

Abstract
Although cerebral microbleeds (CMBs) are frequently associated with traumatic brain injury (TBI), their effects upon clinical outcome after TBI remain controversial and poorly-understood, particularly in older adults. Here we (A) highlight major challenges and opportunities associated with studying the effects of TBI-mediated CMBs, (B) review the evidence on their potential effects upon cognitive and neural outcome as a function of age at injury, and (C) suggest priorities for future research on understanding the clinical implications of CMBs. While TBI-mediated CMBs are likely distinct from those due to cerebral amyloid angiopathy (CAA) or to other neurodegenerative diseases, the effects of these two CMB types upon brain function may share common features. Furthermore, in older TBI victims, the incidence of TBI-mediated CMBs may approximate that of CAA-related CMBs, and thus warrants detailed study. Because the alterations effected by CMBs upon brain structure and function are both unique and age-dependent, it seems likely that novel, age-tailored therapeutic approaches are necessary for the adequate clinical interpretation and treatment of these ubiquitous and under-appreciated TBI sequelae.
Keywords: traumatic brain injury, aging, hemorrhage, microbleed, susceptibility weighted imaging, magnetic resonance imaging
Introduction
Traumatic brain injury (TBI) is a potentially serious condition of substantial epidemiological and clinical significance. The neural and cognitive consequences of TBI can be serious regardless of the victim's age at injury, though sequelae are particularly debilitating in older adults (Harvey and Close, 2012). For illustration, TBIs occur in over 5% of individuals over the age of 60 and carry far greater morbidity and mortality in this age group than in younger cohorts (Ghorbani et al., 2014). Even after adjusting for injury type and severity, every decade of life increases the likelihood of poor clinical outcome after TBI by as much as ∼50% (Hukkelhoven et al., 2003), and older patients are significantly more likely to die from TBI than younger victims (Cheng et al., 2014; Gerber et al., 2009). TBI also accelerates brain aging and the degradation of neural function (Irimia et al., 2013a; Irimia et al., 2013b; Irimia and Van Horn, 2015), with an average difference between survivors' chronological age and their biological brain age of ∼5-years (Cole et al., 2015). Furthermore, a recent large-scale study of nearly 13,000 patients suggests that TBI increases the pathogenic hazard ratio (HR) for neurodegenerative diseases by a factor greater than 3 (Chu et al., 2016).
Cerebral microhemorrhages (or microbleeds, CMBs) constitute a ubiquitous manifestation of TBIs of all severities and their presence is strongly associated with that of traumatic axonal injury (TAI) (Glushakova et al., 2014; Liu et al., 2014). Hay et al. (2015) indicate that 40% of patients dying in the acute phase of TBI and 47% of those who survive TBIs to live for one year or more show multifocal, perivascular and parenchymal CMBs in the gray matter (GM), where (A) long-range axonal connections terminate and (B) brain tissue is subjected to a substantial gradient of physical momentum during traumatic events. Mounting evidence suggests that CMBs are implicated in the pathogenesis of cerebral amyloid angiopathy (CAA) (Fu et al., 2013), an increasingly supported hypothesis whose potential implications are mirrored by epidemiological findings to the effect that CMB occurrence may increase dementia risk by a factor of at least ∼1.7 (Lee et al., 2013). Simultaneously, mild TBI (mTBI) survivors who exhibit CMBs during the acute stage of injury are ∼1.5 times more likely to suffer from Parkinson's disease (PD) (Gardner et al., 2015; Gardner et al., 2014) and their mortality rate is much higher than that of the general population, with an associated HR of ∼1.5 (Fuller et al., 2016).
This review describes the cellular mechanisms and potential consequences of CMBs during senescence and suggests avenues for future inquiry within this significant yet under-prioritized field of research. The challenges of ascertaining the extent to which the CMBs of older TBI victims are associated with either TAI or CAA are highlighted and potential strategies for answering this important question are proposed. Based on existing evidence, we argue that, in older adults, the incidence of TAI-mediated CMBs can be similar to that of CAA-related CMBs, and may therefore be important for detailed future study. Here and throughout, TBI-mediated CMBs refer to white matter (WM) injuries or, more precisely, to SWI-detectable WM hypo-intensities associated with CMBs after TBI.
Mechanisms of CMB occurrence
Intracerebral hemorrhages were known to science as early as the 17th century, when Johann Jacob Wepfer (1620–1695) found fragile vessels in relation to a large cerebral hemorrhage, but was unable to identify a point of rupture (Alg and Werring, 2011). By 1868, Charcot and Bouchard had analyzed the content of micro-aneurysms, and the impact of CMBs became even more widely acknowledged once Ramon y Cajal (1928) had described the neurotoxic effects of blood extravasation. Among these effects is the phenomenon of neural tissue necrosis, which involves the death of neural cells, whose severity increases substantially as the brain ages and whose sequelae last longer as the effectiveness of neural repair mechanisms declines (Morrison and Hof, 1997). The formation of CMBs is thought to involve diapedesis, whereby erythrocytes transiently cross the endothelium of the blood-brain barrier (BBB) to form hemosiderin and/or ferritin deposits within petechiae of the cerebral parenchyma (Zhang et al., 2014). The time rate of diapedesis is strongly dependent upon BBB permeability, which is typically greater in males (Pakulski et al., 2000), begins to increase significantly after age 45 (Blennow et al., 1993), and may be two to three times higher after age 60 compared to age 30 (Rosenberg, 2014). Thus, the higher permeability of the BBB in older TBI patients has long been acknowledged as an important factor contributing to the severity of post-injury brain tissue damage (Farrall and Wardlaw, 2009).
Though CMBs themselves are focal, research indicates that diapedesis effects can extend far outside the penumbra of the microhemorrhage itself, i.e. outside the immediate CMB neighborhood (Patel et al., 2010; Simard et al., 2009). Recent studies additionally suggest that penumbral radii are more extensive in older adults, which may reflect the higher probability that BBB breakage increases the severity of TBI sequelae in this patient group (Hawkins and Davis, 2005). Though penumbral microvasculature may receive damage during impact which is insufficient to rupture the BBB, subsequent molecular response mechanisms can maladaptively cause the subsequent structural failure of these particularly vulnerable capillaries. Such a phenomenon may lead to the delayed formation of petechial hemorrhages, which can coalesce to form iron deposits and then lead to serious complications, including hemorrhagic progression (Kurland et al., 2012). The iron deposits formed after BBB breakage typically result from the phagocytosis of erythrocytes, whereby heme iron in ferritin (i.e., iron bound to heme cofactors within certain proteins, including hemoglobin) is degraded and deposited in the form of hemosiderin. As part of the exogenous inflammatory response to injury, neutrophils may phagocytize and thereby clear cellular debris, but may also release free radicals which harm other parenchymal cells and thereby propagate tissue injury (Whitney et al., 2009).
