

Abstract
Four new eudesmane-type sesquiterpenoids, penicieudesmol A–D (1–4), were isolated from the fermentation broth of the mangrove-derived endophytic fungus Penicillium sp. J-54. Their structures were determined by spectroscopic methods, the in situ dimolybdenum CD method, and modified Mosher’s method. The bioassays results showed that 2 exhibited weak cytotoxicity against K-562 cells.
Keywords: endophytic fungus, Penicillium sp., sesquiterpenoids, cytotoxicity
1. Introduction
Mangrove forests, the unique forest ecosystems distributed in most tropical and subtropical regions, are an important resource of endophytic fungi that have been proved to be an important source of structurally and biologically diverse substances [1,2,3,4,5,6,7,8,9] such as peniphenones A–D, aniquinazolines A–D, phomazines A–C, and so on [10,11,12]. In order to pursue bioactive products from mangrove fungus, the secondary metabolites of mangrove endophytic fungus Penicillium sp. FJ-1 isolated from the stem of Ceriops tagal were studied, and a new drimane-type sesquiterpene [13] with antibacterial activity has been reported in our previous research. In our continuous research, four eudesmane-type new sesquiterpenoids, penicieudesmol A–D (1–4) (Figure 1), were obtained from the culture broth of the Penicillium sp. J-54 isolated from the healthy leaves of Ceriops tagal collected in Dong Zhai Gang Mangrove Reserve in Hainan. Herein, we described the isolation, structure determination, and biological activities of the new sesquiterpenoids 1–4.
Figure 1.
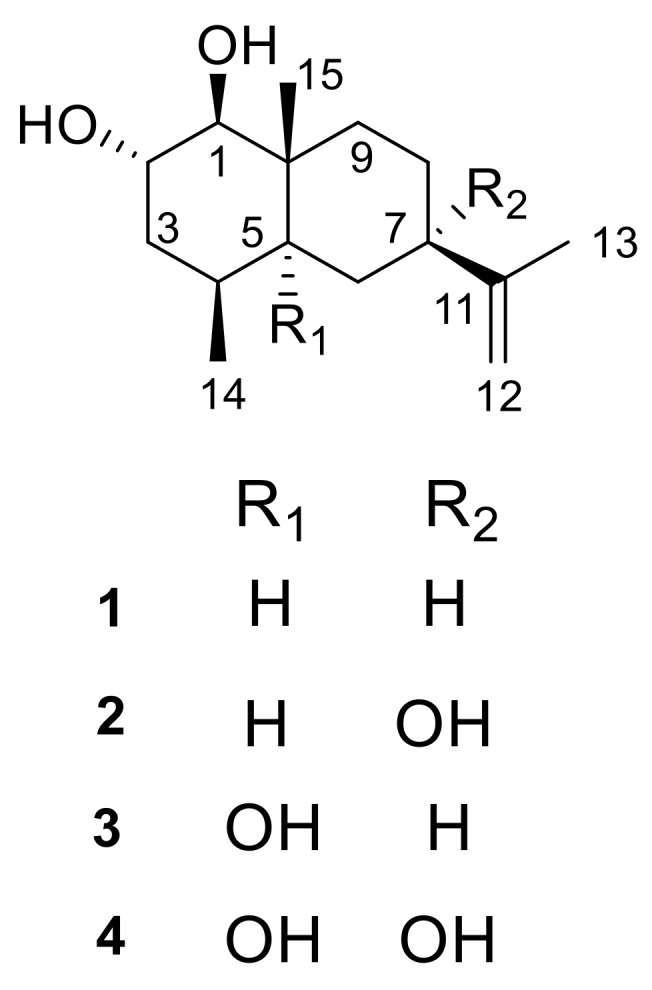
Chemical structures of compounds 1–4 from Penicillium sp. J-54.
