

Abstract
Molecular chirality provides a key challenge in host–guest recognition and other related chemical applications such as asymmetric catalysis. For a molecule to act as an efficient enantioselective receptor, it requires multi‐point interactions between host and chiral guest, which may be achieved by an appropriate chiral 3D scaffold. As a consequence of their interlocked structure, catenanes and rotaxanes may present such a 3D scaffold, and can be chiral by inclusion of a classical chiral element and/or as a consequence of the mechanical bond. This Minireview presents illustrative examples of chiral [2]catenanes and [2]rotaxanes, and discusses where these molecules have been used in chemical applications such as chiral host–guest recognition and asymmetric catalysis.
Keywords: catalysis, catenanes, chirality, host–guest recognition, rotaxanes
1. Introduction
Any undergraduate chemistry student will be familiar with the concept of chirality, and specifically that chiral molecules are non‐superimposable on their mirror image. Many processes in the natural world rely on the ability to recognize chiral molecules, and this, in part at least, inspires chemists to study chiral host–guest recognition and related themes such as asymmetric catalysis.1 It is generally accepted that a 3D arrangement of at least three interactions (one of which needs to be stereochemically dependent) must exist between a chiral host and its chiral guest to achieve chiral recognition.2 As a consequence of their interlocked structures, catenanes3, 4 (molecules consisting of two or more interlocked macrocyclic rings, Figure 1 a) and rotaxanes5 (molecules consisting of stoppered axle(s) components threaded through one or more macrocyclic rings, Figure 1 b) could form the basis of useful 3D scaffolds for chiral hosts.
Figure 1.

Schematic representations of: a) [n+2]catenane, and b) [n+2]rotaxane structures.
It is now well‐established that template synthesis provides a versatile route to interlocked molecules,6 relying on the preorganization of components prior to final covalent bond formation to trap the interlocked species.7 Classical templating strategies using metal cations,8 π–π stacking9 and hydrogen bonding10 have been supplemented by more recent work on anionic,11 radical–radical12 and halogen bond13templation. Although synthetic methodology is still being developed, research is increasingly being focused on exploiting mechanically interlocked molecules in chemical applications,14 such as host‐guest recognition15 or catalysis.16 Some researchers have made use of the 3D geometries created by interlocked molecular structures to achieve guest selectivity, and some have exploited the stimulus‐controlled motion of interlocked components to make examples of molecular machines17 which were celebrated by the 2016 Nobel Prize in Chemistry.18
A catenane or rotaxane may be made chiral by the inclusion of a classical chiral motif. Alternatively, chirality may arise as a consequence of the mechanical bond. Although examples of chiral interlocked molecules have been known for some time (as evidenced by previous reviews),19, 20 it is perhaps only now that this research field has begun to genuinely flourish, as significant progress is not only made in their preparation, but also in the application of these topologically and stereochemically exotic species. This Minireview sets out to provide an informed overview of examples of chiral catenanes and rotaxanes, before detailing their use in emerging chemical applications. As a Minireview, this article is not designed to be an exhaustive collection of every chiral catenane or rotaxane, and instead a range of illustrative examples will be presented, focusing principally on chiral examples of [2]catenanes and [2]rotaxanes, and those that exhibit useful chemical application. For progress in the related class of molecular knots, interested readers are referred to an excellent review that has just been published.21
2. Chirality Arising from Classical Chiral Elements
A straightforward way to create a chiral catenane or rotaxane is by incorporating a classical chiral element, such as a chiral centre, axis or plane into at least one of the components that make up the interlocked molecule. However, care is needed in certain cases. For example, for a homocircuit [2]catenane (a catenane where the two interlocked rings are identical), if one ring has a stereocentre with an R configuration and the other an S, then the catenane is achiral (it is the meso diastereomer of the R,R and S,S enantiomeric pair, see Figure 2).
Figure 2.

Schematic representation of possible stereoisomers of a homocircuit [2]catenane, where each ring contains a single stereogenic centre (the numbers in italics are to illustrate CIP priority assignment).
2.1. Catenanes that possess classical chiral elements
Catenanes possessing chirality arising from a chiral centre,22 axis23 and plane24 may be exemplified by work from the group of Stoddart (Figures 3, 4 and 5). All were prepared by clipping shut a tetra‐pyridinium macrocycle around an electron rich aromatic motif of a crown ether macrocycle—self‐assembled by charge assisted π–π donor‐acceptor templation.
Figure 3.

Stoddart's catenane 1 4+(PF6 −)4 containing a tetra‐pyridinium macrocycle with two stereogenic centres.
Figure 4.
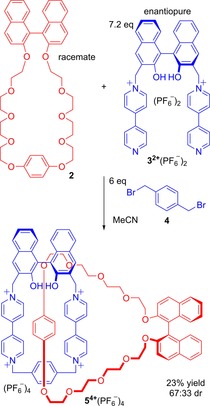
Diastereoselective synthesis of Stoddart's catenane 5 4+(PF6 −)4 containing two rings, both possessing axial chirality.
Figure 5.

