

Abstract
Neurons are dynamic cells that respond and adapt to stimuli throughout their long postmitotic lives. The structural and functional plasticity of neurons requires the regulated transcription of new gene products, and dysregulation of transcription in either developing or adult brains impairs cognition. We discuss how mechanisms of chromatin regulation help to orchestrate the transcriptional programs that underlie maturation of developing neurons and plasticity of adult neurons. We review how chromatin regulation acts locally to modulate the expression of specific genes, and more broadly to coordinate gene expression programs during transitions between cellular states. These data highlight the importance of epigenetic transcriptional mechanisms in postmitotic neurons. We suggest areas where emerging methods may advance understanding in the future.
Keywords: DNA methylation, enhancer, chromatin structure, neuronal maturation, plasticity, neuroepigenetics
Why study chromatin regulation in the brain?
Over the course of vertebrate development, a single fertilized egg with a single genome gives rise to multiple different types of cells that have specialized functions and distinct gene expression programs. It has been well established that the differentiation of cell types is achieved through epigenetic (see Glossary) mechanisms of chromatin regulation, which allow some genes to become transcribed whereas other are permanently silenced. The biochemical mechanisms of gene regulation that underlie the development and maintenance of cellular identity include direct modifications to the DNA itself (e.g. DNA methylation), functional changes in the state of gene regulatory elements, as well as global conformational changes of chromosome structure within the nucleus.
Within the past decade, technological advances in methods for genome-wide sequencing have revolutionized our ability to characterize the epigenome in a wide variety of biological contexts, and these data have brought some significant surprises. Foremost among these is the observation that many of the epigenetic mechanisms once presumed to be highly stable, because they mediate persistent effects on gene regulation, are actually subject to dynamic modulation in specific cellular contexts [1-3]. This paradigm shift has prompted spirited commentary on what does and does not qualify as “epigenetics”, and also generated huge enthusiasm for the idea that the epigenome could serve as a target to improve human health.
Neurons are extremely long-lived cells that, as part of their fundamental function in information processing, undergo robust and dynamic changes in their gene expression repertoires long after they have left the cell cycle and committed to a postmitotic identity. Thus neurons serve as an ideal substrate for studying the biological functions of the dynamic epigenome beyond its role in establishing cellular identity. In this Review, to explore the advances that are emerging from the field of neuroepigenetics, we focus on two different temporal contexts. First we explore the period of postmitotic neuronal maturation in the developing brain, and second, we discuss the induction of stimulus-dependent neuronal plasticity in the adult brain. In each case we consider three different spatial scales at which chromatin regulation can act - from direct methylation of single cytosines in genomic DNA, through functional regulation of histones at promoters and enhancers, to global changes in chromosome structure within the nucleus (Fig. 1). We highlight important insights that have emerged from chromatin profiling studies, and – because many of the advances in this field are driven by the development of new technologies – we close with a discussion of emerging methodologies that will likely push back the boundaries of our knowledge in the near future.
Figure 1.
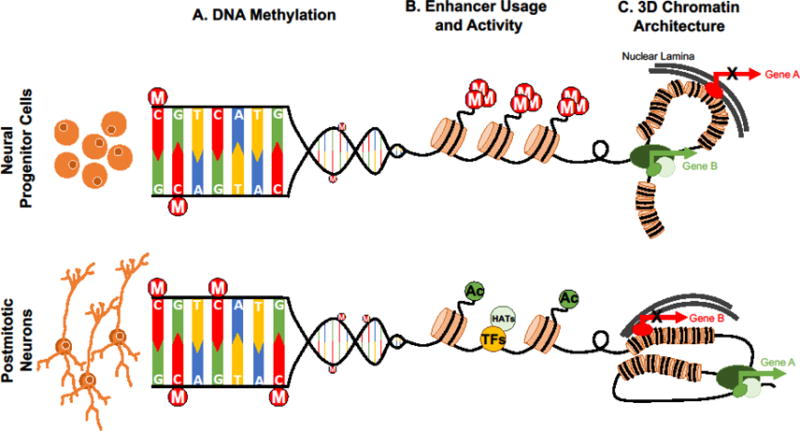
Multiple levels of chromatin regulation interact to coordinate gene expression in developing neurons. As neural progenitor cells make the transition to become mature postmitotic neurons, there is: (A) increased accumulation of DNA methylation (M) mainly in the form of mCpA with no change in mCpG; (B) dynamic modification of post-translational modifications on histones (M, methylation; Ac, acetylation) by enzymes such as histone acetyltransferases (HATs) that allow both increases and decreases in chromatin accessibility and subsequent changes in transcription factor (TF) binding at specific gene regulatory elements; and (C) large-scale changes in chromatin configuration and nuclear architecture that initiate interactions of different DNA sequences with the nuclear lamina (repression, red) or transcriptional factories (activation, green). These three different scales in chromatin regulation can result in unique and cell-specific gene expression programs, appropriate for every stage of neuronal development.
Chromatin regulation and the temporal coordination of neuronal maturation
Fate commitment and cell-cycle exit are key stages of neurogenesis. However they are far from the final steps in the production of a functional nervous system. Newborn postmitotic neurons must still migrate to their final position in the brain, send axons to their appropriate targets, and receive and refine their synaptic connections with other neurons. Gene expression programs change as the cell transitions through these stages of neuronal differentiation, allowing the neuron to exhibit distinct phenotypes without changing its core identity. Furthermore, although fundamental aspects of brain development are hardwired into the genome of neurons, refinement of the functional connectome is highly sensitive to sensory input during critical periods of postnatal life [4]. During the prolonged period of postnatal maturation that is characteristic of vertebrate brain development, neurons need to coordinate both intrinsic and extrinsic mechanisms of gene regulation to achieve optimal levels of brain function, whereas failure to properly regulate these mechanisms can impair brain development and lead to cognitive impairments [5, 6]. Chromatin regulation offers a compelling mechanism to orchestrate these transcriptional programs, and in this section, we review evidence that chromatin regulation coordinates the timing of functional maturation in postmitotic neurons of the developing brain.
A neural-selective form of DNA methylation that tunes gene expression in postnatal neurons
The methylation of genomic DNA at cytosines (C), primarily in the context of CpG dinucleotides (mCpG), is a fundamental epigenetic mechanism that regulates genes in mammalian cells. DNA methylation is essential for the irreversible silencing of specific subsets of genes, including imprinted genes, transposons, and genes on the inactivated X chromosome of female cells [7]. Additionally, DNA methylation also regulates the transcription of expressed genes [8-10]. There are three DNA methyltransferases (Dnmt1, Dnmt3a, and Dnmt3b), all of which are expressed in the developing brain.
During the process of neuronal fate determination, mCpG acts to repress genes that would have been expressed in alternate lineages. Specifically, when embryonic stem (ES) cells are differentiated in culture first to NPCs and then to neurons, a significant number of gene promoters gain mCpG [11, 12]. Many of these promoters are also targets of the polycomb repressive complex (PRC2) and are marked by trimethylation of lysine 27 on histone H3 (H3K27me3) in ES cells. When NPCs are differentiated to neurons, non-neuronal PRC2 target genes gain CpG methylation and become permanently silenced, whereas neuronal PRC2 targets lose this repressive mark and become actively expressed [12], through a recently elucidated mechanism that we discuss in the section on enhancers below. Thus, there exists coordination between different chromatin mechanisms to temporally control aspects of neuronal development such as neuronal fate determination during neurogenesis. More specifically, PRC2 mediates a form of repression that can be reversed during the course of development, whereas CpG methylation is associated with forms of gene repression that persist on a longer time scale.
