] [Google Scholar]
- 32.Boccardo E, Noya F, Broker TR, Chow LT, Villa LL. HPV-18 confers resistance to TNF-alpha in organotypic cultures of human keratinocytes. Virology. 2004;328(2):233–243. doi: 10.1016/j.virol.2004.07.026. [DOI] [PubMed] [Google Scholar]
- 33.Boccardo E, Manzini Baldi CV, Carvalho AF, Rabachini T, Torres C, et al. Expression of human papillomavirus type 16 E7 oncoprotein alters keratinocytes expression profile in response to tumor necrosis factor-alpha. Carcinogenesis. 2010;31(3):521–531. doi: 10.1093/carcin/bgp333. [DOI] [PubMed] [Google Scholar]
- 34.Stelzer MK, Pitot HC, Liem A, Lee D, Kennedy GD, Lambert PF. Rapamycin inhibits anal carcinogenesis in two preclinical animal models. Cancer Prev Res (Phila) 2010;3(12):1542–1551. doi: 10.1158/1940-6207.CAPR-10-0228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li J, Kim SG, Blenis J. Rapamycin: one drug, many effects. Cell Metab. 2014;19(3):373–379. doi: 10.1016/j.cmet.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coppock JD, Vermeer PD, Vermeer DW, Lee KM, Miskimins WK, et al. mTOR inhibition as an adjuvant therapy in a metastatic model of HPV+ HNSCC. Oncotarget. 2016;7(17):24228–24241. doi: 10.18632/oncotarget.8286. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. HPV-16 E6 did not affect PLD activity. (PDF 127 kb)
Figure S2. HPV-16 E6 and E7 expression affect p53 and pRb expression. (PDF 116 kb)
Data Availability Statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.