

Abstract
Live vaccines historically afford superior protection, yet the cellular and molecular mechanisms mediating protective immunity remain unclear. Here we found that vaccination of mice with live, but not dead, Gram-negative bacteria heightened follicular T helper cell (Tfh) differentiation, germinal center formation and protective antibody production through the signaling adaptor TRIF. Complementing the dead vaccine with an innate signature of bacterial viability, bacterial RNA, recapitulated these responses. The interferon (IFN) and inflammasome pathways downstream of TRIF orchestrated Tfh responses extrinsically to B cells and classical dendritic cells. Instead, CX3CR1+CCR2− monocytes instructed Tfh differentiation through interleukin (IL)-1β, a tightly regulated cytokine secreted upon TRIF-dependent IFN licensing of the inflammasome. Hierarchical production of IFN-β and IL-1β dictated Tfh differentiation and elicited the augmented humoral responses characteristic of live vaccines. These findings identify bacterial RNA, an innate signature of microbial viability, as a trigger for Tfh differentiation and suggest new approaches toward vaccine formulations for coordinating augmented Tfh and B cell responses.
eTOC BLURB
Live vaccines typically elicit augmented humoral responses, affording superior protection. Barbet et al. report that innate detection of bacterial RNA, a signature of microbial viability, directs a heightened Tfh cell response. This response is extrinsic to B cells and dendritic cells, and involves CX3CR1+CCR2− monocyte instruction of Tfh differentiation via TRIF-dependent IFN-β licensing of bacterial RNA-driven inflammasome activation.
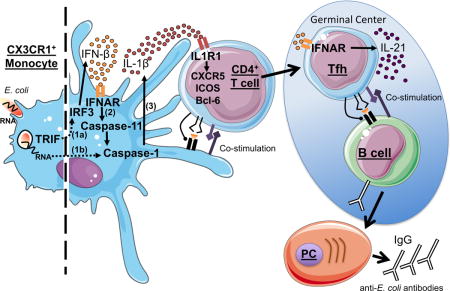
INTRODUCTION
Major illnesses like poliomyelitis, yellow fever, pertussis, diphtheria, measles, mumps, rubella, tetanus, meningitis and smallpox have all been controlled by vaccination (Plotkin and Plotkin, 2011). Live vaccines can best induce long-term protective immunity, but concerns over their safety have caused increasing vaccine reluctance in the population (Detmer and Glenting, 2006; Lin et al., 2015). Numerous vaccine preparations that do not rely on the inoculation of live microorganisms and consist of recombinant highly immunogenic proteins are also efficacious (Finco and Rappuoli, 2014). However, many others are not as certain poorly characterized aspects of natural infection – not incorporated in most vaccines – are particularly effective at inducing the right combination of signals for generating protective immunity. The exact nature of these signals remains elusive.
We previously reported that the innate immune system can discern microbial viability by detecting molecules of microbial origin such as prokaryotic messenger RNA and cyclic-di-adenosine monophosphate, which are present in live bacteria and absent in dead bacteria (Blander and Sander, 2012; Moretti et al., 2017; Sander et al., 2011). To test whether the molecular signatures of microbial viability could enhance the performance of a dead vaccine, we had found that inclusion of bacterial RNA as an adjuvant increased class-switched antibody titers in mice to levels similar to those induced by a counterpart live vaccine (Sander et al., 2011). Innate detection of bacterial RNA in the context of live Gram-negative bacteria mobilized two pathways downstream of the Toll-like receptor (TLR) signaling adaptor TRIF leading to heightened levels of interferon (IFN)-β production as well as inflammasome activation and its associated interleukin (IL)-1β secretion and pyroptosis (Sander et al., 2011). These responses were not made to dead bacteria (Sander et al., 2011).
A critical determinant of protection by many vaccines is the generation of high titers of isotype-switched, high-affinity neutralizing antibodies. The follicular T helper cell (Tfh) subset is specialized at helping B cells proliferate and differentiate into antibody secreting plasma cells and memory B cells (Crotty, 2014). While the nature of cytokines, surface molecules and transcription factors mediating either mouse or human Tfh differentiation have been defined, the physiological triggers remain unclear. We hypothesized that innate immune detection of microbial viability instructs Tfh differentiation.
To test our hypothesis, we evaluated the parameters impacting the antibody response in a vaccination model that engages the same innate pathways we had defined to be important in distinguishing live from dead Gram-negative bacteria (Sander et al., 2011). We found that the live vaccine elicited better Tfh differentiation and germinal center (GC) formation than the dead vaccine, and these responses could be recapitulated by supplementing the dead vaccine with bacterial RNA. Tfh responses were critically dependent on TRIF expression in the non-B cell hematopoietic compartment, and CX3CR1+CCR2− monocytes were responsible for instructing the Tfh response after immunization with the live vaccine. Tfh responses also relied on the interferon regulatory factor 3 (IRF3) and inflammasome pathways responsible for the synthesis of IFN-β and IL-1β, respectively, and mobilized specifically by live bacteria and bacterial RNA. The ability of T cells to respond to IL-1β was critical for their differentiation into Tfh while IFN-β, known to license IL-1β production (Blander, 2014), only promoted Tfh production of IL-21 without impacting Tfh lineage fate commitment. Our work provides the blueprint for designing new generations of vaccines that incorporate the signatures of microbial viability to achieve the superior protection of live vaccines without the associated health risks.
RESULTS
Protective serum antibodies to a live bacterial vaccine require TRIF
We investigated the impact of bacterial viability and TRIF signaling on the ensuing adaptive immune response using an intraperitoneal vaccination model of WT and Trif−/− mice with either live or heat-killed (HK) Gram-negative E. coli (EC). To exclude the effects of replication or virulence factor activity, we chose a replication defective ThyA− avirulent K12 strain of EC (Sander et al., 2011). Consistent with our previous observations (Sander et al., 2011), the live vaccine induced significantly higher titers of EC-specific IgG1, IgG2b and IgG2c than the dead vaccine in WT mice (Figure 1A). TRIF deficiency abrogated these differences (Figure 1A). Both the live and dead vaccines induced similar antibody titers in Trif−/− mice indicating their inability to signal innate detection of live bacteria (Figure S1A). TRIF deficiency also abrogated the IgG3 response, although this response was similar to both live and dead vaccines indicating its independence of bacterial viability (Figure 1A). This is consistent with predominance of IgG3 responses to polysaccharide antigens (Mond et al., 1995) shared between live and dead bacteria. TRIF deficiency had no effect on the IgM response (Figure 1A). Challenging vaccinated mice with live, replication competent, ThyA-sufficient EC revealed lower splenic bacterial burdens in mice vaccinated with the live compared to dead vaccine (Figure 1B). TRIF deficiency negated the protective effects of the live vaccine indicating that TRIF-dependent innate immune pathways are important for protection upon re-infection with Gram-negative bacteria (Figure 1B).
Figure 1. Detection of bacterial viability through TRIF triggers germinal center formation and IgG production.
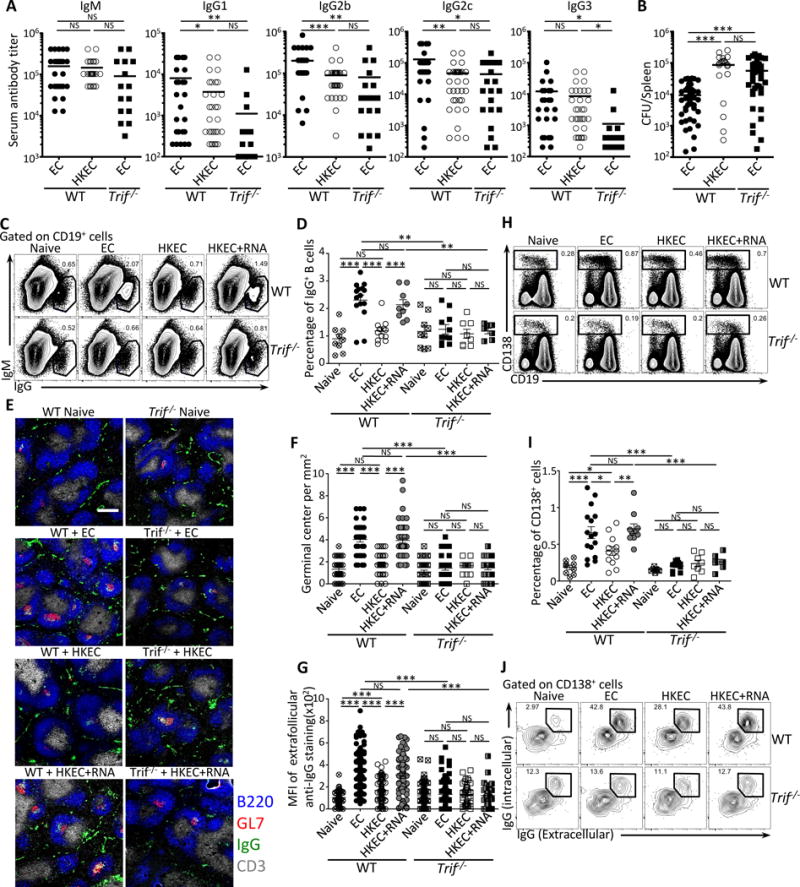
Wild-type (WT) and Trif−/− mice were vaccinated intraperitoneally with 5×107 live ThyA− EC, heat-killed ThyA− EC (HKEC), or HKEC+RNA(30μg).
