

Abstract
Alzheimer's disease (AD) involves progressive deposition of amyloid β-peptide (Aβ), synapse loss and neuronal death, which occur in brain regions critical for learning and memory. Considerable evidence suggests that lipid peroxidation contributes to synaptic dysfunction and neuronal degeneration, both upstream and downstream of Aβ pathology. Recent findings suggest that lipid peroxidation can be inhibited by replacement of polyunsaturated fatty acids (PUFA) with isotope-reinforced (deuterated) PUFA (D-PUFA), and that D-PUFA can protect neurons in experimental models of Parkinson's disease. Here we determined whether dietary D-PUFA would ameliorate Aβ pathology and/or cognitive deficits in a mouse model of AD (APP/PS1 double mutant transgenic mice). The D-PUFA diet did not ameliorate spatial learning and memory deficits in the AD mice. Compared to mice fed an H-PUFA control diet, those fed D-PUFA for 5 months exhibited high levels of incorporation of deuterium into arachidonic acid and docosahexanoic acid, and reduced concentrations of lipid peroxidation products (F2 isoprostanes and neuroprostanes), in brain tissues. Concentrations of Aβ40 and Aβ38 in the hippocampus were significantly lower, with a trend to reduced concentrations of Aβ42, in mice fed D-PUFA compared to those fed H-PUFA. We conclude that a D-PUFA diet reduces the brain tissue concentrations of both ARA and DHA oxidation products, as well as the concentration of Aβs.
Introduction
Alzheimer's disease (AD), the most common neurodegenerative disorder of aging, is characterized by progressive cognitive impairment resulting from degeneration of synapses and neurons that is most pronounced in the hippocampus, entorhinal cortex and functionally connected regions of the parietal, temporal and frontal lobes (Leal and Yassa, 2013). The neurodegenerative process is associated with extracellular accumulation of aggregated forms of amyloid β-peptide (Aβ) and intraneuronal accumulation of hyperphosphorylated forms of the microtubule-associated protein Tau (Raskin et al., 2015). Aβ is generated by cleavages of the β-amyloid precursor protein (APP) by β- and γ-secretases. Mutations in APP, and in presenilin 1 (the catalytic subunit of the γ-secretase enzyme complex) are responsible for rare cases of early-onset dominantly inherited AD. These mutations result in increased production of an aggregation-prone neurotoxic 42 amino acid form of Aβ, strongly suggesting that Aβ pathology is a pivotal event in the disease process (Bertram et al., 2010). The factors that result in Aβ accumulation and neuronal degeneration in the more common late-onset cases of AD are unclear, but are presumably related to mechanisms of aging, because aging is the major risk factor for AD. Studies of experimental cell culture and animal models of AD have shown that aggregating forms of Aβ can cause synaptic dysfunction and neuronal degeneration (Mattson, 2004). Evidence suggests that the cellular and molecular mechanism by which Aβ damages neurons involves oxidative stress and disruption of cellular Ca2+ homeostasis, which results in neuronal network hyperexcitability and excitotoxicity (Mattson et al., 1992; Bezprozvanny and Mattson, 2008).
The brain is susceptible to oxidative stress due to the high metabolic rate of neurons and consequent elevated production of reactive oxygen species (ROS) (Marques-Aleixo et al, 2012). In addition, the brain contains high levels of polyunsaturated fatty acids (PUFA), arachidonic acid (ARA), docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA), which are enzymatically produced from the diet-derived fatty acids linoleic acid (18:2, n-6) and linolenic acid (18:3, n-3), which are prone to attack by ROS, resulting in the non-enzymatic autocatalytic chain reaction of lipid peroxidation (LPO) (Porter et al., 1995). This insidious process requires just one initiating event to get the reaction chain started (Figure 1), leading to extensive oxidative damage of membrane PUFAs, and the generation of multiple classes of LPO products including F2, F3 and F4 isoprostanes are generated from ARA, EPA and DHA, respectively (Montine et al., 2004; Milne et al., 2007; Milne et al, 2011). A particularly sinister group of PUFA decomposition products are the highly reactive cytotoxic carbonyls 4-hydroxynonenal (HNE) and 4-hydroxyhexenal (HHE), which are highly toxic compounds that irreversibly crosslink various proteins (Negre-Salvayre et al., 2008). During aging, brain cells accumulate oxidatively damaged proteins, lipid membrane components, and DNA, and these oxidative modifications are greatly amplified in vulnerable brain regions of AD patients (Butterfield and Boyd-Kimball, 2004; Sultana and Butterfield, 2006; Markesbery and Lovell, 2007; Mattson, 2009). Membrane associated oxidative stress/LPO plays a pivotal role in the synaptic dysfunction and neuronal degeneration of AD, both upstream and downstream of Aβ (Hensley et al., 1994; Mattson 2004; Mark et al, 1997; Gwon et al, 2012, McManus et al, 2011). Considerable evidence implicates LPO in AD pathogenesis including: the accumulation of isoprostanes, neuroprostanes, and proteins covalently modified by HNE in afflicted brain regions and cerebrospinal fluid of AD patients (Lovell et al., 1997; Montine et al., 2004; Williams et al., 2006; Bradley et al., 2010); LPO and HNE accumulation occurs in neurons exposed to neurotoxic Aβ species (Mark et al., 1997a; Mark et al., 1997b); and HNE impairs neuronal Ca2+ homeostasis and energy metabolism, rendering neurons vulnerable to excitotoxicity and apoptosis (Keller et al., 1997; Kruman et al., 1997; Mark et al., 1997a; Bruce-Keller et al., 1998). In addition, data suggest that lifestyle factors that increase AD risk (overweight and a sedentary lifestyle) increase brain oxidative stress, while factors that decrease AD risk (regular exercise) reduce brain oxidative stress (Studzinski et al., 2009; Baumgart et al., 2015; Herring et al., 2016; Camandola and Mattson, 2017).
Figure 1.
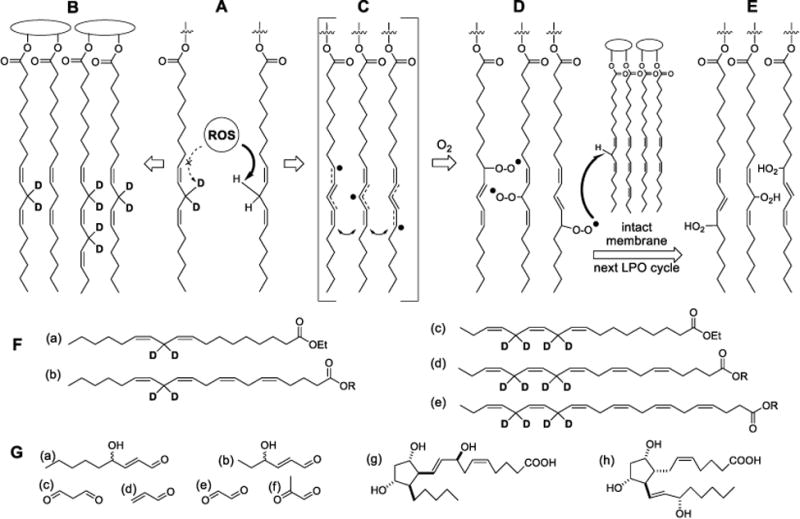
Protective effect of D-PUFAs on lipid peroxidation (LPO). (A) D-PUFAs inhibit the rate-limiting step of reactive oxygen species (ROS)-driven abstraction off a bis-allylic site. (B -E) Lipid bilayer made mostly of D-PUFAs is impervious to LPO. ROS-driven hydrogen abstraction off a bis-allylic site (A) generates resonance-stabilized free radicals (C), which quickly react with abundant molecular oxygen to form lipid peroxyl radicals (D). These newly formed ROS species (L-OO.) abstract hydrogen off a neighboring PUFA molecule (turning themselves into lipid peroxides LOOH (E), thus sustaining the chain reaction of LPO. The chain is eventually terminated by a chain terminating antioxidant or homologous recombination (not shown). (F) 11,11-D2-Lin (a) an omega-6 D-linoleic acid used in this study, is enzymatically converted into 13,13- D2-arachidonic acid (b). 11,11,14,14-D4-Lnn (c), an omega-3 D-linolenic acid used in this study, is enzymatically converted into 13,13,16,16-D4-EPA (d) and 15,15,18,18-D4-DHA (e). (G) lipid peroxides (E), which have greater volume compared to non-oxidized lipids, further decompose through multiple pathways, into numerous species such as reactive carbonyls, for instance 4-HNE (a), 4-HHE (b), malonic dialdehyde (c), acrylic aldehyde (d), oxalic aldehyde (e), methylglyoxal (f), etc. Other classes of products include arachidonic acid-derived isoprostanes (g; iPF2α-IV, or 8-F2-IsoP is one of 64 different isomers), as well as PGF2α (h), a prostaglandin which can be produced both enzymatically and non-enzymatically.
The results of preclinical studies using experimental models of AD provided evidence that antioxidants known to inhibit lipid peroxidation (e.g., vitamin E) or dietary DHA supplementation can ameliorate cognitive deficits and associated Aβ pathology, when administered early in the disease process (Conte et al., 2004; Sung et al., 2004; Lim et al., 2005; Calon and Cole, 2007). However, subsequent trials of vitamin E and DHA in patients with clinical manifestations of AD failed to reveal any clear therapeutic benefit (Quinn et al., 2010; Farina et al., 2012). Possible reasons for this are described elsewhere (Kamat et al., 2008).