2. Results
2.1. Structural Elucidation
Penicieudesmol A (1), a white powder, had the molecular formula of C15H26O2 determined by HREIMS at m/z 238.1931 [M]+ (calcd. for C15H26O2, m/z 238.1933). The 1H-NMR spectrum of 1 clearly exhibited two olefinic protons (δH 4.67, 4.64), three methyl groups (δH 1.68, 0.85, 0.81), and five methine protons (δH 3.54, 2.74, 1.71, 1.90, 1.31). The 13C NMR spectrum combined with the DEPT spectrum (Figure S2) implied a total of 15 carbon resonances including three methyl carbons (δC 21.1, 16.0, 15.6), five methylene carbons (including one sp2 methylene carbon and four sp3 methylene carbons), five methine carbons (including two oxygen bearing methine carbons and three sp3 methine carbons), and two quaternary carbons (δC 150.3, 39.2). The 1D-NMR data of 1 (Table 1) combined with the sequential 1H-1H COSY correlations of H-1/H-2/H-3/H-4/H-5/H-6/ H-7/H-8/H-9, as well as the key HMBC from H3-14 to C-3/C-4/C-5, H3-15 to C-1/C-9/C-5, and H3-13 to C-7/C-11/C-12, suggested an eudesmane-type skeleton for 1. By comparison, above data (Table 1) were very close to that of the known compound nardoeudesmol A [14] with the eudesmane-type skeleton. The major difference between them pointed to the additional of a methine (δC 33.7, C-4) and a methyl (δC 15.6, C-14), as well as the absence of two olefinic carbon (δC 146.4, C-4 and δC 109.7, C-14) in 1 based on the key HMBC from H3-14 to C-3/C-4/C-5. The relative configuration of 1 was identical to the ROESY experiment (Figure 3), such that the observed cross-correlation peaks from H3-15 and H3-14 to H-2, as well as from H-1 and H-7 to H-5, proved H3-15, H3-14, and H-2 were on the same side of the molecular plane and H-1, H-5, and H-7 were on the same side. The large coupling constants (9.2 Hz) between H-1 and H-2 characterised the trans-diaxial relationship. Moreover, the absolute configuration of the 1,2-diol moiety in 1 was determined by the in situ dimolybdenum CD method developed by Snatzke and Frelek [14,15,16]. On the basis of the empirical rule proposed by Snatzke, the positive Cotton effect observed at around 310 and 400 nm, respectively, in the induced CD spectrum (Figure 4a) permitted one to assign the 1S and 2S absolute configuration. Therefore, the absolute configuration of penicieudesmol A was deduced to be 1S, 2S, 4S, 5S, 7R, and 10R.
Table 1.
1H and 13C-NMR Data for 1 and 2 (500 and 125 MHz, DMSO-d6, δ in ppm).
| Position | 1 | 2 | ||
|---|---|---|---|---|
| δC, Type | δH mult. (J in Hz) | δC, Type | δH, mult. (J in Hz) | |
| 1 | 83.9, CH | 2.74, dd, (9.2, 4.0) | 83.8, CH | 2.77, dd, (9.2, 3.9) |
| 2 | 66.7, CH | 3.54, m | 66.6, CH | 3.64, m |
| 3 | 40.5, CH2 | 1.79, m | 40.5, CH2 | 1.68, m |
| 1.43, m | 1.42, m | |||
| 4 | 33.7, CH | 1.71, m | 33.3, CH | 1.59, m |
| 5 | 45.7, CH | 1.31, m | 39.1, CH | 1.76, m |
| 6 | 31.3, CH2 | 1.41, m | 35.8, CH2 | 1.51, m |
| 1.25, m | 1.11, d, (13.0, 2.5) | |||
| 7 | 45.6, CH | 1.90, m | 72.7, qC | |
| 8 | 26.2, CH2 | 1.44, m | 30.4, CH2 | 1.54, m |
| 1.27, m | 1.33, m | |||
| 9 | 40.3, CH2 | 1.67, m | 35.9, CH2 | 1.66, m |
| 0.98, m | 1.35, m | |||