(A) Day 25 serum titers of class-specific anti-E. coli (EC) antibodies.
(B) Spleen EC colony forming units (CFU) at 24 hours post-injection of 5×108 live EC into mice vaccinated 6 months earlier.
(C, D) Flow cytometry dot plots (C) and percentages (D) of gated IgG+CD19+ B cells.
(E–G) Immunofluorescence micrographs (E) at 4X magnification on spleen sections stained for B220, GL-7, IgG and CD3. Scale bar = 300 μm. (F) Numbers of germinal centers (GC) per mm2.
(G) Mean fluorescence intensity (MFI) of extra-follicular IgG staining.
(H, I) Flow cytometry dot plots (H) and percentages (I) of gated CD138+ plasma cells and plasmablasts (inclusive of CD19+ plasmablasts and CD19− plasma cells).
(J) Flow cytometry for intracellular and surface IgG expression by CD138+ plasma cells and plasmablasts. Data represent at least 3 independent experiments.
C–J Data in spleens of indicated genotypes before (naïve) and 7 days post-vaccination. (F, G) Each symbol represents one field counted in the scatter plots. (C, H, J) Numbers adjacent to outlined areas indicate percent of cells in gates. Except for (F) and (G), each symbol represents an individual mouse in the scatter plots.
NS, not significant (P > 0.05); *, P<0.05, **, P≤0.01 and ***, P≤0.001 (two-tailed unpaired t test). All data represent at least 3 experiments pooled. Mouse numbers are in (A) WT+EC, n=32; WT+HKEC, n=38; Trif−/−+EC, n=20; (B) WT+EC, n=49; WT+HKEC, n=22; Trif−/−+EC, n=39; (D) WT (naive, n=9 ;+EC, n=15; +HKEC, n=12; +HKEC+RNA, n=9) and Trif−/− (naive, n=9 ;+EC, n=11; +HKEC, n=8; +HKEC+RNA, n=7); (F) and (G) WT (naive, n=4 ;+EC, n=6; +HKEC, n=5; +HKEC+RNA, n=6) and Trif−/− (naive, n=6 ;+EC, n=5; +HKEC, n=4; +HKEC+RNA, n=4); (I) WT (naive, n=9 ;+EC, n=16; +HKEC, n=12; +HKEC+RNA, n=10) and Trif−/− (naive, n=7 ;+EC, n=11; +HKEC, n=8; +HKEC+RNA, n=8).
See also Figure S1.
TRIF and bacterial RNA orchestrate a robust B cell response
We had previously shown that vaccination with HKEC supplemented with bacterial RNA (HKEC+RNA) increases the serum titers of isotype-switched antibodies in mice to levels similar to those induced by live EC (Sander et al., 2011). The association of both TRIF and bacterial RNA with higher serum antibody titers led us to investigate other parameters of the antibody response. The live EC and HKEC+RNA vaccines led to significantly increased percentages of class-switched IgM−IgG+ B cells compared to the HKEC vaccine, and these increases were not observed in Trif−/− mice (Figure 1C and 1D) regardless of the time of analysis after vaccination (Figure S1B). The percentages of total B cells were not significantly different between WT and Trif−/− naïve and vaccinated mice (Figure S1C). We stained spleen sections for B220, IgG and GL7, a marker for cells in germinal centers (GC) where B cells proliferate, differentiate and undergo antibody class-switch recombination (CSR) and affinity maturation (Swanson et al., 2013). We found significantly increased numbers of GC and extra-follicular IgG production around B220-marked B cell follicles in the spleens of WT mice vaccinated with live compared to dead EC (Figure 1E–1G and Figure S1D). These responses were abrogated in Trif−/− mice and restored with the HKEC+RNA vaccine in WT but not Trif−/− mice (Figure 1E–1G). These results suggested that follicular and extra-follicular IgG production was enhanced by the live and HKEC+RNA vaccines in a manner dependent on the specific TRIF signaling they engaged.
We next examined the emergence of plasmablasts and plasma cells, as assessed by the upregulation of CD138 (Syndecan-1) and lack of GL7 expression (Figure S1E) (Nutt et al., 2015). The combined percentages of CD138+CD19+ plasmablasts and CD138+CD19− plasma cells were significantly increased in WT mice injected with the live or HKEC+RNA vaccines compared to dead vaccine recipient or naïve WT mice, and not in Trif−/− mice (Figure 1H and 1I). We also noted an increase in intracellular IgG in CD138+ cells, characteristic of the high levels of antibody production by these cells (Nutt et al., 2015), in WT but not Trif−/− mice after vaccination, with the strongest increases in response to live EC or HKEC+RNA compared to HKEC (Figure 1J).
TRIF and RNA dependent class-switching in all B cell subsets
B cells comprise a heterogeneous population of specialized cells with highly diverse biology and which contribute differentially to T-dependent and T-independent antibody responses (Swanson et al., 2013). Intraperitoneal vaccination activates B1, B2, and marginal zone (MZ) B cells, all of which can undergo CSR into IgG+ cells (Swanson et al., 2013). We identified these B cell subsets, including B1a and B1b cells, in the spleens of vaccinated mice based on differential expression of CD23, CD21, CD43 and CD5 (Figure 2A)(Baumgarth, 2011). As expected, the most abundant subset is follicular B2 cells (Table S1), which classically require T cell help to promote antibody class-switching within GC (Swanson et al., 2013). We noted no major differences in the percentages of total B cells within each subset in either WT or Trif−/− mice before and after vaccination, with the exception of B1b cells, whose percentage was significantly increased in live and HKEC+RNA vaccine recipient WT compared to Trif−/− mice (Figure S2A). Notably, all B cell subsets class-switched to IgG after vaccination, but only the live and HKEC+RNA vaccines induced statistically significant increases in the percentages of IgG+ B cells in WT mice and not Trif−/− mice, and regardless of subset (Figure 2B and 2C).
Figure 2. Detection of bacterial viability through TRIF induces immunoglobulin class-switching in all B cell subsets.
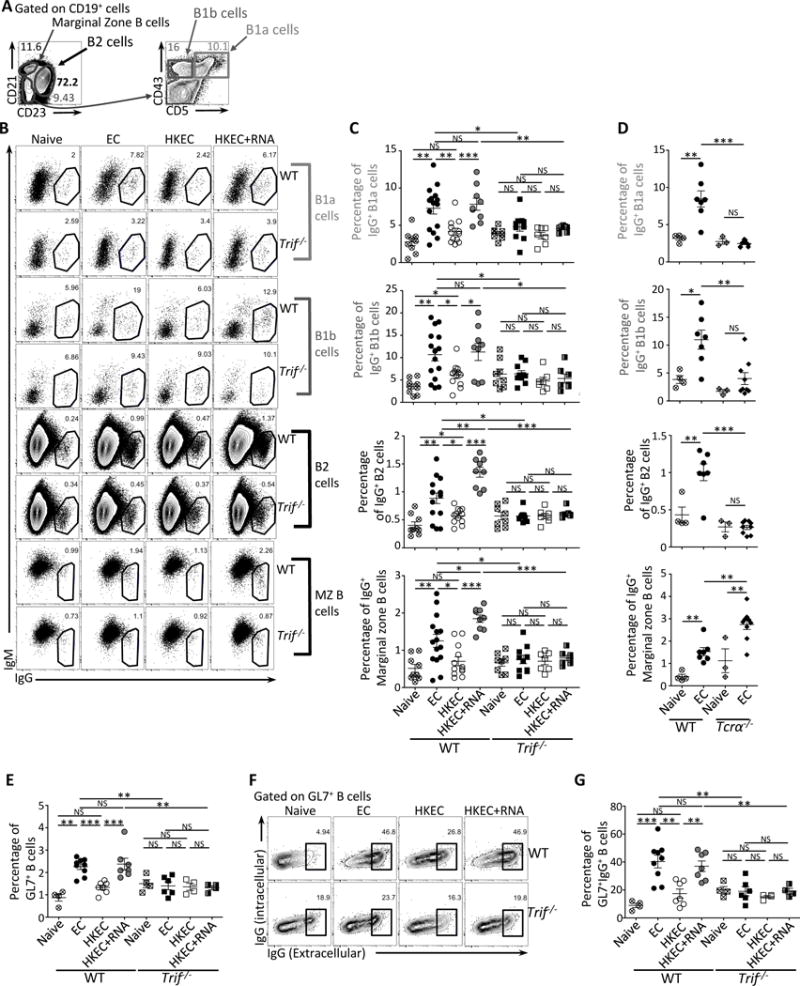
Wild-type (WT), Trif−/− and TCRα−/− mice were vaccinated intraperitoneally as indicated with either 5×107 live ThyA− EC, heat-killed ThyA− EC (HKEC), or HKEC+RNA(30μg).
(A) Flow cytometry gating strategy for different B cell populations.
(B, C) Flow cytometry dot plots (B) and percentages (C) of IgG+ B1a, B1b, B2 or marginal zone (MZ) B cells.
(D) Percentages of IgG+ B1a, B1b, B2 or MZ cells identified as in (A).
(E) Percentages of GL7+CD19+ B cells.
(F) Flow cytometry dot plots gated on GL7+CD19+ GC B cells and stained for intracellular and extracellular IgG.
(G) Percentages of IgG+ cells within GL7+CD19+ B cells.