A novel approach for suppressing membrane lipid peroxidation was proposed based on the replacement of normal dietary PUFAs with deuterated PUFAs (D-PUFAs) (Shchepinov, 2007). Recent studies have shown that such isotope-reinforced PUFAs are relatively resistant to lipid peroxidation. Substitution of just one or two bis-allylic hydrogens with deuterium at carbon 11 in linoleic acid is sufficient to inhibit PUFA autoxidation in yeast cells (Hill et al., 2011; Hill et al., 2012; Andreyev et al., 2015), and can protect cultured mammalian cells against oxidative stress-induced death (Yang et al., 2016). With regards to potential therapeutic applications for neurodegenerative disorders, it was recently reported that D-PUFA can protect dopaminergic neurons against degeneration, can protect cultured neurons against α-synuclein toxicity in experimental models of Parkinson's disease (Shchepinov et al., 2011; Angelova et al., 2015), and suppresses brain lipid peroxidation and restores cognitive deficit in aldehyde dehydrogenase 2 (ALDH2) deficient mice, a model of cognitive impairment caused by lipid peroxidation (Elharram et al., 2017). In the present study, we tested the hypothesis that dietary D-PUFAs can reduce brain cell lipid peroxidation and ameliorate Aβ pathology and associated cognitive deficits in a mouse model of AD.
Materials and Methods
Animals and Diets
Male APP/PS1 double mutant transgenic mice B6.Cg-Tg(APPswe,PSEN1dE9)85Dbo (Borchelt et al., 1997), and age- and gender-matched wild type controls, were obtained from the Jackson Laboratories (stock # 34832), and were maintained in the National Institute on Aging vivarium. Mice had ad libitum access to food and water. All mice were housed with a 12 h light/dark cycle. Mice were fed pellets of AIN-93 diet modified at Research Diets Inc. (Brunswick, NJ) to contain 1% 11,11-D2-Ethyl linoleate:11,11,14,14-D4-ethyl linolenate (1:1) (D-PUFA diet, Figure 1) or 1% ethyl linoleate:ethyl linolenate (1:1) (H-PUFA, control diet) for a period of 5 months beginning when the mice were 4-7 months old. The complete compositions of the diets can be found in Elharram et al. (2017). Methods for synthesis of the D-PUFAs have been described previously (Shchepinov et al., 2011) and H-PUFA were purchased from Sigma (St. Louis, MO). All animal procedures were approved by the Animal Care and Use Committee of the National Institute on Aging Intramural Research Program (protocol #290-LNS-2019) and complied with NIH guidelines for the care and use of laboratory animals.
Behavioral Tests
After nearly 5 months on the diet, the mice completed four behavioral tests in the following order: open field, elevated plus maze, Morris water maze, and fear conditioning. For all behavioral tests the mice were acclimated to the testing room for 1 hour/day for 2 days prior to testing. During the test, the mice roamed freely in a 27.3cm × 27.3cm × 20.3cm mouse open field box (Med Associates Inc.; ENV-510) for 30 minutes. The XY dimensions of the field were electronically segmented into a center square region (16 × 16 cm) and a remaining outer perimeter region using video tracking software (ANY-maze, Wood Dale, IL) which monitored the animals’ movements in each region during the entire testing period.
For the elevated plus maze, mice were placed in the middle of the maze facing one of the open arms and tracked using ANY-Maze tracking software (ANY-maze, Wood Dale, IL). Mice were allowed to roam for 5 minutes and their entrances into, time spent within, and speed of movements in the open arms, closed arms and the middle area of the maze were quantified. Methods for water maze fear conditioning testing have been described previously (Okun et al., 2012). For the water maze testing, mice were trained for 7 days to find the hidden platform in the upper right quadrant of the pool. The water temperature was 24°C and non-toxic white paint was added to the water to obscure the submerged platform. If the mouse found the platform and stayed on it for 2 seconds, the test would end. Otherwise, the trial would continue for 60 seconds, at which time the experimenter guided the mouse to the platform and the mouse was allowed to stay on the platform for 15 seconds. After the seventh day of training, a 4 hour probe trial was performed in which the platform was removed from the pool and the mouse was allowed to swim for 60 seconds; the probe trial was repeated 24, 48, and 72 hours later.
Following the 72 hour probe, a vision test was performed in a pool with clear water with a small blue flag affixed to the platform, and with the platform placed in a location different than the original testing location. Video tracking software (ANY-maze, Wood Dale, IL) was used to quantify goal latencies, swimming distance and swimming speed. For fear conditioning, a neutral stimulus (sound) was paired with an aversive stimulus (electrical shock 0.5 mA) using a commercially available fear conditioning system (Med Associates Inc., Fairfax, VA). The mice were placed in a chamber with an electrifiable wire floor. On the first day of testing the mice were conditioned for 5 minutes to learn that the sound was associated with the shock. The first 2 minutes were for acclimation to the chamber and during each of the remaining 3 minutes there were exposed to a 30 second sound followed by a shock. The second day the mice were placed in the same context for 5 minutes with no sound or shock – the contextual test. Three hours later the environment was changed to have a smooth white floor and a black teepee structure was put in the chamber – the cued test. Mice were placed in the chamber for 10 minutes; for the first 5 minutes the mice roamed freely and during minutes 5-10 there was a 30 second sound every minute but no shock. Mouse movements and freezing were tracked and analyzed with the fear conditioning system software (Med Associates Inc, Fairfax, VT). After all behavioral tests were completed, the mice were allowed to recover in their home cages for 1 week prior to euthanasia.
Immunohistochemistry
Mice were euthanized by isoflurane inhalation and cervical dislocation, blood was collected from the heart, and the brain and liver were removed. The liver tissue was flash frozen and stored at -8°C. One hemisphere of the brain was immersion fixed 4% paraformaldehyde in phosphate buffered saline (PBS) for 24 hours and then cryoprotected by immersion in a solution of 30% sucrose in PBS. The hippocampus, striatum and cerebral cortex were removed from the other brain hemisphere, flash frozen, and stored at -80°C. Frozen sections (30 micrometers thick) were prepared using a Cryostat (Leica CM 3050, Buffalo Grove, IL) and the sections were mounted on charged slides. For antigen retrieval, sections were treated with 70% formic acid in Tris-buffered saline (TBS). Endogenous peroxidases were quenched by incubation with 0.3% hydrogen peroxide in TBS for 30 minutes. The sections were then processed for Aβ immunostaining using antibody 2454 (Cell Signaling Technologies, Danvers, MA; 1:1200 dilution) using methods described previously (Liu et al., 2010). Images of sections were acquired with a Nikon Eclipse E600 microscope with Ivision software (4.5.4. r8) (Biovision Technologies, Chester Springs, PA). For quantification of Aβ immunoreactivity, ImageJ software was used; the images were converted to 8 bit, inverted, and the average integrated intensity was calculated. Average integrated intensity of plaques was calculated by taking a 1088 × 1200 rectangle from the cortex and hippocampus on 10x images. The average integrated intensity was calculated from ImageJ software.
Deuterium Incorporation
At the end of the study, deuterium incorporation was determined as described previously (Shchepinov et al., 2011). In short, tissues were sonicated and aliquots of tissue samples were lyophilized for deuterium evaluation using isotope ratio mass spectrometry (ir-MS). Samples were placed into silver capsules using CCl4 to avoid contamination by hydrogen. Subsequently, samples were loaded into a Costech Zero Blank Auto-sampler connected to a Thermo/Finnigan thermochemical elemental analyzer (TC/EA) (Thermo Fisher, Bremen, Germany) and analyzed in triplicate as described. Each run was standardized using at least two standards of different isotopic values. Data are presented as delta D/H (in permille, ‰), where delta D/H = (Rsample/Rstandard) × 1000; with R being the ratio between the abundances of deuterium and hydrogen (R = D/H).
Measurement of Aβ Concentrations
Cerebral cortical and hippocampal tissues were sonicated in radioimmunoprecipitation assay (RIPA) buffer containing 200 μM Na3VO4. Protein concentration of the supernatant was determined using BSA protein assay. The V-PLEX Plus Aβ Peptide Panel 1 (4G8) Kit (Meso Scale Discovery, Rockville, MD) was used to quantify concentrations of Aβ38, Aβ40, Aβ42 in the samples. After the plate was blocked, and the detection antibody was added, and 0.41 μg of cerebral cortical protein or 1.04 μg of hippocampal protein from each mouse was assayed in duplicate. Concentrations of immunoreactivity were quantified using an MSD plate reader (Meso Scale Discovery, Rockville, MD).
Measurement of F2-isoprostane and F4-neuroprostane concentrations
The frozen tissue was weighed and added to ice-cold Folch solution and homogenized. The sample tube was flushed with a stream of nitrogen or argon for 30–60 seconds to remove air and then capped. The solution was incubated at room temperature (22–25 °C) for 1 hour to allow maximal extraction of lipids from the tissue. The tube was shaken intermittently for several seconds during this period. Next, aqueous NaCl (0.9%) in ultrapure water was added, and the sample was vortexed vigorously for 1 minute. The tube was then centrifuged for 10 minutes in a tabletop centrifuge to separate aqueous and organic layers. After centrifugation, the top aqueous layer was removed and discarded. The organic layer was evaporated under a stream of nitrogen. The lipids were then resuspended in 1 ml of methanol containing 0.005% BHT and vortexed. Next, 1 ml aqueous KOH (15%, wt/vol) was added, vortexed, and the flask was purged with nitrogen and capped. The mixture was incubated at 37°C for 20 minutes. After incubation, the mixture was acidified to pH 3 with 1N HCl, diluted to 0.01N HCl and 1 ng of the internal standard [2H4]-15-F2t-IsoP was added. The sample was then applied to a C-18 Sep-Pak cartridge that had been prewashed with 5 ml methanol and 5 ml 0.01N HCl. The cartridge was then washed with 10 ml 0.01N HCl, followed by 10 ml heptane, and lipids were then eluted with 10 ml ethyl acetate:heptane (50:50, v/v). The eluate was applied to a silica Sep-Pak cartridge prewashed with ethyl acetate (5 ml). It was then rinsed with 5 ml ethyl acetate and compounds eluted with 5 ml ethyl acetate:methanol (50:50, v/v). The eluate was dried under nitrogen. Compounds were converted to the pentafluorobenzyl (PFB) esters by the addition of 40 μl of a 10% solution of pentafluorobenzyl bromide in acetonitrile and 20 μl of a solution of 10% diisopropylethanolamine in acetonitrile and allowed to incubate for 30 min at 37°C.