Structural sequelae and mechanisms of deterioration
Current research indicates that, during senescence, micro-hemorrhages localized to the cerebral parenchyma are associated with focal permeability of the BBB and with potentially-dramatic reorganization of neuronal connectivity, even decades after injury (Glushakova et al., 2014). Because blood vessels are typically more elastic than axons (Spedden et al., 2012; VanBavel et al., 2003), CMB presence has been proposed to be associated with TAI, although the former has not been established unambiguously as primary evidence of the latter. Firstly, higher axonal elasticities do not translate into lower thresholds for their shearing and tearing as a result of acceleration and deceleration forces to which the brain is subjected during physical impact (Arfanakis et al., 2002). Secondly, it is possible that CMB-associated WM damage and/or necrosis may be the result of secondary ischemia rather than the consequence of primary axonal injury (Raghupathi, 2004). Nevertheless, because both the mechanical stiffness and the threshold for capillary rupture increases with age (Sawabe, 2010), the frequency of CMB occurrence after TBI is likely to increase throughout senescence and may additionally be associated with TAI far more often than in youth. Ultimately, histological examination remains the method of choice for ascertaining the biology of WM responses associated with CMBs. Preliminary results from the laboratory of one of the authors (A. I.) suggest that, even in mTBI, CMBs can be associated with substantial macro-scale WM alterations in older adults which may not resolve with time (Figure 1).
Figure 1.
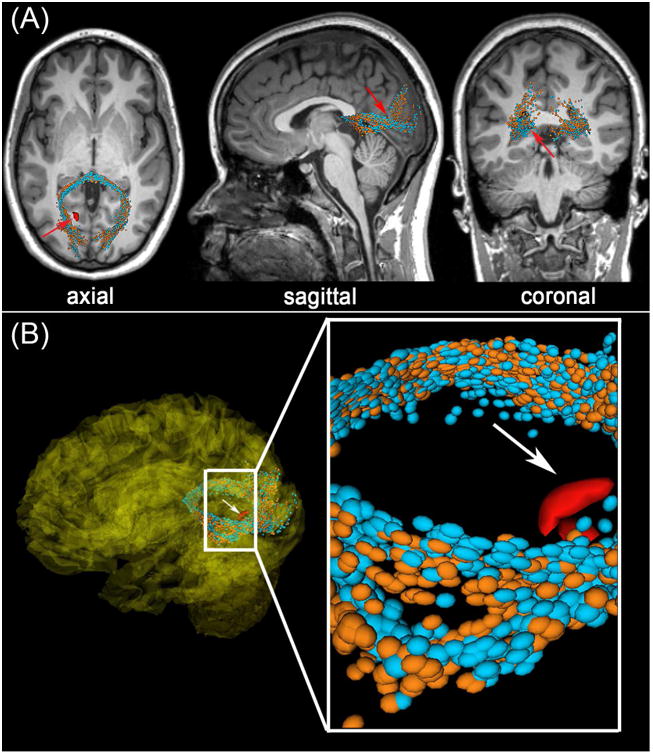
Representative example of DTI streamlines passing through the vicinity of a ∼4 mm3 CMB (red) in an old adult victim of mTBI. Arrows indicate a CMB in the left hemisphere, close to a streamline bundle belonging to the splenium of the corpus callosum. (A) Standard views (coronal, sagittal, axial) of T1-weighted MRI are shown in addition to DTI glyphs associated with perilesional WM streamline bundles imaged acutely (orange) and approximately six months after injury (light blue). The splenium is notably asymmetric at both time points, with the asymmetry being most pronounced close to the CMB (inset). (B) Splenial streamlines ipsilateral to the CMB diverge briefly in its vicinity, and this is not found to occur contralateral to the CMB (inset). This asymmetry is also found at the time of the chronic scan.
An important characteristic of TBI-mediated CMBs is that they frequently occur at the boundary between cortical GM and WM. This is partly due to the distinct mechanical responses of these two tissue types as they are subjected to large physical forces, and partly to differences in venous drainage at the GM/WM boundary (Liu et al., 2014). Because the GM/WM interface is sometimes superficial (superior frontal gyri, middle temporal gyri, etc.) or, other times, deep (insulae, cingulate gyri, etc.) relative to the scalp, one implication of both TBI biomechanics and human neuroanatomy is that the spatial distribution of CMBs throughout the cerebrum can be widespread and/or difficult to anticipate across patients, as confirmed by Hay et al. (2015). Due to the large number of long-range, intra-hemispheric WM connections (e.g. corticospinal tract, arcuate fasciculus, corona radiata), CMBs can be associated with highly-widespread TAI even in mTBI patients (Liu et al., 2014), and their number and size can be greater in older patients compared to younger adults for reasons previously discussed.
Though it has been consistently acknowledged that older age at the time of TBI is usually associated with decreased ability for recovery from CMBs (Miller et al., 2017; Stocchetti et al., 2012), the biological mechanisms involved in this phenomenon remain insufficiently understood. It appears that older individuals' relatively poor ability to recover from insults to the microvasculature is strongly modulated by their frequently-deficient endocrine reactions and by their broader neuroinflammatory responses compared to younger victims. For example, proinflammatory cytokines downregulate physiological responses to a variety of hormones, including insulin, somatotropin, insulin-like growth factor (IGF), thyroid hormone, and estrogens (Ferrucci et al., 2004). On the one hand, the attenuation of these and other neuroendocrine processes which occurs with aging has an appreciable effect upon the reduced ability of the brain to recover from TBI (Cekic and Stein, 2010). On the other hand, whereas microglia play a prominent role in neuroinflammation, their mechanisms of action are greatly affected by aging in numerous ways, which include the alteration of both microglial morphology and of phagocytic activity (Linehan and Fitzgerald, 2015). Together, these phenomena lead to greater oxidative stress and to increased cytokine production (Mosher and Wyss-Coray, 2014). Partly for such reasons, older adults with acute TBI have higher cytokine levels and more phagocytosis-deficient microglia than younger adults (Ritzel et al., 2015). Prior research in older TBI victims describes the down-regulation of numerous genes involved in B-lymphocyte and CD4+ T-cell activity after CMB occurrence–compared to the up-regulation of such genes in younger patients—and partly explains why both B-cell and immunoglobulin counts are lower in older TBI survivors than in younger ones (Ritzel et al., 2015). Additionally, during the acute stage of TBI, regulatory inflammatory genes such as leucine zipper transcription factor 2 (BACH2), leucine-rich repeat neuronal 3 (LRRN3) and lymphoid enhancer-binding factor 1 (LEF1) are expressed at higher levels in younger TBI patients (Ritzel et al., 2015). By contrast, older TBI victims exhibit increased transcriptional activity associated with S100 family genes, including S100 calcium-binding proteins P (S100P) and A8 (S100A8). These two genes are expressed in activated macrophages and microglia and have been linked to poor recovery from TBI due to the increased inflammatory response modulated by these cell types (Beschorner et al., 2000). Unsurprisingly, older adults also exhibit reduced activity of members from the transforming growth factor β family, which are linked to neural recovery and regeneration. These gene expression differences between younger and older adults mirror the higher frequency of positive clinical findings on the chronic brain imaging scans of older TBI survivors (Cho et al., 2016) and suggest a direct association between immune regulation and brain recovery as a function of age at injury.