| 10 | 39.2, qC | 38.9, qC | ||
| 11 | 150.3, qC | 153.2, qC | ||
| 12 | 108.7, CH2 | 4.67, s | 108.5, CH2 | 4.96, d, (1.8) |
| 4.64, s | 4.68 d, (1.8) | |||
| 13 | 21.1, CH3 | 1.68, s | 19.2, CH3 | 1.74, s |
| 14 | 15.6, CH3 | 0.81, s | 14.6, CH3 | 0.79, s |
| 15 | 16.0, CH3 | 0.85, d, (7.6) | 15.9, CH3 | 0.86, d, (7.6) |
| 1-OH | 4.38, d, (3.7) | |||
| 2-OH | 4.41, d, overlap | 4.37, d, (3.7) | ||
| 5-OH | 4.41, d, overlap | |||
| 7-OH | 4.22, s | |||
Penicieudesmol B (2) was isolated as a white powder with a molecular formula C15H26O3 determined by its HREIMS at m/z 254.1878 [M]+ (calcd. for m/z 254.1882). The similarity of 1D and 2D NMR data between 2 (Table 1) and 1 indicated their similar planar structure. The only difference between these two compounds was that H-7 in 1 was substituted by a hydroxyl in 2, which was proved by the obvious downfield shift of C-7 (δC 72.7) and the HMBC correlations from 7-OH to C-7/C-8 and H3-13 to C-7, together with the HREIMS. The relative configuration of 2 was identical with that of 1 by the large coupling constants (9.1 Hz) between H-1 and H-2, as well as the ROESY correlations (Figure 3). In addition, the 2S configuration of compound 2 was clearly defined by the observed chemical shift differences ΔδS−R by the modified Mosher’s method (Figure 4b) [12]. So, the stereogenic centers of penicieudesmol B were determined as 1S, 2S, 4S, 5S, 7S, and 10R.
Penicieudesmol C (3) was obtained as yellow oil with the molecular formula C15H26O3 determined according to the HREIMS peak at m/z 254.1880 [M]+ (calcd. for m/z 254.1882), indicating an isomer of 2. The 1H and 13C NMR data of 3 (Table 2) showed high similarity to those of 2, except for the location of hydroxyl in the two compounds. The sequential 1H-1H COSY correlations of H-6/H-7/H-8/H-9, together with the key HMBC correlations from 5-OH, H3-14, and H3-15, as well as H-7 to C-5, from H3-15 to C-9, and from H3-13 to C-7 displayed that 7-OH in 2 shifted to 5-OH in 3. The relative and absolute configuration of 3 was determined to be consistent with that of 2 through the same method (Figures 3 and 4b). Hence, the stereogenic centers of penicieudesmol C were determined as 1S, 2S, 4S, 5R, 7R, and 10S.
Table 2.
1H and 13C NMR Data for 3 and 4 (500 and 125 MHz, DMSO-d6, δ in ppm).
| Position | 3 | 4 | ||
|---|---|---|---|---|
| δC, Type | δH, mult. (J in Hz) | δC, Type | δH mult. (J in Hz) | |
| 1 | 77.8, CH | 3.45, dd, (9.1, 2.5) | 77.5, CH | 3.40, d, (9.2) |
| 2 | 67.7, CH | 3.66, m | 67.3, CH | 3.60, m |
| 3 | 35.9, CH2 | 2.06, m | 35.0, CH2 | 1.93, m |
| 1.48, m | 1.40, m | |||
| 4 | 41.2, CH | 1.71, m | 40.5, CH | 1.67, m |
| 5 | 75.3, qC | 76.7, qC | ||
| 6 | 37.1, CH2 | 1.79, m | 38.7, CH2 | 1.99, d, (14.0) |
| 1.25, dd, (13.3, 3.0) | 1.15, d, (14.0) | |||
| 7 | 39.2, CH | 2.53, m | 75.1, qC | |
| 8 | 25.4, CH2 | 1.49, m | 30.3, CH2 | 1.46, m |
| 1.34, m | ||||
| 9 | 33.3, CH2 | 1.76, m | 29.8, CH2 | 1.76, m |
| 1.43, m | 1.40, m | |||
| 10 | 41.9, qC | 42.1, qC | ||
| 11 | 150.7, qC | 151.6, qC | ||
| 12 | 108.7, CH2 | 4.73, d, (1.6) | 109.3, CH2 | 4.96, s |
| 4.75, d, (1.6) | 4.74, s | |||