Stoddart's catenane 6 4+(PF6 −)4 containing two rings, both possessing planar chirality.
In cases of [2]catenanes, in which both rings possess chiral elements, researchers have sometimes observed evidence of diastereoselectivity in synthesis. For example, when racemic binaphthyl crown ether 2 and enantiopure (R)‐binapthyl bis‐pyridinium precursor 3 were used, the R,R and S,R stereoisomers of the catenane were obtained in a diastereomeric ratio of 67:33 in favour of R,R (Figure 4).23
For catenane 6 4+(PF6 −)4 (Figure 5), both rings may possess elements of planar chirality, when the pair of binaphthyl spacers in each ring are staggered with respect to one other. In solution, twelve stereoisomers of this catenane are possible (six diastereomers, each existing as pairs of enantiomers), many of which are in exchange with one another through low energy conformational and co‐conformational processes. Although this catenane exhibits complex dynamic stereochemical behaviour in solution, the researchers reported that crystallization afforded enantiopure single crystals of the SS, SS enantiomer.24
2.2. Rotaxanes that possess classical chiral elements
Amongst early work, Vögtle and co‐workers prepared charge assisted π–π donor–acceptor rotaxanes possessing chiral tetraacetyl glucose stoppers (such as 7 4+(PF6 −) in Figure 6 a).25 In addition, the same group prepared examples of hydrogen bond templated rotaxanes stoppered with tetrabenzoyl glucose units (such as 8 in Figure 6 b).26 In the latter case, the rotaxanes exhibited amplified circular dichromism (CD) compared to the free axle.
Figure 6.

Vögtle's glucose stoppered rotaxanes 7 4+(PF6 −)4 and 8, prepared by: a) donor–acceptor and b) hydrogen‐bond templating.
Subsequently, Leigh and co‐workers have prepared a series of hydrogen bond templated rotaxanes incorporating l‐amino acids (Figure 7).27 In chloroform, the rotaxanes 9 exhibit CD signals, but the corresponding non‐interlocked chiral axles do not. Detailed investigations revealed that chirality is transmitted from the chiral centre on the axle via the interlocked macrocycle to the C‐terminal stopper of the rotaxane. Further, this induced circular dichroism (ICD) may be modulated by varying solvent, temperature or size of the chiral substituent. For example, switching the solvent to hydrogen bond competitive methanol leads to a dramatic reduction in CD signal for any given rotaxane as the macrocycle can freely rotate and hence does not communicate the chiral information efficiently.
Figure 7.

Leigh's amino acid containing rotaxanes 9 that demonstrate solvent, temperature and chiral substituent dependent ICD.
In follow‐up work, the same group reported on the use of light‐controlled E/Z isomerism in a related rotaxane 10 to create a chiroptical switch (Figure 8).28 When the fumaramide C=C is trans, then the macrocycle preferentially resides over this functional group. Isomerization of the double bond to cis geometry, causes the macrocycle to shuttle to the glycine‐leucine station, which, incorporating a chiral substituent, allows for the switching on of a chiral optical response (that is the appearance of a signal in the CD spectrum).29
Figure 8.

Leigh's bistable molecular shuttle 10 that demonstrates chiroptical switching.
3. Chirality Arising as a Consequence of the Mechanical Bond
Exciting opportunities are possible because of the mechanical bond, for chirality may arise in catenanes and rotaxanes even when the interlocked components are themselves achiral. This may be described as “mechanical chirality”, a term that has recently been defined specifically by Bruns and Stoddart as “a non‐classical form of chirality resulting from the spatial arrangements of component parts connected by mechanical bonds”.30, 31
Mechanical chirality may arise in [2]catenanes from directionality in both rings (Figure 9 a) or from facially unsymmetric rings (Figure 9 b). Catenanes possessing directionality in both rings may be described as topologically chiral. Mechanical chirality originates in [2]rotaxanes from both the axle and the macrocycle being directional (Figure 9 c) or when a macrocycle is trapped on one side of what would be a prochiral centre of the non‐interlocked axle component—in a manner with parallels to atroposiomerism (Figure 9 d).
Figure 9.

Schematic representations of enantiomers of [2]catenanes and [2]rotaxanes arising as a consequence of the mechanical bond: a) [2]catenane consisting of two directional rings; b) [2]catenane consisting of facially unsymmetric rings; c) [2]rotaxane consisting of directional ring and axle components and d) [2]rotaxanes consisting of a ring trapped on one side of what would be a prochiral centre of the non‐interlocked axle component.
3.1. Topologically chiral catenanes
The first topologically chiral catenane was prepared by Mitchell and Sauvage by use of CuI cation templation.32 Inclusion of a phenyl substituent at position 4 of the phenanthroline group of both rings leads to the creation of enantiomers (Figure 10). The chirality of catenane 11 (which was prepared as a racemate) was confirmed by use of Pirkle's reagent (S‐(+)‐2,2,2‐trifluoro‐1‐(p‐anthryl)ethanol) and careful 1H NMR spectroscopic analysis. Subsequently, partial separation of the enantiomers was achieved by HPLC to allow for recording of mirror image CD spectra (in collaboration with Okamoto and co‐workers).33
Figure 10.