An essential and under-appreciated aspect of TBI in aging adults is the fact that brain responses to it can extend over years and even decades (Van Horn et al., 2017). For example, human neuroimaging scans have confirmed that (A) TBI-activated microglia may persist in the brain for as long as two decades after injury (Jacobowitz et al., 2012; Ramlackhansingh et al., 2011), (B) cerebral inflammation can last for many years, with numerous deleterious effects to brain function (Johnson et al., 2013) and (C) new microvascular abnormalities can appear on common basis even long after injury (Fujita et al., 2012). Together, these phenomena appear to delineate a complex and poorly-understood cascade of cellular events which contribute to delayed neural repair despite potentially deceiving appearances which may suggest that some patients have fully recovered.
Neuroradiological identification
In clinical settings, a common approach to CMB detection involves the use of susceptibility-weighted imaging (SWI), a type of magnetic resonance imaging (MRI) which is highly sensitive to iron accumulation in the body. SWI uses a fully flow-compensated, gradient-recalled echo (GRE) pulse sequence which exploits magnetic susceptibility differences between tissues to produce enhanced-contrast magnitude MRI images of venous blood, hemorrhages and iron storage complexes (Haacke et al., 2009; Mittal et al., 2009). In SWI, CMBs can be defined as ovoid, hypo-intense, neuroanatomic foci which are inconsistent with osseous, vascular or MRI-related artifacts (Liu et al., 2014). MRI techniques such as magnetic field correlation (MFC) imaging, quantitative susceptibility mapping (QSM) and field-dependent relaxation rate increase (FDRI) imaging can additionally quantify diffuse non-heme iron deposition throughout the parenchyma in vivo and can therefore be used to supplement SWI-provided information on CMBs (Haacke et al., 2015; Raz et al., 2011).
In clinical settings, an increasingly prominent strategy for identifying CMB-related TAI involves diffusion MRI techniques such as diffusion weighted, diffusion tensor and diffusion spectrum imaging (DWI, DTI and DSI, respectively). These methods can quantify the preferential direction of water diffusion throughout the brain and thereby identify the locations of physical insults to WM connections. Mapping TAI associated with CMBs can provide clinical and scientific insight above and beyond the ability of more traditional modalities to do so (Scheid et al., 2006). For example, computed tomography (CT) and conventional MRI [including T1-, T2- and T2*-weighted MRI and even GRE or fluid-attenuated inversion recovery (FLAIR)] can routinely isolate relatively-large, intra-parenchymal hemorrhages. These techniques, however, do not allow either short- or long-term changes in WM connectivity to be assessed with quantitative precision, whereas diffusion MRI can greatly assist this task (Irimia et al., 2011).
An important challenge related to the identification of TBI-mediated CMBs in older adults is that a substantial subset of the aging population may have CAA-related CMBs prior to injury. For example, CMBs are present in ∼6% of randomly-selected, asymptomatic individuals over the age of 60 who do not have a history of neuropsychiatric disease (Koennecke, 2006). Though the simultaneous existence of both TBI- and CAA-related CMBs may confound research efforts aimed at quantifying injury-related CMBs, some distinctions exist between these two forms of pathology. For example, CAA-related CMBs occur more often in deep and infratentorial regions if due to hypertensive arteriopathy (Greenberg et al., 2009b), or in posterior brain regions (occipital lobe, in particular) if due to vascular beta-amyloid depositions (Johnson et al., 2007). Additional areas commonly affected by CAA-related CMBs include the mid-subcortical cerebrum and the areas superior to the corpus callosum (Huang et al., 2015; Yates et al., 2014). On the other hand, TBI-mediated CMBs occur more often at the boundary between GM and WM (Liu et al., 2014), in brain regions where primary (e.g. coup and/or contre-coup) injuries are located (Huang et al., 2015), or in midline regions, particularly above the corpus callosum or in medial subcortex (Imaizumi et al., 2011).
Clinical significance
The clinical implications of CMBs in older adults are controversial. Broadly speaking, available scientific and clinical knowledge suggests that post-traumatic CMBs have substantial negative effects upon the aging central nervous system and that the severity of their impact increases with patients' age at injury. Specifically, in older adults with mTBI, the higher permeability of the BBB, the attenuation of neuroendocrine processes (e.g. those responsible for releasing somatostatin and IGF) and the maladaptive neural repair responses of the aging brain to injury are likely to contribute substantially to this population's poorer trajectory of recovery compared to that observed in younger patients. Though neuroradiological examinations of mTBI patients identify CMBs relatively frequently even years after injury—which can facilitate the task of monitoring the temporal dynamics of these phenomena—the long-term consequences of CMBs remain rather obscure (Huang et al., 2015). The association between CMB presence and clinical outcome in mTBI patients remains particularly controversial (Charidimou et al., 2013; Scheid et al., 2006; Talavage et al., 2015), whereas the specific ways in which this type of hemorrhage differentially affects the aging brain remain inadequately explored. Some studies suggest a correlation between CMB presence and TBI-related deficits (Geurts et al., 2012; Toth et al., 2012), whereas others have been ambivalent regarding the clinical relevance of small hemorrhagic lesions (Yuh et al., 2013). Some insight into this matter may be gained from pediatric TBI studies. Specifically, CMBs which arise in typically-developing children after TBI are statistically associated with neural and cognitive deficits (Salehi et al., 2017), and this suggests that the functional effects of CMBs in older adults may be like those in children (though potentially more pronounced due to aging processes).
In the aging brain, where the cerebral microvasculature is increasingly sensitive to mechanical stress (Sawabe, 2010), CMB occurrence after TBI is allegedly more consequential than in younger cohorts (Greenberg et al., 2009a). In patients with moderate-to-severe TBI, the negative effects of isolated CMBs upon clinical outcome may not initially seem to be nearly as dramatic as those of far-larger hemorrhagic and/or non-hemorrhagic lesions. This intuitive argument, however appealing, does not account for the complex and potentially substantial changes effected by CMB-related TAI upon brain regions located far from primary injury sites. Given that such dynamic alterations can substantially affect both cognitive and neural function (Palacios et al., 2011), more research is needed to understand the relationship between the presence of CMBs in the TBI brain and long-term effects upon high-order brain functions.
In older patients with mTBI, the associations between CMBs, histopathology and clinical outcome are particularly difficult to quantify and prognosticate, especially in individuals with negative clinical findings on T1- and/or T2-weighted MRI. One cause of this difficulty is that post mortem neuropathologic examinations are rarely available from mTBI patients who die of other causes. In the very few such cases which have been reported, histopathological examinations have revealed the presence of hemosiderin-laden macrophages in perivascular GM and WM, suggesting that axonal shearing is the primary mechanism of mTBI-related brain damage (Bigler, 2004). In mTBI patients with CMBs, significant correlations have been identified between the number of WM fasciculi damaged by TAI and the magnitudes of delays in cognitive reaction times, which appear to increase with age (Niogi et al., 2008). It seems, thus, that further research is required to (A) assess the prognostic value of CMB quantitation relative to that of DTI-based connectivity analysis, and to (B) disentangle the potential clinical utility of these two approaches as a function of victim age at injury.