| 13 | 21.3, CH3 | 1.67, s | 19.0, CH3 | 1.74, s |
| 14 | 17.8, CH3 | 0.96, d, (7.8) | 17.4, CH3 | 0.95, d, (7.8) |
| 15 | 16.9, CH3 | 0.86, s | 16.7, CH3 | 0.87, s |
| 1-OH | 4.29, d, (3.7) | 4.26, br s | ||
| 2-OH | 4.37, d, (2.8) | 4.20, br s | ||
| 5-OH | 3.74, s | 5.66, s | ||
| 7-OH | 5.63 s | |||
Penicieudesmol D (4) was also obtained as yellow oil. The HREIMS displayed a quasi-molecular ion peak at m/z 270.1833 [M]+ (calcd. for m/z 270.1831), indicating the molecular formula C15H26O4. The 1H and 13C NMR data of compound 4 was very close to those of compound 3. According to the HREIMS of them, hydrogen atoms in 3 were substituted by a hydroxy group in 4. The sequentia 1H–1H COSY correlations of H-1/H-2/H-3/H-4 and H-8/H-9 combined the key HMBC correlations (Figure 2) from 7-OH to C-7 and C-8, as well as H3-13 to C-7, along with the downfiled shifts and 13C multiplicity of C-7 (δC 75.1). Table 2 suggests that the substituent hydrogen atoms were H-7 in 4. The relative and absolute configuration of 4 was determined to be consistent with that of 2 and 3 via the same method (Figure 3 and Figure 4b). Consequently, the stereogenic centers of penicieudesmol D were determined to be 1S, 2S, 4S, 5R, 7S, and 10S.
Figure 2.

The key 2D-NMR correlations for compounds 1−4.
Figure 3.

Key 1H–1H REOSY correlations of compounds 1–4.
Figure 4.

(a) CD spectrum of 1 in DMSO containing Mo2(OAc)4 with the inherent CD spectrum; (b) Δδ (=δS − δR) values for (S)- and (R)-MTPA esters of 2–4.
2.2. The Bioactivities of Compounds 1–4 from Penicillium sp. J-54
All the compounds (1–4) were evaluated for their cytotoxic activity against K-562, SEL-7420, and SGC-7721 cell lines using the MTT method in vitro [17] and antimicrobial activity against Candida albicans and Staphylococcus aureus using the filter paper disc agar diffusion method [18]. The results showed that compound 2 exhibited weak cytotoxicity against K-562 with IC50 value of 90.1 µM, with paclitaxel as the positive control (IC50 = 9.5 µM). Unfortunately, none of these compounds showed antimicrobial activity.
3. Materials and Methods
3.1. General Experimental Procedures
Silica gel (60–80, 200–300 mesh, Qingdao Marine Chemical Co., Ltd., Qingdao, China), ODS gel (20–45 µm, Fuji Silysia Chemical Co., Ltd., Greenville, NC, USA), and Sephadex LH-20 (Merck, Kenilworth, NJ, USA) were used for column chromatography. TLC was conducted on precoated silica gel G plates (Qingdao Marine Chemical Co., Ltd.), and spots were detected by spraying with 5% H2SO4 in EtOH followed by heating. Optical rotation was measured on a Rudolph Autopol III polarimeter. UV spectra were performed on a Shimadzu UV-2550 spectrometer (Beckman, Brea, CA, USA). IR absorptions were obtained on a Nicolet 380 FT-IR instrument (Thermo, Waltham, MA, USA) using KBr pellets. 1D and 2D-NMR spectra were recorded on Bruker AV III spectrometer (Bruker, Billerica, MA, USA) (1H-NMR at 500 MHz and 13C NMR at 125 MHz) using TMS as the internal standard. Chem3D Pro 14.0 (PerkinElmer, Waltham, MA, USA) was used for building these 3D models and calculating energy minimizations.
