

Abstract
Background
Asthmatic and allergic inflammation is mediated by Th2 cytokines (IL-4, IL-5 and IL-13). Though we have learned much about how Th2 cells are differentiated, the Th2 checkpoint mechanisms remain elusive.
Objectives
In this study, we investigate how monocyte chemotactic protein induced protein-1 (MCPIP1, encoded by zc3h12a gene) regulates IL-5-producing Th2 cell differentiation and Th2-mediated inflammation.
Methods
The functions of zc3h12a−/− CD4 T cells were evaluated by checking the expression of Th2 cytokines and transcription factors in vivo and in vitro. Allergic airway inflammation of zc3h12a−/− mice was examined with murine asthma models. In addition, antigen-specific CD4 T cells deficient in MCPIP1 were transferred to WT recipient mice, challenged with OVA or HDM, and accessed for Th2 inflammation.
Results
Zc3h12a−/− mice spontaneously develop severe lung inflammation, with an increase mainly in IL-5- and IL-13-producing but not IL-4-producing Th2 cells in the lung. Mechanistically, the differentiation of IL-5-producing zc3h12a−/− Th2 cells is mediated through Notch signaling and Gata3 independent of IL-4. Gata3 mRNA is stabilized in zc3h12a−/− Th2 cells. MCPIP1 promotes Gata3 mRNA decay via the RNase domain. Furthermore, deletion of MCPIP1 in OVA- or HDM-specific T cells leads to significantly increased Th2-mediated airway inflammation in OVA or HDM murine models of asthma.
Conclusions
Our study reveals that MCPIP1 regulates the development and functions of IL-5-producing Th2 cells through Notch/Gata3 pathway. MCPIP1 represents a new promising target for the treatments of asthma and other Th2-mediated diseases.
Keywords: RNA-binding protein, MCPIP1, mRNA decay, Th2 cells, IL-4, IL-5, IL-13, Gata3, Notch, asthma
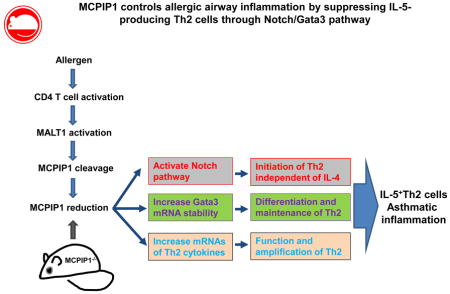
Graphical Abstract
INTRODUCTION
The hallmark of asthma is bronchial hyperreactivity caused by Th2 cell dysregulation1. Th2 cytokines initiate and maintain key pathophysiological features of asthmatic inflammation, with each cytokine exerting different functions in the induction of inflammation2, 3. IL-4 plays a critical role in allergic sensitization and IgE production4, 5; IL-5 is crucial for eosinophil survival6, 7; and IL-13 plays a central role in the development of airway hyperresponsiveness and tissue remodeling8, 9. Expression of Th2 cytokines is controlled by transcription factors through multiple signaling pathways. Th2 cells induce type 2 immunity characterized by high antibody titers and eosinophilia10–12. Th2 cells can also be induced by parasitic helminths13 and are shown to associate with atopic diseases, allergy and asthma11, 14. Though the network of transcription factors required for activation and differentiation of Th2 cells are well understood15–20, how Th2 development and Gata3 expression are negatively regulated, especially through posttranscriptional mechanisms, is largely unknown. Control of gene expression at the posttranscriptional level is a faster event than transcriptional control because of a direct targeting at the protein coding mRNA in the cytoplasm21, 22.
Monocyte chemotactic protein induced protein-1 (MCPIP1) encoded by zc3h12a gene was initially discovered as the most highly induced mRNA by monocyte chemotactic protein-1 (MCP-1) in human peripheral blood monocytes23. MCPIP1 protein is highly expressed in lymph node, thymus and spleen24, 25, and rapidly induced in macrophages upon stimulation with inflammatory stimuli, such as TNF, IL-1β and LPS25–27. MCPIP1 inhibits the expression of proinflammatory cytokines (IL-1, IL-6, and IL-12) by binding to their 3′UTRs for mRNA degradation. MCPIP1 is also named as Regnase-1 based on its RNase activity25. MCPIP1 reduction is required for T activation28. Loss of MCPIP1 in CD4 T cells leads to severe splenomegaly and lymphadenopathy, suggesting that MCPIP1 is essential for inhibiting the development of lethal autoimmune responses24, 25. MCPIP1 functions through regulating mRNA decay and affecting NF-κB signaling25, 29–31, leading to reduced expression of proinflammatory mediators24, 32. Therefore, MCPIP1 is believed to be a key negative regulator involved in the control of inflammation and maintenance of immune homeostasis. Mice deficient of MCPIP1 develop complex phenotypes, including autoimmune disorders, anemia and severe inflammatory responses24, 25. In addition, it was recently reported that MCPIP1 can degrade viral RNA and thus acts as a host defense against virus infection33–35. MCPIP1 is also involved in controlling cytokines-induced endothelial inflammation36 and inducing endothelial dysfunction37. Though MCPIP1 inhibits CD4 T cell activation and cytokine production28, 30, it is largely unknown whether MCPIP1 plays a role in Th2 development and Th2 inflammation.
In this study, we report that zc3h12a−/− mice spontaneously develop severe airway inflammation, with increased airway Th2 cells capable of producing large amounts of IL-5. Zc3h12a−/− CD4+ T cells are mostly effector T cells and produce significant amounts of IL-5 and IL-13 but not IL-4. The early differentiation of IL-5-producing zc3h12a−/− Th2 cells depends on Notch signal. IL-5-producing zc3h12a−/− Th2 cells express high levels of Gata3, and deletion of Gata3 by shRNA/lentivirus results in IL-5 reduction. MCPIP1 suppresses Gata3 expression by promoting its mRNA decay via the RNase domain. Moreover, deletion of MCPIP1 in OVA- or HDM-specific T cells significantly increases Th2-mediated airway inflammation in murine models of asthma. Transferring zc3h12a−/− CD4+ T cells into congenic CD45.1 mice induces severe Th2 inflammation and accumulation of Th2 cells in the airway of the recipient mice. Our study reveals that MCPIP1 regulates the development and functions of IL-5-producing Th2 cells through Notch/Gata3 pathway. MCPIP1 represents a new promising target for the treatments of asthma and other Th2-mediated diseases.
METHODS
Mice
Zc3h12a+/− mice were provided by Dr. Pappachan E. Kolattukudy (University of Central Florida, Orlando, FL). Homozygous zc3h12a−/− mice were bred from zc3h12a+/− breeding pairs and genotyped with PCR by detecting exon 4,5,6, with a band of 300 bp indicating WT and 500 bp for knockout. The primer sequences were listed in Table E1. Zc3h12a−/− mice and wild type littermates were housed in pathogen-free conditions and used at 5–8 weeks old. The CD45.1 congenic WT mice were used at age 6–12 weeks old. All mice were on C57BL/6 background and housed in animal facility at Saint Louis University. All mouse experiments were approved by Institution Animal Care and Use Committee of the Saint Louis University (animal protocol#: 2443).