(B–G) Data in spleens of indicated genotypes before (naïve) and 7 days post vaccination. C, D, E and G, each symbol represents an individual mouse in the scatter plots.
NS, not significant (P > 0.05); *, P<0.05, **, P≤0.01 and ***, P≤0.001 (two-tailed unpaired t test). For each scatter plot, the bar indicates the mean±s.e.m. Numbers adjacent to outlined areas indicate percent of cells in gates. Each symbol represents an individual mouse in scatter plots. Data represent at least 3 experiments pooled. Mouse numbers are in (C) WT(naive, n=9 ;+EC, n=15; +HKEC, n=12; +HKEC+RNA, n=9) and Trif−/−(naive, n=8 ;+EC, n=10; +HKEC, n=7; +HKEC+RNA, n=7); (D) WT(naive, n=4 ;+EC, n=7) and Tcrα−/−(naive, n=3 ;+EC, n=9); (E) and (G) WT(naive, n=4 ;+EC, n=9; +HKEC, n=7; +HKEC+RNA, n=7) and Trif−/−(naive, n=4 ;+EC, n=6; +HKEC, n=4; +HKEC+RNA, n=4).
See also Figure S2 and Tables S1 and S2.
CSR in B cell follicles is impaired in the absence of T cell help (Crotty, 2014). Indeed, vaccination of T cell deficient Tcrα−/− mice with live EC showed impaired antibody class-switching by follicular B2 cells, but also by extra-follicular B1a and B1b cells (Figure 2D), which can undergo CSR in a T-dependent manner (Erickson et al., 2001; Lee et al., 2011). MZ B cells could still express IgG antibody (and at higher percentages) (Figure 2D), likely reflecting neutrophil-dependent help to MZ B cells through BAFF, APRIL and IL-21 (Cerutti et al., 2013). The greatest fold expansion noted in all subsets of splenic IgG+ B cells was in response to the live and HKEC+RNA vaccines, and only in WT but not Trif−/− mice (Table S2). The largest representation of IgG+ B cells in WT mice was by B2 cells, followed by B1b, MZ B, and B1a cells (Table S2). These data suggested that B2 cells, which participate in GC reactions, were the major source of IgG in response to the live and HKEC+RNA vaccines.
We also noted a significant increase in the percentages of these GL7+CD138− GC B cells (Figure S1D) upon vaccination with live but not dead bacteria (Figure 2E). The HKEC+RNA vaccine significantly increased GC B cell percentages to levels comparable to those in live vaccine (Figure 2E). Again, ‘viability-induced’ GC B cell generation was dependent on TRIF (Figure 2E), confirming our observations by immunofluorescence microscopy (Figure 1E–G). Similar percentages were obtained by gating on either GL7+CD138− or CD95+GL7+ B cells (Figures S2B and 2E). In addition, vaccination induced an increase in the percentages of IgG+GL7+ GC B cells in WT but not Trif−/− mice, with statistically significant increases following vaccination with live EC or HKEC+RNA (Figure 2F and 2G). Unlike CD138+ plasma cells and plasmablasts that expressed high levels of intracellular IgG (Figure 1J), post-vaccination increases in IgG expression by GL7+ GC B cells in WT mice were predominantly surface-bound (Figure 2F). Collectively, these results indicated that TRIF-dependent detection of bacterial viability elicited GC B cell responses.
TRIF and bacterial RNA orchestrate a Tfh response
We next investigated how innate detection of microbial viability impacts the generation of a Tfh response. We measured a significant increase in the transcript levels of the Tfh lineage defining transcription factor Bcl6 and signature cytokine IL-21 (Crotty, 2014) in total CD4+ T cells from WT mice vaccinated with live EC compared to HKEC, and compared to CD4+ T cells from Trif−/− mice vaccinated with live EC (Figure 3A). Expression of the Th1 transcription factor T-bet (encoded by Tbx21) was similarly increased in all groups (Figure 3A). Tfh express the chemokine receptor CXCR5, allowing them to enter the B cell zone of lymphoid follicles, as well as PD-1 and ICOS, which promote their close interaction with B cells (Crotty, 2014). Expression of ICOS on CXCR5+CD4+ T cells peaked 5-7 post-vaccination with live EC, while PD-1 expression peaked later around days 7-8 (Figure S3A). Following vaccination with live EC, we noted two distinct populations of CD4+ T cells based on the expression of CD25 and CXCR5 (Figure 3B). The percentages of CD25+CXCR5− CD4+ T cells after all three vaccines were similar in WT and Trif−/− mice (Figure 3B), while those of CD25−CXCR5+CD4+ T cells in WT mice were increased after vaccination with live EC or HKEC+RNA compared to HKEC, and in a TRIF-dependent manner (Figure 3B). By gating on CXCR5+CD4+ T cells, we noted an increased percentage of ICOS+PD1+ cells within this gate in WT mice vaccinated with live EC or HKEC+RNA compared to naïve or HKEC vaccinated mice (Figure 3B). We noted no such increases in Trif−/− mice (Figure 3B). Activated CD4+ T cells from WT mice vaccinated with live EC were also FACS-sorted based on the expression of CXCR5 and the activation marker CD44. Compared to CD44+CXCR5−CD4+ T cells, CD44+CXCR5+CD4+ T cells expressed elevated levels of Bcl6 and Il21 transcripts confirming their differentiation into Tfh (Figure 3C). Moreover, only CD44+CXCR5−CD4+ T cells expressed the Th1 signature transcription factor T-bet (Figure 3C).
Figure 3. TRIF-dependent detection of bacterial viability promotes Tfh cell differentiation.
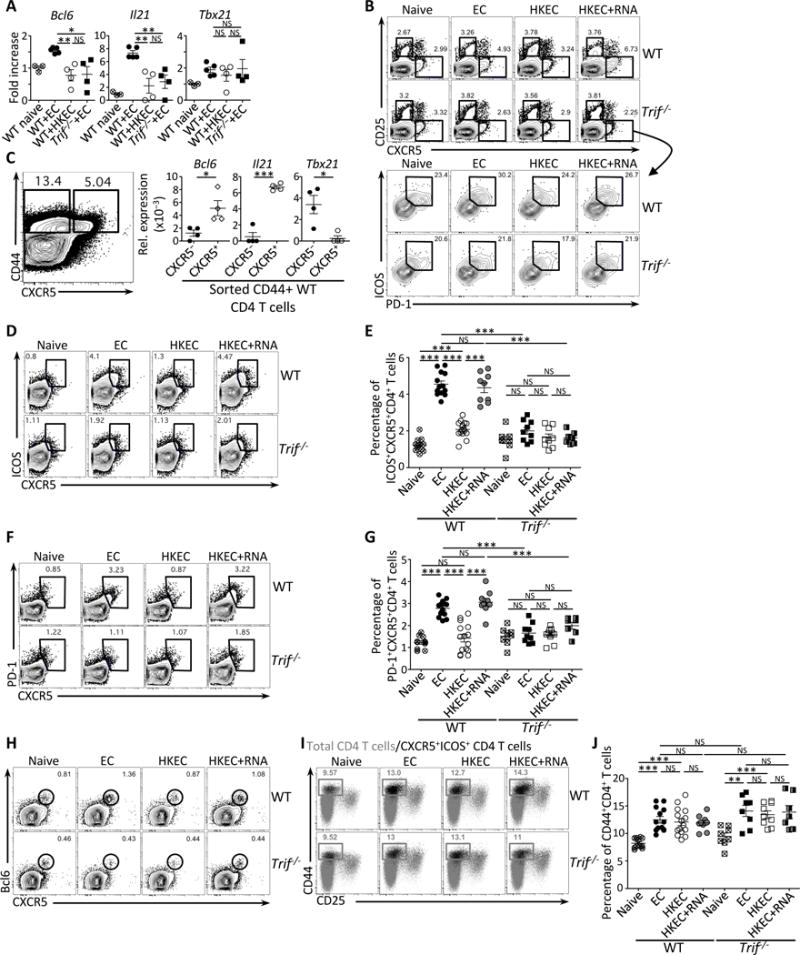
Wild-type (WT) and Trif−/− mice were vaccinated intraperitoneally as indicated with either 5×107 live ThyA− EC, heat-killed ThyA− EC (HKEC), or HKEC+RNA(30μg).
(A) Quantitative RT-PCR for Bcl6, Il21 and Tb×21 transcripts in total CD4+ T cells on day 5 after vaccination. Fold increase over naïve mice is shown.
(B) Flow cytometry dot plots gated on CD4+ T cells showing expression for CD25 and CXCR5 (upper panels), and ICOS and PD-1 within gated CXCR5+ T cells (lower panel).
(C) Quantitative RT-PCR for Bcl6, Il21 and Tbx21 transcripts in sorted CD44+CD4+ T cells either CXCR5− (left contour plot) or CXCR5+ (right contour plot). Data represent relative expression to β-actin.
(D, E) Flow cytometry dot plots (D) and percentages (E) of CXCR5+ICOS+CD4+ T cells.
(F, G) Flow cytometry dot plots (F) and percentages (G) of CXCR5+PD-1+CD4+ T cells.
(H) Flow cytometry dot plots for CXCR5 and Bcl6 gated on CD4+ T cells.
(I) Flow cytometry of CD44+CD25−CD4+ T cells. Gray dots represent the total CD4+ T cell population; black dots represent the CXCR5+ICOS+CD4+ T cell population.