3.2. Fungal Material
Penicillium sp. J-54 was isolated from the healthy leaves of Ceriops tagal, which were collected in Dong Zhai Gang Mangrove Reserve in Hainan province, in July 2011. The endophytic fungus was identified based on the DNA sequences of 18S rDNA gene. For identification of its 18S rDNA gene sequences, the Penicillium sp. J-54 was cultured in potato dextrose agar for five days. The mycelium was ground to a fine powder in liquid N2, then genomic DNA was extracted, and 18S rDNA region was amplified by PCR using primers NS1 (5′-GTAG TCATATGCTTGTCTC-3′) and NS6 (5′-GCATCACAGACCTGTTATTGCCTC-3′). PCR products were sequenced (Applid Biosystems 3730 XL Genetic Analyzer, Applied Biosystems Inc., Foster City, CA, USA). The producing strain was prepared on PDA medium and stored in our Lab. at 4 °C.
3.3. Fermentation and Extraction
Penicillium sp. J-54 was cultured in PDB (the potato liquid media consisting of 200.0 g/L potato, 20.0 g/L glucose, and 1000 mL deionized water) at 29 °C and 130 rpm for 72 h. 20 mL of the seed culture was inoculated into each 1000 mL Erlenmeyer flask of production medium composed of (per litre) 20.0 g potato, 0.4 g glucose, and 400 mL deionized water; the pH was adjusted 7.0. They were cultivated in static for 4 weeks after being incubated at 29 °C for 7 days on a rotary shaker at 130 rpm. The liquid filtrate from 100 L of fermentation broth was collected and extracted four times with ethyl acetate (1000 mL × 4 times) at room temperature.
3.4. Purification and Identification
The obtained EtOAc crude extract (35.5 g), which was separated into 10 fractions (Fr.1–Fr.10) on silica gel (100.0 g, 200–300 mesh) column chromatography (CC) (4 × 60 cm), eluted with a gradient elution of CHCl3-MeOH (v/v, 1:0 to 0:1, each 1000 mL). Fr.2 (3.3 g) was purified by ODS column chromatography (CC) (2.5 × 40 cm) with gradient of Water-MeOH (v/v, 30:70, 40:60, 50:50, 60:40, 70:30, 80:20, 90:10, 100:0, each 1 L) to get five subfractions (Fr.2.1–Fr.2.5). Fr.2.4 was submitted to Sephadex LH-20 (2 × 30 cm), eluted with MeOH (500 mL), then further separated on a silica gel CC (1 × 20 cm) eluted with CHCl3-MeOH step gradient (v/v, 200:1 to 20:1) to yield compound 1 (3.8 mg) and compound 3 (4.1 mg). Fr.3 (2.2 g) was separated on a silica gel CC (2.5 × 40 cm) eluted with CHCl3-MeOH step gradient (v/v, 1:0 to 10:1) to yield eleven subfractions (Fr.3-1–Fr.3-11). Fr.3-4 (451.5 mg) was applied to Sephadex LH-20 (2 × 30 cm) with CHCl3-MeOH (v/v, 1:1, 400 mL) as eluent, and then further purified again by silica gel CC (1 × 20 cm) with eluting of CHCl3-MeOH (v/v, 50:1, 1500 mL) to obtain compound 4 (3.1 mg). Fr.4 (2.5 g) was separated on a silica gel CC (2.5 × 40 cm) eluted with CHCl3-MeOH step gradient (v/v, 1:0 to 0:1) to yield eight subfractions. (Fr.4-1–Fr.4-8). Fr.4-5 (3.7 g) yielded compound 2 (3.5 mg) after purified by silica CC (1 × 20 cm) eluted with CHCl3-MeOH (v/v, 80:1, 1.5 L).
Compound 1: white power; [α]20D + 8.0 (c = 0.5, MeOH); IR (KBr) νmax: 3417.4, 2930.9, 1643.2, 1384.2, 1025.4, 438.9 cm−1; HREIMS: m/z 238.1931 [M]+ (calcd. for C15H26O2, 238.1933); 1H and 13C-NMR data: see Table 1.
Compound 2: white power; [α]20D + 80.0 (c = 0.5, MeOH); IR (KBr) νmax: 3424.5, 2925.7, 1655.4, 1023.4, 582.7 cm−1; HREIMS: m/z 254.1878 [M]+ (calcd. for C15H26O3, 254.1882); 1H and 13C-NMR data: see Table 1.