In vitro T cell differentiation
Naïve CD4+ T cells were purified by EasySep™ Mouse Naïve CD4+ T Cell Negative Isolation Kit (Catalog#: 19765, Stemcell Technologies). The purity of naïve CD4 T cells was up to 95%. Naïve CD4 T cells were treated with plate-coated anti-CD3 (2 μg/ml) and anti-CD28 (2 μg/ml) Abs under the following conditions for three days. Th1 conditions: anti-IL-4 (10 μg/ml), IL-12 (10 U/ml); Th2 conditions: anti-IFN-γ (10 μg/ml), IL-4 (20 ng/ml); Th17 conditions: IL-6 (50 ng/ml), TGF-β (1 ng/ml), IL-23 (20 ng/ml), anti-IL-4 (10 μg/ml), and anti-IFN-γ (10 μg/ml). Then, cells were rested in fresh medium with IL-2 (1 ng/ml) for additional three days. On day 6, cells were stimulated with PMA (50 ng/ml) and Ionomycin (0.5 μg/ml) in the presence of Monensin (GolgiStop; 0.7 μl/ml) for 4 h for analysis of cytokines and surface markers by flow cytometry.
mRNA stability
CD4+ T cells were treated with plate-coated anti-CD3/CD28 Abs for 4 hours and then stimulated with Actinomycin D (5 μg/ml) and 5,6-dichlorobenzimidazole riboside (DRB, 10 μg/ml) for different times. CEM-Tet-on cells expressing WT or mutant MCPIP1 were stimulated with Dox (100 ng/ml) for 24 hours, then treated by PMA and Ionomycin for 3 h, and by Act D and DRB for different times. Total RNAs were collected and cDNA were used to measure the remaining mRNAs for calculating half-life.
Murine models of allergic asthma
OVA (Sigma-Aldrich) was adsorbed onto Imject Alum adjuvant (Pierce) to a final concentration of 500 μg/ml. Mice were immunized i.p. of 100 μg of OVA-alum or 10 μg HDM extract (Dermatophagoides pteronyssinus; Greer Labs) on day 0, and boosted with OVA on day 7 and 14, or with HDM on day 14. One week later, mice were challenged by intranasal injection (i.n.) of 25 μg OVA or HDM extract in PBS per nostril daily for five days. On day 26, mice were euthanized, and lungs were fixed, sectioned and stained with H&E and PAS. Cells from draining lymph nodes were stimulated with OVA (0, 10, 50, 250 μg/ml) or with HDM extract (30 μg/ml) for 72 hours, and then supernatants were collected for measuring cytokines by ELISA. Splenocytes were cultured with 200 μg/ml OVA or 30 μg of HDM extract in the presence of 10 ng/ml recombinant mouse IL-2 (eBioscience). After 72 hours, CD4+ T cells were isolated and resuspended in PBS. 4×106 CD4+ T cells were injected i.v. into recipient mice, followed by giving 50 μg OVA or 50 μg of HDM extract i.n. daily for five times starting on days 3. BAL fluid was collected one day after the last challenge for assessment of inflammatory cell infiltration. Lung sections were stained with H&E and PAS, and examined for pathological changes (see additional Methods information in the Online Repository).
Statistical analysis
Data were analyzed with Prism software 5.0 (GraphPad). For standard data sets, an unpaired two-tailed Student’s t-test was used.
RESULTS
MCPIP1 deficient mice display severe Th2 inflammation in the airway
To test whether MCPIP1 deficient mice have airway inflammation, we stained lung sections with H&E, found a significant accumulation of inflammatory cells and airway remodeling in the lungs of zc3h12a−/− mice compared to WT mice (Fig 1, A). There were more mucin-positive cells (goblet-cell metaplasia) in lung epithelium of zc3h12a−/− mice than WT mice (Fig 1, B). Total number of cells in BAL were also significantly increased in zc3h12a−/− mice compared to WT mice (Fig E1), indicating an asthmatic-like airway inflammation in zc3h12a−/− mice. Since Th2 cytokines are critical for asthmatic inflammation, we measured the levels of Th2 cytokines in BAL and blood. IL-5 and IL-13 in serum (Fig 1, C) and BAL (Fig 1, D) were much higher in zc3h12a−/− mice than in WT mice, while IL-4 was undetectable in both (data not shown). When collecting BALF, we noticed a reduction in “elastic recoil” of the lungs of MCPIP1 knockout mice. Namely, we could hardly retrieve any PBS after 0.7 ml of PBS was injected into lungs of MCPIP1 knockout mice at the first time. For the second flushing, we could only retrieve 0.1–0.3 ml out of 0.7 ml of PBS. For the final third flushing, we could retrieve 0.4–0.5 ml out of 0.7 ml of PBS. Altogether, we could only retrieve about 30% of the total PBS used for BAL flushing, and most of it was from the last flushing which contained the least levels of cytokines among three BALF. We reason that the excessive Th2 inflammation in the lungs of MCPIP1 knockout mice causes a reduction in lung elastic recoil and an increase in airway permeability, resulting in low levels of cytokines in BALF of the MCPIP1 knockout mice. Nevertheless, even with the low levels of cytokines in BALF, the levels of IL-5 and IL-13 in BALF were still significantly increased in MCPIP1 knockout mice compared to WT mice. The expression of Muc5ac and Gob5 mRNA were also increased in the lungs of zc3h12a−/− mice (Fig 1, E). The increased IL-5 and IL-13 in zc3h12a−/− mice compelled us to check Th2 cells. Much more IL-5- and IL-13-produing CD4+ T cells were presented in the lungs of zc3h12a−/− mice than in WT mice; whereas IL-4-producing cells were minimally increased and the cells producing IFN-γ and IL-17A were comparable (Fig 1, F). These data indicate that MCPIP1 deficiency leads to an increase in IL-5/IL-13-producing Th2 cells in the lung, which could cause asthmatic-like airway inflammation.
Fig. 1.

Zc3h12a−/− mice develop asthmatic airway inflammation with increased IL-5+ Th2 cells. Lung tissue sections were stained with H&E (A) and PAS (B) (n=4). Cytokines in sera (C) and in BAL (D) were measured by ELISA (3–4 mice in each group). (E) Muc5ac and Gob5 mRNA expression in lung tissues were measured by qRT-PCR (n=3). (F) Intracellular cytokine staining in lung infiltrates gated on CD4+ T cells and the summary of 3–5 mice per group. *: p< 0.05, ***: p< 0.001.
MCPIP1 deficient CD4 T cells produce significant amounts of Th2 cytokines
In line with the role of MCPIP1 in preventing CD4 T cell activation28, CD4+ T cells deficient in MCPIP1 displayed a significant increase in effector T cells (CD62LloCD44hi) compare with WT mice (Fig E2, A). CD4 and CD8 T cell development were similar between WT and zc3h12a−/− mice (Fig E2, B). To test whether zc3h12a−/− CD4+ T cells produce more Th2 cytokines in vivo, we stimulated splenocytes with PMA/Ionomycin and then measured cytokine production. The percentages of IL-5- and IFN-γ-producing effector CD4+ T cells were increased in zc3h12a−/− CD4+ T cells compared to WT CD4+ T cells (Fig 2, A). Similar to lung CD4 T cells, splenic zc3h12a−/− CD4+ T cells also produced more IL-5 and IL-13 than WT CD4+ T cells when stimulated with PMA/Ionomycin (Fig 2, B&C). Interestingly, when splenic CD4 T cells were activated with plate-bound anti-CD3/CD28 antibodies, the production of IL-4, IFN-γ and IL-17 became comparable (Fig 2, D). Then, we tested the responses of CD4+ T cells to different strengths of TCR signal. Maximal IL-5 and IL-13 production by zc3h12a−/− CD4+ T cells were induced even by low TCR signaling, while IL-4 production was caught up by WT CD4+ T cells and IFN-γ production remained comparable (Fig 2, E and Fig E2, C). These data indicate that once becoming effector cells, WT and zc3h12a−/− CD4+ T cells have similar capacity to produce cytokines except IL-5 and IL-13 that are still overproduced in zc3h12a−/− CD4+ T cells. Indeed, IFN-γ-producing cells were comparable between WT and zc3h12a−/− effector T cells, while IL-5-producing cells remained increased in zc3h12a−/− cells (Fig E2, D). Taken together, these data indicate that MCPIP1 specifically regulates IL-5/IL-13-producing Th2 cells.
Fig. 2.