(J) Percentages of total CD44+CD4+ T cells.
(B–J) Data for CD4+ T cells in spleens of indicated genotypes before and 5 days post vaccination. Each symbol represents an individual mouse in scatter plots.
NS, not significant (P > 0.05); *, P<0.05, **, P≤0.01and ***, P≤0.001 (two-tailed unpaired t test). Data are mean±s.e.m. Numbers adjacent to outlined areas indicate percent of cells in gates. Data are representative of at least 3 independent experiments. Mouse numbers are in (E), (G) and (J) WT (naive, n=11; +EC, n=13; +HKEC, n=15; +HKEC+RNA, n=9) and Trif−/− (naive, n=8; +EC, n=9; +HKEC, n=9; +HKEC+RNA, n=7).
See also Figure S3.
Based on these characterizations, we determined the actual percentages of Tfh within the CD4+ T cell gates by assessing dual ICOS and CXCR5 expression, characteristic of Tfh (Crotty, 2014). We observed a statistically significant increase in ICOS+CXCR5+CD4+ T cells in live and HKEC+RNA vaccine recipient WT and not Trif−/− mice compared to naïve controls (Figure 3D and 3E). Similar results were observed by gating on PD-1 and CXCR5 double positive CD4+ T cells (Figure 3F and 3G). Staining for Bcl-6 protein confirmed highest expression in WT mice vaccinated with live EC or HKEC+RNA compared to HKEC or Trif−/− vaccinated mice (Figure 3H). Defective Tfh differentiation in the absence of TRIF was not caused by delayed responses, as their numbers remained unaltered 10 days after vaccination (Figure S3B), and the percentages of total CD4+ T cells were equivalent in WT and Trif−/− mice in all groups (Figure S3C). Compared to naïve mice, CD4+ T cells from WT and Trif−/− mice receiving all three vaccines expressed similar levels of CD44 (Figure 3I and 3J). CD44+CD4+ T cells harbored ICOS+CXCR5+ Tfh cells (Figure 3I). Furthermore, similar percentages of T-bet expressing Th1 cells were present in WT and Trif−/− mice (Figure S3D), and anti-CD3 re-stimulation of total splenocytes showed no difference in IFN-γ production between cells from WT mice vaccinated with live or dead EC and Trif−/− mice vaccinated with live EC (Figure S3E). These results indicated a specific defect in Tfh and not Th1 differentiation in response to the live or HKEC+RNA vaccines when TRIF was absent.
In accordance with these observations in vivo, we noted no differences in vitro in the maturation of WT and Trif−/− bone marrow (BM)-derived dendritic cells (DC) after bacterial stimulation (Figure S3F), nor in their capacity to activate T cell receptor (TCR) transgenic CD4+ T cells to specific antigen (Figure S3G). Re-stimulation of CD4+ T cells primed with either WT or Trif−/− BMDC led to similar levels of IFN-γ production (Figure S3H). Adoptive transfer of OVA-specific TCR transgenic OT-II CD4+ T cells into WT mice vaccinated with either HKEC+RNA or OVA-expressing HKEC (HKEC-OVA) with or without bacterial RNA, showed Tfh differentiation of OT-II T cells exclusively in WT mice vaccinated with HKEC-OVA+RNA whereas endogenous polyclonal CD4+ T cells underwent Tfh differentiation with both HKEC+RNA and HKEC-OVA+RNA vaccines, as expected (Figure S3I). Therefore, Tfh differentiation was driven by cognate antigen, and an inflammatory Tfh skewing milieu was not sufficient to drive Tfh differentiation of bystander CD4+ T cells. Additionally, RNA acted as an adjuvant in promoting antigen-dependent Tfh differentiation (Figure S3I). Collectively, these data indicated that within the context of a response to live Gram-negative bacteria, TRIF signaling was critical for Tfh differentiation and dispensable for Th1 differentiation.
TRIF signaling in B cells is dispensable for the Tfh response
During Tfh priming, naïve CD4+ T cells receive signals from DC and emerge as Bcl6+CXCR5+ cells (Crotty, 2014). We transiently depleted myeloid cells based on CD11c expression by administering diphtheria toxin (DT) to chimeric lethally irradiated C57BL/6J mice reconstituted with BM from donor CD11c-DTR mice where the CD11c promoter controls expression of the DT receptor (Figure S4A). Depletion of CD11c+ cells impaired the appearance of CXCR5+ICOS+ Tfh cells following vaccination with live bacteria (Figure S4B and S4C) indicating a critical role for a CD11c+ myeloid cell type. The absolute numbers of total CD4 T cells were not altered (Figure S4D).
After the initiation of Tfh differentiation, additional signals from B cells complete and maintain the Tfh differentiation program (Crotty, 2014). To address the specific contribution of TRIF signaling in B cells versus non-B cell hematopoietic cells, we crossed B cell-deficient μMT mice to Trif−/− mice and subsequently generated mixed chimeric mice by reconstituting lethally irradiated C57BL/6J mice with 80% μMT BM (lacking B cells) – either on a WT (μMT) or Trif−/− (μMT × Trif−/−) background – and 20% BM from either WT or Trif−/− mice (Figure 4A). Chimeric mice thus bear Trif deficiency either in B cells or in other hematopoietic cells allowing assessment of the impact of TRIF signaling in each cellular compartment on Tfh differentiation. Consistent with our observations in non-chimeric WT mice (Figure 3B and 3D–3G), the percentages of Tfh cells in mice where TRIF expression was intact on both B cells and non-hematopoietic cells were highest after vaccination with EC or HKEC+RNA compared to HKEC (Figure 4B and 4C). Compared to these mice, confining TRIF deficiency to B cells did not significantly impair Tfh generation in response to the live or HKEC+RNA vaccines (Figure 4B and 4C). By contrast, the percentages of Tfh cells were significantly decreased when TRIF deficiency was limited to the non-B cell hematopoietic compartment indicating a role for B cell-extrinsic TRIF signaling in the Tfh response to bacterial viability (Figure 4B and 4C). These data excluded a role for B cell intrinsic TRIF signaling in Tfh differentiation, and pointed instead to a role for TRIF in myeloid antigen-presenting cells (APC) during the early phase of Tfh differentiation.
Figure 4. Tfh cell differentiation requires TRIF-mediated type-I interferon and inflammasome signals and CX3CR1+CCR2− monocytes.
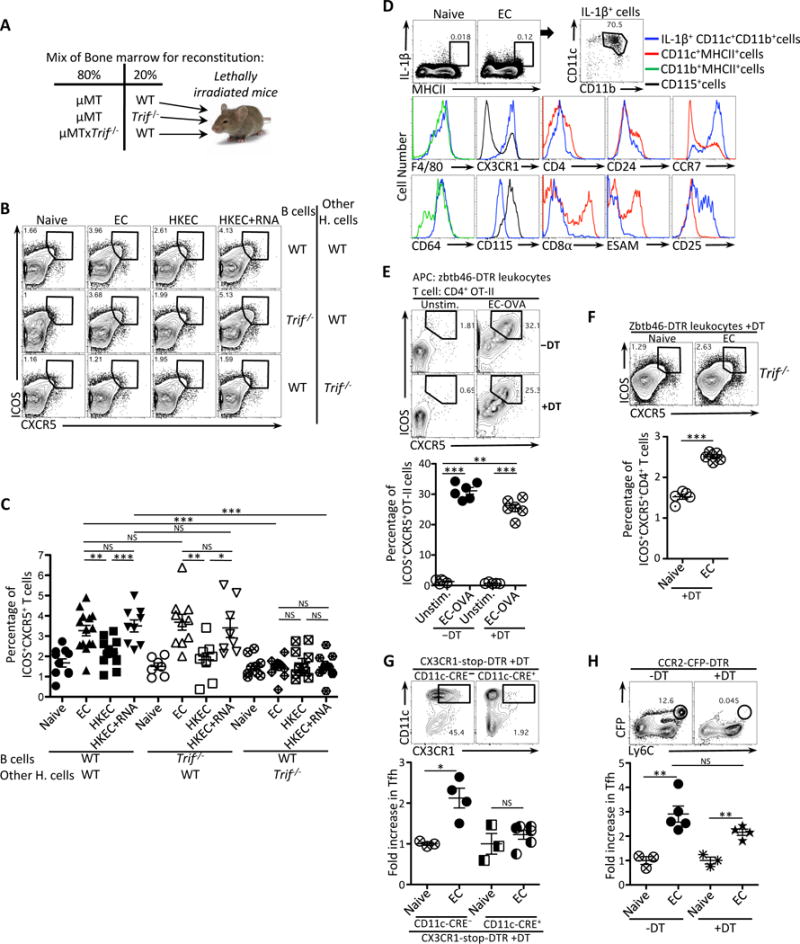
(A) Experimental strategy to generate bone marrow chimeric mice.
(B, C) Flow cytometry dot plots (B) and percentages (C) of CXCR5+ICOS+CD4+ T cells in chimeric mice generated as in (A) and vaccinated with either 5×107 live ThyA− EC, heat-killed ThyA− EC (HKEC), or HKEC+RNA(30μg).
(D) Flow cytometry analyses of splenic MHC-II+ cells stained intracellularly for IL-1β and for indicated surface markers in gated cell populations in naïve or vaccinated WT mice at 5 hours post-injection of live EC. Brefeldin A was injected at 1 hour post injection of live EC.