Reagents were dried under nitrogen and the residue reconstituted in 30 μl chloroform and 20 μl methanol and divided equally into two 25 ml aliquots. Aliquot 1 was chromatographed on a silica TLC plate to 13 cm in a solvent system of chloroform:methanol (93:7, v/v). The methyl ester of PGF2α was chromatographed on a separate lane and visualized with 10% phosphomolybdic acid in ethanol by heating. The Rf of PGF2α methyl ester in this solvent system was 0.15. Derivatized F2- and F3-IsoPs migrating in the region 1 cm below the PGF2α standard to 3.5 cm above the standard were scraped from the TLC plate, extracted with 1 ml ethyl acetate, and dried under nitrogen. Aliquot 2 was chromatographed on a silica TLC plate to 15 cm in a solvent system of chloroform:ethanol (90:10, v/v). The methyl ester of 17-F4c-NP was chromatographed on a separate lane and visualized with 10% phosphomolybdic acid in ethanol by heating. Derivatized F4-NPs migrating immediately below the TLC standard to 2 cm above the standard were scraped from the TLC plate, extracted with 1 ml ethyl acetate, and dried under nitrogen. Following TLC purification, compounds were converted to trimethylsilyl (TMS) ether derivatives by addition of 20 μl N,O-bis(trimethylsilyl)trifluoroacetamide and 10 μl dimethylformamide. The sample was incubated at 37°C for 10 minutes and then dried under nitrogen. The residue was re-dissolved for GC/MS analysis in 20 μl undecane that had been stored over a bed of calcium hydride.
Mass spectrometric (MS) analysis was performed using an Agilent 5973 Inert Mass Selective Detector that is coupled with an Agilent 6890n Network gas chromatography (GC) system (Agilent Labs, Torrance, CA) that is interfaced with an Agilent computer (Milne et al., 2007). GC was performed using a 15 m, 0.25 μm film thickness, DB-1701 fused silica capillary column (J and W Scientific, Folsom CA). The column temperature was programmed from 190°C to 300°C at 20° per minute. The major ion generated in the negative ion chemical ionization (NICI) mass spectrum of the PFB ester, TMS ether derivative of F2-IsoPs, was the m/z 569 carboxylate anion [M-181 (M-CH2C6F5) ]. The major ion generated in the NICI mass spectrum of the PFB ester, TMS ether derivative of F4-NPs, was the corresponding m/z 593 carboxylate anion. The ion generated by the [2H4]-15-F2t-IsoP internal standard was m/z 573.
Concentrations of endogenous F2-IsoPs/F4-NPs in a biological sample were calculated from the ratio of intensities of the ions m/z 569 to m/z 573. By employing this assay, the lower limit of detection of F2-IsoPs was in the range of 4pg using an internal standard with a blank of 3 parts per thousand. The precision of this assay in biological fluids was +6% and the accuracy 94%.
Measurement of PUFA Concentrations
Extraction was performed using a modified Bligh-Dyer (BD) procedure (Axelsen and Murphy, 2010). Tissue samples (∼10 mg) were weighed and homogenized with a tip sonicator (F60 Sonic Dismembrator, Fisher Scientific, Pittsburgh, PA) for 60 s in 760 μl of the BD monophase (400 μl methanol, 200 μl dichloromethane, and 160 μl of 5 mM ammonium chloride) in 1.8 ml high recovery vial (Microsolv Eatontown, NJ). 10 μl volumes of an internal standard (0.1 mM d8-ARA) and 10 μl of a butylated hydroxytoluene solution (100 μM in methanol) were added prior to sonication. After sonication, 200 μl of dichloromethane and 160 μl of water were added to break the monophase and the vial was vortexed.
The three resulting phases were separated by 2 minutes of low speed centrifugation. The lower (dichloromethane) phase (∼1-2 ml) was withdrawn, transferred to a 5 ml glass tube (Fisher Scientific, Pittsburgh, PA), and dried under argon. The residue was resuspended in 85% MeOH in water with 0.5 M Na2OH (1 ml final volume) and saponified by incubating at 80°C for one hour. Following saponification, samples were cooled to room temperature, acidified with 150 μl of 5M HCl and evaporated under argon. The residue was resuspended in 150 μl methanol for analysis.
Samples (5 μl) were injected onto a 1 × 250 mm Zorbax 300SB 5 μm C18 column (Agilent, Santa Clara, CA). Solvent A was 8% acetonitrile, 92% water, and 0.1% formic acid. Solvent B was 100% acetonitrile and 0.1% formic acid. The mobile phase was pumped at 0.1 ml/min as the composition was changed linearly from 8% to 80% solvent B over 25 min, and held at 80% B for 9.5 min. The eluent was alkalinized post-column with 0.15 M ammonium hydroxide in methanol flowing at 50 μl/min before being introduced via electrospray ionization into an ABI 4000 mass spectrometer (Sciex, Toronto, Canada) operating in negative ion mode with a declustering potential of –70V, an ionization energy of –4500V, and a 200°C drying gas. The collision gas was nitrogen at 7 psi when operating in multiple reaction monitoring (MRM) or product ion scanning mode, with the transitions and collision energy values listed in Table 1. The collection time for each transition was 100 msec.
Quantification of peaks was done using Analyst software (AB Sciex). The peak area for each fatty acid transition was divided by the peak area of the d8-ARA internal standard. A calibration curve for d8-ARA was prepared using unlabeled neat ARA, and the final results were divided by tissue weight.
Data and Statistical Analysis
Statistical analysis was run on GraphPad Prism and either t-tests or one- or two-way ANOVAs were run on the data. Neuman-Keuls post-hoc tests were performed for t-tests and one-way ANOVAs and Bonferroni post hoc tests were performed for two-way ANOVAs. A p value < 0.05 was considered statistically significant.
Results
Analyses were performed on wild type (WT) mice that had been maintained for 5 months on the H-PUFA diet (n = 11), APP/PS1 double mutant transgenic (AD) mice that been maintained for 5 months on either the H-PUFA diet (n = 14) or D-PUFA diet (n = 13). At the end of the study, efficient D-PUFA incorporation was confirmed by measuring deuterium content on sections of the cortex and hippocampus as well as liver by irMS. The tissue deuterium contents were: in the cortex, D-PUFA (32070 ± 3126‰) versus H-PUFA (-143.1 ± 18.21‰); in the hippocampus, D-PUFA (33230 ± 1843‰) versus H-PUFA (-149.2 ± 12.3‰); in the liver, D-PUFA (29082 ± 2958‰) versus H-PUFA (-149.9 ± 32.77‰). For all measurements, p < 0.0001 (t-test, unpaired, nonparametric, Mann Whitney correction). The difference between the D-PUFA and H-PUFA groups corresponds to a level of D-PUFA incorporation in the total PUFA pool of approximately 40% (Shchepinov et al., 2014; Elharram et al., 2017), and correlates with PUFA-specific incorporation measurements (vide infra). This level of D-PUFA substitution is biologically relevant as approximately 10-20% is sufficient to terminate LPO (Hill et al., 2012).
For behavioral analyses, mice were subjected to two tests of anxiety-like behavior (open field and elevated plus maze) and two tests of learning and memory (water maze and fear conditioning). In the open field test, there were no significant differences between AD mice in the H-PUFA and D-PUFA diet groups in distance traveled (Figure 2A), number of entries into the center zone (Figure 2B) or time spent in the center zone (Figure 2C). There were also no significant differences between WT mice, and AD mice in either diet group, except for the first 5 minutes in the open field, during which time AD mice in the D-PUFA group traveled further than WT mice. In the elevated plus maze there were no significant differences between the three groups of mice in time spent in open and closed arms (Figure 2D) or time spent in the open arms (Figure 2E), although there was a non-significant trend for AD mice on the D-PUFA diet to spend more time in the open arms.
Figure 2.
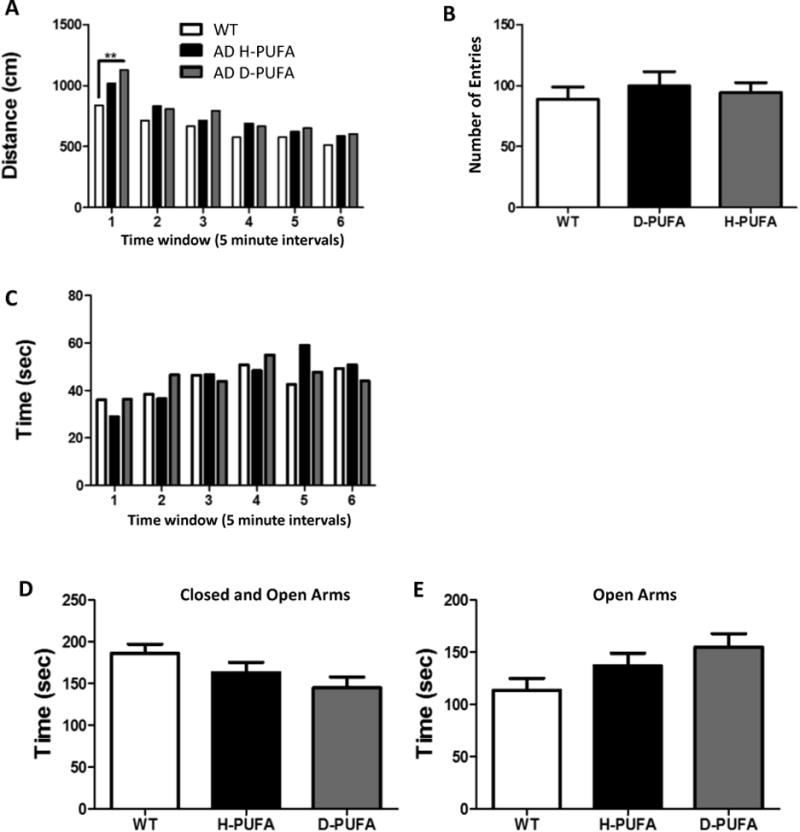
A D-PUFA diet does not affect levels of anxiety-related behaviors in AD mice in the open field and elevated plus maze tests. (A-C) Results of open field test showing total distance traveled (A), number of entries into the center zone (B) and the time spent in the center zone (C). (D and E) Results of the elevated plus maze test showing time spent in both the closed and open arms (D) and time spent in the open arms (E). Values are the mean and SEM of 11 wild type (WT) mice on the H-PUFA diet, 14 AD mice on the H-PUFA diet and 13 AD mice on the D-PUFA diet. *p<0.05.