In contrast to the marked accumulation of mCpG over the course of neuronal lineage commitment, the total level of mCpG in postmitotic neurons remains remarkably constant across postnatal development and adult life [13]. Surprisingly however, this postnatal period of neuronal maturation in both rodents and humans is marked by a rapid accumulation of methylation at CpH dinucleotides (H represents any base other than G), mainly in the form of mCpA [13, 14]. These developmental increases in neuronal mCpA coincide with the peak expression of Dnmt3a, and ablation of Dnmt3a results in loss of mCpA, suggesting that Dnmt3a is the primary methyltransferase responsible for mCpA sites across the genome during development [14, 15]. Surprisingly, this postnatal increase in mCpA is so substantial that adult neurons contain nearly as many mCpA sites as mCpG sites [13]. This distribution is very different from other cell types that have been profiled to date, and it is specifically distinct from the mCpG-predominant profile found in non-neuronal cells in the brain. The evidence that neurons have a unique epigenomic landscape relative to other cell types in the CNS raises the possibility that mCpA could contribute to neural-specific aspects of gene regulation both in development and disease.
Consistent with this idea, mCpA has been suggested to play an experience-dependent role in modulation of a subset of neural-selective genes in postmitotic neurons, and disruption of this regulation has been implicated in neurological disease [15-17]. At 2 weeks after birth in the mouse brain, even though Dnmt3a is maximally expressed and distributed broadly across the genome, it is largely excluded from both highly transcribed and actively silenced, heterochromatic genes [15]. Nonetheless, Dnmt3a is bound across the transcribed regions of neuronal genes that are expressed at relatively low levels, and this binding correlates with the deposition of mCpA across these genes. Most interestingly, increasing the transcription of activity-inducible genes in young neurons by neurochemical or sensory stimulation immediately decreases Dnmt3a binding following stimulation and persistently decreases the levels of mCpA found on these genes in the adult brain [15]. These data raise the possibility that early life experience could impact later neuronal function by fine-tuning the levels of mCpA deposited across a subset of genes.
In addition to being found associated with genes that are expressed at low levels, mCpA is also concentrated in the gene bodies of genes that are particularly long. These long genes are enriched for gene products that are neural-selective and that mediate aspects of synaptic function and/or cell adhesion [14, 16, 18]. At these sites, mCpA selectively recruits binding of the methyl-DNA binding protein MeCP2 [16, 17]. This is important because loss of function mutations in MECP2 causes the human neurodevelopmental disorder Rett Syndrome (RTT). RTT is characterized by defects in synapse formation and neuronal function that manifest as neurological symptoms in the postnatal period of brain development [19, 20]. The neurological specificity of RTT has long been a conundrum because MeCP2 and mCpG are found in most cell types. However, this specificity could be explained by the selective accumulation of mCpA in maturing postmitotic neurons, which would then recruit MeCP2 to tune the expression of long genes to modulate synaptic function. Indeed, mutations in Mecp2 cause aberrant upregulation of mCpA-rich long genes in neurons, and overexpression of this gene as well as some MeCP2-dependent cellular phenotypes, can be partially rescued with pharmacological inhibitors of topoisomerases, an enzyme selectively required for long gene expression [16, 21, 22]. Whether manipulating long gene expression can improve brain function in RTT animal models and eventually patients still remains to be determined.
Developmental stage-specific regulation of enhancer activity
Chromatin is the macromolecular structural complex composed of genomic DNA and its tightly associated histone proteins. Just as DNA can be covalently modified, histones are also subject to extensive post-translational modifications that impact gene transcription by modulating the accessibility and activation state of gene regulatory elements such as distal enhancers [23]. Recent studies show how dynamic modification of chromatin at enhancers during periods of neurogenesis and postmitotic maturation of fate-committed neurons contributes to developmental stage-specific expression of genes in the developing brain [24].
Enhancers are a class of DNA regulatory elements whose chromatin is accessible to transcription factor binding. Enhancers share many genomic, biochemical, and functional similarity with promoters but can be found at long distances from genes and are frequent substrates for cell-type specific and developmental regulation [25, 26]. Although enhancers are functionally defined by their ability to promote gene transcription, these elements can be predicted biochemically by a combination of chromatin features and classified into three main types: active, primed, or poised [27]. Specifically, accessible regions that are enriched for histone H3K27 acetylation (H3K27ac), histone H3K4 monomethylation (H3K4me1), and bound by the histone acetyltransferases p300/CBP are highly likely to function as active enhancers when tested in reporter gene assays [23, 28, 29]. Regulatory elements marked by H3K4me1 but not H3K27ac are considered “primed” and are associated with lower levels of transcription [30]. Whether these histone modifications themselves are functionally important for the activity of enhancers or simply an indication of enzyme recruitment to these elements remains to be fully established [31]. However modified histones can provide docking sites for transcriptional regulatory complexes and in this way have the potential to modulate their target promoters. For example, H3K4me1 facilitates recruitment of the cohesion complex, which plays a major role in promoting chromatin looping between promoters and enhancers [32], whereas acetylated lysines at enhancers recruit the bromodomain protein Brd4, which promotes transcriptional elongation [33]. CRISPR-based epigenome editing offers the potential to directly test the function of enhancer histone marks, as we discuss in the Concluding Remarks and Future Perspectives section below.
Regardless of what role enhancer chromatin plays in transcription, the strong correlation between histone marks and enhancer activity provides a convenient means for discovering gene regulatory elements via comparative epigenomic profiling between cell types and developmental stages. To discover enhancers that function in neuronal development, several groups have profiled enhancer chromatin marks (e.g., accessibility, H3K4me1, H3K27ac, p300) across different stages of brain development, from the beginning of neurogenesis, through regional specification, to neuronal maturation [26, 34-36]. Regions that show differential chromatin regulation between stages comprise only a small fraction of the total genome, permitting bioinformatic analyses that offer insight into the molecular mechanisms of brain development.
For example, because enhancers are transcription factor binding sites, the sequence data derived from a genome-wide survey can be mined to discover transcription factor codes that mediate neuronal differentiation. Nord et al. [34] profiled H3K27ac in forebrain samples from embryonic day 11.5 (E11.5) though adulthood and then conducted in vivo validation with LacZ reporter transgenics to demonstrate that these elements promote gene expression at specific times and in specific regions of the developing brain. Of more than 40 enhancers that showed distinct regional patterns of reporter gene expression in the developing cortex at E11.5, fate mapping with a subset showed that these enhancers define subregions of the developing pallium that are fated to develop into distinct subdivisions of the frontal cortex [37]. Computational comparisons between the enhancers that were active in different pallial subdivisions revealed common transcription factor binding motifs among enhancers that were functionally regulated, suggesting new clues to the transcription factor code that specifies cortical patterning [37].