Cytokines are overproduced in zc3h12a−/− CD4 T cells in vivo. Frequency of cytokine+ and CD44+ splenic cells gated on CD4 after stimulation with P/I (A and B) (n=5). (C) Summary of cytokine-expressing cells as shown in (B) (three independent experiments with 3–6 mice per group). (D) Intracellular cytokine expression in total CD4 T cells after stimulation with anti-CD3/CD28 Abs for 4 days, resting for 2 days and then re-stimulation with P/I (6 mice per group). (E) Total CD4+ T cells were stimulated by anti-CD3 Ab with or without anti-CD28 Ab for 48 h and supernatants were used to detect cytokines by ELISA (3 mice per group). *: p< 0.05, **: p<0.01, ***: p< 0.001.
MCPIP1 regulates Th2 cytokine production in a cell-intrinsic manner
Zc3h12a−/− CD4+ T cells produce significant amounts of Th2 cytokines which might have an autocrine effect on Th2 cytokine production. Therefore, we cultured the WT and zc3h12a−/− CD4+ T cells at a ratio of 1:1 in the same well to let them first be exposed to the same levels of cytokines. This setting will exclude the possible effects of different levels of cytokines on Th2 cytokine production in comparisons between WT and KO cells. IL-5- and IL-13-producing cells were significantly higher in zc3h12a−/− CD4+ T cells (CD45.2+) than in WT CD4+ T cells (CD45.2−), whereas IL-4-, IFN-γ- and IL-17-producing cells were comparable (Fig 3, A&B). Next we measured the mRNAs of Th2 cytokines and transcription factors important for Th2 cell differentiation. Th2-related transcription factor Gata3, IRF4, and Gfi mRNA were higher in zc3h12a−/− CD4+ T cells than in WT CD4+ T cells (Fig 3, C). The master transcription factors important for Th17 cells (Rorc) and for Th1 (Tbx21) and Treg (Foxp3) cells were reduced and slightly increased in zc3h12a−/− cells, respectively (Fig 3, C). The mRNA expression of Th2 cytokines (IL-4, IL-5 and IL-13) was increased in zc3h12a−/− CD4+ T cells, so as the expression of IFN-γ and IL-17A mRNAs, while TGF-β transcript remained unchanged in zc3h12a−/− CD4+ T cells, compared to WT CD4+ T cells in response to anti-CD3/CD28 stimulation (Fig 3, D). In addition, mRNAs encoding costimulatory molecules (ox40l and icosl) and chemokine receptors (ccr3 and cxcr4) associated with Th2 cells were also highly expressed in zc3h12a−/− CD4+ T cells (Fig. E3). Taken together, these data indicate an intrinsic effect for MCPIP1 on Th2 cytokine production, especially IL-5 and IL-13.
Fig. 3.

Loss of MCPIP1 leads to enhanced Th2 cytokine production in a cell-intrinsic manner. Naïve WT (CD45.1+) and zc3h12a−/− (CD45.2+) CD4+ T cells were co-cultured with anti-CD3/CD28 Abs in the same well for 3 d, rested for 3 d with IL-2, and then treated with P/I for 4 h. Numbers in outlined areas indicate the percentages of cytokine-producing cells (A) and a summary of 3–4 mice per group (B). (C) The mRNA expression in naïve WT and zc3h12a2−/− CD4+ T cells stimulated with anti-CD3/CD28 Abs for 3 d (n=3). (D) The mRNA expression of cytokines in naïve WT and zc3h12a2−/− CD4+ T cells stimulated with anti-CD3/CD28 Abs for 3 days, rested for 3 days, and re-stimulated with P/I for 4 h (n=3). *: p< 0.05, **: p<0.01, ***: p< 0.001.
MCPIP1 regulates IL-5-producing Th2 cell differentiation under unfavorable condition
To determine the effects of MCPIP1 on Th2 development, we differentiated naïve CD4+ T cells from zc3h12a−/− mice and wild-type littermates toward Th1 and Th2 cells, and then assessed lineage-specific cytokines. Compared with WT cells, the percentages of IL-4-producing CD4+ T cells were increased in zc3h12a−/− CD4+ T cells under non-polarizing condition, while this difference was abolished under Th2-polarizing condition (Fig 4, A–C). However, naïve zc3h12a−/− CD4+ T cells expressed much higher levels of IL-5 and IL-13 under both non-polarizing and Th2 polarizing conditions (Fig 4, B&C), indicating that naïve zc3h12a−/− CD4+ T cells were programed to produce IL-5 and IL-13 once activated even under unfavorable conditions. Indeed, higher levels of IL-5, IL-13 and Gata3 mRNA were detected in zc3h12a−/− cells than in WT T cells under unfavorable Th1- and Th17-polarizing conditions (Fig 4, D).
Fig. 4.

Loss of MCPIP1 results in enhanced IL-5+T cell development under unfavorable conditions. Intracellular cytokines were stained in naïve WT and zc3h12a−/− CD4+ T cells under Th0, Th1, or Th2 polarizing conditions (A and B). The percentages of cytokine-producing CD4 T cells (C) and the mRNA levels (D) as in (A and B) were measured and summarized (n=3–4). (E) Naïve zc3h12a−/− CD4+ T cells were stimulated with anti-CD3/CD28 Abs with anti-IL-4 (20 ng/ml), medium, or recombinant IL-4 (10 ng/ml), and stained for Gata3, IL-4 and IL-5 after P/I treatment for 4 h. (F) A summary of three independent experiments as in (E) was shown. *: p< 0.05, **: p<0.01, ***: p< 0.001.
To define the role of IL-4 for IL-5 production in zc3h12a−/− CD4+ T cells, we respectively added recombinant IL-4 and IL-4 neutralizing antibody during expansion of zc3h12a−/− CD4+ T cells, and found that neither adding IL-4 nor neutralizing IL-4 had any effects on IL-5 expression (Fig 4, E&F). Neutralizing IL-4 inhibited the expression of IL-4 and Gata3 in non-IL-5-producing zc3h12a−/− T cells (Fig 4, E&F), suggesting that IL-4 is not required for the differentiation of IL-5-producing zc3h12a−/− CD4 T cells. In addition, to confirm the role of MCPIP1 in Th2 cytokine expression, we treated WT and zc3h12a−/− CD4+ T cells with MI-2, a specific inhibitor of MALT1 known to increase MCPIP1 levels by blocking its degradation28, and measured cytokine responses. Neither increasing MCPIP1 by MI-2 treatment nor deleting MCPIP1 affected IL-4 expression in CD4+ T cells (Fig E4, A), further suggests the independence of MCPIP1 in IL-4 expression. Increasing MCPIP1 by MI-2 treatment inhibited IL-5 and IL-13 expression in WT CD4+ T cells but not in zc3h12a−/− CD4+ T cells (Fig E4, B&C). These data further indicate that MCPIP1 selectively regulates the development of IL-5/IL-13-producing Th2 cells but not the IL-4-producing Th2 cells.
Notch signaling mediates the initiation of IL-5-producing Th2 cells by MCPIP1 deficiency
Notch-mediated Th2 cell differentiation and Th2 immunity38–40 are also IL-4 independent. Notch signaling is initiated by binding of Notch ligands to its receptors. There are five mammalian Notch ligands, three of which belong to the Delta-like family (DLL1, DLL3 and DLL4) and two belong to the Jagged family (Jag1 and Jag2)41. We found that the expression of Jag1, Jag2 and DLL4 were increased in naïve zc3h12a−/− CD4+ T cells compared with WT T cells (Fig 5, A). To determine whether Notch signaling is involved, we pretreated naïve zc3h12a−/− CD4+ T cells with GSI, a γ-secretase inhibitor that blocks cleavage of the Notch intracellular domain (NICD) from the membrane, stimulated the cells with anti-CD3/CD28 antibodies, and then measured Th2 cytokines. IL-5 and IL-4 production were significantly inhibited while IFN-γ production enhanced after blocking Notch signaling by GSI (Fig 5, B&C), demonstrating the involvement of Notch pathway in MCPIP1-mediated Th2 inhibition. Next, we tested whether Notch signaling is required for cytokine production in effector T cells. We purified total CD4+ T cells (most are effector cells) from zc3h12a−/− mice, added GSI, and then measured Th2 cytokines and Gata3. Blockade of Notch pathway did not affect IL-5 and IL-4 production as well as Gata3 expression in zc3h12a−/− effector CD4 T cells (Fig 5, D and Fig E5, A). Furthermore, we co-cultured total WT and zc3h12a−/− CD4+ T cells in the same wells with or without GSI. Blockade of Notch signaling by GSI inhibited the production of IL-4 and IL-13 in WT CD4+ T cells but not in zc3h12a−/− CD4+ T cells, while IL-5 production was not affected by GSI in zc3h12a−/− CD4+ T cells. (Fig 5, E and Fig E5, B). These data indicate that Notch mediates the initiation but not the maintenance of IL-5-producing Th2 cells with MCPIP1 deficiency.