Enantiomers of Sauvage's first topologically chiral catenane 11. The stereochemical labels R and S are assigned thus: in the direction of an arrow pointing from the highest priority atom (labelled 1) to its highest priority neighbour (labelled 2), interlocked rings that are disposed in a clockwise manner are R, those in an anticlockwise manner are S.
An alternative source of directionality in metal templated mechanically chiral catenanes has been illustrated in an example of a bimetallic [2]catenane (Figure 11).34 In the solid state, crystal structures reveal that only one of the imine nitrogen atoms is coordinating to the ZnII cation, thus generating directionality in each of the interlocked rings. However, in solution, catenane 12 is fluxional, with cleavage and formation of coordination bonds occurring. Such processes are forbidden when considering topology, and so in solution (at least) this catenane cannot be considered topologically chiral.
Figure 11.

Enantiomers of Charbonnière and Tranolsi's catenane 12. The numbers and arrows are to illustrate how stereochemical labels are determined.
3.2. Facially unsymmetric chiral catenanes
The first facially unsymmetric chiral catenanes were reported by Puddephatt in 2002 (Figure 12).35, 36 A set of [2]catenanes 13 were prepared using aurophilic templation. Chirality arises from the orientation of the bromo‐aromatic substituents, in a manner similar to how chirality arises in appropriately substituted allenes. Bruns and Stoddart have therefore proposed the use of the term “mechanically axial chirality” and the stereochemical labels R ma and S ma to describe the chirality in such catenanes.30
Figure 12.
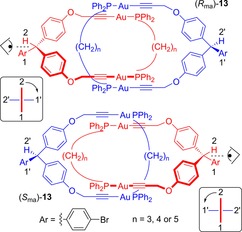
Puddephatt's catenanes, where chirality arises from facially unsymmetrical rings. The stereochemical labels R ma and S ma are assigned as one would assign R and S to allenes.
A CuI‐templated catenane has been prepared by Marinetti and co‐workers that also exhibits facially unsymmetric mechanical chirality (Figure 13).37 In this case, facial dissymmetry arises from incorporation of phosphine oxides in each ring. In addition to the mechanical chirality there are stereogenic carbon atoms. By use of an enantiopure macrocycle to generate the catenane, the resulting diastereomers of demetallated catenane 14 were separable by preparative HPLC to give optically pure diastereomers.
Figure 13.

Diastereomers of Marinetti's facially unsymmetric mechanically chiral catenane 14. The numbers and arrows are to illustrate how the R ma and S ma stereochemical labels are determined.
3.3. Mechanically planar chiral rotaxanes
In 1997, Vögtle and Okamoto reported upon the preparation and HPLC resolution of an amide‐sulfonamide [2]rotaxane, that is chiral by consisting of directional axle and macrocyclic components (Figure 14).38 Although the researchers successfully separated the enantiomers of rotaxane 15 and recorded mirror‐image CD spectra, they were unable to assign the absolute configuration of the two enantiomeric samples.
Figure 14.

Enantiomers of Vögtle's mechanically planar chiral [2]rotaxane 15. The stereochemical labels R mp and S mp are assigned thus: View the rotaxane along its axle from the highest priority atom in the axle (A1) to that atom's highest priority neighbour (A2). If in the macrocycle the highest priority neighbour (M2) is disposed clockwise from the highest priority atom (M1), then the stereochemical label is R mp; if it is disposed anticlockwise then the label is S mp.
The authors of this paper used the term “cycloenantiomeric” to describe the chirality of 15, but it has since been proposed that the term “mechanically planar chiral” is possibly a more appropriate descriptor.39 Goldup has also suggested a nomenclature for describing the enantiomers of a mechanically planar chiral rotaxane, as illustrated for rotaxane 15 in Figure 14.
The key synthetic challenge in exploiting mechanically planar chiral rotaxanes is to prepare enantiopure examples on a preparative scale. An illustration of the challenge involved is reflected by Takata's report on attempts to achieve catalytic asymmetric synthesis of a planar chiral rotaxane consisting of a substituted 18‐dibenzocrown‐6 macrocycle and secondary ammonium salt axle (Figure 15).40, 41 The rotaxanes were prepared by an acylative end‐capping using a chiral bisphosphine catalyst. Unfortunately, the maximum observed enantiomer excess was only 4.4 %.
Figure 15.