These data show that different enhancers are used in different kinds of neurons across the developing forebrain, but the question arises – do changes in chromatin regulation also control programs of gene expression in single neuronal subtypes over time? To address this question, Frank et al. [26] took advantage of the relative cellular homogeneity of the rodent cerebellum as a model system to study the role of enhancer activity in the temporal development of cerebellar granule neurons (CGNs). Genome-wide comparisons of enhancer accessibility and H3K27ac correlated with gene expression changes again identified a large set of developmentally regulated enhancers. Computational analysis of transcription factor binding sites in these enhancers led to the surprising finding that the Zic family of transcription factors both inhibit maturation of CGN progenitors and promote maturation of postmitotic CGNs. The distinct functions of the Zic family are mediated via their differential recruitment to distinct groups of enhancers within progenitors and CGNs that have developmentally-regulated changes in enhancer accessibility. These data show how interactions between chromatin landscapes and transcription factor expression patterns work together to determine gene expression programs in developing neurons.
Enhancer identification through comparative epigenomics also has the potential to advance understanding of neurodevelopmental disorders. Genetic variants associated with a number of complex diseases are found primarily within non-coding regions of the genome. To determine whether these variants might alter enhancer function, de la Torre-Ubieta et al [35] asked whether genetic variants that are associated with a variety of disorders were found within a set of enhancers that show differential accessibility when comparing germinal zone progenitors with cortical plate neurons in fetal human brains. These analyses revealed significant enrichment of variants associated with both cognitive function and psychiatric disorders within the set of enhancers that are active during neurogenesis, raising the possibility that impairments in gene regulation during early stages of neural development could contribute to disorders of the adult brain.
The examples we have highlighted above rely on analysis of enhancers that are differentially active between developmental stages, and this raises the question of what mechanisms control the timing of enhancer activation. New clues to these mechanisms have been found in the study of a distinct biochemically defined class of enhancers called poised (or bivalent) enhancers, which are marked by H3K4me1 as well as the repressive histone modification H3K27me3. Poised enhancers were first defined in ES cells, where they are preferentially associated with genes that become active upon differentiation [38]. The loss of H3K27me3 at these enhancers is correlated with transcriptional activation of their target genes, thus it was presumed that the presence of the repressive mark at the enhancers helped to keep these genes inactive during earlier stages of cellular differentiation. However, emerging evidence suggests that poised enhancers also play a direct role in preparing genes for active transcription. In cultured mouse ES cells, poised enhancers of anterior neural progenitor genes physically interact through looping with their target gene promoters prior to differentiation [39]. Surprisingly, CRISPR-mediated disruption of poised enhancers did not lead to premature expression of neural genes in ES cells, but did impair their induction upon neuronal differentiation. These data suggest that poised enhancers act in ES cells to create a permissive regulatory structure for the induction of neuronal genes, showing how enhancer elements interact with higher-level chromatin structure to integrate regulatory information that coordinates gene expression in differentiating cells.
Dynamic reorganization of chromatin in the nucleus
In addition to local changes in chromatin state, the way that chromatin is folded and looped in the nucleus into complex secondary and tertiary structures also influences gene expression [40]. Differential looping between promoters and enhancers is a well-established mechanism of developmental gene expression that, for example, controls the switch in globin gene expression during erythropoiesis [41]. Similarly, large scale changes in chromatin architecture, such as the regulated associations of chromatin with the nuclear lamina and chromatin looping mediated by chromatin binding of architectural proteins like CCCTC-binding factor (CTCF) and cohesion, has the potential to offer a mechanism that could coordinate the regulation of large groups of genes simultaneously in developing neurons [42].
Yet given the huge difference in spatial scale between any single gene and the volume of the nucleus, it remains challenging at the experimental level to delve into the mechanisms by which conformational changes in chromosome structure contribute to functional differences in gene regulation during neural differentiation. Fortunately this is an area of research in which methods are rapidly evolving (see Future Perspectives below) and current data offer broad strokes of the types of regulatory processes that are likely to be regulated by global chromatin structure. For example, the physical localization of chromatin domains next to the nuclear lamina that lines the inside of the nuclear envelope is usually associated with gene repression, whereas relocalization away from the lamina can permit gene activation [43]. Biochemical identification of lamina-associated domains (LADs) has revealed that the specific DNA sequences localized to the lamina change globally as ES cells differentiate sequentially into NPCs and then astrocytes [44]. Though only about two-thirds of the genes that lose their laminar association during the ESC to NPC transition show upregulated expression in NPCs, many of the others become expressed at later stages of differentiation. These data suggest that release from the nuclear lamina is permissive and primes specific genes for regulation by additional mechanisms.
Rather than focusing on interactions between genomic DNA and the nuclear envelope, the HiC sequencing method is designed to detect all physical interactions that occur between different DNA sequences in the genome. HiC data have revealed that genomic DNA is divided structurally at intermediate scales (up to 1Mb) into topologically associating domains (TADs) that show stage- and cell-type specific organization. By comparing high resolution HiC interaction maps of ES cells, NPCs, and cortical neurons, Bonev et al. [45] reaffirmed core principles of chromatin organization elucidated previously, such as TAD structure and the role of CTCF in domain insulation, but they also discovered dynamic aspects of chromatin organization that change over neurogenesis, such as the dynamic emergence of chromatin interactions between loci bound by the neuronal cell-type specific transcription factors Pax6, Neurod2, and Tbr1.
One striking example of how global chromatin reorganization can coordinate gene regulation in neurons has emerged from study of the rodent olfactory system. Olfactory receptor choice is one of the most extreme examples of gene regulation, in which each individual sensory neuron stochastically comes to express only one of the thousands of olfactory receptors (ORs) that are encoded within the genome. Soft X-ray tomography showed that across the olfactory epithelium, as stem cells develop into mature neurons, there is a gradual relocation of heterochromatin away from the nuclear periphery and toward to the nuclear core [46]. This global reconfiguration of chromatin architecture is associated with a clustering of silenced ORs into heterochromatic foci, and is regulated by decreased expression of the lamin B receptor and the heterochromatin protein HP1beta, both of which are involved in tethering chromatin domains to the nuclear lamina [46, 47]. It is hypothesized that this chromatin reorganization allows multiple OR enhancers to engage in extensive interchromosomal interactions with one specific OR locus in order to derepress local chromatin and drive robust, singular OR expression. The downregulation of lamin B receptor during differentiation is common to retinal cells [42] and cortical neurons [45], suggesting a potential conserved mechanism of global chromatin reorganization. Future studies that apply these and other methods to additional neuronal cell types, developmental stages, and neurological disease models [48] will advance understanding of how spatial information in the nucleus contributes to the temporal regulation of gene expression during neuronal differentiation.
Chromatin Regulation of Neuronal Activity-Dependent Transcription
Epigenetic modifications to chromatin are instrumental in regulating the transcription of cell-type specific and developmentally-governed gene programs in cells across the body. However, differentiated, post-mitotic neurons represent a unique class of “stable” cells wherein existing gene programs are dynamically regulated to allow these developmentally-static cells to change themselves in response to varied environmental stimuli. This begs the question: how can ostensibly stable epigenetic modifications to chromatin mediate this activity-dependent dynamic regulation in a way that tunes changes to gene expression and cellular physiology in response to experiential stimuli? Animals interact with their environments and learn from them by virtue of their ability to convert patterns of sensory-driven neuronal activity into long-lasting changes in brain structure and function, The cellular foundations of these long-term changes in the brain are known to depend, at least in part, on the neuronal activity-regulated transcription of new gene products [4]. The first hint that chromatin regulation might contribute to neuronal activity-dependent transcriptional plasticity came from the discovery that phosphorylation of the canonical activity-regulated transcription factor CREB functions to recruit the histone acetyltransferase CBP to CREB target genes [49]. Subsequently, numerous chromatin regulatory processes have been shown to undergo activity-dependent regulation, and the importance of these processes in learning and memory [50-52] as well as their dysregulation in the cognitive impairment that accompanies aging [53] is an explosive area of research.