Fig. 5.

Notch signal is required for the initiation of IL-5-producing Th2 cells mediated by MCPIP1. (A) The mRNA levels of Notch ligands in naïve WT and zc3h12a2−/− CD4 T cells stimulated with anti-CD3/CD28 Abs were measured by qRT-PCR (n=3). (B) Naïve zc3h12a−/− CD4 T cells were treated with GSI and anti-CD3/CD28 Abs for 3 days, and then stained for Gata3, IL-4 and IL-5 (n=3). (C) A summary of three independent experiments as in (B) was shown. (D) Total zc3h12a−/− CD4 T cells were treated with GSI and anti-CD3/CD28 Abs for 3 days and then stained for Gata3, IL-4 and IL-5. (E) Total CD4 T cells of WT (CD45.2−) and zc3h12a−/− (CD45.2+) mice were cultured in the same well at 1:1 ratio with anti-CD3/CD28 Abs and GSI for 3 d, rested for 3 d, and then stimulated by P/I for 4 h. Cytokine-producing cells were measured by FACS gated on either CD45.2+ (zc3h12a−/−) or CD45.2− cells (WT). *: p< 0.05, **: p<0.01, ***: p< 0.001.
MCPIP1 targets Gata3 to control IL-5 production by Th2 cells
Since Gata3 is the master transcription factor for Th2 cell development, we checked Gata3 mean fluorescence intensity (MFI) and found that Gata3 protein was highly expressed in zc3h12a−/− CD4+ T cells compared to WT CD4+ T cells (Fig 6, A). Based on Gata3 levels, we divided zc3h12a−/− CD4+ T cells into three populations, Gata3hi, Gata3mid and Gata3lo. Gata3hi cells produced higher levels of IL-5 than Gata3mid and Gata3low cells (Fig 6, B). To confirm whether Gata3 accounts for IL-5 overproduction in zc3h12a−/− CD4+ T cells, we knocked down Gata3 with Gata3-shRNA/lentivirus and found that Gata3 knockdown resulted in reduced IL-5 expression in a dose-dependent manner (Fig 6, C and Fig E6, A), indicating that Gata3 mediates IL-5 production in zc3h12a−/− T cells. In addition, we generated a human T cell line (CEM) with an inducible MCPIP1 expression Tet-On system. Addition of Doxycycline (Dox) dose-dependently induced GFP/MCPIP1 fusion protein expression in CEM/MCPIP1-Tet-On cells (Fig E6, B). As a consequence, the expression of Gata3 protein (Fig 6, D) and mRNA (Fig 6, E and Fig E6, C) was reduced significantly. Since MCPIP1 is an RNase 25, we measured the half-life of Gata3 mRNA and found that the half-life of Gata3 mRNA was increased in zc3h12a−/− CD4+ T cells compared to WT CD4+ T cells (Fig 6, F) and decreased in CEM/MCPIP1-Tet-On cells after inducing MCPIP1 (Fig 6, G). The Th2-related transcription factor IRF4 mRNA was also stabilized, and STAT6 mRNA unaffected in zc3h12a−/− CD4+ T cells (Fig E6, D). These data indicate that MCPIP1 inhibits Gata3 mRNA expression through promoting its mRNA decay.
Fig. 6.

MCPIP1 promotes Gata3 mRNA decay. (A) Gata3 was measured in naïve WT and zc3h12a−/− CD4+ T cells stimulated with anti-CD3/CD28 Abs. (B) IL-5 in zc3h12a−/− CD4+ T cells was measured in groups based on Gata3 levels: high (up); intermediate (middle); and low (low) and summarized. (C) Zc3h12a−/− CD4 T cells were transduced with Gata3/lentivirus or scramble/lentivirus and the percentages of Gata3+ and IL-5+ were measured by FACS. (D) Gata3+ and GFP+ CEM-MCPIP1-Tet-on cells were measured after Dox treatment (100 ng/ml). (E) Gata3 and MCPIP1 mRNA levels in CEM-Tet-on cells were measured after treatment with different amounts of Dox. (F) CD4+ T cells were stimulated with anti-CD3/CD28 Abs for 4 h, and then added ActD and DRB. Remaining Gata3 mRNA was measured at different times. (G) CEM-Tet-on cells were treated with and without Dox (100 ng/ml) for 24 h and then added ActD and DRB. The remaining Gata3 mRNA was measured as in (F). Gata3 3 ′UTR luciferase reporters or empty vector (Control) were transiently transfected into CEM-Tet-On cells expressing WT MCPIP1 (H) or expressing mutant MCPIP1 (I), and then added Dox. Luciferase activity was determined in cell lysates (n=3). *: p< 0.05, **: p<0.01, ***: p< 0.001.
Fully differentiated Th2 cells expressed high levels of Gata3 and low levels of MCPIP1 (Fig E6, E). To test whether MCPIP1 binds to Gata3 mRNA, we purified the MCPIP1-binding complex with anti-MCPIP1 antibody from cytoplasmic extracts of the Th2 cells and then detected the bound mRNAs by RT-PCR. Compared with controls, Gata3 mRNA was enriched in the mRNAs pulled down with anti-MCPIP1 antibody (Fig E6, F), indicating a direct binding of MCPIP1 to Gata3 mRNA. Next, we transfected a Gata3 3′UTR luciferase construct into CME-Tet-On cells, induced MCPIP1 by adding dox, and then measured luciferase activity. Induction of MCPIP1 suppressed luciferase activity (Fig 6, H), demonstrating that MCPIP1 promotes Gata3 mRNA decay through 3′UTR. As illustrated in Fig E6, G, there are several domains in MCPIP1 protein. To determine which domain mediates Gata3 mRNA decay, we generated CEM-Tet-On stable clones expressing several MCPIP1 mutants, transfected the Gata3–3′UTR luciferase construct into these stable CEM-Tet-On cells, and then induced their expression by adding Dox and measured luciferase activity. Induction of both WT MCPIP1 and mutant MCPIP1 except the mutant D141N inhibited luciferase activity (Fig 6, I). We also measured the levels of Gata3 mRNA in CEM-Tet-On cells after adding Dox. Consistent with the luciferase results, both WT and mutant MCPIP1 except the mutant D141N inhibited the endogenous Gata3 mRNA expression (Fig E6, H). The expression of Gata3 protein displayed similar patterns after expressing WT and mutant MCPIP1 (Fig E6, I). To further confirm whether mutant D141N mediate Gata3 mRNA decay, we measured half-life of Gata3 mRNA in the above CEM-Tet-On cells. The half-lives of Gata3 mRNA were shortened in cells expressing WT MCPIP1 but not in cells expressing the D141N mutants (Fig E6, J), indicating that the RNase domain is required for Gata3 mRNA degradation by MCPIP1. Taken together, these data demonstrate that MCPIP1 via RNase activity destabilizes Gata3 mRNA, therefore inhibits IL-5 and IL-13 production by CD4 T cells.