Some argue (Yates et al., 2014) that there is no definite way to determine CMB chronicity in TBI patients. However, progress in this direction could be made by using neuroimaging to monitor populations at high risk for TBI (e.g. contact-sports athletes). Firstly, this could afford comparison of their baseline scans to their post-mTBI MRI readings and thereby distinguish recently-acquired from older CMBs. Secondly, because TBI-mediated CMBs may not be readily distinguishable from micro-hemorrhages associated with CAA, factorial design studies (factors: CAA, TBI) may be useful for understanding the associative relationship and interaction between these two pathology types, as well as the putative differences in their effects upon neural function. We propose that, barring severe neurovascular or neurodegenerative pathology, TBI-mediated CMBs can be equally—if not more—common in older TBI victims than those associated with CAA. Confirmation of this hypothesis would suggest that renewed efforts should be dedicated to understanding CMB effects upon the TBI-affected brain, preferably in the context of studies where TBI victims and TBI-free volunteers can be stratified based on their risk factors for CAA. This strategy could allow researchers to control for the confounding effects of such risk factors when assessing differences in CMB count, spatial distribution and prevalence between the two populations. Importantly, the extent to which cognitive reserve modulates post-traumatic neurodegeneration and/or recovery—whether in the presence or absence of CAA—has not been explored sufficiently (Griesbach et al., 2018), and future research should accommodate and examine this important neuropsychological measure.
Relationship to neurodegeneration
Microvascular damage has long been associated with neurodegeneration (Cordonnier and van der Flier, 2011). For example, the brains of aging adults with a history of repeated TBIs exhibit hemosiderin deposits and accumulations of tau protein in the direct vicinity of blood capillaries (McKee et al., 2009). Such patients have been found to experience accelerated demyelination, extensive accumulation of tau-positive neurofibrillary tangles (NFTs), non-heme iron deposition as well as widespread vascular damage (Nisenbaum et al., 2014). In many TBI patients, the extent and spatial pattern of these phenomena are often reminiscent of those observed in TBI-free individuals affected by cognitive impairment (Schrag et al., 2010). Amyloid deposits are also commonly found within micro-hemorrhagic foci in mouse models and human familial AD (Cacciottolo et al., 2016; Finch and Shams, 2016).
TBI-mediated CMBs are clearly distinct from those due to CAA or to other neurodegenerative diseases, if only based on the criterion of their causative factors. Nevertheless, the effects of these two CMB types upon brain function may share common features beyond those related to the mechanisms of their occurrence, and may additionally implicate their effects upon brain function. In this respect, the extent to which TBI-mediated CMBs can contribute to or even aggravate CAA and neurodegeneration in general has not been quantified adequately. To this end, future studies should compare mTBI survivors to TBI-free control volunteers while stratifying all participants based on their CAA-related environmental risk factors. This approach could provide valuable insights into the differential effects of CAA- vs. TBI-mediated CMBs upon attention, executive control and memory, all of which are frequently impacted in both TBI and CAA-related neurodegeneration and cognitive degradation, though potentially in distinct ways. It is reasonable to hypothesize that knowledge of cognitive reserve at the time of injury can assist in determining the probability that SWI-detectable CMBs are related to either CAA or TBI. If this is indeed the case, this important neuropsychological measure could allow researchers to examine the relationship between CAA and older adults' vulnerability to CMBs after TBI.
Apolipoprotein E (apoE) polymorphisms modulate many neuroinflammatory responses and to accelerate neurodegenerative pathology after TBI (Laskowitz et al., 2010). These factors may modulate CMB evolution as well; in animal models, DTI measures of WM damage have been linked to physical insults effected upon axons stained with amyloid precursor protein, to loss of myelination, to increased permeability of neuronal membranes and to other forms of pathology which are commonly observed in neurodegenerative diseases (Mac Donald et al., 2007; Povlishock and Katz, 2005). As in such conditions, the survivors of repeated TBI exhibit substantial, redox-active non-heme iron deposition in the hippocampus and within inferior temporal cortex (Bouras et al., 1997). These patients often also feature chronic upregulation of heme-oxygenase 1 (HO-1), an enzyme which degrades heme-bound iron into free iron and which contributes to iron overload (Wu et al., 2003). HO-1 is well documented as a macromolecule involved in the pathologies of several neurodegenerative disorders of aging, where its upregulation promotes astrocytic iron accumulation, oxidative stress and mitochondrial iron sequestration (Schipper, 2004).
Older adults' unique structural and functional changes prompted by mTBI-mediated CMBs, however, may indicate that tailored therapeutic approaches are likely necessary for their adequate treatment. To acquire detailed understanding of how micro-hemorrhages can cause or aggravate secondary brain injury in older adults, future studies should investigate the mechanisms whereby HO-1 mediates such phenomena as a function of age. Furthermore, because neural responses to mTBI may persist for decades after injury and contribute to patients' susceptibility to neurodegenerative diseases such as Alzheimer's disease (AD) and PD, understanding why and how the brain ages faster after mTBI should be prioritized as a significant research goal.
Conclusions
The literature on the topic reviewed here suggests several important directions for future research. Firstly, more basic science studies should attempt to alleviate CMB sequelae in animal models of the TBI-affected, aging brain. We identify eight major specific aims in this respect, namely (1) promoting therapeutic revascularization, (2) reducing cytokine levels in CMB-affected regions, (3) interrupting the degradation of heme-bound iron, (4) alleviating oxidative stress at BBB breakage locations, (5) stimulating macrophages' phagocytic ability, (6) increasing immunoglobulin production, (7) decelerating NFT deposition in the CMB (pen)umbra, and (8) stimulating the re-myelination of TAI-affected axons in the CMB (pen)umbra.
Older adults' relatively-high vulnerability to TBI-mediated CMBs indicates that next-generation therapeutic interventions should be tailored to the specific needs of this particularly vulnerable population. Such interventions should aim to reduce aging-related deficiencies in the brain's systemic response to injury and to accommodate the potentially serious implications of CMBs. Historically, no clinical trial evaluating neuroprotective compounds for the treatment of TBI has been successful (Marklund and Hillered, 2011), partly because the clinical efficacy of many such compounds hinges on their administration before—rather than after—TBI (Menon, 2009). Nevertheless, research aimed at reversing microvascular damage in the aging brain has identified compounds which can promote therapeutic revascularization even if administered after TBI. One such compound is fucoidan, a fucose-based sulfated polysaccharide which can reduce neuronal apoptosis, lipid peroxidation, reactive oxygen species (ROS) generation and mitochondrial dysfunction (Wang et al., 2016). These observed benefic effects are possibly mediated by the activation of Sirt3, a deacetylase from the sirtuin class of proteins which is localized within the mitochondrion and which is well known for its involvement in aging processes (Preyat and Leo, 2013). Fucoidan-treated old rats exhibit significant reductions in lesion volumes as well as improvements in sensorimotor function, in spatial learning and memory formation. Given the paucity of currently-available lifestyle interventions which can specifically target hemorrhagic lesions in TBI patients, the efficacy of this and other compounds with similar benefic effects should be studied in detail.