Mice in all three groups learned the location of the hidden platform in the Morris water maze as indicated by decreases in swimming distance (Figure 4A) and time required to locate the platform (Figure 3B) over the 7-day training period. WT mice learned the platform location more quickly (by day 4) than AD mice (Figure 3A, 3B). AD mice in the D-PUFA group exhibited a 1-2 day delay in learning the location of the hidden platform (on training days 4 and 5), but by training day seven AD mice in both diet groups exhibited identical times required to find the platform and distance traveled. In the probe trials performed at 4 hours and 24 hours after the last day of training, AD mice on the H-PUFA and D-PUFA diets spent less time in the target quadrant compared to WT mice, indicating poor retention of the memory of the platform location (Figure 3C). Similarly, in the 4-hour probe trial the AD mice in the H-PUFA and D-PUFA diet groups entered the platform area less frequently compared to the WT mice (Figure 3D). There were no significant differences between the AD mice in the H-PUFA and D-PUFA groups in the probe trials (Figure 3C, D). For the fear conditioning tests, there were no differences between the three groups for the training day (Figure 4A), contextual test (Figure 4B), and the cued test (Figure 4C).
Figure 4.
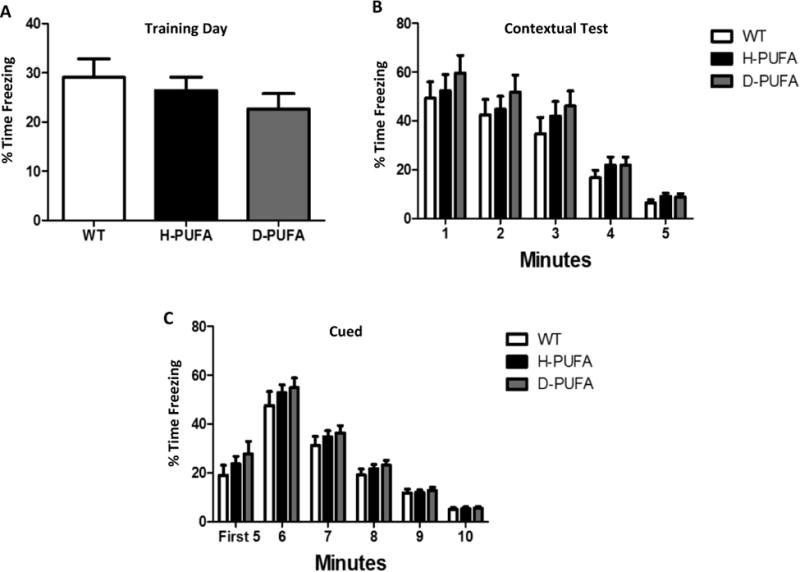
Wild type mice and AD mice on either H-PUFA or D-PUFA diets perform similarly in fear conditioning tests. (A) Percentage of time that the mice spent freezing on the training day. (B) Percentage of time that the mice spent freezing on day 2 in the absence of a shock (context setting). (C) Percentage of time that the mice spent freezing on day 2 during and after exposure to the same sound used as the cue on the preceding training day. Values are the mean and SEM of 11 wild type (WT) mice on the H-PUFA diet, 14 AD mice on the H-PUFA diet and 13 AD mice on the D-PUFA diet.
Figure 3.
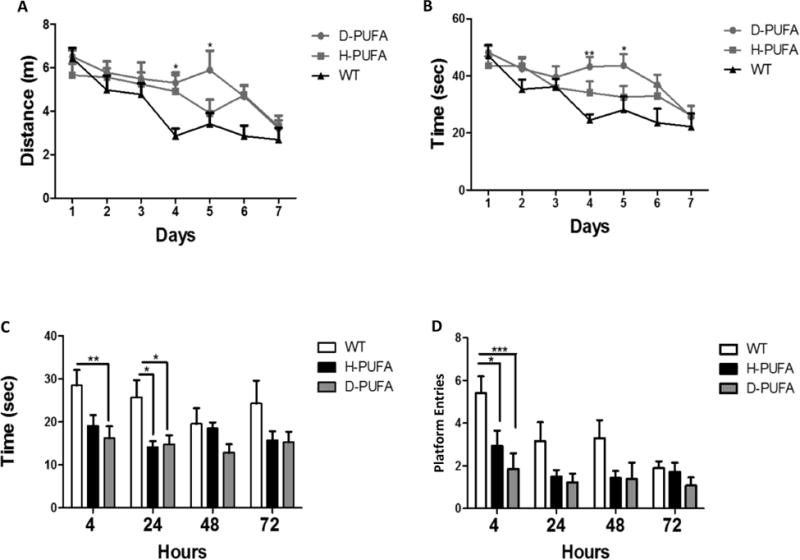
A D-PUFA diet does not ameliorate spatial learning deficits in AD mice. (A and B) Results of water maze testing during the 7 days of learning trials showing distance traveled to reach the hidden platform (A) and time required to locate the hidden platform (B). (C and D) Results of water maze probe trials performed at 4, 24, 48 and 72 hours after the last (day 7) learning trial showing time spent in the quadrant of the pool where the platform had been during the training trials (C) and number of entries into the exact location of the platform (D). Values are the mean and SEM of 11 wild type (WT) mice on the H-PUFA diet, 14 AD mice on the H-PUFA diet and 13 AD mice on the D-PUFA diet. *p<0.05, **p<0.01, ***p<0.001 compared to the values for WT mice.
After the behavioral testing and a week rest period, the mice were euthanized at 9-12 months of age and brain tissue processed for either histological or biochemical analyses. We immunostained brain sections from AD mice with an Aβ antibody and evaluated the number of plaque-like depositions in the hippocampus and cerebral cortex (Figure 5A-C). As a negative control, we immunostained brain sections from a WT mouse, and no Aβ deposits were evident (Figure 5A). Relative Aβ plaque loads were not significantly different in either brain region in AD mice in the H-PUFA and D-PUFA diet groups (Figure 5D, 5E). To determine whether the D-PUFA diet might affect APP processing, we quantified concentrations of Aβ38, Aβ40 and Aβ42 in the cerebral cortex and hippocampus (Figure 6) of AD mice on the H-PUFA and D-PUFA diets. In the cerebral cortex, there were no significant differences in any of the Aβ species between the H-PUFA and D-PUFA diet groups (Figure 6A-D). In the hippocampus, the amounts of Aβ40 and Aβ38 were significantly lower in the AD mice maintained on the D-PUFA diet compared to those maintained on the H-PUFA diet (Figure 6F). There was a nonsignificant trend towards reduced Aβ42 levels in the hippocampus of AD mice fed D-PUFA compared to those fed H-PUFA (Figure 6G).
Figure 5.
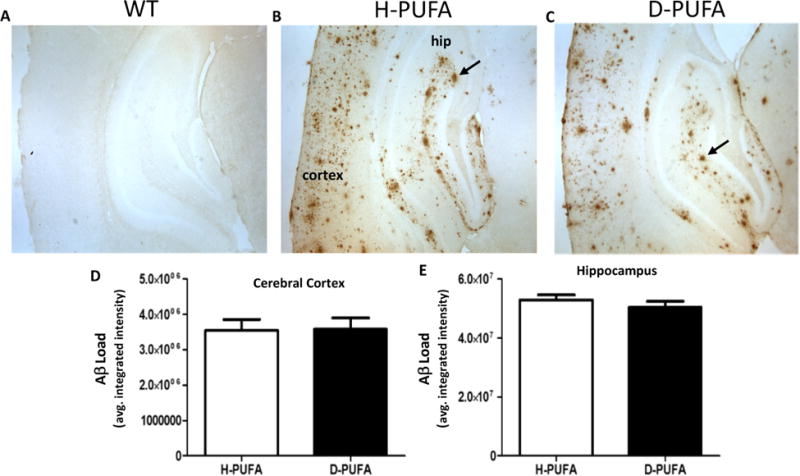
The amounts of Aβ deposits in the cerebral cortex and hippocampus are similar in AD mice fed the D-PUFA diet and AD mice fed the H-PUFA diet. (A-C) Representative images of Aβ immunoreactivity in the cerebral cortex and hippocampus of a wild type mouse (A), an AD mouse that had been maintained on the H-PUFA diet for 5 months (B), and an AD mouse that had been maintained on the D-PUFA diet for 5 months (C). Arrows point to plaque-like Aβ deposits. (D and E) Results of quantification of Aβ load in the cerebral cortex and hippocampus. Values are the mean and SEM of 14 AD mice on the H-PUFA diet and 13 AD mice on the D-PUFA diet.
Figure 6.