MCPIP1 deficiency in CD4 T cells enhances Th2-mediated inflammation in allergen-induced asthma models
To determine the physiological consequence of MCPIP1-mediated Th2 inhibition, we immunized WT and zc3h12a−/− mice with OVA mixed with adjuvant aluminum hydroxide, and then challenged them with intranasal OVA (Fig 7, A). One day after the last challenge, we stained lung tissue by H&E and PAS. Compared to WT mice, there were increased infiltrating inflammatory cells and mucin-positive cells (goblet-cell metaplasia) in lungs of the zc3h12a−/− mice (Fig E7, A&B). Effector CD4+ T cells were also increased after OVA-immunization and challenge in both mice (Fig E7, C). In addition, IL-4- and IL-5-producing effector CD4 T cells were highly increased in the lungs of zc3h12a−/− mice compared with WT mice (Fig E7, D). In line with the results shown in Fig 2, D, zc3h12a−/− CD4+ T cells expressed higher IL-5 and IL-13 but not IL-4 than WT CD4+ T cells in response to OVA stimulation in vitro (Fig E7, E). These data indicate that MCPIP1 inhibits OVA-specific CD4 T cell responses in vivo, through down-regulating allergen-specific Th2 inflammation mediated by IL-5 and IL-13.
Fig. 7.

Zc3h12a−/− CD4 T cells cause increased Th2 cell-mediated disease. (A and B) Zc3h12a−/− mice and WT littermates were sensitized and then challenged by OVA and sacrificed on day 26. Cells isolated from DLN were stimulated with different amounts of OVA for 72 h and cytokines in the supernatants were measured by ELISA (n=3). (C) CD45.1 recipient mice were transferred with OVA-specific CD4 T cells as in (B) and then challenged daily with OVA for 5 times. H&E-stained lung sections of the recipient mice were shown and inflammation was scored. (D) The percentages of eosinophil and neutrophil in BAL of the CD45.1+ recipient mice after T cell transfer were gated on SiglecF and Ly6G (n=3). (E) mRNA levels in lungs of the recipient mice were measured by qRT-PCR (n=3). (F) The percentages of CD44+CD45.1− (donor) CD4 T cells were measured in the CD45.1+ recipient mice. (G) Intracellular IL-5 and IL-4 in the donor cells were stained as in (F) gated on CD44 (n=3). *: p< 0.05, **: p<0.01, ***: p< 0.001.
To define whether zc3h12a−/− Th2 cells are pathogenic, we first expanded OVA-specific T cells by culturing splenocytes from the above OVA-immunized mice with OVA and IL-2. The OVA-specific zc3h12a−/− CD4+ T cells secreted large amounts of IL-4, IL-5 and IL-13 in response to OVA stimulation, while IFN-γ production was reduced (Fig 7, B). Next, we purified the OVA-specific CD4+ T cells and transferred them intravenously into congenic CD45.1+ recipient mice. Three days after T cell transfer, recipient mice were challenged with OVA for 5 days and then analyzed. Mice that received OVA-specific zc3h12a−/− CD4+ T cells displayed an increase in infiltrating immune cells and inflammation in the airways compared to mice received WT OVA-specific CD4 T cells (Fig 7, C). Lung eosinophils (CD11b+SiglecF+) were also increased in the mice receiving OVA-specific zc3h12a−/− CD4 T cells compared with mice transferred with OVA-specific WT CD4 T cells (Fig 7, D and Fig E7, F), indicating that zc3h12a−/− CD4 T cells are pathogenic and can induce asthmatic-like airway inflammation. Analyses of Th2 cytokine transcripts revealed that IL-5 and IL-13 but not IL-4 mRNA were increased in the lungs of the mice receiving OVA-specific zc3h12a−/− CD4 T cells (Fig 7, E). The purity of donor cells was confirmed by checking CD3 and CD4 expression gated on CD45.1 and CD45.2 (Fig E7, G). In addition, zc3h12a−/− donor CD4 T cells displayed enhanced proliferation and effector phenotypes (Fig 7, F), and produced increased IL-5 and IL-4 in vivo (Fig 7, G).
Furthermore, we tested the effects of zc3h12a−/− CD4 T cells with physiopathologically relevant house dust mite extracts (HDM). Zc3h12a−/− mice and WT littermates were sensitized and challenged with HDM as illustrated in Fig 8, A. Lung inflammation and PAS positive cells were increased in zc3h12a−/− mice compared to WT mice (Fig 8, B). We next expanded HDM-specific CD4 T cells with HDM and IL-2, and checked the HDM-specific responses. Zc3h12a−/− CD4+ T cells produced significantly more IL-5 and IL-13 than WT CD4+ T cells in response to HDM stimulation (Fig 8, C). Then, we transferred the HDM-specific CD4+ T cells into WT B6 mice and challenged them with HDM. The mice receiving the HDM-specific zc3h12a−/− CD4+ T cells displayed more severe lung inflammation and more PAS positive cells than the mice transferred with WT cells (Fig 8, D). The expression of IL-5 and IL-13 were also increased in lungs of the mice received KO HDM-specific CD4 T cells (Fig 8, E). Taken together, the results of the HDM model are consistent with the OVA model, indicating that CD4 T cells deficient in MCPIP1 exaggerate type 2 biased immune responses and cause Th2 inflammation in the airway.
Fig. 8.

HDM-induced Th2 inflammation is increased in zc3h12a−/− mice. (A) Zc3h12a−/− mice and WT littermates were sensitized and then challenged by HDM. (B) Mice were sacrificed on day 26. Lung sections were stained with H&E and PAS and inflammation was scored (n=3). (C) Cells isolated from DLN as in (B) were stimulated with HDM for 72 h. IL-5 and IL-13 in supernatants were measured by ELISA (n=3). (D) WT B6 mice were transferred with HDM-specific CD4 T cells as in (C) and then sensitized with HDM. Representative H&E- and PAS-stained lung sections were shown from the recipients received HDM-treated WT or zc3h12a−/− cells and inflammation was scored (five mice each group). (E) Cells from lungs of the recipient mice as in (D) were stimulated with P/I for 4 h and then RNA was extracted to measure mRNAs by qRT-PCR (n=3). *: p< 0.05, **: p<0.01, ***: p< 0.001.
DISCUSSION
We provide several lines of evidence identifying MCPIP1 as a major negative regulator for Th2 cell differentiation and effector function, especially IL-5/IL-13-producing Th2 cells. We discovered enhanced IL-5+Th2 differentiation and airway inflammation in MCPIP1 deficient mice. Mechanistically, we identified Notch pathway and Gata3 as critical targets of MCPIP1 for regulating IL-5/IL-13-producing Th2 cell development. CD4+ T cells are polarized and differentiated into IL-5-producing Th2 cells in the absence of MCPIP1. MCPIP1 also directly targets Th2 cytokines for mRNA decay (data not shown). The preferential differentiation of naïve zc3h12a−/− CD4 T cells towards Th2 but not Th17 or Treg cells in vitro may be due to targeting several key transcription factors important for Th2 cell differentiation, including Gata3, Gfi, and Irf4. The mRNAs of Rorc, Tbx21 and Foxp3 were not altered significantly in zc3h12a−/− CD4 T cells. IL-4 is a key cytokine for Th2 differentiation. T cells deficient in MCPIP1 lead to IL-4 overproduction that might have an autocrine effect on Th2 differentiation. However, we found that neutralizing IL-4 only inhibited IL-4 expression and had no effects on IL-5 expression in zc3h12a−/− CD4 T cells, indicating that the development of IL-5-producing zc3h12a−/− Th2 cells is independent of IL-4. MCPIP1 also targets ICOS and OX4028 which have been shown to be important for Th2 differentiation42, 43. Most importantly, our data show that MCPIP1 directly targets the Th2 specific transcript factor, Gata3. Naïve T cells express low levels of Gata3 induced by TCR and IL-4R44. Zheng and colleagues18 show that Gata3 was present in naïve CD4 T cells and increased during Th2 development and extinguished during Th1 development. Gata3 can be induced by both IL-4-dependent and independent way44. MCPIP1-deficient CD4+ T cells express high levels of Gata3 even under non-polarizing conditions, indicating that MCPIP1 is critical for naïve CD4 T cells to maintain their non-polarized phenotypes. When the levels of MCPIP1 are reduced, CD4 T cells are more likely to polarize and differentiate into IL-5-producing Th2 cells, leading to Th2-associated inflammation. Type 2 innate lymphoid cells (ILC2) share many features with zc3h12a−/− Th2 cells and also produce IL-13 and IL-545–47. ILC2 is regulated by Gata3 and plays an important role in allergic airway inflammation48–50. It will be of interest to see whether MCPIP1 regulates ILC2 as well.