From a neuroimaging standpoint, the ability to distinguish between CAA- and TBI-mediated CMBs using currently-available MRI sequences remains a significant goal and challenge. The differences between these two types of hemorrhages should be investigated more actively to understand (A) the differential effects of these two CMB types upon the aging brain, (B) the manner in which cognitive reserve modulates recovery, as well as (C) their potential interaction in modulating clinical outcome. Future MRI sequences for imaging the TBI-affected brain in the acute stage of injury could exploit differences in structural neuropathology between ‘older’ (CAA-mediated) and ‘newer’ (TBI-mediated) CMBs to distinguish between the two. Such an ability could substantially enhance our ability to study how underlying CAA can modulate brain responses to TBI, and may even lead to better understanding of CAA itself.
Finally, it is important to emphasize that future studies should preferentially target older TBI patients above and beyond the scope of other ongoing efforts in the field of basic, translational and clinical brain injury research. The elderly represent a prominent and growing demographic segment of the population affected by TBI (Stocchetti et al., 2012), such that future interventions which take into account the distinct needs of older TBI survivors should be incorporated into geriatric care protocols. This could synergistically alter TBI trajectories in older victims, with potentially substantial effects upon patients' well-being and quality of life.
Highlights.
Traumatic brain injury (TBIs) frequently result in cerebral microbleeds (CMBs)
The effects of TBI-related CMBs upon the ageing brain remain controversial
Incidences of CMBs due to TBI vs. chronic amyloid angiopathy (CAA) are comparable
Major opportunities for studying the effects of TBI-related CMBs using MRI exist
Ageing-tailored therapies are necessary for adequate clinical treatment of CMBs
Acknowledgments
This work was supported by the National Institutes of Health [grant numbers R01 NS 100973 to A. I. and R44 NS 081792 to J. D. V. H.]. A. I. wishes to thank Prof. Caleb E. Finch for his excellent and detailed suggestions, comments and contributions, as well as for valuable discussions on the topic of this review. He also wishes to thank Kenneth A. Rostowsky and Nahian F. Chowdhury for their assistance with preparing the figure.
Footnotes
Competing Interests Statement: The authors have no competing interests to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alg VS, Werring DJ. Historical overview: microaneurysms, cerebral microbleeds and intracerebral hemorrhage. In: Werring DJ, editor. Cerebral microbleeds: pathophysiology to clinical practice. Cambridge University Press; New York, USA: 2011. pp. 1–12. [Google Scholar]
- Arfanakis K, Haughton VM, Carew JD, Rogers BP, Dempsey RJ, Meyerand ME. Diffusion tensor MR imaging in diffuse axonal injury. AJNR Am J Neuroradiol. 2002;23(5):794–802. [PMC free article] [PubMed] [Google Scholar]
- Beschorner R, Engel S, Mittelbronn M, Adjodah D, Dietz K, Schluesener HJ, Meyermann R. Differential regulation of the monocytic calcium-binding peptides macrophage-inhibiting factor related protein-8 (MRP8/S100A8) and allograft inflammatory factor-1 (AIF-1) following human traumatic brain injury. Acta Neuropathol. 2000;100(6):627–634. doi: 10.1007/s004010000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigler ED. Neuropsychological results and neuropathological findings at autopsy in a case of mild traumatic brain injury. J Int Neuropsychol Soc. 2004;10(5):794–806. doi: 10.1017/S1355617704105146. [DOI] [PubMed] [Google Scholar]
- Blennow K, Fredman P, Wallin A, Gottfries CG, Karlsson I, Langstrom G, Skoog I, Svennerholm L, Wikkelso C. Protein analysis in cerebrospinal fluid. II. Reference values derived from healthy individuals 18-88 years of age. Eur Neurol. 1993;33(2):129–133. doi: 10.1159/000116919. [DOI] [PubMed] [Google Scholar]
- Bouras C, Giannakopoulos P, Good PF, Hsu A, Hof PR, Perl DP. A laser microprobe mass analysis of brain aluminum and iron in dementia pugilistica: comparison with Alzheimer's disease. Eur Neurol. 1997;38(1):53–58. doi: 10.1159/000112903. [DOI] [PubMed] [Google Scholar]
- Cacciottolo M, Christensen A, Moser A, Liu JH, Pike CJ, Smith C, Ladu MJ, Sullivan PM, Morgan TE, Dolzhenko E, Charidimou A, Wahlund LO, Wiberg MK, Shams S, Chiang GCY, Finch CE, Neuroimaging AsD. The APOE4 allele shows opposite sex bias in microbleeds and Alzheimer's disease of humans and mice. Neurobiol Aging. 2016;37:47–57. doi: 10.1016/j.neurobiolaging.2015.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cekic M, Stein DG. Traumatic brain injury and aging: is a combination of progesterone and vitamin D hormone a simple solution to a complex problem? Neurotherapeutics. 2010;7(1):81–90. doi: 10.1016/j.nurt.2009.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charidimou A, Krishnan A, Werring DJ, Rolf Jager H. Cerebral microbleeds: a guide to detection and clinical relevance in different disease settings. Neuroradiology. 2013;55(6):655–674. doi: 10.1007/s00234-013-1175-4. [DOI] [PubMed] [Google Scholar]
- Cheng PL, Lin HY, Lee YK, Hsu CY, Lee CC, Su YC. Higher mortality rates among the elderly with mild traumatic brain injury: a nationwide cohort study. Scand J Trauma Resus. 2014;22 doi: 10.1186/1757-7241-22-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho YE, Latour LL, Kim H, Turtzo LC, Olivera A, Livingston WS, Wang D, Martin C, Lai C, Cashion A, Gill J. Older Age Results in Differential Gene Expression after Mild Traumatic Brain Injury and Is Linked to Imaging Differences at Acute Follow-up. Front Aging Neurosci. 2016;8:168. doi: 10.3389/fnagi.2016.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu SF, Chiu WT, Lin HW, Chiang YH, Liou TH. Hazard Ratio and Repeat Injury for Dementia in Patients With and Without a History of Traumatic Brain Injury: A Population-Based Secondary Data Analysis in Taiwan. Asia-Pac J Public He. 2016;28(6):519–527. doi: 10.1177/1010539516662956. [DOI] [PubMed] [Google Scholar]
- Cole JH, Leech R, Sharp DJ Alzheimer's Disease Neuroimaging, I. Prediction of brain age suggests accelerated atrophy after traumatic brain injury. Ann Neurol. 2015;77(4):571–581. doi: 10.1002/ana.24367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordonnier C, van der Flier WM. Brain microbleeds and Alzheimer's disease: innocent observation or key player? Brain. 2011;134(Pt 2):335–344. doi: 10.1093/brain/awq321. [DOI] [PubMed] [Google Scholar]