(E) Flow cytometry (upper panel) and percentages (lower panel) of CXCR5+ICOS+CD4+ T cells obtained in vitro after 4 days of co-culture of Zbtb46-DTR leukocytes and OVA-specific OT-II CD4+ T cells. DT was added at 100ng/ml to leukocytes 2 hours before infection with EC.
(F) Flow cytometry (upper panel) and percentages (lower panel) of CXCR5+ICOS+CD4+ T cells in vivo 7 days after vaccination with EC in Trif−/− mice that had been adoptively transferred with zbtb46-DTR leukocytes 26 hours before vaccination and treated with DT 2 hours later.
(G) Flow cytometry analyses of CD11c+CX3CR1+ cells in the spleen before and after depletion (upper panels). Fold increase of CXCR5+ICOS+CD4+ T cells in the spleens of DT-treated CX3CR1-stop-DTR (either CD11c-CRE− or CD11c-CRE+) 5 days after vaccination with EC (lower panel).
(H) Flow cytometry analyses of CFP+Ly6C+ monocytes in the spleens before and after depletion (upper panel). Fold increase of CXCR5+ICOS+CD4+ T cells in the spleens of PBS or DT treated CCR2-CFP-DTR mice 5 days after vaccination with EC (lower panel).
NS, not significant (P > 0.05); *, P<0.05; **, P≤0.01; ***, P≤0.001 (two-tailed unpaired t test). Data are mean±s.e.m. Numbers adjacent to outlined areas indicate percent of cells in gates. Each symbol represents an individual mouse. Mouse numbers are in (C) WT(WT B cells)(naive, n=11 ;+EC, n=14; +HKEC, n=12; +HKEC+RNA, n=9), WT(Trif−/− B cells)(naive, n=7 ;+EC, n=10; +HKEC, n=8; +HKEC+RNA, n=8) and Trif−/−(WT B cells)(naive, n=9 ;+EC, n=11; +HKEC, n=10; +HKEC+RNA, n=12); (F) naïve, n=5, +EC, n=5; (G) CCR2-CFP-DTR−DT(naïve, n=3, +EC, n=5) CCR2-CFP-DTR+DT(naïve, n=3, +EC, n=4); (H) CD11c-CRE−(naïve, n=3, +EC, n=4) CD11c-CRE+(naïve, n=3, +EC, n=6).
See also Figure S4
Monocyte-derived antigen presenting cells induce Tfh differentiation via TRIF
Our results so far demonstrated a reliance of Tfh differentiation on TRIF and the hematopoietic CD11c+ compartment, which encompasses different subsets of mononuclear phagocytes (MP) including DC, macrophages and monocytes. We next addressed the role of TRIF signaling in classical DC (cDC) whose differentiation relies on the zinc finger transcription factor, Zbtb46 (Meredith et al., 2012). We generated mixed chimeric mice by reconstituting lethally irradiated C57BL/6J mice with 50% Zbtb46-DTR BM (Satpathy et al., 2012) and 50% Trif−/− BM. Injection of DT achieved partial (≈60%) depletion of CD11c+MHC-II+ cells as expected (Figure S4E) whereby after depletion of WT cDC, only Trif−/− cDC remained amongst other TRIF-deficient and TRIF-sufficient non-cDC. Vaccination of chimeric DT-treated mice with live EC still induced upregulation of Tfh markers ICOS and CXCR5 on CD4 T cells (Figure S4E) demonstrating no role for cDC-intrinsic TRIF signaling in Tfh differentiation.
Macrophages and DC secrete IL-1β uniquely in response to live and not dead Gram-negative bacteria (Sander et al., 2011), thus we used IL-1β as a readout of the myeloid cell type that instructs Tfh differentiation. Intracellular staining of splenocytes for IL-1β at 5 hours post-injection of live EC revealed a small population of IL-1β-producing cells – not present in naïve mice – that expressed high levels of MHC-II (Figure 4D). Further characterization showed that these IL-1β+MHC-IIhigh cells were CD11c+CD11b+ and expressed CD64, CX3CR1, heterogeneous levels of F4/80, and intermediate levels of CD115, but not CD4, CD8α, CD24 or ESAM (Figure 4D). IL-1β+MHC-IIhigh cells also expressed CCR7 suggesting their localization to the T cell zone, as well as CD25, which quenches the Tfh inhibitory cytokine IL-2 and marks Tfh inducing cells (Figure 4D)(Li et al., 2016). This expression profile was characteristic of monocytes and/or macrophages consistent with their lack of Zbtb46 expression (Satpathy et al., 2012), and suggested the involvement of those cells in mediating the persistent Tfh response when Zbtb46+ cDC were depleted (Figure S4E).
In vitro stimulation of circulating leukocytes from Zbtb46-DTR mice with live EC in the presence of DT (to deplete cDC) led to the detection of a population of CD11b+ cells which similar to that in vivo (Figure 4D) expressed CD11c, CD64 and CCR7 (Figure S4F). After 4 days of co-culture with OT-II CD4+ T cells, Zbtb46-DTR leukocytes that had been stimulated with live EC-OVA induced OT-II T cell upregulation of CXCR5 and ICOS, and this induction persisted after cDC depletion with DT (Figure 4E). To show the capacity of non-cDC MP to induce Tfh differentiation in vivo after vaccination, we adoptively transferred circulating leukocytes from Zbtb46-DTR mice into Trif−/− mice, treated the mice with DT, and vaccinated them with live EC. Given that Trif−/− mice are impaired in their ability to induce Tfh differentiation (Figure 3), this strategy allowed us to determine that TRIF-sufficient non-cDC, remaining after depletion of TRIF-sufficient Zbtb46+ cDC, could rescue the impaired Tfh response in vaccinated Trif−/− mice (Figure 4F). Restoration of the Tfh response also correlated with partial restoration of a GC B cell response (Figure S4G). Altogether these data indicated that a non-cDC cell type of monocyte and/or macrophage origin induced a Tfh response to live bacteria, and further pinpointed TRIF signaling specifically in these cells as the driver of Tfh differentiation.
CX3CR1+CCR2− monocytes drive the Tfh response to live bacteria
The chemokine receptor CX3CR1 is broadly expressed by MP and its expression patterns had led to the characterization of two monocyte subsets in mice characterized as ‘inflammatory’ CX3CR1intLy6C+ and ‘patrolling’ CX3CR1hiLy6C− cells (Geissmann et al., 2003). Because of the involvement of CD11c+ cells in the Tfh response to live EC (Figure S4A–S4D), we depleted CX3CR1-expressing CD11c+ cells using a mouse model where expression of CD11c promoter-driven Cre recombinase excises a loxP-flanked stop cassette upstream of the DTR-coding region knocked into the Cx3cr1 locus. To determine the impact on the Tfh response after vaccination with live EC, we assessed fold increases in Tfh cells to account for any differences in baseline ICOS+CXCR5+CD4+ T cells in naïve mice before and after DT treatment. Mice depleted of CD11c+CX3CR1+ cells showed a significant decrease in Tfh induction compared to non-depleted mice after vaccination with live EC, and no significant increase over that in naive mice (Figure 4G and percentages shown in S4H). Given the lack of cDC involvement in the Tfh response to live EC (Figure 4E, 4F and S4E), these data indicated the importance of CX3CR1-expressing monocytes in the Tfh response.
To discriminate between the roles of CX3CR1intLy6C+ and CX3CR1hiLy6C− cells, we took advantage of the differential expression of CCR2 by Ly6C+ and not Ly6C− monocytes (Jakubzick et al., 2017). After vaccination with live EC, mice depleted of CCR2 monocytes were still able to induce a Tfh response compared to naïve CCR2-depleted mice, and with a fold increase in Tfh cells over naïve that did not achieve a statistically significant difference from that in non-depleted mice (Figure 4H and S4I). These data showed that CCR2+Ly6C+ monocytes, including those that express intermediate levels of CX3CR1 (CX3CR1intCCR2+Ly6C+), are dispensable for the Tfh response to live EC. Altogether, results from these genetic depletion models show that a CD11c+CX3CR1hiCCR2−Ly6C− monocyte mediates the Tfh response to a live vaccine.
Innate inflammasome and type-I IFN pathways mobilized by live bacteria dictate the Tfh response
IL-6 and IL-21 play redundant roles in inducing optimal Tfh differentiation (Crotty, 2014; Eto et al., 2011; Schmitt and Ueno, 2015). However, we could not detect IL-21 induction either at the transcript or protein level, while IL-6 levels were similar in either WT or Trif−/− DC and irrespective of the viability of EC (Figure S5A and S5B), consistent with our previous findings (Sander et al., 2011). Substantial Tfh differentiation has been reported in the combined absence of IL-6 and IL-21 indicating the existence of IL-6 and IL-21 independent pathways (Eto et al., 2011). We considered the role of the IRF3 and inflammasome innate pathways mobilized downstream of TRIF in response to live EC.
To assess the role of the TRIF-dependent type I IFN and inflammasome pathways in Tfh differentiation, we used type I IFN receptor 1-deficient (Ifnar−/−) and IRF3-deficient (Irf3−/−) mice, as well as Casp1−/−Casp11129mt/129mt mice where both the canonical and non-canonical pathways of inflammasome activation are impaired (Lamkanfi and Dixit, 2014). Like Trif−/− mice (Figure 3D–G), Tfh differentiation was impaired in Ifnar−/−, Irf3−/−, and Casp1−/−Casp11129mt/129mt mice compared to WT mice (Figure 5A), indicating an important role for both the type-I IFN and inflammasome pathways in mediating Tfh differentiation.