The Notch pathway is important in Th2 cell differentiation and activation51–55. Notch-mediated Th2 cell differentiation and Th2 immunity38–40 are quite similar to the effects of MCPIP1 on IL-5/IL-13-producing Th2 cells, namely both are independent of IL-4 and STAT6 (data not shown). Blockade of Notch signaling inhibits Th2 cytokine production only in naïve but not in effector zc3h12a−/− CD4 T cells, suggests that Notch signaling only controls the initiation of Th2 cell differentiation mediated by MCPIP1. Since GSI blocks NICD cleavage from the membrane, it suggests that ligand-receptor interaction upstream of the intracellular Notch pathway mediates the effects of MCPIP1 on Th2 cells. Indeed, the expression of Notch ligands, Jag1/2 and DLL4 were increased in MCPIP1-deficient CD4 T cells (Fig 5, A), suggesting that Jag1/2 and DLL4 may involve in activation of surrounding zc3h12a−/− CD4 T cells to attain the Th2 phenotype.
Several studies reported that MCPIP1 protein levels in T cells were decreased in response to anti-CD3/CD28 antibody stimulation28, 34. The degradation and cleavage of MCPIP1 were abolished in the presence of a Malt1 protease inhibitor, as well as in Bcl10 or Malt1 deficient CD4+ T cells, indicating that MCPIP1 is cleaved by Malt128. Inhibition of Malt1 activity leads to reduction of IL-5/IL-13 expression in WT CD4 T cells, but not in zc3h12a−/− CD4 T cells, indicating that Malt1/MCPIP1 axis mainly regulates the development of IL-5/IL-13-producing Th2 cells. IL-5 expression is known to be more dependent on the expression levels of Gata3 than IL-456, 57. In T cell transfer experiments, MCPIP1-deficient Th2 cells were capable of causing enhanced airway inflammation and accumulation of Th2 cytokine-producing cells with a memory phenotype. It was reported that treatment of MCPIP1 deficient mice with antibiotics increased survival and reduced mRNA levels of inflammatory cytokines58, suggesting a role of microbiota in MCPIP1-mediated inflammation. MCPIP1 selectively targets the mRNAs encoding Th2 cytokine IL-5 and IL-13 for degradation (data not shown). Why MCPIP1 selectively targets specific cytokines, especially those associated with Th2 cell differentiation and function needs further investigation.
Though the role of Gata3 in regulating Th2 cytokines is well established59, the mechanisms that suppress Gata3 expression have not been fully defined. Gata3 can be induced through TCR, IL-4R/STAT6, IL-2R/STAT5 and Notch signaling during Th2 differentiation. Our work reveals high levels of Gata3 in zc3h12a−/− CD4 T cells, and MCPIP1 promotes Gata3 mRNA decay via the RNase domain. We provide strong evidence indicating that MCPIP1 directly regulates Gata3 by binding to its 3′UTR for mRNA destabilization. Blonska et al60 showed that CARMA1 up-regulates Gata3 on the transcriptional level independent of NF-κB activation. Given the fact that MCPIP1 is cleaved by Malt1, up-regulation of Gata3 by CARMA1 may involve in cleavage of MCPIP1 by the CARMA1–BCL10-MALT1 complex.
Based on our results, we propose the following model of Th2 cell development regulated by MCPIP1. MCPIP1 inhibits Th2 cell development by binding to the mRNAs encoding multiple Th2 transcription factors, including Gata3, and by inhibiting Th2-associated Notch signaling. Upon T cell activation, MCPIP1 level is reduced and therefore releases its suppression on Th2 transcription factor and Notch pathway, which allows Gata3 to bind to the IL-5 and IL-13 gene loci, resulting in IL-5+/IL-13+Th2 cell differentiation. Meanwhile, Reduction of MCPIP1 increases the mRNA stability of IL-5 and IL-13. Combination of the increased Th2 differentiation and Th2 cytokine transcripts leads to enhanced Th2 development and Th2 effector function. Therefore, strategies that increase the levels of MCPIP1 will inhibit IL-5+/IL-13+Th2 cell differentiation and effector function, resulting in suppression of Th2-mediated inflammation.
Supplementary Material
Key Messages.
MCPIP1 specifically inhibits IL-5/IL-13-producing Th2 cell differentiation and function.
MCPIP1 suppresses the early differentiation of IL-5-producing Th2 cells through Notch.
MCPIP1 inhibits IL-5-producing Th2 development by targeting Gata3 and Th2 cytokine transcripts for degradation.
Acknowledgments
This work was supported by the National Cancer Institute of the National Institutes of Health under Award Number R01CA163808 (J Liu). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We thank Jonathan M. Kurie (The University of Texas MD Anderson Cancer Center) for providing the Gata3 3′UTR luciferase constructs and Mingui Fu (University of Missouri Kansas City) for providing the MCPIP1 expression constructs.
Abbreviations
- BALF
bronchoalveolar lavage fluid
- EGFP
enhanced green fluorescent protein
- KO
knockout
- WT
wild type
- MCPIP-1
monocytes chemoattractant induced protein 1
- MCP-1
monocytes chemoattractant protein 1
- FSH
forward scatter
- HDM
house dust mites
- OVA
ovalbumin
- P/I
PMA and ionomycin
- Ab
antibody
- DLN
draining lymph nodes
- MFI
median fluorescence intensity
- H&E
hematoxylin and eosin
- PAS
periodic acid-Schiff)
Footnotes
Disclosure of potential conflicts of interest: All authors declare no financial conflict of interest in this study.
Supplemental information includes seven figures and two tables.
AUTHOR CONTRIBUTIONS
Concept and design: J. Liu
Development of methodology: J. Liu, H. Peng, H. Ning
Acquisition of data (provided animals acquired and managed patients, provided facilities, etc.): H. Peng, H. Ning, W. Lu, Q. Wang, J. Liu
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): J. Liu, H. Peng, H. Ning
Writing, review, and/or revision of the manuscript: J. Liu, H. Peng, D. Hoft, R. Hou, M. Dykewicz
Administrative, technical, or material support (i.e., reporting or organizing data, construction database): T. Wang, Y. Chang, P. K.