- Farrall AJ, Wardlaw JM. Blood-brain barrier: ageing and microvascular disease--systematic review and meta-analysis. Neurobiol Aging. 2009;30(3):337–352. doi: 10.1016/j.neurobiolaging.2007.07.015. [DOI] [PubMed] [Google Scholar]
- Ferrucci L, Ble A, Bandinelli S, Lauretani F, Suthers K, Guralnik JM. A flame burning within. Aging Clin Exp Res. 2004;16(3):240–243. doi: 10.1007/BF03327390. [DOI] [PubMed] [Google Scholar]
- Finch CE, Shams S. Apolipoprotein E and Sex Bias in Cerebrovascular Aging of Men and Mice. Trends Neurosci. 2016;39(9):625–637. doi: 10.1016/j.tins.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, He Q, Zhu D, Wang Y, Gao Y, Cao H, Cheng J. A BODIPY dye as a reactive chromophoric/fluorogenic probe for selective and quick detection of vapors of secondary amines. Chem Commun (Camb) 2013;49(96):11266–11268. doi: 10.1039/c3cc46571c. [DOI] [PubMed] [Google Scholar]
- Fujita M, Wei EP, Povlishock JT. Intensity- and interval-specific repetitive traumatic brain injury can evoke both axonal and microvascular damage. J Neurotrauma. 2012;29(12):2172–2180. doi: 10.1089/neu.2012.2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller GW, Ransom J, Mandrekar J, Brown AW. Long-Term Survival Following Traumatic Brain Injury: A Population-Based Parametric Survival Analysis. Neuroepidemiology. 2016;47(1):1–10. doi: 10.1159/000445997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RC, Burke JF, Nettiksimmons J, Goldman S, Tanner CM, Yaffe K. Traumatic brain injury in later life increases risk for Parkinson disease. Ann Neurol. 2015;77(6):987–995. doi: 10.1002/ana.24396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RC, Burke JF, Nettiksimmons J, Kaup A, Barnes DE, Yaffe K. Dementia risk after traumatic brain injury vs nonbrain trauma: the role of age and severity. JAMA Neurol. 2014;71(12):1490–1497. doi: 10.1001/jamaneurol.2014.2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerber LM, Ni Q, Hartl R, Ghajar J. Impact of falls on early mortality from severe traumatic brain injury. J Trauma Manag Outcomes. 2009;3:9. doi: 10.1186/1752-2897-3-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geurts BHJ, Andriessen TMJC, Goraj BM, Vos PE. The reliability of magnetic resonance imaging in traumatic brain injury lesion detection. Brain Injury. 2012;26(12):1439–1450. doi: 10.3109/02699052.2012.694563. [DOI] [PubMed] [Google Scholar]
- Ghorbani P, Falken M, Riddez L, Sundelof M, Oldner A, Strommer L. Clinical review is essential to evaluate 30-day mortality after trauma. Scand J Trauma Resusc Emerg Med. 2014;22:18. doi: 10.1186/1757-7241-22-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glushakova OY, Johnson D, Hayes RL. Delayed increases in microvascular pathology after experimental traumatic brain injury are associated with prolonged inflammation, blood-brain barrier disruption, and progressive white matter damage. J Neurotrauma. 2014;31(13):1180–1193. doi: 10.1089/neu.2013.3080. [DOI] [PubMed] [Google Scholar]
- Greenberg SM, Vernooij MW, Cordonnier C, Viswanathan A, Al-Shahi Salman R, Warach S, Launer LJ, Van Buchem MA, Breteler MM, Microbleed Study G. Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol. 2009a;8(2):165–174. doi: 10.1016/S1474-4422(09)70013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SM, Vernooij MW, Cordonnier C, Viswanathan A, Salman RAS, Warach S, Launer LJ, Van Buchem MA, Breteler MMB, Grp MS. Cerebral microbleeds: a guide to detection and interpretation. Lancet Neurol. 2009b;8(2):165–174. doi: 10.1016/S1474-4422(09)70013-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griesbach GS, Masel BE, Helvie RE, Ashley MJ. The Impact of Traumatic Brain Injury on Later Life: Effects on Normal Aging and Neurodegenerative Diseases. J Neurotrauma. 2018;35(1):17–24. doi: 10.1089/neu.2017.5103. [DOI] [PubMed] [Google Scholar]
- Haacke EM, Liu S, Buch S, Zheng W, Wu D, Ye Y. Quantitative susceptibility mapping: current status and future directions. Magn Reson Imaging. 2015;33(1):1–25. doi: 10.1016/j.mri.2014.09.004. [DOI] [PubMed] [Google Scholar]
- Haacke EM, Mittal S, Wu Z, Neelavalli J, Cheng YC. Susceptibility-weighted imaging: technical aspects and clinical applications, part 1. AJNR Am J Neuroradiol. 2009;30(1):19–30. doi: 10.3174/ajnr.A1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey LA, Close JC. Traumatic brain injury in older adults: characteristics, causes and consequences. Injury. 2012;43(11):1821–1826. doi: 10.1016/j.injury.2012.07.188. [DOI] [PubMed] [Google Scholar]
- Hawkins BT, Davis TP. The blood-brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57(2):173–185. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- Hay JR, Johnson VE, Young AMH, Smith DH, Stewart W. Blood-Brain Barrier Disruption Is an Early Event That May Persist for Many Years After Traumatic Brain Injury in Humans. J Neuropath Exp Neur. 2015;74(12):1147–1157. doi: 10.1097/NEN.0000000000000261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YL, Kuo YS, Tseng YC, Chen DYT, Chiu WT, Chen CJ. Susceptibility-weighted MRI in mild traumatic brain injury. Neurology. 2015;84(6):580–585. doi: 10.1212/WNL.0000000000001237. [DOI] [PubMed] [Google Scholar]
- Hukkelhoven CW, Steyerberg EW, Rampen AJ, Farace E, Habbema JD, Marshall LF, Murray GD, Maas AI. Patient age and outcome following severe traumatic brain injury: an analysis of 5600 patients. J Neurosurg. 2003;99(4):666–673. doi: 10.3171/jns.2003.99.4.0666. [DOI] [PubMed] [Google Scholar]
- Imaizumi T, Miyata K, Inamura S, Kohama I, Nyon KS, Nomura T. The Difference in Location between Traumatic Cerebral Microbleeds and Microangiopathic Microbleeds Associated with Stroke. J Neuroimaging. 2011;21(4):359–364. doi: 10.1111/j.1552-6569.2011.00593.x. [DOI] [PubMed] [Google Scholar]