Figure 5. The TRIF-dependent effector cytokines IFN-β and IL-1β augment the Tfh and antibody responses to the killed vaccine.
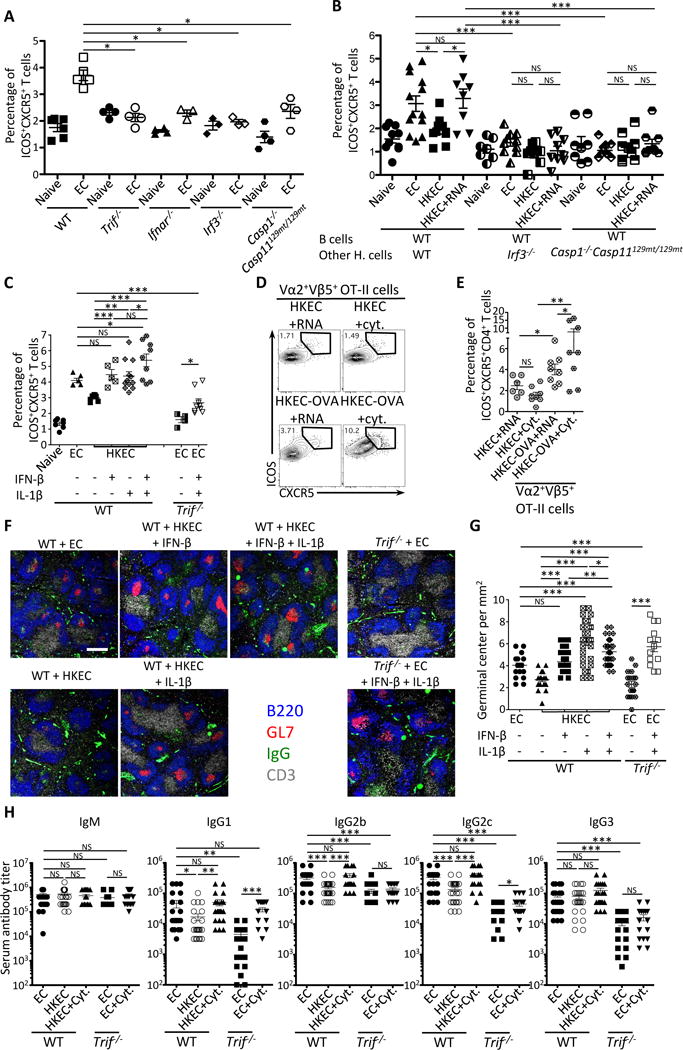
(A) Percentages of CXCR5+ICOS+CD4+ T cells in naïve and live EC vaccinated mice indicated on the X-axis.
(B) Percentages of CXCR5+ICOS+CD4+ T cells in indicated naïve or vaccinated chimeric mice generated as in Figure 4A, except here lethally irradiated mice received 20% B cell-sufficient WT BM and 80% B cell-deficient μMT BM on either WT, Irf3−/− or Casp1−/−Casp129mt/129mt backgrounds.
(C) Percentages of ICOS+CXCR5+CD4+ T cells in WT or Trif−/− mice that had received indicated vaccines.
(D, E) Flow cytometry dot plots (D) and percentages (E) of CXCR5+ICOS+ cells within gated Vα2+Vβ5+ transgenic OT-II TCR expressing CD4+ T cells adoptively transferred into mice 48 hours before receiving indicated vaccines.
(F, G) Immunofluorescence micrographs at 4X magnification on spleen sections from indicated mice stained for B220, GL-7, IgG and CD3. Scale bar = 300μm (F), and quantification of GC per mm2 (G) where each symbol represents an individual field of view.
(H) Day 25 serum titers of class-specific anti-EC antibodies.
Tfh and GC responses were measured in spleens on days 5 and 7, respectively, after vaccination of indicated mice with 5×107 live EC, HKEC or HKEC+RNA(30μg). 50 U IFN-β and/or 1μg IL-1β were injected intravenously 20 hours after vaccination.
NS, not significant (P > 0.05); *, P<0.05; **, P≤0.01 and ***, P≤0.001 (two-tailed unpaired t test). Data are mean±s.e.m. Numbers adjacent to outlined areas indicate percent of cells in gates. Mouse numbers are in (A) WT(naive, n=6 ;+EC, n=5), Trif−/−(naive, n=4 ;+EC, n=4), Ifnar−/−(naive, n=4 ;+EC, n=3), Irf3−/−(naive, n=3 ;+EC, n=3) Casp1−/−Casp11129mt/129mt(naive, n=4 ;+EC, n=4); (B) WT(WT B cells)(naive, n=10 ;+EC, n=13; +HKEC, n=10; +HKEC+RNA, n=8), Irf3−/−(WT B cells)(naive, n=7 ;+EC, n=10; +HKEC, n=9; +HKEC+RNA, n=9), Casp1−/−Casp11129mt/129mt(WT B cells)(naive, n=8 ;+EC, n=10; +HKEC, n=9; +HKEC+RNA, n=9); (C) WT(naive, n=7 ; +EC, n=6; +HKEC, n=7; +HKEC+IFN-β, n=5; +HKEC+IL-1β, n=12; +HKEC+IFN-β+IL-1β, n=9) and Trif−/−(+EC, n=4; +EC+IFN-β+IL-1β, n=9); (E) WT+OTII T cells(+HKEC+RNA, n=6; +HKEC+IFN-β+IL-1β, n=8+HKEC+RNA, n=8; +HKEC+IFN-β+IL-1β, n=8); (G) WT(+EC, n=5; +HKEC, n=4; +HKEC+IFN-β, n=6; +HKEC+IL-1β, n=6; +HKEC+IFN-β+IL-1β, n=6) and Trif−/− (+EC, n=3; +EC+IFN-β+IL-1β, n=4); (H) WT(+EC, n=25; +HKEC, n=28; +HKEC+IFN-β+IL-1β, n=23) and Trif (+EC, n=22; +HKEC, n=15; +HKEC+IFN-β+IL-1β, n=18).
See also Figure S5.
We next dissected the differential requirement for IRF3 and caspases 1 and 11 in B cells versus non-B cell hematopoietic cells using mixed chimeric mice. As in total knock-out mice (Figure 5A), we found a complete abrogation of Tfh differentiation in response to the live and HKEC+RNA vaccines in the absence of IRF3 or caspases 1 and 11 in non-B cell hematopoietic cells (Figure 5B and S5C). The ability of TRIF-sufficient non-cDC MP to restore the Tfh response in Trif−/− mice (Figure 4F) indicated that the Tfh relevant IRF3 and caspase 1 and 11 pathways downstream of TRIF operated in MP and not B cells, consistent with the innate immune recognition of live EC via TRIF. Collectively, these data echoed the reported impairment of Tfh responses in the absence of IFNAR signaling in DC (Cucak et al., 2009), and further unveiled a role for innate inflammasome activation in Tfh induction.
IFN-β and IL-1β mediate the Tfh and B cell response to bacterial viability
We next reasoned that the provision of IFN-β and/or IL-1β during T cell priming would rescue Tfh differentiation in response to dead bacteria and override the inability of Trif−/− mice to mount a Tfh response to live bacteria. Injection of IFN-β or IL-1β significantly increased Tfh differentiation in response to the HKEC vaccine compared to non-injected mice, and to levels comparable to those induced by the live vaccine (Figure 5C and S5D). Injection of both IFN-β and IL-1β after vaccination induced a Tfh response that exceeded that induced by live bacteria (Figure 5C and S5D), and might reflect the higher effective concentrations of these cytokines achieved by injection as compared to production during infection. Injection of bacterial RNA or the combination of IFN-β and IL-1β alone without killed bacteria did not elicit Tfh differentiation (Figure S5E). Notably, IFN-β and IL-1β rescued the ability of Trif−/− mice to mount a Tfh response to the live vaccine although this rescue was partial suggesting a contribution from additional TRIF-dependent signals other than IFN-β and IL-1β at the priming stage of the response (Figure 5C and S5D). Antigen-specific Tfh differentiation could also be induced by supplementation of the killed vaccine with either RNA or the combination of IFN-β and IL-1β as evidenced by Tfh differentiation of adoptively transferred OT-II CD4+ T cells specifically when the killed vaccine expressed the cognate antigen OVA (Figure 5D and 5E).
The augmented Tfh response with IFN-β and IL-1β correlated with an increase in GC formation under the same conditions (Figure 5F and 5G). The concentration of recombinant IL-1β injected with HKEC led to significantly more GC compared to live EC, and likely exceeded IL-1β levels made endogenously to live EC (Figure 5F and 5G). The importance of IFN-β and IL-1β for GC formation was highlighted by the ability to rescue GC formation in Trif−/− mice in response to the live vaccine, and to levels comparable to those in WT mice (Figure 5F and 5G).