Compound 3: yell ow oil; [α]20D + 31.0 (c = 0.5, MeOH); IR (KBr) νmax: 3423.2,2923.9,1636.7, 1384.4, 1044.5, 668.2 cm−1; HREIMS: m/z 254.1880 [M]+ (calcd. for C15H26O3, 254.1882); 1H and13C-NMR data: see Table 2.
Compound 4: yellow oil; [α]20D + 12.0 (c = 0.5, MeOH); IR (KBr) νmax: 3415.8, 2923.9, 1636.7, 1384.1, 1029.5, 462.3 cm−1; HREIMS: m/z 270.1833 [M]+ (calcd. for C15H26O4, 270.1831); 1H and 13C-NMR data: see Table 2.
3.5. Preparation of S-MTPA and R-MTPA Esters 1a, 1b, 2a, 2b, 3a, and 3b of Compounds 1, 2, and 3
Compound 2 (1 mg) was dissolved in 1 mL CH2Cl2, and 4-dimethylaminopyridine (3 mg) and (R)-MTPACl (10 μL) were added. The reaction was stirred for 5 h at room temperature. Then, 1 mL of H2O was added to stop the reaction and to extract the solution three times with CH2Cl2 (5 mL each). Finally, the residue was purified by semipreparative HPLC (80% MeOH-H2O) after removal of CH2Cl2 under reduced pressure to obtain (S)-MTPA ester 2a (1 mg, tR = 7.84 min). By the same procedure, (R)-MTPA ester 2b (1 mg, tR = 8.17 min), (S)-MTPA ester 3a (1 mg, tR = 8.55 min), (R)-MTPA ester 3b (1 mg, tR = 8.64 min), (S)-MTPA ester 4a (1 mg, tR = 6.79 min), and (R)-MTPA ester 4b (1 mg, tR = 6.98 min) were got via the reaction of 2, 3, and 4 (1 mg, each) with (S)-MTPACl, (R)-MTPACl, (S)-MTPACl, (R)-MTPACl, and (S)-MTPACl, respectively [12].
3.6. Absolute Configuration of the 1, 2-Diol Moiety in 1
A mixture of diol-Mo2(OAc)4 (1:1.3) for 1 was subjected to CD measurements at a concentration of 0.5 mg/mL in HPLC grade DMSO dried with 4 Å molecular sieves, according the literature report [19]. The first CD spectrum was recorded after mixing immediately, and the CD spectrum was recorded again after mixing for 10 min. The inherent CD was subtracted. The observed signs of the diagnostic bands at about 310 and 400 nm in the induced CD spectrum were correlated to the absolute configuration of the 1, 2-diol moiety.
3.7. Bioassays
The cytotoxic activity for compounds 1–4 were tested against three cell lines including human hepatic carcinoma cell lines (SEL-7420), gastric cell lines (SGC-7721), and leukemia cell lines (K-562). These cell lines were purchased from Shang Hai Cell Bank of Chinese Academy of Sciences. The purity of the tested compounds and paclitaxel (PTX) was determined to be over 95% using the chromatography. The cytotoxic effects on these tests cell were assessed by the IC50 values and determined by the MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide] colometric method as described in reference [17]. Each set of tests was conducted three times to confirm reproducibility of the results. These compounds were dissolved in DMSO, PTX was used as a positive control, and the medium without test compound was used as a negative control in the bioassay.
The antimicrobial activity of compounds 1–4 against C. albicans and S. aureus were also evaluated using the 2-fold dilution method [18]. The tested strains were cultivated in YPD broth for C. albicans and LB broth for bacteria at 28 °C. The test compounds were dissolved in DMSO at different concentrations from DMSO at different concentrations from 1000 to 7.8 μg/mL (from 6.25 to 0.025 μg/mL for the positive controls) by the continuous 2-fold dilution methods in 96-well plates. Each well contains 100 μL of contents composed of 20 μL of inoculums (5 × 105 CFU/mL), test compounds, and YPD or LB media. The microtiter plates were incubated at 28 °C for 24 h and were examined for microbes’ growth by turbidity in daylight. Chlorhexidine acetate and kanamycin sulfate were used as positive controls for C. albicans and S. aureus, respectively.