Connecting the dots from DNA methylation to genes involved in learning
Because DNA methylation can mediate very long-lasting processes like X-chromosome inactivation and genomic imprinting, it was a natural favorite to be considered as a possible persistent biochemical mechanism for memory. However if DNA methylation is to contribute to neuronal plasticity, the canonical perspective of its lasting stability needs to be challenged by data showing that the process can be dynamically induced or repressed by neuronal activity. Genome-wide sampling of DNA methylation confirmed that the distribution of mCpG can be dynamically modified by neuronal activity in vivo [54]. In fact, these data revealed a surprisingly large number of both gains and losses of DNA methylation broadly across the genome following neuronal firing, revealing the dynamism of this mark in contrast to the common conception that it is highly persistent. Nonetheless, at least a subset of the activity-dependent changes in DNA methylation state persisted 24 hours post-stimulation, at which time they would have the potential to underlie long-lasting changes in gene expression [54].
At the cellular level, activity-dependent changes in DNA methylation have been linked to the transcriptional regulation of gene products that have functions in neuronal plasticity. For example, associative memory is correlated with significant changes in DNA methylation state at sets of genes encoding transcription factors and ion-gated transmembrane channels, both of which can modulate synaptic plasticity [50]. Pharmacological and genetic inhibition of DNA methyltransferase activity has also been shown to significantly increase the amplitude of miniature excitatory post-synaptic currents, which is a major cellular mechanism of learning and memory [55]. These changes in synaptic strength are dependent on gene transcription, consistent with a cause arising from methylation-dependent transcriptional regulation. Nonetheless, drawing clear lines from DNA methylation to behavior through cellular plasticity remains tenuous at best, and testing this hypothesis will benefit from the epigenome editing methods we discuss in the Future Perspective section below.
Histones: modifications, position, and variants influence activity-inducible transcription
As discussed above, the N-terminal tails of histone proteins can incur post-translational modifications at various amino acid residues. As predicted, given the neuronal activity-dependent recruitment of the histone acetyltransferase CBP to CREB and other activity-regulated transcription factors, histone acetylation is acutely induced at promoters and enhancers of activity-inducible genes in coordination with the induction of their transcription by neuronal activity (Fig. 2A) [56]. Reporter gene assays demonstrate that this set of enhancers is highly enriched for sequences that possess the capacity to stimulate activity-regulated transcription [56]. Furthermore, at least for the Fos and Arc genes, inhibiting the function of enhancers that show membrane depolarization-inducible accumulation of H3K27ac is an effective means to block activity-inducible gene expression [57, 58].
Figure 2.
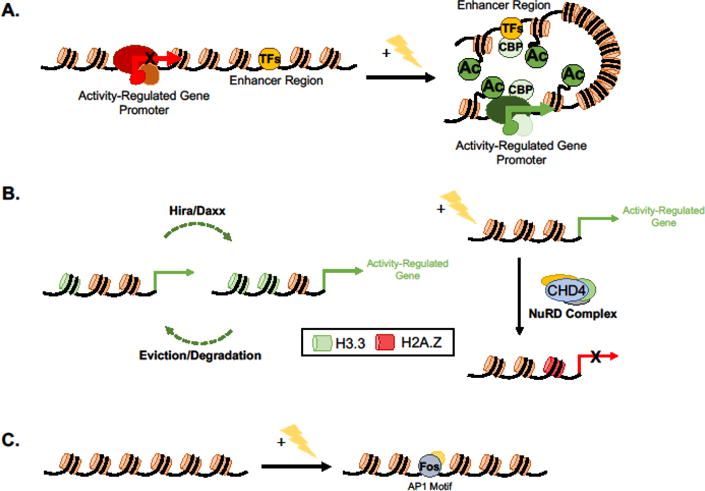
Histone modifications that modulate neuronal activity-dependent gene transcription. (A) Membrane depolarization-induced H3K27ac at distal enhancer regions and gene promoters is associated with the induction of activity-dependent gene transcription and the formation of stable loops between active enhancer regions and the promoters of their target genes. (B) Turnover of histone proteins results in regulated deposition of the variant histones H3.3 via Hira/DaXX and H2A.Z via the NuRD complex. H3.3 turnover facilitates the transcription of membrane depolarization-induced late-response genes whereas H2A.Z deposition at gene promoters represses the transcription of activity-dependent plasticity genes. (C) Neuronal activity induces changes in chromatin accessibility. Activity-induced opening of chromatin at distal regulatory elements is mediated via the recruitment of Fos and other AP-1 element binding proteins. TF, transcription factor; Ac, histone acetylation; CBP, Creb Binding Protein; CHD4, Chromodomain protein 4.
An alternate way to add dynamic range to histone function is through the differentiation expression and chromatin incorporation of histone variants. Attention has focused in particular on the H2A.Z and H3.3 variants, both of which show activity-regulated deposition into genomic DNA but which have opposite effects on activity-inducible transcription (Fig. 2B) [52, 59, 60]. Compared to the H3.1 and H3.2 isoforms, histone H3.3 shows replication-independent incorporation into DNA, such that it accumulates in long-lived postmitotic neurons to cover >94% of the genome [59]. Despite its prevalence, histone H3.3 retains its dynamic nature in mature neurons, cycling in and out of nucleosomes in a manner that is induced by neuronal activity and regulated by the chaperone proteins HIRA and/or Daxx [59, 61]. Disruption of H3.3 turnover in cultured neurons impairs the activity-dependent induction of late primary-response genes suggesting this variant promotes activity-inducible transcription [59]. By contrast activity-dependent incorporation of histone H2A.Z, which is mediated by NuRD/ChD4, is associated with the termination of transcription [62]. In the CA1 region of the hippocampus following the administration of an aversive foot shock in vivo, levels of H2A.Z decrease [52]. In this context, H2A.Z depletion significantly enhanced the expression of plasticity-relevant genes such as Fos and Npas4 [52].
Finally, histones can be repositioned or evicted by nucleosome remodeling complexes to regulate the accessibility of gene regulatory elements for transcription factor binding (Fig. 2C). Using ATAC-seq [63] to identify accessible chromatin regions, Su et al.[64] showed that ECT-induced firing of dentate granule neurons of the hippocampus in vivo acutely causes widespread changes in chromatin accessibility. The newly opened or closed regions correlated with the regulation of gene expression and many newly opened regions overlapped with known gene enhancers. Interestingly, computational analysis of the opening sties revealed enrichment for binding sites for AP-1 complex transcription factors such Fos, and loss of function experiments showed the requirement for Fos in the activity-dependent opening of these chromatin regions [64]. How does Fos induction drive chromatin accessibility? Recent studies in fibroblasts show that Fos collaborates with cell-type specific transcription factors to recruit the SWI/SNF family BAF chromatin-remodeling complex and establish accessible chromatin at enhancers during cellular differentiation [65]. Whether similar mechanisms function in neurons is not known, but this model suggests an intriguing avenue of intersection between activity-dependent Fos induction and the stimulus-dependent modulation of chromatin accessibility at enhancers of secondary response genes.