Injection of IFN-β and IL-1β with HKEC significantly increased serum titers of E. coli-specific IgG1, IgG2b and IgG2c in WT mice compared to HKEC alone, and these titers were comparable to those in WT mice vaccinated with live EC (Figure 5H). Anti-E. coli IgM and IgG3 titers were similar among all groups of vaccinated WT mice (Figure 5H). When vaccination of Trif−/− mice with live EC was supplemented with IFN-β and IL-1β, the serum titers of IgG1and IgG2c, but not IgG2b and IgG3, were significantly increased over those to live EC alone (Figure 5H). While the cytokine-driven increase in IgG2c titers remained significantly lower than those in WT live EC-vaccinated mice, IFN-β and IL-1β supplementation increased IgG1 titers to levels that were not significantly different than those in WT live EC-vaccinated mice (Figure 5H). Altogether, these data showed that the IFN-β and IL-1β effectors of the IRF3 and inflammasome pathways could augment vaccine-induced Tfh and GC responses, and to some extent, CSR of select IgG isotypes.
T cell IL-1R1 and IFNAR play leading and supporting roles, respectively, in the Tfh response to live bacteria
To investigate the roles of IFN-β and IL-1β in Tfh differentiation, we adoptively transferred purified CD45.2+CD4+ T cells from Ifnar−/− or Il1r1−/− mice into CD45.1+ recipient mice and vaccinated them with live EC (Figure 6A, left panel). CXCR5 and ICOS expression was intact in Ifnar−/− T cells and impaired in Il1r1−/− T cells (Figure 6A and 6B). The percentages of Ifnar−/− and Il1r1−/− CD45.2+ T cells were similar after vaccination and CD44 expression was increased to similar levels, excluding differences in activation (Figure S6A).
Figure 6. T cell intrinsic role of TRIF-dependent cytokines in Tfh cell differentiation.
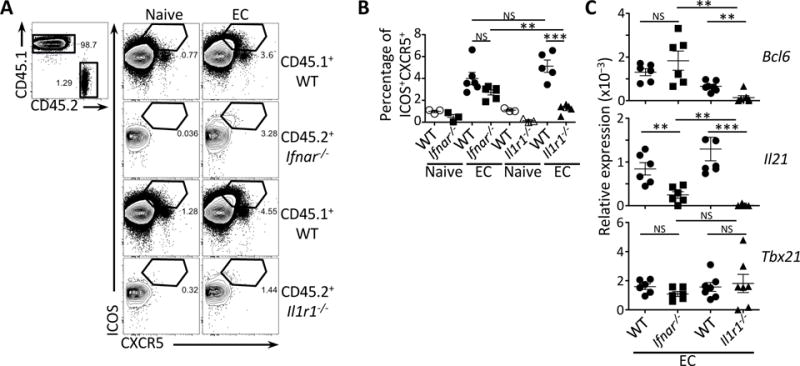
(A) Flow cytometry dot plots for ICOS and CXCR5 on CD45.1+ or CD45.2+CD4+ T cells that were either Ifnar−/− or Il1r1−/−.
(B) Percentages of ICOS+CXCR5+CD4+ T cells within the CD45.1+ or CD45.2+ populations from (A).
(C) Quantitative RT-PCR analysis for Bcl6, Il21 and Tbx21 transcripts in sorted CD44+CD4+ T cells either CD45.1+ (WT) or CD45.2+ (Ifnar−/− or Il1r1−/−). Data represent expression relative to β-actin.
(A, B, C) Data on pre-gated CD4+ T cells showing endogenous CD45.1+ cells or adoptively transferred CD45.2+ (Ifnar−/− or Il1r1−/−) CD4+ T cells isolated from spleens 5 days after injection of 5×107 live EC.
NS, not significant (P > 0.05); *, P<0.05; **, P≤0.01 and ***, P≤0.001 (two-tailed unpaired t test). Data are mean±s.e.m. Numbers adjacent to outlined areas indicate percent of cells in gates. Mouse numbers are in (B) WT+ Ifnar−/− T cells (naive, n= 3; +EC, n=6) and WT+ Il1r1−/− T cells (naive, n= 3; +EC, n=5); (C) WT+ Ifnar−/− T cells (+EC, n=6) and WT+ Il1r1−/− T cells (+EC, n=7).
See also Figure S6.
The levels of Tbx21 and Bcl6 transcripts in activated Ifnar−/− T cells were similar to those in WT T cells on day 5, whereas Il21 transcripts were significantly reduced indicating a role for IFNAR in promoting IL-21 production (Figure 6C). Indeed, IFN-β promotes IL-21 production in human T cells (Strengell et al., 2004), but also in murine T cells when in conjunction with IL-6 as the case here in the response to live EC (Nakayamada et al., 2014). On the other hand, activated Il1r1−/− T cells expressed significantly reduced Bcl6 and Il21 transcripts compared to WT T cells, whereas Tbx21 transcript levels were similar (Figure 6C). The critical role for IL1R1 in Tfh differentiation was supported by the inability of adoptively transferred MyD88−/− CD45.2+CD4+ T cells, deficient in the IL1R1 signaling adaptor MyD88, to increase expression of ICOS and CXCR5 upon vaccination with live EC (Figure S6B). This finding was consistent with a previous reported role for MyD88 in Tfh differentiation (Kubinak et al., 2015).
Collectively, these data indicated that during the response to live EC, IL-1β played a leading role in the Tfh response through T cell IL1R1-MyD88 signaling to induce expression of the master Tfh transcription factor Bcl6 and signature surface markers (ICOS, CXCR5). On the other hand, IFN-β played a supporting role through T cell IFNAR signaling to promote IL-21 production by already differentiated Tfh.
DISCUSSION
Here we show that bacterial viability constitutes a physiological trigger for instruction of Tfh differentiation. In a mouse model of vaccination, specific detection of bacterial RNA within live Gram-negative bacteria, elicited Tfh cell differentiation and by consequence an isotype switched antibody response. Human monocytes also discriminated between live and dead bacteria through detection of bacterial RNA, and produced a distinct set of cytokines specifically in response to live bacteria including IL-12 and IL-1β (L.S. and M. Ugolini, unpublished data). Notably, the same innate trigger, bacterial RNA, led to Tfh differentiation in humans and mice, although the pathways involved and their cytokine outputs were different (L.S. and M. Ugolini, unpublished data) reflecting the known similarities and differences in the cytokine requirements for murine versus human Tfh differentiation (Ueno et al., 2015). These data collectively indicate that the physiological triggers for Tfh cell differentiation are evolutionarily conserved in mice and humans despite divergence of the signaling pathways that shape Tfh differentiation in each species. Furthermore, distinct innate signaling modules that detect microbial viability might have evolved in these species to ensure pairing with the adaptive response most appropriate for the microbial threat encountered. The findings in both species provide a mechanistic basis for the superior performance of live vaccines. They solidify the promising potential of exploiting the molecular signatures of microbial viability as new adjuvants in vaccine design.
Within the context of a murine response to Gram-negative bacteria, innate detection of bacterial RNA engages the signaling adaptor TRIF upstream of a heightened IFN-β response and inflammasome-dependent IL-1β production. Cytosolic translocation of bacterial RNA upon phagocytosis of Gram-negative bacteria also engages the canonical NLRP3 inflammasome (Blander, 2014). We found that the same pathways critically orchestrate the ensuing adaptive Tfh response in a series of sequential events that culminate in the production of cytokines that favor Tfh differentiation. These cytokines include IL-6, which plays a more dominant role in murine compared to human Tfh differentiation (Ueno et al., 2015) and is commonly induced by both live and dead bacteria, but also IFN-β and IL-1β (Blander and Sander, 2012; Sander et al., 2011). The latter cytokines were important for murine Tfh differentiation as evidenced by their ability – when provided exogenously – to rescue the Tfh response of Trif−/− mice to the live vaccine and of WT mice to the dead vaccine.
The ability of each IFN-β and IL-1β to restore the Tfh response when injected alone into mice contrasted with the severe Tfh defect we noted in Ifnar−/−, Irf3−/− and Casp1−/−Casp11129mt/129mt mice, and indicated that IFN-β and IL-1β did not play redundant roles in Tfh differentiation. These roles relate to the hierarchical relationship between IFN-β and IL-1β during the earliest stages of an innate immune response to infection with Gram-negative bacteria. Bacterial RNA-mediated NLRP3 inflammasome activation (Sander et al., 2011) alongside LPS-mediated caspase-11 activation (Broz and Dixit, 2016) leads to mobilization of both the canonical and non-canonical inflammasomes in response to live EC (Blander, 2014). In vitro studies have shown that DC and macrophage production of biologically active IL-1β in this case is contingent upon engagement of IFNAR on those cells by the IFN-β made as a direct result of TRIF signaling (Blander, 2014). Indeed, TRIF licenses NLRP3 inflammasome and caspase-1 activation by enabling caspase-11 expression and activation via type-I IFN signaling (Blander, 2014). Therefore, IFN-β production necessarily precedes that of IL-1β, and IL-1β production is dependent on the availability of IFN-β. This hierarchy explains our observations and reconciles the dichotomy of the impaired Tfh response in mice deficient in IFNAR, IRF3 or caspases 1 and 11, with the restoration of Tfh responses upon provision of either IFN-β or IL-1β.