Takata's catalytic asymmetric synthesis of a mechanically planar chiral rotaxane (only one rotaxane enantiomer depicted, note the configuration of the major enantiomer was not established).
A critical breakthrough in accessing enantiopure mechanically chiral rotaxanes was achieved in the elegant work of Bordoli and Goldup (Figure 16).39, 42 Using a CuAAC “click”‐active metal template synthesis,43 they constructed rotaxane 24 from directional macrocycle 21 and two half threads—achiral alkyne 22 and the other enantiopure azide 23. The diastereomers of 24 were formed in an essentially 1:1 ratio, but crucially they could be separated by standard flash chromatography. Substitution of the chiral stopper for an achiral one then generated enantiopure samples of the enantiomers of the mechanically planar chiral rotaxane 26. By obtaining crystal structures of the two diastereomers of rotaxane 24, the researchers were also able to assign the absolute configurations of both diastereomers of 24 and the resulting enantiopure samples of mechanically chiral rotaxane 26.
Figure 16.

Goldup's preparation of enantiopure mechanically planar chiral rotaxanes. The labels (A1, etc) and arrows are to illustrate how the R mp and S mp stereochemical labels are determined.
3.4. Point mechanical chiral rotaxanes
Mechanical chirality may arise in rotaxanes—which is more appropriately termed point mechanical chirality—when a macrocycle is trapped on one side of what would be a prochiral centre of the non‐interlocked axle component. This has been demonstrated in a rotaxane prepared by Leigh and co‐workers (Figure 17).44 At room temperature, the macrocycle of the achiral [2]rotaxane 27 can move between the two fumaramide functional groups on a symmetrical axle. However, if the alcohol at the centre of the axle is benzoylated, the macrocycle becomes trapped at one end of the axle, and the carbon atom attached to the benzoylated alcohol group becomes a stereogenic centre. If DMAP is used to catalyze the benzoylation reaction, a racemate of the point mechanically chiral rotaxane 29 is generated. However, use of a chiral catalyst allows for enantioselectivity, with rotaxane 29 being isolated with an enantiomeric ratio of 67:33 (S:R).
Figure 17.

Leigh's conversion of achiral rotaxane 27 into rotaxane 29 that possesses point mechanical chirality. The asterisks mark the stereocentre in rotaxane 29, which may be assigned according to standard CIP rules.
4. Chiral Catenanes and Rotaxanes in Application
Mechanically interlocked molecules are increasingly being put towards some form of functional application.14 For example, the three dimensional structures of certain catenanes and rotaxanes have been used to achieve selectivity in the binding and sensing of ionic and small molecular guest species.15 Meanwhile, others have been shown to act as catalysts, including examples for which controlled motion of the interlocked components is used to switch catalytic activity on and off.16 Examples of chiral catenanes and rotaxanes being used in such applications have been somewhat rare, but are now growing in number.
4.1. Chiral host–guest recognition
In 2006, Kameta and Hiratani reported upon the chiral sensing of phenylalaninol by a mechanically planar chiral rotaxane (Figure 18).45 The racemate of rotaxane 30 was prepared by a covalent bond formation (rather than template synthesis) approach.46 1H NMR and fluorescence spectroscopic experiments provided evidence that l‐phenylalaninol was selectively bound through hydrogen bonding by one of the enantiomers of the rotaxane, and d‐phenylalaninol by the other in chloroform.
Figure 18.

Structure of one enantiomer of Kameta and Hiratani's mechanically planar chiral rotaxane 30, showing the tentative proposed mode of binding of phenylalaninol as suggested by the authors.
Being studied as the racemate, the Kameta and Hiratani system is somewhat limited as an enantioselective host. To avoid this issue, Niemeyer and co‐workers prepared an enantiopure catenane by use of macrocyclic components containing an axially chiral binaphthyl‐phosphoric acid unit (Figure 19).47 Following removal of the calcium cation used to template the formation of the catenane, the researchers demonstrated that catenane 31 (as the bis‐tetrabutylammonium salt) possessed enantioselectivity for bis‐HCl salts of chiral diamine guests in DMSO. While the levels of enantioselective guest recognition were modest (K fav/K disfav=1.4–1.6), they were greater than for the non‐interlocked macrocycle.
Figure 19.
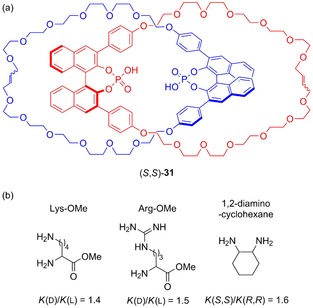
Niemeyers's catenane (S,S)‐31 host prepared from enantiopure axially chiral binaphthyl‐phosphoric acid units: a) structure of catenane, and b) ratios of association constants for the binding of bis‐HCl salts of enantiopure diamine guests by the tetrabutylammonium salt of the catenane in [D6]‐DMSO.
Very recently, Beer and co‐workers have reported a detailed experimental and computational study of a set of chiral rotaxanes and their enantioselective anion binding properties (Figure 20).48 Prepared by active‐metal templating, both macrocycle and axle components possess iodotriazoles that can halogen bond to anionic guests in solution. By 1H NMR titrations, they demonstrated that rotaxanes 32 +PF6 − and 33 +PF6 − can bind chiral anions with enantioselectivities (K fav/K disfav) of up to 2.9 and 3.4 respectively (Table 1). Notably rotaxane 34 +PF6 − exhibits negligible enantioselective behaviour towards any of the chiral anions investigated (K fav/K disfav <1.2), meaning, for these rotaxanes, that the presence of a chiral macrocyclic component is essential to achieve reasonable levels of guest enantiodiscrimination.
Figure 20.