Study supervision: J. Liu
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Lambrecht BN, Hammad H. The immunology of asthma. Nat Immunol. 2014;16:45–56. doi: 10.1038/ni.3049. [DOI] [PubMed] [Google Scholar]
- 2.Cohn L, Elias JA, Chupp GL. Asthma: mechanisms of disease persistence and progression. Annu Rev Immunol. 2004;22:789–815. doi: 10.1146/annurev.immunol.22.012703.104716. [DOI] [PubMed] [Google Scholar]
- 3.Finkelman FD, Hogan SP, Hershey GK, Rothenberg ME, Wills-Karp M. Importance of cytokines in murine allergic airway disease and human asthma. J Immunol. 2010;184:1663–74. doi: 10.4049/jimmunol.0902185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coffman RL, Ohara J, Bond MW, Carty J, Zlotnik A, Paul WE. B cell stimulatory factor-1 enhances the IgE response of lipopolysaccharide-activated B cells. J Immunol. 1986;136:4538–41. [PubMed] [Google Scholar]
- 5.Finkelman FD, Katona IM, Urban JF, Jr, Holmes J, Ohara J, Tung AS, et al. IL-4 is required to generate and sustain in vivo IgE responses. J Immunol. 1988;141:2335–41. [PubMed] [Google Scholar]
- 6.Ohnishi T, Kita H, Weiler D, Sur S, Sedgwick JB, Calhoun WJ, et al. IL-5 is the predominant eosinophil-active cytokine in the antigen-induced pulmonary late-phase reaction. Am Rev Respir Dis. 1993;147:901–7. doi: 10.1164/ajrccm/147.4.901. [DOI] [PubMed] [Google Scholar]
- 7.Yamaguchi Y, Suda T, Suda J, Eguchi M, Miura Y, Harada N, et al. Purified interleukin 5 supports the terminal differentiation and proliferation of murine eosinophilic precursors. J Exp Med. 1988;167:43–56. doi: 10.1084/jem.167.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhu Z, Homer RJ, Wang Z, Chen Q, Geba GP, Wang J, et al. Pulmonary expression of interleukin-13 causes inflammation, mucus hypersecretion, subepithelial fibrosis, physiologic abnormalities, and eotaxin production. J Clin Invest. 1999;103:779–88. doi: 10.1172/JCI5909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McKenzie GJ, Emson CL, Bell SE, Anderson S, Fallon P, Zurawski G, et al. Impaired development of Th2 cells in IL-13-deficient mice. Immunity. 1998;9:423–32. doi: 10.1016/s1074-7613(00)80625-1. [DOI] [PubMed] [Google Scholar]
- 10.Amsen D, Spilianakis CG, Flavell RA. How are T(H)1 and T(H)2 effector cells made? Curr Opin Immunol. 2009;21:153–60. doi: 10.1016/j.coi.2009.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lloyd CM, Hessel EM. Functions of T cells in asthma: more than just T(H)2 cells. Nat Rev Immunol. 2010;10:838–48. doi: 10.1038/nri2870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paul WE, Zhu J. How are T(H)2-type immune responses initiated and amplified? Nat Rev Immunol. 2010;10:225–35. doi: 10.1038/nri2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pearce EJ, Caspar P, Grzych JM, Lewis FA, Sher A. Downregulation of Th1 cytokine production accompanies induction of Th2 responses by a parasitic helminth, Schistosoma mansoni. J Exp Med. 1991;173:159–66. doi: 10.1084/jem.173.1.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Licona-Limon P, Kim LK, Palm NW, Flavell RA. TH2, allergy and group 2 innate lymphoid cells. Nat Immunol. 2013;14:536–42. doi: 10.1038/ni.2617. [DOI] [PubMed] [Google Scholar]
- 15.Tumes DJ, Onodera A, Suzuki A, Shinoda K, Endo Y, Iwamura C, et al. The polycomb protein Ezh2 regulates differentiation and plasticity of CD4(+) T helper type 1 and type 2 cells. Immunity. 2013;39:819–32. doi: 10.1016/j.immuni.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 16.Yang XO, Angkasekwinai P, Zhu J, Peng J, Liu Z, Nurieva R, et al. Requirement for the basic helix-loop-helix transcription factor Dec2 in initial TH2 lineage commitment. Nat Immunol. 2009;10:1260–6. doi: 10.1038/ni.1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seki Y, Inoue H, Nagata N, Hayashi K, Fukuyama S, Matsumoto K, et al. SOCS-3 regulates onset and maintenance of T(H)2-mediated allergic responses. Nat Med. 2003;9:1047–54. doi: 10.1038/nm896. [DOI] [PubMed] [Google Scholar]
- 18.Zheng W, Flavell RA. The transcription factor GATA-3 is necessary and sufficient for Th2 cytokine gene expression in CD4 T cells. Cell. 1997;89:587–96. doi: 10.1016/s0092-8674(00)80240-8. [DOI] [PubMed] [Google Scholar]
- 19.Zhu J, Jankovic D, Grinberg A, Guo L, Paul WE. Gfi-1 plays an important role in IL-2-mediated Th2 cell expansion. Proc Natl Acad Sci U S A. 2006;103:18214–9. doi: 10.1073/pnas.0608981103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu J, Min B, Hu-Li J, Watson CJ, Grinberg A, Wang Q, et al. Conditional deletion of Gata3 shows its essential function in T(H)1-T(H)2 responses. Nat Immunol. 2004;5:1157–65. doi: 10.1038/ni1128. [DOI] [PubMed] [Google Scholar]
- 21.Kafasla P, Skliris A, Kontoyiannis DL. Post-transcriptional coordination of immunological responses by RNA-binding proteins. Nat Immunol. 2014;15:492–502. doi: 10.1038/ni.2884. [DOI] [PubMed] [Google Scholar]
- 22.Anderson P. Post-transcriptional control of cytokine production. Nat Immunol. 2008;9:353–9. doi: 10.1038/ni1584. [DOI] [PubMed] [Google Scholar]
- 23.Zhou L, Azfer A, Niu J, Graham S, Choudhury M, Adamski FM, et al. Monocyte chemoattractant protein-1 induces a novel transcription factor that causes cardiac myocyte apoptosis and ventricular dysfunction. Circ Res. 2006;98:1177–85. doi: 10.1161/01.RES.0000220106.64661.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liang J, Saad Y, Lei T, Wang J, Qi D, Yang Q, et al. MCP-induced protein 1 deubiquitinates TRAF proteins and negatively regulates JNK and NF-kappaB signaling. J Exp Med. 2010;207:2959–73. doi: 10.1084/jem.20092641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matsushita K, Takeuchi O, Standley DM, Kumagai Y, Kawagoe T, Miyake T, et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature. 2009;458:1185–90. doi: 10.1038/nature07924. [DOI] [PubMed] [Google Scholar]
- 26.Liang J, Wang J, Azfer A, Song W, Tromp G, Kolattukudy PE, et al. A novel CCCH-zinc finger protein family regulates proinflammatory activation of macrophages. J Biol Chem. 2008;283:6337–46. doi: 10.1074/jbc.M707861200. [DOI] [PubMed] [Google Scholar]
- 27.Huang S, Miao R, Zhou Z, Wang T, Liu J, Liu G, et al. MCPIP1 negatively regulates toll-like receptor 4 signaling and protects mice from LPS-induced septic shock. Cell Signal. 2013;25:1228–34. doi: 10.1016/j.cellsig.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uehata T, Iwasaki H, Vandenbon A, Matsushita K, Hernandez-Cuellar E, Kuniyoshi K, et al. Malt1-induced cleavage of regnase-1 in CD4(+) helper T cells regulates immune activation. Cell. 2013;153:1036–49. doi: 10.1016/j.cell.2013.04.034. [DOI] [PubMed] [Google Scholar]
- 29.Mino T, Murakawa Y, Fukao A, Vandenbon A, Wessels HH, Ori D, et al. Regnase-1 and Roquin Regulate a Common Element in Inflammatory mRNAs by Spatiotemporally Distinct Mechanisms. Cell. 2015;161:1058–73. doi: 10.1016/j.cell.2015.04.029. [DOI] [PubMed] [Google Scholar]
- 30.Jeltsch KM, Hu D, Brenner S, Zoller J, Heinz GA, Nagel D, et al. Cleavage of roquin and regnase-1 by the paracaspase MALT1 releases their cooperatively repressed targets to promote T(H)17 differentiation. Nat Immunol. 2014;15:1079–89. doi: 10.1038/ni.3008. [DOI] [PubMed] [Google Scholar]