4. Conclusions
Four new eudesmane-type sequiterpenes (1–4) were isolated from the PDB fermentation broth of the mangrove-derived endophytic fungus Penicillium sp. J-54 originated from the healthy leaves of Ceriops tagal collected in Dong Zhai Gang Mangrove Reserve in Hainan. Their structures were determined by spectroscopic methods, the in situ dimolybdenum CD method, and the modified Mosher’s method. Compound 2 exhibited weak cytotoxicity against K-562 with an IC50 value of 90.1 μM. The results proved that mangrove endophytic fungi are the source of new bioactive substances.
Acknowledgments
This work was financially supported by International Science and Technology Cooperation Project of Hainan Province (GJHZ2013-17), National Natural Science Foundation of China (Nos. 41506096, 41406083 and 41776093), Natural Science Foundation of Hainan (No. 20163117 and No. 217254), Key Laboratory of Tropical Medicinal Plant Chemistry of Ministry of Education, and Hainan Normal University (No. 201601).
Supplementary Materials
The NMR and HREIMS spectra for 1–4 and the 1H-NMR spectra for S-MTPA and R-MTPA esters are available online at http://www.mdpi.com/1660-3397/16/4/108/s1.
Author Contributions
For research articles with 10 authors. Wenli Mei, Haofu Dai, and Pei Wang conceived and designed the experiments; Liuming Qiu and Gei Liao performed the isolation of the fungus, the fermentation, isolation of the compounds, and preparation of S-MTPA and R-MTPA esters. Caihong Cai and Pei Wang performed the biological tests; The CD measurements was subjected by Fandong Kong; Zhikai Guo contributed to the acquirement of the NMR data; Pei Wang, Liuming Qiu, Wenli Mei, Haofu Dai, Yanbo Zeng, and Peter Proksch analyzed the data; Liuming Qiu, Pei Wang, Wenli Mei, and Haofu Dai wrote the paper.
Conflicts of Interest
The authors declare no conflict of interest.
References
- 1.Gunatilaka A.A.L. Natural products from plant-associatedmicroorganisms: Distribution, structural diversity, bioactivity, and implications of their occurrence. J. Nat. Prod. 2006;69:509–526. doi: 10.1021/np058128n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guo B., Wang Y., Sun X., Tang K. Bioactive natural products from endophytes: A review. Appl. Biochem. Microbiol. 2008;44:136–142. doi: 10.1134/S0003683808020026. [DOI] [PubMed] [Google Scholar]
- 3.Aly A.H., Debbab A., Proksch P. Fungal endophytes: Unique plant inhabitants with great promises. Appl. Microbiol. Biotechnol. 2011;90:1829–1845. doi: 10.1007/s00253-011-3270-y. [DOI] [PubMed] [Google Scholar]
- 4.Blunt J.W., Copp B.R., Keyzers R.A., Munroa M.H.G., Prinsepd M.R. Marine natural products. Nat. Prod. Rep. 2012;29:144–222. doi: 10.1039/C2NP00090C. [DOI] [PubMed] [Google Scholar]
- 5.Blunt J.W., Copp B.R., Keyzers R.A., Munroa M.H.G., Prinsepd M.R. Marine natural products. Nat. Prod. Rep. 2013;30:237–323. doi: 10.1039/C2NP20112G. [DOI] [PubMed] [Google Scholar]
- 6.Blunt J.W., Copp B.R., Keyzers R.A., Munroa M.H.G., Prinsepd M.R. Marine natural products. Nat. Prod. Rep. 2014;31:160–258. doi: 10.1039/c3np70117d. [DOI] [PubMed] [Google Scholar]