Beer's chiral halogen bonding rotaxanes 32–34+PF6 − capable of enantioselective anion binding a) structures of rotaxanes and b) structures of chiral anions investigated.
Table 1.
Ratios of association constants for the binding of enantiopure anionic guests (as tetrabutylammonium salts) by rotaxanes 32–34+PF6 −.
| Boc‐Leu | Boc‐Pro | Boc‐Try | BINOL‐PO4 | |
|---|---|---|---|---|
| 32 +PF6 −[a] | K S/K R=1.62 | K S/K R=2.93 | K S/K R=1.73 | K R/K S=2.14 |
| 33 +PF6 −[b] | K R/K S=3.41 | K R/K S=1.50 | K R/K S=1.61 | K R/K S=1.78 |
| 34 +PF6 −[a] | K R/K S=1.07 | K R/K S=1.11 | K R/K S=1.18 | K S/K R=1.19 |
[a] In 98:2 [D6]‐acetone/D2O. [b] In 99:1 [D6]‐acetone/D2O.
4.2. Asymmetric catalysis
In 2004, Takata reported upon the use of rotaxanes possessing axially chiral binaphthyls to create a chiral environment to asymmetrically catalyze benzoin condensations (Figure 21).49 Good yields (up to 90 %), but with rather modest enantiomeric excesses (<32 % ee), were observed for rotaxanes, for which the chiral group was either part of the axle, or more impressively when the chiral group was part of the macrocycle (as depicted in Figure 21), thus transmitting the chiral information between the interlocked components to the thioazolium on the axle.
Figure 21.

Takata's chiral rotaxanes for asymmetric benzoin condensations: a) example of one of the chiral rotaxanes studied, and b) example of benzoin condensation reaction where rotaxane (R)‐35 +Cl− acted as a catalyst.
Leigh and co‐workers have since reported on a number of chiral rotaxanes for asymmetric catalysis.50, 51, 52 For instance, an active metal template synthesized rotaxane possessing a chiral C 2 symmetric trans‐cyclohexanediamine macrocycle, for use in enantioselective nickel‐catalyzed conjugate addition reactions (Figure 22).50 In comparison to an analogous acyclic ligand, rotaxane (R,R)‐38 exhibited a much better enantiomeric ratio of product (93:7 compared to 68:32), but considerably slower reaction times (27 vs. 2 days for full conversion as determined by 1H NMR spectroscopy). These observations are consistent with the rotaxane improving expression of chirality arising from the two stereogenic carbon atoms (by reducing degrees of freedom), but also restricting access to the cation (since it is buried within the rotaxane structure while coordinated to the nitrogen amine atoms), thus reducing the rate of reaction.
Figure 22.

Leigh's chiral rotaxane for asymmetric transition metal catalysis: a) structure of rotaxane (R,R)‐38, and b) example of nickel‐catalysed reaction where rotaxane (R,R)‐38 induces enantioselective bond formation.
The same group has also exploited a point mechanical chiral rotaxane (S)‐42 in catalysis (Figure 23).51 A secondary amine on the axle component may participate in enantioselective Michael addition and enamine reactions. The recorded enantiomeric ratio of products was somewhat low (68:32 and 71:29 were the best reported for the two types of reaction)—this is partly due to rotaxane (S)‐42 being prepared in only 84 % ee—but the proof of principle was clearly demonstrated (enantiomeric ratios of 50:50 being observed in all cases where the achiral axle was used in place of the rotaxane).
Figure 23.

Leigh's point mechanically chiral rotaxane for asymmetric organocatalysis: a) structure of rotaxane (S)‐42, and b) example of enamine reaction where rotaxane (S)‐42 induces enantioselective bond formation.
The researchers have also demonstrated that a switchable rotaxane may asymmetrically catalyze reactions in a controlled fashion (Figure 24).52 Rotaxane (R)‐46 +PF6 − includes two stations: in acidic conditions, the central secondary amine of the axle is protonated and the crown ether macrocycle resides over the resulting ammonium group, preventing the substrate from accessing the N atom. Once the amine is deprotonated, the crown ether moves to an alternative methyl triazolium station; substrates can then access the amine (which is bonded to a stereogenic carbon atom) and catalytic reactivity is thus turned on. This process may be reversed by addition of acid to reprotonate the amine, leading to translation of the macrocycle back over the reformed ammonium group. In its deprotonated state, rotaxane (R)‐46 +PF6 − can catalyze asymmetric Michael additions with reasonable conversion (60–70 %) and high enantiomeric ratios of products (up to 94:6).
Figure 24.