- 31.Garg AV, Amatya N, Chen K, Cruz JA, Grover P, Whibley N, et al. MCPIP1 Endoribonuclease Activity Negatively Regulates Interleukin-17-Mediated Signaling and Inflammation. Immunity. 2015;43:475–87. doi: 10.1016/j.immuni.2015.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Niu J, Shi Y, Xue J, Miao R, Huang S, Wang T, et al. USP10 inhibits genotoxic NF-kappaB activation by MCPIP1-facilitated deubiquitination of NEMO. EMBO J. 2013;32:3206–19. doi: 10.1038/emboj.2013.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lin RJ, Chien HL, Lin SY, Chang BL, Yu HP, Tang WC, et al. MCPIP1 ribonuclease exhibits broad-spectrum antiviral effects through viral RNA binding and degradation. Nucleic Acids Res. 2013;41:3314–26. doi: 10.1093/nar/gkt019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu S, Qiu C, Miao R, Zhou J, Lee A, Liu B, et al. MCPIP1 restricts HIV infection and is rapidly degraded in activated CD4+ T cells. Proc Natl Acad Sci U S A. 2013;110:19083–8. doi: 10.1073/pnas.1316208110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lin RJ, Chu JS, Chien HL, Tseng CH, Ko PC, Mei YY, et al. MCPIP1 suppresses hepatitis C virus replication and negatively regulates virus-induced proinflammatory cytokine responses. J Immunol. 2014;193:4159–68. doi: 10.4049/jimmunol.1400337. [DOI] [PubMed] [Google Scholar]
- 36.Qi Y, Liang J, She ZG, Cai Y, Wang J, Lei T, et al. MCP-induced protein 1 suppresses TNFalpha-induced VCAM-1 expression in human endothelial cells. FEBS Lett. 2010;584:3065–72. doi: 10.1016/j.febslet.2010.05.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.He M, Liang X, He L, Wen W, Zhao S, Wen L, et al. Endothelial dysfunction in rheumatoid arthritis: the role of monocyte chemotactic protein-1-induced protein. Arterioscler Thromb Vasc Biol. 2013;33:1384–91. doi: 10.1161/ATVBAHA.113.301490. [DOI] [PubMed] [Google Scholar]
- 38.Amsen D, Antov A, Jankovic D, Sher A, Radtke F, Souabni A, et al. Direct regulation of Gata3 expression determines the T helper differentiation potential of Notch. Immunity. 2007;27:89–99. doi: 10.1016/j.immuni.2007.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fang TC, Yashiro-Ohtani Y, Del Bianco C, Knoblock DM, Blacklow SC, Pear WS. Notch directly regulates Gata3 expression during T helper 2 cell differentiation. Immunity. 2007;27:100–10. doi: 10.1016/j.immuni.2007.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ouyang W, Lohning M, Gao Z, Assenmacher M, Ranganath S, Radbruch A, et al. Stat6-independent GATA-3 autoactivation directs IL-4-independent Th2 development and commitment. Immunity. 2000;12:27–37. doi: 10.1016/s1074-7613(00)80156-9. [DOI] [PubMed] [Google Scholar]
- 41.Radtke F, MacDonald HR, Tacchini-Cottier F. Regulation of innate and adaptive immunity by Notch. Nat Rev Immunol. 2013;13:427–37. doi: 10.1038/nri3445. [DOI] [PubMed] [Google Scholar]
- 42.Salek-Ardakani S, Song J, Halteman BS, Jember AG, Akiba H, Yagita H, et al. OX40 (CD134) controls memory T helper 2 cells that drive lung inflammation. J Exp Med. 2003;198:315–24. doi: 10.1084/jem.20021937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Simpson TR, Quezada SA, Allison JP. Regulation of CD4 T cell activation and effector function by inducible costimulator (ICOS) Curr Opin Immunol. 2010;22:326–32. doi: 10.1016/j.coi.2010.01.001. [DOI] [PubMed] [Google Scholar]
- 44.Pot C, Jin H, Awasthi A, Liu SM, Lai CY, Madan R, et al. Cutting edge: IL-27 induces the transcription factor c-Maf, cytokine IL-21, and the costimulatory receptor ICOS that coordinately act together to promote differentiation of IL-10-producing Tr1 cells. J Immunol. 2009;183:797–801. doi: 10.4049/jimmunol.0901233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fort MM, Cheung J, Yen D, Li J, Zurawski SM, Lo S, et al. IL-25 induces IL-4, IL-5, and IL-13 and Th2-associated pathologies in vivo. Immunity. 2001;15:985–95. doi: 10.1016/s1074-7613(01)00243-6. [DOI] [PubMed] [Google Scholar]
- 46.Fallon PG, Ballantyne SJ, Mangan NE, Barlow JL, Dasvarma A, Hewett DR, et al. Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J Exp Med. 2006;203:1105–16. doi: 10.1084/jem.20051615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kang Z, Swaidani S, Yin W, Wang C, Barlow JL, Gulen MF, et al. Epithelial cell-specific Act1 adaptor mediates interleukin-25-dependent helminth expulsion through expansion of Lin(−)c-Kit(+) innate cell population. Immunity. 2012;36:821–33. doi: 10.1016/j.immuni.2012.03.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Barlow JL, Bellosi A, Hardman CS, Drynan LF, Wong SH, Cruickshank JP, et al. Innate IL-13-producing nuocytes arise during allergic lung inflammation and contribute to airways hyperreactivity. J Allergy Clin Immunol. 2012;129:191–8. e1–4. doi: 10.1016/j.jaci.2011.09.041. [DOI] [PubMed] [Google Scholar]
- 49.Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-responsive lineage-CD25+ CD44(hi) lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol. 2012;188:1503–13. doi: 10.4049/jimmunol.1102832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hoyler T, Klose CS, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity. 2012;37:634–48. doi: 10.1016/j.immuni.2012.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tu L, Fang TC, Artis D, Shestova O, Pross SE, Maillard I, et al. Notch signaling is an important regulator of type 2 immunity. J Exp Med. 2005;202:1037–42. doi: 10.1084/jem.20050923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kubo M. Notch: filling a hole in T helper 2 cell differentiation. Immunity. 2007;27:3–5. doi: 10.1016/j.immuni.2007.07.005. [DOI] [PubMed] [Google Scholar]
- 53.Mowen KA, Glimcher LH. Signaling pathways in Th2 development. Immunol Rev. 2004;202:203–22. doi: 10.1111/j.0105-2896.2004.00209.x. [DOI] [PubMed] [Google Scholar]
- 54.Maekawa Y, Tsukumo S, Chiba S, Hirai H, Hayashi Y, Okada H, et al. Delta1-Notch3 interactions bias the functional differentiation of activated CD4+ T cells. Immunity. 2003;19:549–59. doi: 10.1016/s1074-7613(03)00270-x. [DOI] [PubMed] [Google Scholar]
- 55.Amsen D, Blander JM, Lee GR, Tanigaki K, Honjo T, Flavell RA. Instruction of distinct CD4 T helper cell fates by different notch ligands on antigen-presenting cells. Cell. 2004;117:515–26. doi: 10.1016/s0092-8674(04)00451-9. [DOI] [PubMed] [Google Scholar]
- 56.Lee HJ, O’Garra A, Arai K, Arai N. Characterization of cis-regulatory elements and nuclear factors conferring Th2-specific expression of the IL-5 gene: a role for a GATA-binding protein. J Immunol. 1998;160:2343–52. [PubMed] [Google Scholar]
- 57.Zhang J, Kuvelkar R, Cheewatrakoolpong B, Williams S, Egan RW, Billah MM. Evidence for multiple promoters of the human IL-5 receptor alpha subunit gene: a novel 6-base pair element determines cell-specific promoter function. J Immunol. 1997;159:5412–21. [PubMed] [Google Scholar]
- 58.Miao R, Huang S, Zhou Z, Quinn T, Van Treeck B, Nayyar T, et al. Targeted disruption of MCPIP1/Zc3h12a results in fatal inflammatory disease. Immunol Cell Biol. 2013;91:368–76. doi: 10.1038/icb.2013.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tindemans I, Serafini N, Di Santo JP, Hendriks RW. GATA-3 function in innate and adaptive immunity. Immunity. 2014;41:191–206. doi: 10.1016/j.immuni.2014.06.006. [DOI] [PubMed] [Google Scholar]
- 60.Blonska M, Joo D, Zweidler-McKay PA, Zhao Q, Lin X. CARMA1 controls Th2 cell-specific cytokine expression through regulating JunB and GATA3 transcription factors. J Immunol. 2012;188:3160–8. doi: 10.4049/jimmunol.1102943. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.