- 7.Blunt J.W., Copp B.R., Keyzers R.A., Munroa M.H.G., Prinsepd M.R. Marine natural products. Nat. Prod. Rep. 2015;32:116–211. doi: 10.1039/C4NP00144C. [DOI] [PubMed] [Google Scholar]
- 8.Blunt J.W., Copp B.R., Keyzers R.A., Munroa M.H.G., Prinsepd M.R. Marine natural products. Nat. Prod. Rep. 2016;33:382–431. doi: 10.1039/C5NP00156K. [DOI] [PubMed] [Google Scholar]
- 9.Blunt J.W., Copp B.R., Keyzers R.A., Munroa M.H.G., Prinsepd M.R. Marine natural products. Nat. Prod. Rep. 2017;34:235–294. doi: 10.1039/C6NP00124F. [DOI] [PubMed] [Google Scholar]
- 10.Li H.X., Jiang J.Y., Liu Z.M., Lin S., Xia G.P., Xia X.K., Dingm B., He L., Lu Y.J., She Z.G. Peniphenones A–D from the mangrove fungus Penicillium dipodomyicola HN4-3A as inhibitors of mycobacterium tuberculosis phosphatase (MptpB) J. Nat. Prod. 2014;77:800–806. doi: 10.1021/np400880w. [DOI] [PubMed] [Google Scholar]
- 11.An C.Y., Li X.M., Li C.S., Wang M.H., Xu G.M., Wang B.G. 4-Phenyl-3,4-dihydroquinolone derivatives from Aspergillus nidulans MA-143 an endophytic fungus isolated from the mangrove plant Rhizophora stylosa. J. Nat. Prod. 2013;76:1896–1901. doi: 10.1021/np4004646. [DOI] [PubMed] [Google Scholar]
- 12.Kong F.D., Wang Y., Liu P.P., Tian H.D., Zhu W.M. Thiodiketopiperazines from the marine-derived Fungus Phoma sp. OUCMDZ-1847. J. Nat. Prod. 2014;77:132–137. doi: 10.1021/np400802d. [DOI] [PubMed] [Google Scholar]
- 13.Jin P.F., Zuo W.J., Guo Z.K., Mei W.L., Dai H.F. Metabolites from the endophytic fungus Penicillium sp. FJ-1 of Ceriops tagal. Acta Pharm. Sin. 2013;48:1688–1691. [PubMed] [Google Scholar]
- 14.Liu M.L., Duan Y.H., Zhang J.B., Yu Y., Dai Y., Yao X.S. Novel sesquiterpenes from Nardostachys chinensis Batal. Tetrahedron. 2013;69:6574–6578. doi: 10.1016/j.tet.2013.05.134. [DOI] [Google Scholar]
- 15.Di B.L., Pescitelli G., Pratelli C., Pini D., Salvadori P. Determination of absolute configuration of acyclic 1,2-diols with Mo2(OAc)4.1.Snatzke’s method revisited. J. Org. Chem. 2001;66:4819–4825. doi: 10.1021/jo010136v. [DOI] [PubMed] [Google Scholar]
- 16.Gorecki M., Jablonska E., Kruszewska A., Suszczynska A., Urbanczyk-Lipkowska Z., Gerards M., Morzycki J.W., Szczepek W.J., Frelek J. Practical method for the absolute configuration assignment of tert/tert 1,2-diols using their complexes with Mo2(OAc)4. J. Org. Chem. 2007;72:2906–2916. doi: 10.1021/jo062445x. [DOI] [PubMed] [Google Scholar]
- 17.Mosmann T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods. 1983;65:55–63. doi: 10.1016/0022-1759(83)90303-4. [DOI] [PubMed] [Google Scholar]
- 18.Fu P., Kong F.D., Wang Y.F., Wang Y., Liu P.P., Zuo G.Y., Zhu W.M. Antibiotic metabolites from the coral-associated actinomycete Streptomyces sp. OUCMDZ-1703. Chin. J. Chem. 2013;31:100–104. doi: 10.1002/cjoc.201201062. [DOI] [Google Scholar]
- 19.Chen S.X., Ren F.X., Niu S.B., Liu X.Z., Che Y.S. Dioxatricyclic and oxabicyclic polyketides from Trichocladium opacum. J. Nat. Prod. 2014;77:9–14. doi: 10.1021/np4004799. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.