Leigh's switchable rotaxane for controlled asymmetric Michael additions: a) structure of rotaxane (R)‐46 +PF6 −, and b) example of Michael addition asymmetrically catalysed by deprotonated rotaxane (R)‐46 +PF6 −.
Very recently, Niemeyer and co‐workers have reported upon the use of binaphthyl‐phosphoric acid catenane (S,S)‐31 (depicted in Figure 19) as a catalyst in asymmetric transfer hydrogenation reactions (Figure 25).53 Impressively, using catenane (S,S)‐31 as the catalyst led to dramatically increased stereoselectivity compared to using the non‐interlocked macrocycle, with comparable yields (although notably slower reaction times). Detailed computational calculations provide evidence that the high stereoselectivities observed with the catenane are a direct result of its interlocked nature.
Figure 25.

Example of transfer hydrogenation reaction catalyzed by catenane (S,S)‐31 investigated by Niemeyer and co‐workers.
5. Conclusions
Future investigations into the preparation and study of chiral catenanes and rotaxanes have great potential. As highlighted in the Introduction, much progress has been made in developing new synthetic methodologies to prepare interlocked molecules since the early work on chiral catenanes and rotaxanes by the groups of Sauvage, Stoddart and Vögtle. Recent demonstrations of the application of chiral interlocked molecules in host–guest recognition and catalysis provide significant encouragement for researchers to work towards overcoming unresolved challenges in the field such as directly accessing enantiopure mechanically chiral catenanes and rotaxanes through enantioselective synthesis. By overcoming such challenges, we can look forward to realizing the full potential of interlocked molecules as useful three‐dimensional scaffolds in real‐world chiral applications.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Nick Evans graduated from Wadham College, University of Oxford with a First Class Masters in Chemistry (2006), before obtaining a DPhil in Inorganic Chemistry (2011), having worked on anion‐sensing rotaxanes and catenanes in the group of Prof. Paul Beer. After undertaking postdoctoral research on chiral lanthanide complexes with Prof. David Parker (Durham University), he took up a Lectureship in Chemistry at Lancaster University in 2013. His current research interests are in the area of functional supramolecular chemistry, including the synthesis and study of chiral interlocked host molecules for enantioselective guest recognition.