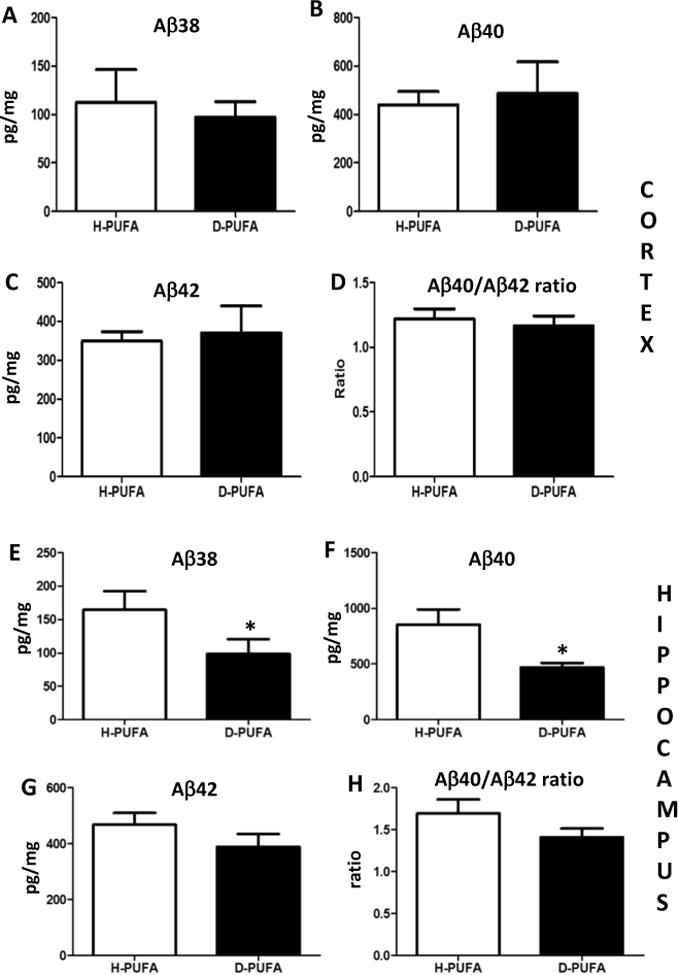
Levels of soluble Aβ40 in the hippocampus are lower in AD mice fed D-PUFA compared to AD mice fed H-PUFA. (A-D) Levels of Aβ38, Aβ40, Aβ42 and the Aβ40/Ab42 ratio in samples of cerebral cortex tissue from AD mice that had been maintained for 5 months on either H-PUFA or D-PUFA diets. (E-H) Levels of Aβ38, Aβ40, Aβ42 and the Aβ40/Aβ42 ratio in samples of hippocampal tissue from AD mice that had been maintained for 5 months on either H-PUFA or D-PUFA diets. Values are the mean and SEM of 14 AD mice on the H-PUFA diet and 13 AD mice on the D-PUFA diet. *p<0.05.
We previously reported that when mice are fed D-PUFA for 3 months, D-PUFA are incorporated into major neuronal membrane lipid species known to be susceptible to lipid peroxidation including ARA and DHA (Shchepinov et al., 2014). To confirm that D-PUFA were incorporated into these lipid species throughout the brains of the APP/PS1 double mutant transgenic (AD) mouse line used in the present study, we measured total and D-containing levels of ARA, DHA, 18:2, 18:3n-3, 16:0 and 18:0 in cerebral cortex, hippocampus, striatum, cerebellum, liver and adipose tissues. D-PUFA accounted for approximately 30-40% of ARA and 50-70% of DHA in all four brain regions (Figure 7A-D). In the liver and adipose tissues there were high levels of incorporation of D-PUFA into DHA, 18:2 and 18:3n-3 (Figure 7E, F).
Figure 7.
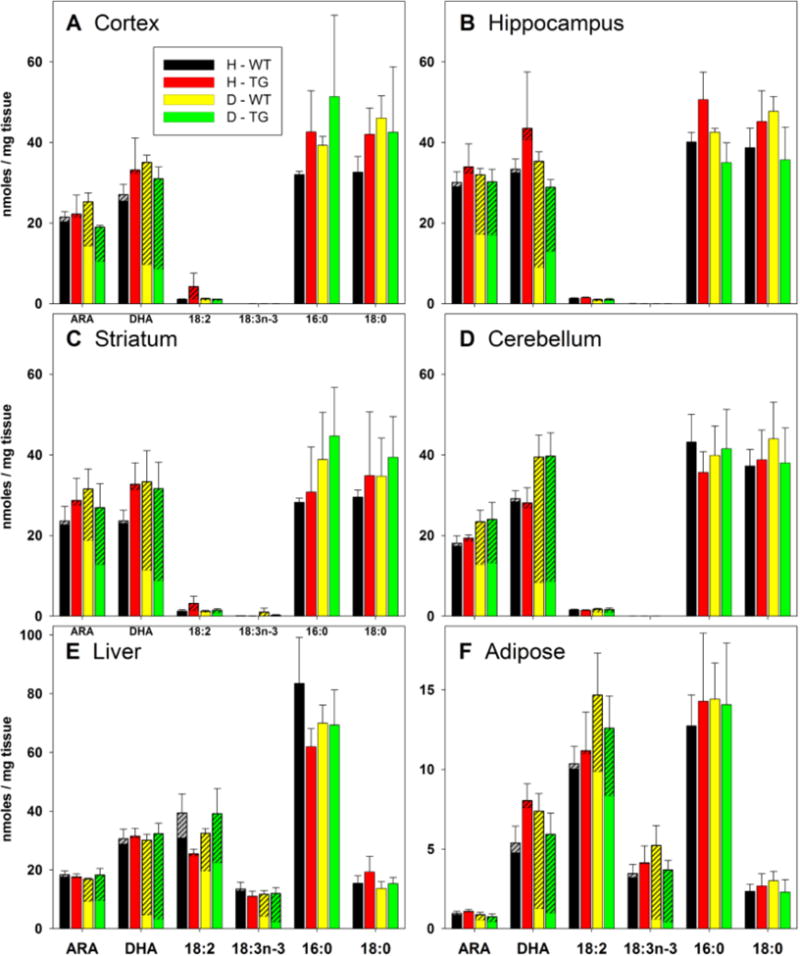
Concentrations of H- and D- fatty acids in brain, liver and adipose tissues of wild type mice (WT) and APP/PS1 double mutant transgenic mice (TG) that had been fed either H-PUFA (H) or D-PUFA (D) diets for 5 months. The overall bar height represents the mean total concentration of the fatty acid and error bars represent standard errors for the total concentration. The hatched portion of each bar represents the portion of the total fatty acid concentration comprised of deuterated isomers. Note that small portions of some bars in the H-WT and H-TG bars are hatched; these reflect baseline noise, and are invariably smaller than the standard errors of the means (shown as error bars). n = 5 for the D-WT group; n = 4 for the other groups.
Because the D-PUFA diet had little or no impact on cognition or Aβ plaque pathology in the AD mice, we next determined whether the mice on the D-PUFA diet did, in fact, exhibit reduced lipid peroxidation in their brain tissues. Accordingly, we quantified concentrations of F4-neuroprostanes (generated by peroxidation of DHA) and F2-isoprostanes (generated by peroxidation of ARA) in cerebral cortical and liver tissues from AD and WT mice that had been maintained for 5 months on H-PUFA or D-PUFA diets (4 or 5 mice/group). Concentrations of F4-neuroprostanes were significantly lower in cerebral cortex tissues from WT and AD mice in the D-PUFA groups compared to mice in the H-PUFA groups (Figure 8A). Concentrations of F2-isoprostanes were significantly lower in the livers of WT and AD mice on the D-PUFA diet compared to those on the H-PUFA diet (Figure 8B). There were non-significant trends towards reduced concentrations of F2-isoprostanes in the cerebral cortex of WT and AD mice in the D-PUFA groups compared to those in the H-PUFA groups (Figure 8C).
Figure 8.
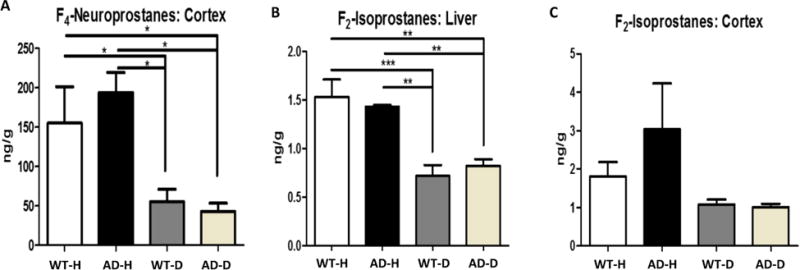
Mice fed D-PUFA exhibit reductions in levels of lipid peroxidation products in the brain and liver compared to mice fed H-PUFA. (A) Levels of F4-neuroprostanes in cerebral cortical tissue samples from WT and AD mice that had been maintained on H-PUFA or D-PUFA diets for 5 months. (B) Levels of F2-isoprostanes in liver tissue samples from WT and AD mice that had been maintained on H-PUFA or D-PUFA diets for 5 months. (C) Levels of F2-isoprostanes in cerebral cortical tissue samples from WT and AD mice that had been maintained on H-PUFA or D-PUFA diets for 5 months. Values are the mean and SEM of 4 WT mice fed H-PUFA, 5 WT mice fed D-PUFA, 4 AD mice fed H-PUFA and 4 AD mice fed D-PUFA. AD mice on the H-PUFA diet and 13 AD mice on the D-PUFA diet. *p<0.05, **p<0.01, ***p<0.001.
Discussion
Because aggregating Aβ damages and kills neurons by a mechanism involving membrane lipid peroxidation (Hensley et al., 1994; Mark et al., 1997a; Mark et al., 1997b; Murray et al., 2005; Murray et al., 2007; Liu et al., 2008; Axelsen et al., 2011; Eskici and Axelsen, 2012), and because D-PUFA can suppress membrane lipid peroxidation (Hill et al., 2011; Hill et al., 2012; Andreyev et al., 2015), we tested the hypothesis that dietary D-PUFA would ameliorate Aβ pathology and cognitive deficits in the APP/PS1 double mutant transgenic mouse model of AD.