Activity-induced global reconfiguration of nuclear architecture
One might think that global nuclear architecture would be relatively stable in postmitotic, fate-committed cells. Nonetheless, at the level of single genes, neuronal activity has been shown to reconfigure loops between distal enhancers and the promoters of stimulus-regulated genes including Fos and Arc [57, 58]. Furthermore for the Fos gene, which is flanked by multiple activity-regulated enhancers, the specific nature of the promoter-enhancer loops induced depends on the nature of the upstream stimulus [57]. This suggests a novel level of transcriptional specificity mediated by stimulus-specific 3D chromatin interactions. More broadly, neuronal activity has been shown to have effects on global chromatin architecture. The general transcription factor TFIIIC has been suggested to act as a gate on the membrane depolarization-driven activation of genes by restricting the relocation of activity-dependent gene promoters into so-called transcriptional factories (TF), which are subregions of the nucleus enriched for the transcriptional machinery [66]. Activity-dependent re-localization of genes away from the repressive nuclear lamina has also been demonstrated in adult neurons. Specifically, re-localization of the locus encoding the neurotrophic factor Bdnf has been observed in adult neurons of the hippocampus following experimentally induced status epilepticus in vivo, which promotes robust Bdnf transcription [3]. Finally membrane depolarization of hippocampal neurons in culture has been shown to promote the formation of massive nuclear infoldings, which would be expected to radically remodel chromosome-laminar interactions [67]. New methods for the evaluation and disruption of nuclear architecture discussed below [68] will be key to understanding the functional importance and general relevance of these observations.
Concluding remarks and future perspectives
The widespread availability of cheap, high quality methods for genome-wide sequencing has driven an explosion of epigenome profiling studies. As a result, we now have a massive amount of epigenomic data from all sorts of cells under a wide variety of conditions. In postmitotic neurons of the postnatal and adult brain, these data have revealed that chromatin regulatory mechanisms contribute to functionally important changes in neuronal state long after neuronal fate is fully established. Some of these data have refined hypotheses of the molecular mechanisms by which specific neuronal proteins function, such as the evidence that MeCP2 interacts with the neuron-selective methyl-CpA mark, or the elucidation of new transcription factor codes that mediate frontal cortical development and CGN maturation. However our ability to either integrate these datasets into a more conceptual understanding of transcriptional regulation or to evaluate the mechanistic contributions of chromatin regulation to neuronal function at the circuit and behavioral level remains embryonic (see Outstanding Questions).
Outstanding Questions Box.
Is there coordination between the different types of chromatin mechanisms that mediate specific gene expression programs during neuronal development and plasticity? If so, what are the advantages of using chromatin to coordinately modulate cell state? Do disturbances in chromatin coordination of gene expression lead to aberrant changes in physiology and disease onset?
Is non-CpG methylation (e.g. mCpA) selectively enriched in neurons, or does this chromatin mark accumulate in other long-lived, postmitotic cells as well? Is the interaction between mCpA and MeCP2 critical for the development of Rett Syndrome phenotypes? Will rescue of long gene expression in MeCP2 mutant cells improve neurological dysfunction in Rett Syndrome?
Do histone modifications (e.g. acetylation, methylation) play a causative role in the regulation of gene transcription or do they merely reflect the presence of chromatin regulatory enzymes, which may more directly impact gene transcription through non-enzymatic mechanisms?
Do global changes in nuclear structure orchestrate programs of gene expression that underlie neuronal maturation and plasticity? If so, which proteins mediate these structural changes? Is chromatin architecture cell type specific, and if so, how are these differences in nuclear architecture established?
Are there novel or better ways to detect chromatin state and structure at the single-cell level and in live cells such that the dynamics of chromatin regulation and its relationship to temporal changes in neuronal function can be better understood?
We see five promising areas of methodological progress that will help to overcome these limitations. First is the development of advanced statistical methods for the analysis of epigenomic datasets, These improved analyses will permit insights to be drawn from high dimensional data. For example, ChromHMM is a Bayesian statistical method that integrates the analysis of multiple histone marks simultaneously across chromosomal space, and this method allows the characterization of regulatory elements across the genome as active promoters, poised enhancers, silencers, etc [69]. More complex Bayesian models that add developmental stage as an orthogonal variable have begun to model how chromatin changes over time, to analyze how gene regulatory networks mediate neuronal differentiation [70]. Developmental stage can also be computationally reconstructed from single-cell RNA-seq data, and has been used, for example, to identify intermediate stages of neural stem/progenitor cell differentiation in the adult hippocampus [71].
A second area of technological change, which we see as one of the most exciting near-term frontiers for growth in the field, is the development and application of new methods for the study of global chromatin and nuclear architecture [68]. Genome-wide chromosome interaction maps using Hi-C, Chia-PET, DamID, and related methods are defining gene organization in the nucleus in a growing number of contexts [45, 72-74]. Molecular improvements in the methods have made single-cell HiC [75] and single-cell DamID [76] possible, revealing cell-to-cell heterogeneity in global chromatin architecture. Perhaps most exciting is the new development of a ligase-independent, sequencing-based method for determining 3D genome structure called genome architecture mapping (GAM), which uses the likelihood that two sequences co-appear in libraries derived from thin cryosections through the nucleus to calculate the statistical probability of their physical interaction in 3D [77]. In addition to providing an orthogonal way to confirm results of traditional chromatin conformation studies, this method has the potential to be used for structural analysis of highly limited samples, such as sections of human brain tissues, from which genome structure could be studied in situ without the need for cellular isolation.
Third, the scaling down of chromatin methods is essential for permitting characterization of the epigenome in specific neuronal populations isolated from the heterogeneous and complex circuits of the brain. Beyond the current highly useful Cre-dependent methods for cell-type specific nuclear purification [78, 79], the increasing popularity of single cell RNA sequencing is now being paralleled by the development of improved single cell chromatin assays. For example, single cell methylomes have been successfully used to identify unique neuronal sub-classes in both mouse and human brain [80]. Single-cell chromatin accessibility profiles have been generated using ATAC-seq either from cells isolated by microfluidics [81] or via combinatorial cellular indexing and single cell data reconstruction [82] though the method has not yet been broadly applied. Despite publication of a proof-of-principle study [83], histone modifications remain challenging to detect in single cells using standard protocols and will likely benefit from conceptually new methodologies.
A fourth area of methodological progress relates to imaging-based methods, which provide a means to study chromatin and transcriptional regulation at the single-cell level in situ, rather than removing cells from their context in the brain. For RNA, highly multiplexed fluorescent hybridization methods such as merFISH [84] and FISSEQ [85] allow for simultaneous quantitative in situ sequencing of large numbers of transcripts in single cells. For chromatin, combining labeling of DNA by multiplexed FISH or labeling of histone marks by immunostaining, together with super-resolution imaging methods such as 3D-STORM and array tomography, allows global aspects of chromatin structure to be visualized at the light level [48, 86]. Light-level imaging also offers the opportunity to visualize chromatin regulation in real time, as has been done using FAb-based live cell endogenous modification labeling, which can relate chromatin dynamics to transcriptional kinetics [87].