Acknowledgements
N.H.E. would like to thank Lancaster University (PhD studentship), EPSRC (Equality and Diversity internship) and the Royal Society of Chemistry (Research Fund grant) for supporting projects working on chiral rotaxanes.
N. H. Evans, Chem. Eur. J. 2018, 24, 3101.
References
- 1. Chirality in Supramolecular Assemblies: Causes and Consequences (Ed.: F. R. Keene), Wiley, Chichester, 2016. [Google Scholar]
- 2. Berthod A., Anal. Chem. 2006, 78, 2093–2099. [DOI] [PubMed] [Google Scholar]
- 3. Evans N. H., Beer P. D., Chem. Soc. Rev. 2014, 43, 4658–4683. [DOI] [PubMed] [Google Scholar]
- 4. Gil-Ramírez G., Leigh D. A., Stephens A. J., Angew. Chem. Int. Ed. 2015, 54, 6110–6150; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 6208–6249. [Google Scholar]
- 5. Xue M., Yang Y., Chi X., Yan X., Huang F., Chem. Rev. 2015, 115, 7398–7501. [DOI] [PubMed] [Google Scholar]
- 6.Alternative statistical and directed covalent synthetic approaches for the preparation of catenanes and rotaxanes are known but are much less frequently used.
- 7.A [2]catenane is typically generated by cyclization of one or both rings (clipping), whereas a [2]rotaxane is typically prepared by either: a) stoppering a pseudo-rotaxane, b) clipping shut a ring around a stoppered axle, or c) snapping two stoppered half axles through the cavity of a ring.
- 8. Beves J. E., Blight B. A., Campbell C. J., Leigh D. A., McBurney R. T., Angew. Chem. Int. Ed. 2011, 50, 9260–9327; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 9428–9499. [Google Scholar]
- 9. Barin G., Coskun A., Fouda M. M. G., Stoddart J. F., ChemPlusChem 2012, 77, 159–185. [Google Scholar]
- 10. Schalley C. A., Weildandt T., Brüggemann J., Vögtle F., Top. Curr. Chem. 2004, 248, 141–200. [Google Scholar]
- 11. Spence G. T., Beer P. D., Acc. Chem. Res. 2013, 46, 571–586. [DOI] [PubMed] [Google Scholar]
- 12. Li H., Fahrenbach A. C., Dey S. K., Basu S., Trabolsi A., Zhu Z., Botros Y. Y., Stoddart J. F., Angew. Chem. Int. Ed. 2010, 49, 8260–8265; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 8436–8441. [Google Scholar]
- 13. Gilday L. C., Lang T., Caballero A., Costa P. J., Félix V., Beer P. D., Angew. Chem. Int. Ed. 2013, 52, 4356–4360; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 4452–4456. [Google Scholar]
- 14. van Dongen S. F. M., Cantekin S., Elemans J. A. A. W., Rowan A. E., Nolte R. J. M., Chem. Soc. Rev. 2014, 43, 99–122. [DOI] [PubMed] [Google Scholar]
- 15. Langton M. J., Beer P. D., Acc. Chem. Res. 2014, 47, 1935–1949. [DOI] [PubMed] [Google Scholar]
- 16. Leigh D. A., Marcos V., Wilson M. R., ACS Catal. 2014, 4, 4490–4497. [Google Scholar]
- 17. Erbas-Cakmak S., Leigh D. A., McTernan C. T., Nussbaumer A. L., Chem. Rev. 2015, 115, 10081–10206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. https://www.nobelprize.org/nobel_prizes/chemistry/laureates/2016/; (accessed August 2017).
- 19. Chambron J.-C., Dietrich-Buchecker C., Sauvage J.-P., Top. Curr. Chem. 1993, 165, 131–162. [Google Scholar]
- 20. Wolf C., in Dynamic Stereochemistry of Chiral Compounds: Principles and Applications, Chapter 9, Topological Isomerism and Chirality, RSC, Cambridge, 2007, pp. 444–479. [Google Scholar]
- 21. Fielden S. D. P., Leigh D. A., Woltering S. L., Angew. Chem. Int. Ed. 2017, 56, 11166–11194; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11318–11347. [Google Scholar]
- 22. Ashton P. R., Iriepa I., Reddington M. V., Spencer N., Slawin A. M. Z., Stoddart J. F., Williams D. J., Tetrahedron Lett. 1994, 35, 4835–4838. [Google Scholar]
- 23. Ashton P. R., Heiss A. M., Pasini D., Raymo F. M., Shipway A. N., Stoddart J. F., Spencer N., Eur. J. Org. Chem. 1999, 995–1004. [Google Scholar]
- 24. Ashton P. R., Boyd S. E., Menzer S., Pasini D., Raymo F. M., Spencer N., Stoddart J. F., White A. J. P., Williams D. J., Wyatt P. G., Chem. Eur. J. 1998, 4, 299–310. [Google Scholar]
- 25. Archut A., Müller W. M., Baumann S., Habel M., Vögtle F., Liebigs. Ann./Recl. 1997, 495–499. [Google Scholar]
- 26. Schmidt T., Schmieder R., Müller W. M., Kiupel B., Vögtle F., Eur. J. Org. Chem. 1998, 2003–2007. [Google Scholar]
- 27. Asakawa M., Brancato G., Fanti M., Leigh D. A., Shimizu T., Slawin A. M. Z., Wong J. K. Y., Zerbetto F., Zhang S., J. Am. Chem. Soc. 2002, 124, 2939–2950. [DOI] [PubMed] [Google Scholar]
- 28. Bottari G., Leigh D. A., Pérez E. M., J. Am. Chem. Soc. 2003, 125, 13360–13361. [DOI] [PubMed] [Google Scholar]
- 29.For examples of rotaxanes incorporating components with chiral axes and planar chirality see:
- 29a. Cantrill S. J., Fyfe M. C. T., Heiss A. M., Stoddart J. F., White A. J. P., Williams D. J., Chem. Commun. 1999, 1251–1252; [Google Scholar]