Deuterium was incorporated into DHA and ARA in the hippocampus and cerebral cortex in AD mice on the D-PUFA diet, and the D-PUFA diet resulted in significant reductions in concentrations of lipid peroxidation products in the brain (Ara- derived F2-isoprostanes and DHA-derived F4-neuroprostanes), demonstrating that the D-PUFA did in fact enter the brain where it was incorporated into membrane lipids and suppressed neuronal lipid peroxidation. Moreover, levels of Aβ40 and Aβ38 were significantly lower and Aβ42 marginally lower in the hippocampus of AD mice fed D-PUFA, suggesting a role for lipid peroxidation upstream of Aβ production in this mouse model. The AD mice exhibited impaired learning and memory retention in a water maze test compared to WT mice, but the D-PUFA diet had no significant impact on these cognitive deficits. Because previous studies have shown that Aβ accumulation and lipid peroxidation can impair synaptic plasticity and learning and memory in experimental models of AD (Mattson, 2004, 2009), it is unclear why the D-PUFA diet did not ameliorate behavioral deficits in the present study despite reducing lipid peroxidation and Aβ levels. One possibility is that the time of initiation and/or duration of the D-PUFA diet intervention were not sufficient to reduce lipid peroxidation and Aβ accumulation to levels sufficient to ameliorate cognitive deficits. In addition to inducing membrane-associated oxidative stress (Mattson, 2009), it has been proposed that Aβ oligomers impair cognition by binding to specific synaptic proteins and impairing their function (Viola and Klein, 2015). The D-PUFA may not impact the latter mechanism.
Although we did not discern significant effects of the D-PUFA on learning and memory in the APP/PS1 double mutant transgenic mice, this does not preclude potential beneficial effects of the D-PUFA on cognition in other mouse models. In the APP/PS1 mice we used there is considerable Aβ deposition, but the fact that the mutant human APP and PS1 are overexpressed in an unregulated manner may result in masking of potential therapeutic effects that would be discernable in more relevant models such as APP/PS1 double mutant knock-in mice (Abdul et al., 2008) and APP mutant knockin mice (Masuda et al., 2016). The APP/PS1 double mutant knock-in mice have been shown to exhibit increased lipid peroxidation and oxidative protein modification in their brains, and cognitive deficits that can be ameliorated by administration of the antioxidant N-acetyl-L-cysteine (Abdul et al., 2008; Huang et al., 2010; Webster et al., 2013). It would therefore be of considerable interest to evaluate D-PUFA in the latter mouse model of AD. Also, in addition to increasing Aβ production, increased generation of N- and C-terminal APP fragments can trigger neuronal degeneration (Saganich et al., 2006; Nhan et al., 2015) by mechanisms that may be independent of lipid peroxidation. If such APP fragments contribute to cognitive impairment in APP/PS1 double mutant transgenic mice used in the present study, then D-PUFA may not counteract that neurotoxic mechanism.
Previous findings have suggested roles for membrane lipid peroxidation in the pathogenesis of AD both downstream and upstream of accumulation of neurotoxic forms of Aβ. During the process of aggregation, Aβ generates reactive oxygen species (Hensley et al., 1994) and, when the peptide aggregation occurs on cell membranes, the ROS induce membrane lipid peroxidation (Mark et al., 1997a). By impairing the function of ion-motive ATPases and glutamate and glucose transporters, the lipid peroxidation product 4-hydroxynonenal may play a particularly prominent role in the synaptic dysfunction and neuronal degeneration caused by Aβ (Mark et al., 1997a, 1997b; Keller et al., 1997a, 1997b). In addition to HNE, HHE is another highly reactive and potentially neurotoxic lipid peroxidation product implicated in AD pathogenesis (Bradley et al., 2012). However, HHE, and other aldehydes, as well as their conjugates, are less often assessed due to the unreliable analytical techniques available for their detection (Long et al., 2010). Our finding that dietary D-PUFA can reduce brain lipid peroxidation in a mouse model of AD in which Aβ accumulates in plaque-like deposits suggests a potential for this therapeutic approach in protecting neurons against Aβ-induced degeneration. It has been reported that Aβ can induce LPO (Mark et al., 1997a, 1997b; Mattson, 2009). Moreover, previous studies have shown that lipid peroxidation can alter the proteolytic processing of the β-amyloid precursor protein in a manner that increases the production of Aβ. Specifically, oxidative lipid modification the γ-secretase complex protein nicastrin enhances activity of γ-secretase resulting in increased Aβ production (Gwon et al., 2012). Thus, by suppressing lipid peroxidation, D-PUFA would be expected to reduce the levels of Aβ, a possibility consistent with our finding that Aβ40 and Aβ38 levels were significantly reduced in the hippocampus of APP/PS1 mutant transgenic mice that consumed D-PUFA. LPO is also implicated in the pathogenesis of Parkinson's disease, both upstream and downstream of intracellular accumulation of α-synuclein aggregates (Angelova et al., 2015; Zhang et al., 2017).
Mice lacking ALDH2 exhibit accumulation of lipid peroxidation products in various tissues including the brain. The increased cerebral lipid peroxidation in ALDH2 deficient mice is associated with cognitive impairment that can be ameliorated by feeding the mice D-PUFA (Elharram et al., 2017). It is not clear why the D-PUFA diet did not improve cognition in APP/PS1 mice with Aβ pathology in the present study, despite reducing lipid peroxidation. It will therefore be of interest to determine the relative contributions of lipid peroxidation and other molecular pathways of Aβ-associated neuropathology in AD.
Highlights.
Dietary isotope-reinforced (deuterated) PUFA (D-PUFA) are incorporated into arachidonic acid and docosohexanoic acid in the brain of APP/PS1 mutant AD mice.
AD mice fed D-PUFA exhibit reduced cerebral lipid peroxidation compared to AD mice fed H-PUFA.
AD mice fed D-PUFA exhibit reduced levels of amyloid β-peptides in the hippocampus compared to AD mice fed H-PUFA.
The D-PUFA diet did not ameliorate spatial learning and memory deficits in the AD mice.
Acknowledgments
This work was supported by the Intramural Research Program of the National Institute on Aging and by Retrotope Inc.
Sources of financial support related to the manuscript being submitted are included in the Acknowledgements statement.
Footnotes
Conflict of interest statement: M. S. S. is an employee of Retrotope Inc. No other authors have any conflicts of interest to declare.
3. The data contained in the manuscript being submitted have not been previously published, have not been submitted elsewhere and will not be submitted elsewhere while under consideration at Neurobiology of Aging.
4. A statement is included in the Methods section verifying that appropriate approval and procedures were used concerning animals.
5. All authors have reviewed the contents of the manuscript being submitted, approve of its contents and validate the accuracy of the data.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abdul HM, Sultana R, St Clair DK, Markesbery WR, Butterfield DA. (2008) Oxidative damage in brain from human mutant APP/PS-1double knock-in mice as a function of age. Free Radic Biol Med. 2008;45:1420–1425. doi: 10.1016/j.freeradbiomed.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreyev AY, Tsui HS, Milne GL, Shmanai VV, Bekish AV, Fomich MA, Pham MN, Nong Y, Murphy AN, Clarke CF, Shchepinov MS. Isotope-reinforced polyunsaturated fatty acids protect mitochondria from oxidative stress. Free Radic Biol Med. 2015;82:63–72. doi: 10.1016/j.freeradbiomed.2014.12.023. [DOI] [PubMed] [Google Scholar]
- Angelova PR, Horrocks MH, Klenerman D, Gandhi S, Abramov AY, Shchepinov MS. Lipid peroxidation is essential for α-synuclein-induced cell death. J Neurochem. 2015;133:582–589. doi: 10.1111/jnc.13024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axelsen PH, Komatsu H, Murray IVJ. Oxidative Stress and Cell Membranes in the Pathogenesis of Alzheimer's Disease. Physiol. 2011;26:54–69. doi: 10.1152/physiol.00024.2010. [DOI] [PubMed] [Google Scholar]
- Baumgart M, Snyder HM, Carrillo MC, Fazio S, Kim H, Johns H. Summary of the evidence on modifiable risk factors for cognitive decline and dementia: A population-based perspective. Alzheimers Dement. 2015;11:718–726. doi: 10.1016/j.jalz.2015.05.016. [DOI] [PubMed] [Google Scholar]
- Bertram L, Lill CM, Tanzi RE. The genetics of Alzheimer disease: back to the future. Neuron. 2010;68:270–281. doi: 10.1016/j.neuron.2010.10.013. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Mattson MP. Neuronal calcium mishandling and the pathogenesis of Alzheimer's disease. Trends Neurosci. 2008;31:454–463. doi: 10.1016/j.tins.2008.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borchelt DR, Ratovitski T, van Lare J, Lee MK, Gonzales V, Jenkins NA, Copeland NG, Price DL, Sisodia SS. Accelerated amyloid deposition in the brains of transgenic mice expressing mutant presenilin 1 and amyloid precursor proteins. Neuron. 1997;19:939–945. doi: 10.1016/s0896-6273(00)80974-5. [DOI] [PubMed] [Google Scholar]