Finally, the use of CRISPR/Cas9-based methods for genome and epigenome editing are essential for testing the causative relationship between chromatin and its cellular functions [88]. The fusion of enzymatically dead Cas9 (dCas9) tethered to transcriptional activators or repressors is a rapid and efficient way to conduct functional annotation of gene regulatory elements in their endogenous context [26, 57] and offers the potential to conduct genome-scale screens for regulatory functions of non-coding regions of the genome [89]. Moreover dCas9 fusions with chromatin regulatory enzymes (e.g. G9a, p300, Dnmt3a, Ezh2) are emerging as an explosive new wave of tools that offer the means to directly test the functional (e.g. transcriptional, cellular, behavioral) consequences of chromatin modifications in a site-specific manner [90-93]. Biotinylated dCas9 has been used to develop an alternative to ligase-based methods for capturing cis-DNA regulatory elements and protein interactions with specific genomic loci in an unbiased manner [94]. dCas9 has also been employed to force long-distance loops in a site-specific and reversible manner in order to functionally test the transcriptional consequences of chromatin architecture [95]. Given the flexibility of the CRISPR platform and the low barrier to entry for deploying the methodology, we expect to see continued explosive and creative use of this technology expanding our understanding of the functional relevance of genome regulation.
Highlights.
The widespread availability of low-cost and high-quality methods for genome-wide sequencing has driven a massive accumulation of data regarding the state of the epigenome in neurons over a wide variety of conditions.
Changes in methylation of cytosines in DNA, regulation of chromatin state at gene regulatory elements, and shifts in global nuclear architecture can all mediate dynamic gene expression changes.
Chromatin regulation contributes to functional changes in the biology of postmitotic neurons both during postnatal stages of brain development and in adult neural plasticity.
Proper gene expression during development is tightly controlled by mechanisms of chromatin regulation, and abnormalities in these mechanisms contribute to neurodevelopmental and neuropsychiatric disorders.
Acknowledgments
This work was supported by National Institutes of Health grants R21DA041878 and R01NS098804 to A.E.W.
Glossary
- Chromatin
The macromolecular complex of genomic DNA and its tightly associated proteins (e.g. histones) in eukaryotic cells that confer structure and ultimately function upon DNA
- Chromatin accessibility
The clearing of histones away from a DNA sequence such that it is available for binding by sequence-specific DNA-binding transcription factors
- Chromatin state
The combination of DNA and histone modifications as well as nuclear structure and nuclear position that occur at a given gene regulatory element
- Enhancer
A gene regulatory element that increases transcription of a gene. Enhancers are functionally defined and may be at any distance from the gene and in any orientation. Enhancers are presumed to be transcription factor binding sites and they are thought to physically interact via looping with the promoters that they activate
- Epigenetic
The term “epigenetics” was first coined by the developmental biologist Conrad Waddington, who used the term to refer to causal interactions between genotype and phenotype. However over time the meaning of the term has evolved. In its strictest definition, epigenetics refers to inheritable changes in gene expression that do not involve changes in the underlying DNA sequence. The “heritability” part emerged from the study of mechanisms of gene regulation such as DNA methylation that could be inherited across cell divisions. However for long-lived, non-dividing cells, such as neurons, in which heritability is irrelevant, the term has come to encompass a set of biochemical chromatin mechanisms (e.g. DNA methylation, chromatin accessibility, nuclear structure) that can be highly persistent over time and underlie the expression of phenotypic variations from a common genotype
- Epigenome engineering
Using artificial protein tethers to site-specifically recruit histone-modifying enzymes to the genome in order to induce or remove chromatin marks
- Gene regulatory element
A genomic DNA sequence bound by DNA binding factors (transcription factors, insulators, silencers) that confer transcriptional regulatory function on the sequence
- Heterochromatin
Highly condensed, nucleosome-rich regions of chromatin that appear dark in micrographs and are generally transcriptionally inactive
- Histone
A family of proteins that wrap genomic DNA into nucleosomal structures. The four histones of the canonical nucleosome octamer are H2A, H2B, H3 and H4. H1 is a linker histone found between nucleosomes
- Histone modifications
Posttranslational chemical groups covalently added to the histone proteins by regulatory enzymes. These modifications change histone structure or alter histone function by providing docking sites for other regulatory proteins
- Nuclear lamina
The dense protein network on the inside of the nuclear envelope that provides mechanical structure to the nucleus and exerts regulatory control over gene expression
- Nucleosome
The core repeating unit of chromatin, which consists of ~150 base pairs of DNA wrapped around an octamer of histone proteins
- Polycomb repressive complex (PRC)
A complex of proteins first discovered as developmental regulators in Drosophila that mediate transcriptional repression and the methylation of H3K27
- Transcription factories
Subcompartments of the nucleus enriched for the transcriptional machinery. These are thought to be sites where active genes co-cluster
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Guan Z, et al. Integration of long-term-memory-related synaptic plasticity involves bidirectional regulation of gene expression and chromatin structure. Cell. 2002;111(4):483–93. doi: 10.1016/s0092-8674(02)01074-7. [DOI] [PubMed] [Google Scholar]
- 2.Miller CA, Sweatt JD. Covalent modification of DNA regulates memory formation. Neuron. 2007;53(6):857–69. doi: 10.1016/j.neuron.2007.02.022. [DOI] [PubMed] [Google Scholar]
- 3.Walczak A, et al. Novel higher-order epigenetic regulation of the Bdnf gene upon seizures. J Neurosci. 2013;33(6):2507–11. doi: 10.1523/JNEUROSCI.1085-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chen LF, et al. Transcribing the connectome: roles for transcription factors and chromatin regulators in activity-dependent synapse development. J Neurophysiol. 2017;118(2):755–770. doi: 10.1152/jn.00067.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Iwase S, et al. Epigenetic Etiology of Intellectual Disability. J Neurosci. 2017;37(45):10773–10782. doi: 10.1523/JNEUROSCI.1840-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gabriele M, et al. The chromatin basis of neurodevelopmental disorders: Rethinking dysfunction along the molecular and temporal axes. Prog Neuropsychopharmacol Biol Psychiatry. 2018 doi: 10.1016/j.pnpbp.2017.12.013. [DOI] [PubMed] [Google Scholar]
- 7.Bestor TH, et al. Notes on the role of dynamic DNA methylation in mammalian development. Proc Natl Acad Sci U S A. 2015;112(22):6796–9. doi: 10.1073/pnas.1415301111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wu H, et al. Dnmt3a-dependent nonpromoter DNA methylation facilitates transcription of neurogenic genes. Science. 2010;329(5990):444–8. doi: 10.1126/science.1190485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kinde B, et al. DNA methylation in the gene body influences MeCP2-mediated gene repression. Proc Natl Acad Sci U S A. 2016;113(52):15114–15119. doi: 10.1073/pnas.1618737114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007;315(5815):1141–3. doi: 10.1126/science.1136352. [DOI] [PubMed] [Google Scholar]