- Irimia A, Chambers MC, Alger JR, Filippou M, Prastawa MW, Wang B, Hovda DA, Gerig G, Toga AW, Kikinis R, Vespa PM, Van Horn JD. Comparison of acute and chronic traumatic brain injury using semi-automatic multimodal segmentation of MR volumes. J Neurotrauma. 2011;28(11):2287–2306. doi: 10.1089/neu.2011.1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia A, Goh SY, Torgerson CM, Chambers MC, Kikinis R, Van Horn JD. Forward and inverse electroencephalographic modeling in health and in acute traumatic brain injury. Clinical Neurophysiology. 2013a;124(11):2129–2145. doi: 10.1016/j.clinph.2013.04.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia A, Goh SY, Torgerson CM, Stein NR, Chambers MC, Vespa PM, Van Horn JD. Electroencephalographic inverse localization of brain activity in acute traumatic brain injury as a guide to surgery, monitoring and treatment. Clinical Neurology & Neurosurgery. 2013b;115(10):2159–2165. doi: 10.1016/j.clineuro.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irimia A, Van Horn JD. Epileptogenic focus localization in treatment-resistant post-traumatic epilepsy. Journal of Clinical Neuroscience. 2015;22(4):627–631. doi: 10.1016/j.jocn.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobowitz DM, Cole JT, McDaniel DP, Pollard HB, Watson WD. Microglia activation along the corticospinal tract following traumatic brain injury in the rat: a neuroanatomical study. Brain Res. 2012;1465:80–89. doi: 10.1016/j.brainres.2012.05.008. [DOI] [PubMed] [Google Scholar]
- Johnson KA, Gregas M, Becker JA, Kinnecom C, Salat DH, Moran EK, Smith EE, Rosand J, Rentz DM, Klunk WE, Mathis CA, Price JC, DeKosky ST, Fischman AJ, Greenberg SM. Imaging of amyloid burden and distribution in cerebral amyloid angiopathy. Ann Neurol. 2007;62(3):229–234. doi: 10.1002/ana.21164. [DOI] [PubMed] [Google Scholar]
- Johnson VE, Stewart JE, Begbie FD, Trojanowski JQ, Smith DH, Stewart W. Inflammation and white matter degeneration persist for years after a single traumatic brain injury. Brain. 2013;136(Pt 1):28–42. doi: 10.1093/brain/aws322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koennecke HC. Cerebral microbleeds on MRI: prevalence, associations, and potential clinical implications. Neurology. 2006;66(2):165–171. doi: 10.1212/01.wnl.0000194266.55694.1e. [DOI] [PubMed] [Google Scholar]
- Kurland D, Hong C, Aarabi B, Gerzanich V, Simard JM. Hemorrhagic progression of a contusion after traumatic brain injury: a review. J Neurotrauma. 2012;29(1):19–31. doi: 10.1089/neu.2011.2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskowitz DT, Song P, Wang H, Mace B, Sullivan PM, Vitek MP, Dawson HN. Traumatic brain injury exacerbates neurodegenerative pathology: improvement with an apolipoprotein E-based therapeutic. J Neurotrauma. 2010;27(11):1983–1995. doi: 10.1089/neu.2010.1396. [DOI] [PubMed] [Google Scholar]
- Lee YK, Hou SW, Lee CC, Hsu CY, Huang YS, Su YC. Increased risk of dementia in patients with mild traumatic brain injury: a nationwide cohort study. PLoS One. 2013;8(5):e62422. doi: 10.1371/journal.pone.0062422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linehan E, Fitzgerald DC. Ageing and the Immune System: Focus on Macrophages. Eur J Microbiol Immu. 2015;5(1):14–24. doi: 10.1556/EUJMI-D-14-00035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Kou Z, Tian Y. Diffuse axonal injury after traumatic cerebral microbleeds: an evaluation of imaging techniques. Neural Regen Res. 2014;9(12):1222–1230. doi: 10.4103/1673-5374.135330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mac Donald CL, Dikranian K, Song SK, Bayly PV, Holtzman DM, Brody DL. Detection of traumatic axonal injury with diffusion tensor imaging in a mouse model of traumatic brain injury. Exp Neurol. 2007;205(1):116–131. doi: 10.1016/j.expneurol.2007.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marklund N, Hillered L. Animal modelling of traumatic brain injury in preclinical drug development: where do we go from here? Br J Pharmacol. 2011;164(4):1207–1229. doi: 10.1111/j.1476-5381.2010.01163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Cantu RC, Nowinski CJ, Hedley-Whyte ET, Gavett BE, Budson AE, Santini VE, Lee HS, Kubilus CA, Stern RA. Chronic traumatic encephalopathy in athletes: progressive tauopathy after repetitive head injury. J Neuropathol Exp Neurol. 2009;68(7):709–735. doi: 10.1097/NEN.0b013e3181a9d503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon DK. Unique challenges in clinical trials in traumatic brain injury. Crit Care Med. 2009;37(1 Suppl):S129–135. doi: 10.1097/CCM.0b013e3181921225. [DOI] [PubMed] [Google Scholar]
- Miller PR, Chang MC, Hoth JJ, Hildreth AN, Wolfe SQ, Gross JL, Martin RS, Carter JE, Meredith JW, D'Agostino R., Jr Predicting Mortality and Independence at Discharge in the Aging Traumatic Brain Injury Population Using Data Available at Admission. J Am Coll Surg. 2017;224(4):680–685. doi: 10.1016/j.jamcollsurg.2016.12.053. [DOI] [PubMed] [Google Scholar]
- Mittal S, Wu Z, Neelavalli J, Haacke EM. Susceptibility-weighted imaging: technical aspects and clinical applications, part 2. AJNR Am J Neuroradiol. 2009;30(2):232–252. doi: 10.3174/ajnr.A1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JH, Hof PR. Life and death of neurons in the aging brain. Science. 1997;278(5337):412–419. doi: 10.1126/science.278.5337.412. [DOI] [PubMed] [Google Scholar]
- Mosher KI, Wyss-Coray T. Microglial dysfunction in brain aging and Alzheimer's disease. Biochem Pharmacol. 2014;88(4):594–604. doi: 10.1016/j.bcp.2014.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niogi SN, Mukherjee P, Ghajar J, Johnson C, Kolster RA, Sarkar R, Lee H, Meeker M, Zimmerman RD, Manley GT, McCandliss BD. Extent of microstructural white matter injury in postconcussive syndrome correlates with impaired cognitive reaction time: a 3T diffusion tensor imaging study of mild traumatic brain injury. AJNR Am J Neuroradiol. 2008;29(5):967–973. doi: 10.3174/ajnr.A0970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nisenbaum EJ, Novikov DS, Lui YW. The presence and role of iron in mild traumatic brain injury: an imaging perspective. J Neurotrauma. 2014;31(4):301–307. doi: 10.1089/neu.2013.3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pakulski C, Drobnik L, Millo B. Age and sex as factors modifying the function of the blood-cerebrospinal fluid barrier. Medical Science Monitor. 2000;6(2):314–318. [PubMed] [Google Scholar]
- Palacios EM, Fernandez-Espejo D, Junque C, Sanchez-Carrion R, Roig T, Tormos JM, Bargallo N, Vendrell P. Diffusion tensor imaging differences relate to memory deficits in diffuse traumatic brain injury. BMC Neurol. 2011;11:24. doi: 10.1186/1471-2377-11-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AD, Gerzanich V, Geng Z, Simard JM. Glibenclamide reduces hippocampal injury and preserves rapid spatial learning in a model of traumatic brain injury. J Neuropathol Exp Neurol. 2010;69(12):1177–1190. doi: 10.1097/NEN.0b013e3181fbf6d6. [DOI] [PubMed] [Google Scholar]
- Povlishock JT, Katz DI. Update of neuropathology and neurological recovery after traumatic brain injury. J Head Trauma Rehabil. 2005;20(1):76–94. doi: 10.1097/00001199-200501000-00008. [DOI] [PubMed] [Google Scholar]