- 29b. Ashton P. R., Bravo J. A., Raymo F. M., Stoddart J. F., White A. J. P., Williams D. J., Eur. J. Org. Chem. 1999, 899–908. [Google Scholar]
- 30. Bruns C. J., Stoddart J. F., in The Nature of the Mechanical Bond: From Molecules to Machines, Chapter 5, The Stereochemistry of the Mechanical Bond, Wiley, Hoboken, 2016. [Google Scholar]
- 31.As proposed by a reviewer of a draft of this Minireview, the term “inherent chirality” could be used in place of “mechanical chirality”. The term “inherently chirality” was first introduced with respect to concave molecules (Cort A., Mandolini L., Pasquini C., Schiaffino L., New J. Chem. 2004, 28, 1198–1199), and has been used to describe chirality arising from the mechanical bond of catenanes, rotaxanes and knots (see ref. [30]). An important point to note is that [2]catenanes and [2]rotaxanes can only be inherently chiral conditionally, whereas trefoil knots are inherently chiral unconditionally. [Google Scholar]
- 32. Mitchell D. K., Sauvage J.-P., Angew. Chem. Int. Ed. Engl. 1988, 27, 930–931; [Google Scholar]; Angew. Chem. 1988, 100, 985–987. [Google Scholar]
- 33. Kaida Y., Okamoto Y., Chambron J.-C., Mitchell D. K., Sauvage J.-P., Tetrahedron Lett. 1993, 34, 1019–1022. [Google Scholar]
- 34. Prakasam T., Lusi M., Nauha E., Olsen J. C., Sy M., Platas-Iglesias C., Charbonnière L. J., Trabolsi A., Chem. Commun. 2015, 51, 5840–5843. [DOI] [PubMed] [Google Scholar]
- 35. McArdle C. P., Van S., Jennings M. C., Puddephatt R. J., J. Am. Chem. Soc. 2002, 124, 3959–3965. [DOI] [PubMed] [Google Scholar]
- 36.As pointed out by two reviewers of a draft of this Minireview, these catenanes, although chiral (due to possessing facially unsymmetric rings), are not topologically chiral as stated in ref. [35]. This is because the topologically allowed inversion of the carbon atom bearing the bromo-aromatic substituent will exchange the enantiomers.
- 37. Theil A., Mauve C., Adeline M.-T., Marinetti A., Sauvage J.-P., Angew. Chem. Int. Ed. 2006, 45, 2104–2107; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 2158–2161. [Google Scholar]
- 38. Yamamoto C., Okamoto Y., Schmidt T., Jäger R., Vögtle F., J. Am. Chem. Soc. 1997, 119, 10547–10548. [Google Scholar]
- 39. Bordoli R. J., Goldup S. M., J. Am. Chem. Soc. 2014, 136, 4817–4820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Makita Y., Kihara N., Nakakoji N., Takata T., Inagaki S., Yamamoto C., Okamoto Y., Chem. Lett. 2007, 36, 162–163. [Google Scholar]
- 41.Takata used the term “planar chiral” to describe the chirality of these rotaxanes. Goldup's more specific “mechanically planar chiral” clarifies that the chirality arises as a consequence of the mechanical bond, not another source of planar chirality.
- 42.A similar approach to that of Bordoli and Goldup to prepare enantiopure mechanically chiral rotaxanes, was attempted by Lee and co-workers: Glen P. E., O'Neill J. A. T., Lee A.-L., Tetrahedron 2013, 69, 57–68. In this case, the researchers were aiming to achieve diastereoselective rotaxane formation using a C 1-symmetric macrocycle possessing bulky chiral substituents designed to direct incoming half-thread components. Unfortunately, the macrocycle did not support rotaxane formation for a range of active metal template conditions investigated. [Google Scholar]
- 43. Denis M., Goldup S. M., Nature Rev. Chem. 2017, 1, 0061. [Google Scholar]
- 44. Alvarez-Perez M., Goldup S. M., Leigh D. A., Slawin A. M. Z., J. Am. Chem. Soc. 2008, 130, 1836–1838. [DOI] [PubMed] [Google Scholar]
- 45. Kameta N., Nagawa Y., Karikomi M., Hiratani K., Chem. Commun. 2006, 3714–3716. [DOI] [PubMed] [Google Scholar]
- 46. Kameta N., Hiratani A., Nagawa Y., Chem. Commun. 2004, 466–467. [DOI] [PubMed] [Google Scholar]
- 47. Mitra R., Thiele M., Octa-Smolin F., Letzel M. C., Niemeyer J., Chem. Commun. 2016, 52, 5977–5980. [DOI] [PubMed] [Google Scholar]
- 48. Lim J. Y. C., Marques I., Félix V., Beer P. D., J. Am. Chem. Soc. 2017, 139, 12228–12239. [DOI] [PubMed] [Google Scholar]
- 49. Tachibana Y., Kihara N., Takata T., J. Am. Chem. Soc. 2004, 126, 3438–3439. [DOI] [PubMed] [Google Scholar]
- 50. Hoekman S., Kitching M. O., Leigh D. A., Papmeyer M., Roke D., J. Am. Chem. Soc. 2015, 137, 7656–7659. [DOI] [PubMed] [Google Scholar]
- 51. Cakmuk Y., Erbas-Cakmak S., Leigh D. A., J. Am. Chem. Soc. 2016, 138, 1749–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Blanco V., Leigh D. A., Marcos V., Morales-Serna J. A., Nussbaumer A. L., J. Am. Chem. Soc. 2014, 136, 4905–4908. [DOI] [PubMed] [Google Scholar]
- 53. Mitra R., Zhu H., Grimme S., Niemeyer J., Angew. Chem. Int. Ed. 2017, 56, 11456–11459; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 11614–11617. [Google Scholar]