- 11.Meissner A, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454(7205):766–70. doi: 10.1038/nature07107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohn F, et al. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol Cell. 2008;30(6):755–66. doi: 10.1016/j.molcel.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 13.Lister R, et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341(6146):1237905. doi: 10.1126/science.1237905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo JU, et al. Distribution, recognition and regulation of non-CpG methylation in the adult mammalian brain. Nat Neurosci. 2014;17(2):215–22. doi: 10.1038/nn.3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Stroud H, et al. Early-Life Gene Expression in Neurons Modulates Lasting Epigenetic States. Cell. 2017;171(5):1151–1164.e16. doi: 10.1016/j.cell.2017.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gabel HW, et al. Disruption of DNA-methylation-dependent long gene repression in Rett syndrome. Nature. 2015;522(7554):89–93. doi: 10.1038/nature14319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen L, et al. MeCP2 binds to non-CG methylated DNA as neurons mature, influencing transcription and the timing of onset for Rett syndrome. Proc Natl Acad Sci U S A. 2015;112(17):5509–14. doi: 10.1073/pnas.1505909112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zylka MJ, et al. Gene length matters in neurons. Neuron. 2015;86(2):353–5. doi: 10.1016/j.neuron.2015.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amir RE, et al. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23(2):185–8. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 20.Leonard H, et al. Clinical and biological progress over 50 years in Rett syndrome. Nat Rev Neurol. 2017;13(1):37–51. doi: 10.1038/nrneurol.2016.186. [DOI] [PubMed] [Google Scholar]
- 21.Sugino K, et al. Cell-type-specific repression by methyl-CpG-binding protein 2 is biased toward long genes. J Neurosci. 2014;34(38):12877–83. doi: 10.1523/JNEUROSCI.2674-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.King IF, et al. Topoisomerases facilitate transcription of long genes linked to autism. Nature. 2013;501(7465):58–62. doi: 10.1038/nature12504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heintzman ND, et al. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009;459(7243):108–12. doi: 10.1038/nature07829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gray JM, et al. Genomic Views of Transcriptional Enhancers: Essential Determinants of Cellular Identity and Activity-Dependent Responses in the CNS. J Neurosci. 2015;35(41):13819–26. doi: 10.1523/JNEUROSCI.2622-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Song L, et al. Open chromatin defined by DNaseI and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 2011;21(10):1757–67. doi: 10.1101/gr.121541.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Frank CL, et al. Regulation of chromatin accessibility and Zic binding at enhancers in the developing cerebellum. Nat Neurosci. 2015;18(5):647–56. doi: 10.1038/nn.3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heinz S, et al. The selection and function of cell type-specific enhancers. Nat Rev Mol Cell Biol. 2015;16(3):144–54. doi: 10.1038/nrm3949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ernst J, Kellis M. Discovery and characterization of chromatin states for systematic annotation of the human genome. Nat Biotechnol. 2010;28(8):817–25. doi: 10.1038/nbt.1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Visel A, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers. Nature. 2009;457(7231):854–8. doi: 10.1038/nature07730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Creyghton MP, et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc Natl Acad Sci U S A. 2010;107(50):21931–6. doi: 10.1073/pnas.1016071107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dorighi KM, et al. Mll3 and Mll4 Facilitate Enhancer RNA Synthesis and Transcription from Promoters Independently of H3K4 Monomethylation. Mol Cell. 2017;66(4):568–576.e4. doi: 10.1016/j.molcel.2017.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Local A, et al. Identification of H3K4me1-associated proteins at mammalian enhancers. Nat Genet. 2018;50(1):73–82. doi: 10.1038/s41588-017-0015-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zippo A, et al. Histone crosstalk between H3S10ph and H4K16ac generates a histone code that mediates transcription elongation. Cell. 2009;138(6):1122–36. doi: 10.1016/j.cell.2009.07.031. [DOI] [PubMed] [Google Scholar]
- 34.Nord AS, et al. Rapid and pervasive changes in genome-wide enhancer usage during mammalian development. Cell. 2013;155(7):1521–31. doi: 10.1016/j.cell.2013.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de la Torre-Ubieta L, et al. The Dynamic Landscape of Open Chromatin during Human Cortical Neurogenesis. Cell. 2018;172(1–2):289–304.e18. doi: 10.1016/j.cell.2017.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thakurela S, et al. Dynamics and function of distal regulatory elements during neurogenesis and neuroplasticity. Genome Res. 2015;25(9):1309–24. doi: 10.1101/gr.190926.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pattabiraman K, et al. Transcriptional regulation of enhancers active in protodomains of the developing cerebral cortex. Neuron. 2014;82(5):989–1003. doi: 10.1016/j.neuron.2014.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rada-Iglesias A, et al. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011;470(7333):279–83. doi: 10.1038/nature09692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Cruz-Molina S, et al. PRC2 Facilitates the Regulatory Topology Required for Poised Enhancer Function during Pluripotent Stem Cell Differentiation. Cell Stem Cell. 2017;20(5):689–705e9. doi: 10.1016/j.stem.2017.02.004. [DOI] [PubMed] [Google Scholar]
- 40.Rajarajan P, et al. Spatial genome organization and cognition. Nat Rev Neurosci. 2016;17(11):681–691. doi: 10.1038/nrn.2016.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Higgs DR, Wood WG. Long-range regulation of alpha globin gene expression during erythropoiesis. Curr Opin Hematol. 2008;15(3):176–83. doi: 10.1097/MOH.0b013e3282f734c4. [DOI] [PubMed] [Google Scholar]
- 42.Solovei I, et al. LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell. 2013;152(3):584–98. doi: 10.1016/j.cell.2013.01.009. [DOI] [PubMed] [Google Scholar]
- 43.Therizols P, et al. Chromatin decondensation is sufficient to alter nuclear organization in embryonic stem cells. Science. 2014;346(6214):1238–42. doi: 10.1126/science.1259587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peric-Hupkes D, et al. Molecular maps of the reorganization of genome-nuclear lamina interactions during differentiation. Mol Cell. 2010;38(4):603–13. doi: 10.1016/j.molcel.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bonev B, et al. Multiscale 3D Genome Rewiring during Mouse Neural Development. Cell. 2017;171(3):557–572 e24. doi: 10.1016/j.cell.2017.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Le Gros MA, et al. Soft X-Ray Tomography Reveals Gradual Chromatin Compaction and Reorganization during Neurogenesis In Vivo. Cell Rep. 2016;17(8):2125–2136. doi: 10.1016/j.celrep.2016.10.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Clowney EJ, et al. Nuclear aggregation of olfactory receptor genes governs their monogenic expression. Cell. 2012;151(4):724–37. doi: 10.1016/j.cell.2012.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Linhoff MW, et al. A high-resolution imaging approach to investigate chromatin architecture in complex tissues. Cell. 2015;163(1):246–55. doi: 10.1016/j.cell.2015.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chrivia JC, et al. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 50.Halder R, et al. DNA methylation changes in plasticity genes accompany the formation and maintenance of memory. Nat Neurosci. 2016;19(1):102–10. doi: 10.1038/nn.4194. [DOI] [PubMed] [Google Scholar]
- 51.Rudenko A, Tsai LH. Epigenetic regulation in memory and cognitive disorders. Neuroscience. 2014;264:51–63. doi: 10.1016/j.neuroscience.2012.12.034. [DOI] [PubMed] [Google Scholar]