- Bradley MA, Markesbery WR, Lovell MA. Increased levels of 4-hydroxynonenal and acrolein in the brain in preclinical Alzheimer disease. Free Radic Biol Med. 2010;48:1570–1576. doi: 10.1016/j.freeradbiomed.2010.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley MA, Xiong-Fister S, Markesbery WR, Lovell MA. Elevated 4-hydroxyhexenal in Alzheimer's disease (AD) progression. Neurobiol Aging. 2012;33:1034–1044. doi: 10.1016/j.neurobiolaging.2010.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Li YJ, Lovell MA, Kraemer PJ, Gary DS, Brown RR, Markesbery WR, Mattson MP. 4-hydroxynonenal, a product of lipid peroxidation, damages cholinergic neurons and impairs visuospatial memory in rats. J Neuropathol Exp Neurol. 1998;57:257–267. doi: 10.1097/00005072-199803000-00007. [DOI] [PubMed] [Google Scholar]
- Butterfield DA, Boyd-Kimball D. Amyloid beta-peptide(1-42) contributes to the oxidative stress and neurodegeneration found in Alzheimer disease brain. Brain Pathol. 2004;14:426–432. doi: 10.1111/j.1750-3639.2004.tb00087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camandola S, Mattson MP. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017 Apr 24; doi: 10.15252/embj.201695810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole GM, Frautschy SA. Docosahexaenoic acid protects from amyloid and dendritic pathology in an Alzheimer's disease mouse model. Nutr Health. 2006;18:249–259. doi: 10.1177/026010600601800307. [DOI] [PubMed] [Google Scholar]
- Conte V, Uryu K, Fujimoto S, Yao Y, Rokach J, Longhi L, Trojanowski JQ, Lee VM, McIntosh TK, Praticò D. Vitamin E reduces amyloidosis and improves cognitive function in Tg2576 mice following repetitive concussive brain injury. J Neurochem. 2004;90:758–764. doi: 10.1111/j.1471-4159.2004.02560.x. [DOI] [PubMed] [Google Scholar]
- Dobretsov GE, Borschevskaya TA, Petrov VA, Vladimirov YA. The increase of phospholipid bilayer rigidity after lipid peroxidation. FEBS Lett. 1977;84:125–128. doi: 10.1016/0014-5793(77)81071-5. [DOI] [PubMed] [Google Scholar]
- Elharram A, Czegledy NM, Golod M, Milne GL, Pollock E, Bennett BM, Shchepinov MS. Deuterium-reinforced polyunsaturated fatty acids improve cognition in a mouse model of sporadic Alzheimer's disease. FEBS J. 2017 Oct 12; doi: 10.1111/febs.14291. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskici G, Axelsen PH. Copper and Oxidative Stress in the Pathogenesis of Alzheimer's Disease. Biochem. 2012;51:6289–6311. doi: 10.1021/bi3006169. 2012. [DOI] [PubMed] [Google Scholar]
- Farina N, Isaac MG, Clark AR, Rusted J, Tabet N. Vitamin E for Alzheimer's dementia and mild cognitive impairment. Cochrane Database Syst Rev. 2012;11:CD002854. doi: 10.1002/14651858.CD002854.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueraud F, Atalay M, Bresqen N, Cipak A, Eckl PM, Huc L, Jouanin I, Siems W, Uchida K. Chemistry and biochemistry/of lipid peroxidation products. Free Radic Res. 2010;44:1098–1124. doi: 10.3109/10715762.2010.498477. [DOI] [PubMed] [Google Scholar]
- Greenberg ME, Li XM, Guqui BG, Gu X, Qin J, Salomon RG, Hazen SL. The lipid whisker model of the structure of oxidized cell membranes. J Biol Chem. 2008;283:2385–2396. doi: 10.1074/jbc.M707348200. [DOI] [PubMed] [Google Scholar]
- Gwon AR, Park JS, Arumugam TV, Kwon YK, Chan SL, Kim SH, Baik SH, Yang S, Yun YK, Choi Y, Kim S, Tang SC, Hyun DH, Cheng A, Dann CE, 3rd, Bernier M, Lee J, Markesbery WR, Mattson MP, Jo DG. Oxidative lipid modification of nicastrin enhances amyloidogenic gamma-secretase activity in Alzheimer's disease. Aging Cell. 2012;11:559–568. doi: 10.1111/j.1474-9726.2012.00817.x. http://www.ncbi.nlm.nihgov/pubmed/22404891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley K, Carney JM, Mattson MP, Aksenova M, Harris M, Wu JF, Floyd RA, Butterfield DA. A model for beta-amyloid aggregation and neurotoxicity based on free radical generation by the peptide: relevance to Alzheimer disease. Proc Natl Acad Sci U S A. 1994;91:3270–3274. doi: 10.1073/pnas.91.8.3270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herring A, Münster Y, Metzdorf J, Bolczek B, Krüssel S, Krieter D, Yavuz I, Karim F, Roggendorf C, Stang A, Wang Y, Hermann DM, Teuber-Hanselmann S, Keyvani K. Late running is not too late against Alzheimer's pathology. Neurobiol Dis. 2016;94:44–54. doi: 10.1016/j.nbd.2016.06.003. [DOI] [PubMed] [Google Scholar]
- Hill S, Hirano K, Shmanai VV, Marbois BN, Vidovic D, Bekish AV, Kay B, Tse V, Fine J, Clarke CF, Shchepinov MS. Isotope-reinforced polyunsaturated fatty acids protect yeast cells from oxidative stress. Free Radic Biol Med. 2011;50:130–138. doi: 10.1016/j.freeradbiomed.2010.10.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill S, Lamberson CR, Xu L, To R, Tsui HS, Shmanai VV, Bekish AV, Awad AM, Marbois BN, Cantor CR, Porter NA, Clarke CF, Shchepinov MS. Small amounts of isotope-reinforced polyunsaturated fatty acids suppress lipid autoxidation. Free Radic Biol Med. 2012;53:893–906. doi: 10.1016/j.freeradbiomed.2012.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Q, Aluise CD, Joshi G, Sultana R, St Clair DK, Markesbery WR, Butterfield DA. Potential in vivo amelioration by N-acetyl-L-cysteine of oxidative stress in brain in human double mutant APP/PS-1 knock-inmice: toward therapeutic modulation of mild cognitive impairment. J Neurosci Res. 2010;88:2618–2629. doi: 10.1002/jnr.22422. [DOI] [PubMed] [Google Scholar]
- Jicha GA, Markesbery WR. Omega-3 fatty acids: potential role in the management of early Alzheimer'sdisease. Clin Interv Aging. 2010;5:45–61. doi: 10.2147/cia.s5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan VE, Borisenko GG, Tyurina YY, Tyurin VA, Jiang J, Potapovich AI, Kini V, Amoscato AA, Fujii Y. Oxidative lipidomics of apoptosis: redox catalytic interactions of cytochrome c with cardiolipin and phosphatidylserine. Free Rad Biol Med. 2004;37:1963–1985. doi: 10.1016/j.freeradbiomed.2004.08.016. [DOI] [PubMed] [Google Scholar]
- Kamat CD, Gadal S, Mhatre M, Williamson KS, Pye QN, Hensley K. Antioxidants in central nervous system diseases: preclinical promise and translational challenges. J Alzheimers Dis. 2008;15:473–493. doi: 10.3233/jad-2008-15314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller JN, Pang Z, Geddes JW, Begley JG, Germeyer A, Waeg G, Mattson MP. Impairment of glucose and glutamate transport and induction of mitochondrial oxidative stress and dysfunction in synaptosomes by the lipid peroxidation product 4-hydroxynonenal. J Neurochem. 1997a;69:273–284. doi: 10.1046/j.1471-4159.1997.69010273.x. [DOI] [PubMed] [Google Scholar]
- Keller JN, Mark RJ, Bruce AJ, Blanc E, Rothstein JD, Uchida K, Waeg G, Mattson MP. 4-Hydroxynonenal, an aldehydic product of membrane lipid peroxidation, impairs glutamate transport and mitochondrial function in synaptosomes. Neuroscience. 1997b;80:685–696. doi: 10.1016/s0306-4522(97)00065-1. [DOI] [PubMed] [Google Scholar]
- Kruman I, Bruce-Keller AJ, Bredesen D, Waeg G, Mattson MP. Evidence that 4-hydroxynonenal mediates oxidative stress-induced neuronal apoptosis. J Neurosci. 1997;17:5089–5100. doi: 10.1523/JNEUROSCI.17-13-05089.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Lan, Michelsen Klaus, Elena N, Kitova, Schnier Paul D, Brown Alex, Klassen John S. Deuterium Kinetic Isotope Effects on the Dissociation of a Protein-Fatty Acid Complex in the Gas Phase. J Am Chem Soc. 2012;134:5931–5937. doi: 10.1021/ja300092w. [DOI] [PubMed] [Google Scholar]
- Leal SL, Yassa MA. Perturbations of neural circuitry in aging, mild cognitive impairment, and Alzheimer's disease. Ageing Res Rev. 2013;12:823–831. doi: 10.1016/j.arr.2013.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim P, Wuenschell GE, Holland V, Lee DH, Pfeifer GP, Rodriguez H, Termini J. Peroxyl radical mediated oxidative DNA base damage: implications for lipid peroxidation induced mutagenesis. Biochemistry. 2004;43:15339–15348. doi: 10.1021/bi048276x. [DOI] [PubMed] [Google Scholar]
- Liu D, Pitta M, Lee JH, Ray B, Lahiri DK, Furukawa K, Mughal M, Jiang H, Villarreal J, Cutler RG, Greig NH, Mattson MP. The KATP channel activator diazoxide ameliorates amyloid-β and tau pathologies and improves memory in the 3xTgAD mouse model of Alzheimer's disease. J Alzheimers Dis. 2010;22:443–457. doi: 10.3233/JAD-2010-101017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Komatsu H, Murray IVJ, Axelsen PH. Promotion of Amyloid □ Protein Misfolding and Fibrillogenesis by a Lipid Oxidation Product. J Mol Biol. 2008;377:1236–1250. doi: 10.1016/j.jmb.2008.01.057. [DOI] [PubMed] [Google Scholar]