- Preyat N, Leo O. Sirtuin deacylases: a molecular link between metabolism and immunity. J Leukocyte Biol. 2013;93(5):669–680. doi: 10.1189/jlb.1112557. [DOI] [PubMed] [Google Scholar]
- Raghupathi R. Cell death mechanisms following traumatic brain injury. Brain Pathol. 2004;14(2):215–222. doi: 10.1111/j.1750-3639.2004.tb00056.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramlackhansingh AF, Brooks DJ, Greenwood RJ, Bose SK, Turkheimer FE, Kinnunen KM, Gentleman S, Heckemann RA, Gunanayagam K, Gelosa G, Sharp DJ. Inflammation after trauma: microglial activation and traumatic brain injury. Ann Neurol. 2011;70(3):374–383. doi: 10.1002/ana.22455. [DOI] [PubMed] [Google Scholar]
- Ramon y, Cajal S. Degeneration and regeneration of the nervous system. Oxford University Press; London: 1928. [Google Scholar]
- Raz E, Jensen JH, Ge Y, Babb JS, Miles L, Reaume J, Grossman RI, Inglese M. Brain iron quantification in mild traumatic brain injury: a magnetic field correlation study. AJNR Am J Neuroradiol. 2011;32(10):1851–1856. doi: 10.3174/ajnr.A2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritzel RM, Patel AR, Pan S, Crapser J, Hammond M, Jellison E, McCullough LD. Age- and location-related changes in microglial function. Neurobiol Aging. 2015;36(6):2153–2163. doi: 10.1016/j.neurobiolaging.2015.02.016. [DOI] [PubMed] [Google Scholar]
- Rosenberg GA. Blood-Brain Barrier Permeability in Aging and Alzheimer's Disease. J Prev Alzheimers Dis. 2014;1(3):138–139. doi: 10.14283/jpad.2014.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salehi A, Zhang JH, Obenaus A. Response of the cerebral vasculature following traumatic brain injury. J Cerebr Blood F Met. 2017;37(7):2320–2339. doi: 10.1177/0271678X17701460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawabe M. Vascular aging: from molecular mechanism to clinical significance. Geriatr Gerontol Int. 2010;10(1):S213–220. doi: 10.1111/j.1447-0594.2010.00603.x. [DOI] [PubMed] [Google Scholar]
- Scheid R, Walther K, Guthke T, Preul C, von Cramon DY. Cognitive sequelae of diffuse axonal injury. Arch Neurol. 2006;63(3):418–424. doi: 10.1001/archneur.63.3.418. [DOI] [PubMed] [Google Scholar]
- Schipper HM. Heme oxygenase expression in human central nervous system disorders. Free Radic Biol Med. 2004;37(12):1995–2011. doi: 10.1016/j.freeradbiomed.2004.09.015. [DOI] [PubMed] [Google Scholar]
- Schrag M, McAuley G, Pomakian J, Jiffry A, Tung S, Mueller C, Vinters HV, Haacke EM, Holshouser B, Kido D, Kirsch WM. Correlation of hypointensities in susceptibility-weighted images to tissue histology in dementia patients with cerebral amyloid angiopathy: a postmortem MRI study. Acta Neuropathol. 2010;119(3):291–302. doi: 10.1007/s00401-009-0615-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard JM, Kilbourne M, Tsymbalyuk O, Tosun C, Caridi J, Ivanova S, Keledjian K, Bochicchio G, Gerzanich V. Key role of sulfonylurea receptor 1 in progressive secondary hemorrhage after brain contusion. J Neurotrauma. 2009;26(12):2257–2267. doi: 10.1089/neu.2009.1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spedden E, White JD, Naumova EN, Kaplan DL, Staii C. Elasticity Maps of Living Neurons Measured by Combined Fluorescence and Atomic Force Microscopy. Biophys J. 2012;103(5):868–877. doi: 10.1016/j.bpj.2012.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stocchetti N, Paterno R, Citerio G, Beretta L, Colombo A. Traumatic brain injury in an aging population. J Neurotrauma. 2012;29(6):1119–1125. doi: 10.1089/neu.2011.1995. [DOI] [PubMed] [Google Scholar]
- Talavage TM, Nauman EA, Leverenz LJ. The Role of Medical Imaging in the Recharacterization of Mild Traumatic Brain Injury Using Youth Sports as a Laboratory. Front Neurol. 2015;6:273. doi: 10.3389/fneur.2015.00273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth A, Kovacs N, Perlaki G, Orsi G, Aradi M, Komaromy H, Bukovics P, Farkas O, Doczi T, Janszky J, Schwarcz A, Buki A. Advanced Magnetic Resonance Imaging in the Acute and Subacute Phase of Mild Traumatic Brain Injury: Can We See the Difference? Journal of Neurotrauma. 2012;29(10):A41–A42. doi: 10.1089/neu.2012.2486. [DOI] [PubMed] [Google Scholar]
- Van Horn JD, Irimia A, Torgerson CM, Bhattrai A, Jacokes Z, Vespa PM. Mild cognitive impairment and structural brain abnormalities in a sexagenarian with a history of childhood traumatic brain injury. J Neurosci Res. 2017 doi: 10.1002/jnr.24084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanBavel E, Siersma P, Spaan JA. Elasticity of passive blood vessels: a new concept. Am J Physiol Heart Circ Physiol. 2003;285(5):H1986–2000. doi: 10.1152/ajpheart.00248.2003. [DOI] [PubMed] [Google Scholar]
- Wang T, Zhu M, He ZZ. Low-Molecular-Weight Fucoidan Attenuates Mitochondrial Dysfunction and Improves Neurological Outcome After Traumatic Brain Injury in Aged Mice: Involvement of Sirt3. Cell Mol Neurobiol. 2016;36(8):1257–1268. doi: 10.1007/s10571-015-0323-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitney NP, Eidem TM, Peng H, Huang Y, Zheng JC. Inflammation mediates varying effects in neurogenesis: relevance to the pathogenesis of brain injury and neurodegenerative disorders. J Neurochem. 2009;108(6):1343–1359. doi: 10.1111/j.1471-4159.2009.05886.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Hua Y, Keep RF, Nakamura T, Hoff JT, Xi G. Iron and iron-handling proteins in the brain after intracerebral hemorrhage. Stroke. 2003;34(12):2964–2969. doi: 10.1161/01.STR.0000103140.52838.45. [DOI] [PubMed] [Google Scholar]
- Yates PA, Villemagne VL, Ellis KA, Desmond PM, Masters CL, Rowe CC. Cerebral microbleeds: a review of clinical, genetic, and neuroimaging associations. Front Neurol. 2014;4:205. doi: 10.3389/fneur.2013.00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuh EL, Mukherjee P, Lingsma HF, Yue JK, Ferguson AR, Gordon WA, Valadka AB, Schnyer DM, Okonkwo DO, Maas AIR, Manley GT, Investigators TT. Magnetic resonance imaging improves 3-month outcome prediction in mild traumatic brain injury. Ann Neurol. 2013;73(2):224–235. doi: 10.1002/ana.23783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Li X, Li C, Lian Z, Huang X, Zhong G, Zhu D, Li K, Jin C, Hu X, Han J, Guo L, Hu X, Li L, Liu T. Inferring functional interaction and transition patterns via dynamic Bayesian variable partition models. Hum Brain Mapp. 2014;35(7):3314–3331. doi: 10.1002/hbm.22404. [DOI] [PMC free article] [PubMed] [Google Scholar]