- 52.Zovkic IB, et al. Histone H2A.Z subunit exchange controls consolidation of recent and remote memory. Nature. 2014;515(7528):582–6. doi: 10.1038/nature13707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Benayoun BA, et al. Epigenetic regulation of ageing: linking environmental inputs to genomic stability. Nat Rev Mol Cell Biol. 2015;16(10):593–610. doi: 10.1038/nrm4048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guo JU, et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat Neurosci. 2011 doi: 10.1038/nn.2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sweatt JD. Dynamic DNA methylation controls glutamate receptor trafficking and synaptic scaling. J Neurochem. 2016;137(3):312–30. doi: 10.1111/jnc.13564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Malik AN, et al. Genome-wide identification and characterization of functional neuronal activity-dependent enhancers. Nat Neurosci. 2014;17(10):1330–9. doi: 10.1038/nn.3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Joo JY, et al. Stimulus-specific combinatorial functionality of neuronal c-fos enhancers. Nat Neurosci. 2016;19(1):75–83. doi: 10.1038/nn.4170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Schaukowitch K, et al. Enhancer RNA facilitates NELF release from immediate early genes. Mol Cell. 2014;56(1):29–42. doi: 10.1016/j.molcel.2014.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Maze I, et al. Critical Role of Histone Turnover in Neuronal Transcription and Plasticity. Neuron. 2015;87(1):77–94. doi: 10.1016/j.neuron.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dunn CJ, et al. Histone Hypervariants H2A.Z.1 and H2A.Z.2 Play Independent and Context-Specific Roles in Neuronal Activity-Induced Transcription of Arc/Arg3.1 and Other Immediate Early Genes. eNeuro. 2017;4(4) doi: 10.1523/ENEURO.0040-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Michod D, et al. Calcium-dependent dephosphorylation of the histone chaperone DAXX regulates H3.3 loading and transcription upon neuronal activation. Neuron. 2012;74(1):122–35. doi: 10.1016/j.neuron.2012.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang Y, et al. Chromatin remodeling inactivates activity genes and regulates neural coding. Science. 2016;353(6296):300–5. doi: 10.1126/science.aad4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Buenrostro JD, et al. ATAC-seq: A Method for Assaying Chromatin Accessibility Genome-Wide. Curr Protoc Mol Biol. 2015;109:21 29 1–9. doi: 10.1002/0471142727.mb2129s109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Su Y, et al. Neuronal activity modifies the chromatin accessibility landscape in the adult brain. Nat Neurosci. 2017;20(3):476–483. doi: 10.1038/nn.4494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vierbuchen T, et al. AP-1 Transcription Factors and the BAF Complex Mediate Signal-Dependent Enhancer Selection. Mol Cell. 2017;68(6):1067–1082 e12. doi: 10.1016/j.molcel.2017.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Crepaldi L, et al. Binding of TFIIIC to sine elements controls the relocation of activity-dependent neuronal genes to transcription factories. PLoS Genet. 2013;9(8):e1003699. doi: 10.1371/journal.pgen.1003699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wittmann M, et al. Synaptic activity induces dramatic changes in the geometry of the cell nucleus: interplay between nuclear structure, histone H3 phosphorylation, and nuclear calcium signaling. J Neurosci. 2009;29(47):14687–700. doi: 10.1523/JNEUROSCI.1160-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dekker J, et al. The 4D Nucleome Project. Nature. 2017;549:219–226. doi: 10.1038/nature23884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ernst J, Kellis M. Chromatin-state discovery and genome annotation with ChromHMM. Nat Protoc. 2017;12(12):2478–2492. doi: 10.1038/nprot.2017.124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Velasco S, et al. A Multi-step Transcriptional and Chromatin State Cascade Underlies Motor Neuron Programming from Embryonic Stem Cells. Cell Stem Cell. 2017;20(2):205–217 e8. doi: 10.1016/j.stem.2016.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shin J, et al. Single-Cell RNA-Seq with Waterfall Reveals Molecular Cascades underlying Adult Neurogenesis. Cell Stem Cell. 2015;17(3):360–72. doi: 10.1016/j.stem.2015.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tang Z, et al. CTCF-Mediated Human 3D Genome Architecture Reveals Chromatin Topology for Transcription. Cell. 2015;163(7):1611–27. doi: 10.1016/j.cell.2015.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fraser J, et al. Hierarchical folding and reorganization of chromosomes are linked to transcriptional changes in cellular differentiation. Mol Syst Biol. 2015;11(12):852. doi: 10.15252/msb.20156492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mitchell AC, et al. Longitudinal assessment of neuronal 3D genomes in mouse prefrontal cortex. Nat Commun. 2016;7:12743. doi: 10.1038/ncomms12743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Nagano T, et al. Single-cell Hi-C reveals cell-to-cell variability in chromosome structure. Nature. 2013;502(7469):59–64. doi: 10.1038/nature12593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kind J, et al. Genome-wide maps of nuclear lamina interactions in single human cells. Cell. 2015;163(1):134–47. doi: 10.1016/j.cell.2015.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Beagrie RA, et al. Complex multi-enhancer contacts captured by genome architecture mapping. Nature. 2017;543(7646):519–524. doi: 10.1038/nature21411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mo A, et al. Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron. 2015;86(6):1369–84. doi: 10.1016/j.neuron.2015.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mellen M, et al. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151(7):1417–30. doi: 10.1016/j.cell.2012.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Luo C, et al. Single-cell methylomes identify neuronal subtypes and regulatory elements in mammalian cortex. Science. 2017;357(6351):600–604. doi: 10.1126/science.aan3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Buenrostro JD, et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523(7561):486–90. doi: 10.1038/nature14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cusanovich DA, et al. Multiplex single cell profiling of chromatin accessibility by combinatorial cellular indexing. Science. 2015;348(6237):910–4. doi: 10.1126/science.aab1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rotem A, et al. Single-cell ChIP-seq reveals cell subpopulations defined by chromatin state. Nat Biotechnol. 2015;33(11):1165–72. doi: 10.1038/nbt.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen KH, et al. RNA imaging. Spatially resolved, highly multiplexed RNA profiling in single cells. Science. 2015;348(6233):aaa6090. doi: 10.1126/science.aaa6090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lee JH, et al. Highly multiplexed subcellular RNA sequencing in situ. Science. 2014;343(6177):1360–3. doi: 10.1126/science.1250212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boettiger AN, et al. Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature. 2016;529(7586):418–22. doi: 10.1038/nature16496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Stasevich TJ, et al. Regulation of RNA polymerase II activation by histone acetylation in single living cells. Nature. 2014;516(7530):272–5. doi: 10.1038/nature13714. [DOI] [PubMed] [Google Scholar]
- 88.Yang MG, West AE. Editing the Neuronal Genome: a CRISPR View of Chromatin Regulation in Neuronal Development, Function, and Plasticity. Yale J Biol Med. 2016;89(4):457–470. [PMC free article] [PubMed] [Google Scholar]
- 89.Klann TS, et al. CRISPR-Cas9 epigenome editing enables high-throughput screening for functional regulatory elements in the human genome. Nat Biotechnol. 2017;35(6):561–568. doi: 10.1038/nbt.3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Heller EA, et al. Locus-specific epigenetic remodeling controls addiction- and depression-related behaviors. Nat Neurosci. 2014;17(12):1720–7. doi: 10.1038/nn.3871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Liu XS, et al. Editing DNA Methylation in the Mammalian Genome. Cell. 2016;167(1):233–247 e17. doi: 10.1016/j.cell.2016.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Hilton IB, et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol. 2015;33(5):510–517. doi: 10.1038/nbt.3199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Albert M, et al. Epigenome profiling and editing of neocortical progenitor cells during development. EMBO J. 2017;36(17):2642–2658. doi: 10.15252/embj.201796764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Liu X, et al. In Situ Capture of Chromatin Interactions by Biotinylated dCas9. Cell. 2017;170(5):1028–1043 e19. doi: 10.1016/j.cell.2017.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Morgan SL, et al. Manipulation of nuclear architecture through CRISPR-mediated chromosomal looping. Nat Commun. 2017;8:15993. doi: 10.1038/ncomms15993. [DOI] [PMC free article] [PubMed] [Google Scholar]