- Long EK, Picklo MJ. Trans-4-Hydroxy-2-hexenal, a product of n-3 fatty acid peroxidation: make some room HNE…. Free Rad Biol Med. 2010;49:1–8. doi: 10.1016/j.freeradbiomed.2010.03.015. [DOI] [PubMed] [Google Scholar]
- Lovell MA, Ehmann WD, Mattson MP, Markesbery WR. Elevated 4-hydroxynonenal in ventricular fluid in Alzheimer's disease. Neurobiol Aging. 1997;18:457–461. doi: 10.1016/s0197-4580(97)00108-5. [DOI] [PubMed] [Google Scholar]
- Mark RJ, Pang Z, Geddes JW, Uchida K, Mattson MP. Amyloid beta-peptide impairs glucose transport in hippocampal and cortical neurons: involvement of membrane lipid peroxidation. J Neurosci. 1997a;17:1046–1054. doi: 10.1523/JNEUROSCI.17-03-01046.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mark RJ, Lovell MA, Markesbery WR, Uchida K, Mattson MP. A role for 4-hydroxynonenal, an aldehydic product of lipid peroxidation, in disruption of ion homeostasis and neuronal death induced by amyloid beta-peptide. J Neurochem. 1997b;68:255–264. doi: 10.1046/j.1471-4159.1997.68010255.x. [DOI] [PubMed] [Google Scholar]
- Markesbery WR, Lovell MA. Damage to lipids, proteins, DNA, and RNA in mild cognitive impairment. Arch Neurol. 2007;64:954–956. doi: 10.1001/archneur.64.7.954. [DOI] [PubMed] [Google Scholar]
- Masuda A, Kobayashi Y, Kogo N, Saito T, Saido TC, Itohara S. Cognitive deficits in single APP knock-in mouse models. Neurobiol Learn Mem. 2016;135:73–82. doi: 10.1016/j.nlm.2016.07.001. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. beta-Amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Pathways towards and away from Alzheimer's disease. Nature. 2004;430:631–639. doi: 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson MP. Roles of the lipid peroxidation product 4-hydroxynonenal in obesity, the metabolic syndrome, and associated vascular and neurodegenerative disorders. Exp Gerontol. 2009;44:625–633. doi: 10.1016/j.exger.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McManus MJ, Murphy MP, Franklin JL. The mitochondria-targeted antioxidant MitoQ prevents loss of spatial memory retention and early neuropathology in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2011;31:15703–15715. doi: 10.1523/JNEUROSCI.0552-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne GL, Sanchez SC, Musiek ES, Morrow JD. Quantification of F2-isoprostanes as a biomarker of oxidative stress. Nat Protoc. 2007;2:221–226. doi: 10.1038/nprot.2006.375. [DOI] [PubMed] [Google Scholar]
- Milne GL, Yin H, Hardy KD, Davies SS, Roberts LJ. Isoprostane generation and function. Chem Rev. 2011;111:5973–5996. doi: 10.1021/cr200160h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montine KS, Quinn JF, Zhang J, Fessel JP, Roberts LJ, 2nd, Morrow JD, Montine TJ. Isoprostanes and related products of lipid peroxidation in neurodegenerative diseases. Chem Phys Lipids. 2004;128:117–124. doi: 10.1016/j.chemphyslip.2003.10.010. [DOI] [PubMed] [Google Scholar]
- Morrow JD, Roberts LJ., 2nd Mass spectrometric quantification of F2-isoprostanes in biological fluids and tissues as measure of oxidant stress. Methods Enzymol. 1999;300:3–12. doi: 10.1016/s0076-6879(99)00106-8. [DOI] [PubMed] [Google Scholar]
- Murray IVJ, Sindoni ME, Axelsen PH. Promotion of oxidative lipid membrane damage by amyloid beta proteins. Biochem. 2005;44:12606–12613. doi: 10.1021/bi050926p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray IVJ, Liu L, Komatsu H, Uryu K, Xiao G, Lawson JA, Axelsen PH. Membrane mediated amyloidogenesis and the promotion of oxidative lipid damage by amyloid beta proteins. J Biol Chem. 2007;282:9335–9345. doi: 10.1074/jbc.M608589200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nhan HS, Chiang K, Koo EH. The multifaceted nature of amyloid precursor protein and its proteolytic fragments: friends and foes. Acta Neuropathol. 2015;129:1–19. doi: 10.1007/s00401-014-1347-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negre-Salvayre A, Coatrieux C, Inguenau C, Salvayre R. Advanced lipid peroxidation endproducts in oxidative damage to proteins: potential role in diseases and therapeutic prospects for the inhibitors. Br J Pharmacol. 2008;153:6–20. doi: 10.1038/sj.bjp.0707395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okun E, Barak B, Saada-Madar R, Rothman SM, Griffioen KJ, Roberts N, Castro K, Mughal MR, Pita MA, Stranahan AM, Arumugam TV, Mattson MP. Evidence for a developmental role for TLR4 in learning and memory. PLoS One. 2012;7(10):e47522. doi: 10.1371/journal.pone.0047522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter NA, Caldwell SE, Mills KA. Mechanisms of free radical oxidation of unsaturated lipids. Lipids. 1995;30:277–290. doi: 10.1007/BF02536034. [DOI] [PubMed] [Google Scholar]
- Quinn JF, Raman R, Thomas RG, Yurko-Mauro K, Nelson EB, Van Dyck C, Galvin JE, Emond J, Jack CR, Jr, Weiner M, Shinto L, Aisen PS. Docosahexaenoic acid supplementation and cognitive decline in Alzheimer disease: a randomized trial. JAMA. 2010;304:1903–1911. doi: 10.1001/jama.2010.1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raskin J, Cummings J, Hardy J, Schuh K, Dean RA. Neurobiology of Alzheimer's disease: integrated molecular, physiological, anatomical, biomarker, and cognitive dimensions. Curr Alzheimer Res. 2015;12:712–722. doi: 10.2174/1567205012666150701103107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts LJ, 2nd, Montine TJ, Markesbery WR, Tapper AR, Hardy P, Chemtob S, Dettbarn WD, Morrow JD. Formation of isoprostane-like compounds (neuroprostanes) in vivo from docosahexaenoic acid. J Biol Chem. 1998;273:13605–13612. doi: 10.1074/jbc.273.22.13605. [DOI] [PubMed] [Google Scholar]
- Saganich MJ, Schroeder BE, Galvan V, Bredesen DE, Koo EH, Heinemann SF. Deficits in synaptic transmission and learning in amyloid precursor protein (APP) transgenic mice require C-terminal cleavage of APP. J Neurosci. 2006;26:13428–13436. doi: 10.1523/JNEUROSCI.4180-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shchepinov MS. Reactive oxygen species, isotope effect, essential nutrients, and enhanced longevity. Rejuvenation Res. 2007;10:47–59. doi: 10.1089/rej.2006.0506. [DOI] [PubMed] [Google Scholar]
- Shchepinov MS, Chou VP, Pollock E, Langston JW, Cantor CR, Molinari RJ, Manning-Boğ AB. Isotopic reinforcement of essential polyunsaturated fatty acids diminishes nigrostriatal degeneration in a mouse model of Parkinson's disease. Toxicol Lett. 2011;207:97–103. doi: 10.1016/j.toxlet.2011.07.020. [DOI] [PubMed] [Google Scholar]
- Shchepinov MS, Roginsky VA, Brenna JT, Molinari RJ, To R, Tsui H, Clarke CF, Manning-Bog AB. Chapter 31: Deuterium protection of polyunsaturated fatty acis against lipid peroxidation: a novel approach to mitigating mitochondrial neurological diseases. In: Watson RR, editor. Omega 3 fatty acids in brain and neurologic health. Elsevier; Amsterdam: 2014. p. 496. [Google Scholar]
- Studzinski CM, Li F, Bruce-Keller AJ, Fernandez-Kim SO, Zhang L, Weidner AM, Markesbery WR, Murphy MP, Keller JN. Effects of short-term Western diet on cerebral oxidative stress and diabetes related factors in APP x PS1 knock-in mice. J Neurochem. 2009;108:860–866. doi: 10.1111/j.1471-4159.2008.05798.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana R, Perluigi M, Butterfield DA. Protein oxidation and lipid peroxidation in brain of subjects with Alzheimer's disease: insights into mechanism of neurodegeneration from redox proteomics. Antioxid Redox Signal. 2006;8:2021–2037. doi: 10.1089/ars.2006.8.2021. [DOI] [PubMed] [Google Scholar]
- Sung S, Yao Y, Uryu K, Yang H, Lee VM, Trojanowski JQ, Praticò D. Early vitamin E supplementation in young but not aged mice reduces Abeta levels and amyloid deposition in a transgenic model of Alzheimer's disease. FASEB J. 2004;18:323–325. doi: 10.1096/fj.03-0961fje. [DOI] [PubMed] [Google Scholar]
- Viola KL, Klein WL. Amyloid β oligomers in Alzheimer's disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015;129:183–206. doi: 10.1007/s00401-015-1386-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster SJ, Bachstetter AD, Van Eldik LJ. (2013) Comprehensive behavioral characterization of an APP/PS-1 double knock-in mouse model of Alzheimer's disease. Alzheimers Res Ther. 2013;5(3):28. doi: 10.1186/alzrt182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams TI, Lynn BC, Markesbery WR, Lovell MA. Increased levels of 4-hydroxynonenal and acrolein, neurotoxic markers of lipid peroxidation, in the brain in Mild Cognitive Impairment and early Alzheimer's disease. Neurobiol Aging. 2006;27:1094–1099. doi: 10.1016/j.neurobiolaging.2005.06.004. [DOI] [PubMed] [Google Scholar]
- Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, Stockwell BR. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A. 2016;113:E4966–4975. doi: 10.1073/pnas.1603244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Eitan E, Wu TY, Mattson MP. Intercellular transfer of pathogenic α-synuclein by extracellular vesicles is induced by the lipid peroxidation product 4-hydroxynonenal. Neurobiol Aging. 2017;61:52–65. doi: 10.1016/j.neurobiolaging.2017.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]