Adoptive transfer experiments of Ifnar−/− or Il1r1−/− CD4 T cells additionally showed T cell intrinsic effects of IL-1β and type-I IFN. IL-1β signaling through IL-1R1 plays a dominant role in Tfh differentiation controlling Bcl6 and Il21 transcription and CXCR5 and ICOS expression. IFN-β signaling through IFNAR plays a partial role regulating IL-21 expression without controlling Bcl6 or CXCR5 and ICOS. Type-I IFN maintains Bcl6 expression at later stages of the response, and in conjunction with IL-6, which is also produced by APC in response to live EC, enhances IL-21 production consistent with our results (Nakayamada et al., 2014). Therefore, IFN-β licenses IL-1β production by APC during the innate response and promotes IL-21 production by Tfh cells during the adaptive response.
The roles of type-I IFN and IL-1β in Tfh cell differentiation have also been investigated in human cells. When added to human CD4+ T cells together with a IL-6, IL-12 or IL-23, and TGF-β, IL-1β promoted the expression of various Tfh cell molecules including Bcl-6, while decreasing expression of the Bcl-6 antagonistic transcription factor Blimp-1 (Schmitt et al., 2014; Ueno et al., 2015). When added directly to human CD4+ T cells, type-I IFN suppresses Bcl-6, CXCR5 and ICOS expression but promotes Blimp-1 and IL-21 expression (Schmitt et al., 2014; Wong et al., 2010). Direct stimulation of naïve CD4 T cells with different members of the type-I IFN cytokine family also suppressed CXCR5 and PD-1 expression (Locci et al., 2016). Notably, type-I IFN-induced IL-27 expression through IFNAR signaling is paramount for the ability of dermal human CD14+ DC to instruct Tfh differentiation upon RIG-I signaling during Dengue virus infection (Sprokholt et al., 2017). In a similar manner, type-I IFN signaling through IFNAR on mouse DC is critical for Tfh differentiation (Cucak et al., 2009). These findings in both human and mouse are consistent with ours in highlighting a primary role for type-I IFN in licensing innate cells for the instruction of Tfh differentiation. Therefore, type-I IFNs are important for inducing expression of the cytokines most conducive to Tfh differentiation with a lesser role on CD4 T cell directly.
The increased adjuvanticity of bacterial RNA may stem from its ability to induce both IFN-β and IL-1β to optimize APC licensing for Tfh differentiation. Agents which directly activate the inflammasome could conceivably also mobilize a Tfh response. However, not all of these agents, like flagellin for example, induce a type-I IFN response, which as discussed above, is an important mediator of Tfh-promoting cytokines such as IL-27. It will be necessary to determine the relative contributions of the type-I IFN and inflammasome pathways to Tfh responses especially in humans.
The first signals for Tfh differentiation are provided at the stage of CD4+ T cell priming (Crotty, 2014). Although our expectation was that a DC would be involved, the combination of Zbtb46, CCR2 and CD11c-CX3CR1 depletion experiments pointed towards a dominant role for CX3CR1+CCR2− monocytes in the Tfh response to live bacteria. CD11c depletion also impaired Tfh generation in line with the reported expression of CD11c by CX3CR1+ monocytes (Sunderkotter et al., 2004). CX3CR1+ monocytes circulate in the blood and promptly extravasate into sites of infection to mediate the early inflammatory response – including the expression of IL-1β – and to differentiate into macrophages (Auffray et al., 2007). IL-1β was produced by MHC-II+CD11b+CD11c+CX3CR1+ monocytes within the first few hours after injection of live EC, and CD64 and F4/80 expression indicated further differentiation into macrophages. These cells also expressed CCR7 and CD25 linking them to T cell priming (Li et al., 2016).
Different APCs and pathways have been reported to drive mucosal antibody responses (Kanagavelu et al., 2016; Krishnaswamy et al., 2017; Leonardi et al., 2018). In vaccination models, subcutaneous injection of incomplete Freund’s adjuvant drives a Tfh response in an IFNAR and MyD88 dependent but IL-1β or IL-18 independent manner (Riteau et al., 2016), whereas intraperitoneal injection of Alum induces strictly IL-1β-dependent Tfh differentiation (Ritvo et al., 2017). Our data point to the necessity of the TRIF-IFNβ-IL1β axis specifically for immunization in the context of innate signals from both bacterial RNA and LPS. These studies indicate that the site of infection, class and viability of the microbe, or composition of the vaccine are critical factors to consider when optimizing an antibody response.
Our studies point to the molecular signatures of bacterial viability and the unique innate pathways they elicit as critical determinants of the augmented humoral response to live bacterial vaccines. In the context of live Gram-negative bacteria, bacterial RNA and TRIF-dependent IRF3 and inflammasome activation effectively instruct murine Tfh and GC responses. New vaccine formulations that incorporate molecular signatures of microbial viability have the potential to effectively control infection and establish protective immunity against re-infection.
STAR METHODS
Detailed methods are provided in the online version of this paper and include the following:
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-mouse CD11b, PE-Cy7, Clone M1/70 | eBioscience | Cat#25-0112-81 |
| Anti-mouse CD11c, Alexa647, Clone N418 | BioLegend | Cat#117312 |
| Anti-mouse CD11c, Alexa700, Clone N418 | eBioscience | Cat#56-0114-80 |
| Anti-mouse CD115, PerCP-eFluor710, Clone AF598 | eBioscience | Cat#46-1152-82 |
| Anti-mouse CD138, BV605, Clone 281-2 | BioLegend | Cat#142516 |
| Anti-mouse CD138, PE, Clone 281-2 | BD Biosciences | Cat#553714 |
| Anti-mouse CD162 (PSGL1), Alexa647, Clone 2PH1 | BD Biosciences | Cat#562806 |
| Anti-mouse CD185 (CXCR5), Biotin, Clone 2G8 | BD Biosciences | Cat#551960 |
| Anti-mouse CD19, FITC, Clone MB119-1 | eBioscience | Cat#11-0191-85 |
| Anti-mouse CD19, eFluor450, Clone eBio1D3 | eBioscience | Cat#48-0193-82 |
| Anti-mouse CD19, APC-Cy7, Clone 6D5 | BioLegend | Cat#115530 |
| Anti-mouse CD19, BV510, Clone 6D5 | BioLegend | Cat#115545 |
| Anti-mouse CD192 (CCR2), Alexa647, Clone SA203G11 | BioLegend | Cat#150604 |
| Anti-mouse CD197 (CCR7), Biotin, Clone 4B12 | eBioscience | Cat#13-1971-82 |
| Anti-mouse CD21/35, APC-eFluor780, Clone eBio8D9 | eBioscience | Cat#47-0211-82 |
| Anti-mouse CD23, PE, Clone B3B4 | eBioscience | Cat#12-0232-82 |
| Anti-mouse CD24, PerCP-eFluor710, Clone M1/69 | eBioscience | Cat#46-0242-82 |
| Anti-mouse CD25, FITC, Clone PC61.5 | eBioscience | Cat#53-0251-82 |
| Anti-mouse CD25, APC, Clone PC61 | BioLegend | Cat#102012 |
| Anti-mouse CD252 (OX40L), Biotin, Clone RM134L | eBioscience | Cat#13-5905-82 |
| Anti-mouse CD274 (PD-L1), APC, Clone 10F.9G2 | BioLegend | Cat#124312 |
| Anti-mouse CD275 (ICOSL), PE, Clone HK5.3 | BioLegend | Cat#107405 |
| Anti-mouse CD278 (ICOS), PE-Cy7, Clone C398.4A | BioLegend | Cat#313520 |
| Anti-mouse CD279 (PD-1), PerCP-Cy5.5, clone 29F.1A12 | BioLegend | Cat#135208 |
| Anti-mouse CD3e, APC-Cy7, Clone 145-2C11 | BioLegend | Cat#100330 |
| Anti-mouse CD4, Alexa700, Clone GK1.5 | eBioscience | Cat#56-0041-82 |
| Anti-mouse CD40, PE, Clone IL10 | BioLegend | Cat#102806 |
| Anti-mouse CD43, APC, Clone S7 | BD Biosciences | Cat#560663 |
| Anti-mouse CD44, eFluor450, Clone IM7 | eBioscience | Cat#48-0441-82 |
| Anti-mouse CD45.1 (Ly5.1), PE, clone A20 | eBioscience | Cat#12-0453-82 |
| Anti-mouse CD45.1 (Ly5.1), APC, clone A20 | eBioscience | Cat#17-0453-82 |
| Anti-mouse CD45.2 (Ly5.2), APC, clone 104 | eBioscience | Cat#17-0454-82 |
| Anti-mouse/human CD45R/B220, BV510, clone RA3-6B2 | BioLegend | Cat#103247 |
| Anti-mouse CD5, APC, clone 53-73 | eBioscience | Cat#17-00051-81 |
| Anti-mouse CD64, APC, clone 54-5/7.1 | BioLegend | Cat#139306 |
| Anti-mouse CD80, PerCP-Cy5.5, clone 16.10A1 | BioLegend | Cat#104722 |
| Anti-mouse CD86, PE, clone GL-1 | BioLegend | Cat#105008 |
| Anti-mouse CD95, PE, clone 15A7 | eBioscience | Cat#12-0951-81 |
| Anti-mouse CX3CR1, BV711, clone SA011F11 | BioLegend | Cat#149031 |
| Anti-mouse ESAM, PE, clone 1G8/ESAM | BioLegend | Cat#136203 |
| Anti-mouse F4/80, BV421, clone BM8 | BioLegend | Cat#123131 |
| Anti-mouse F4/80, APC, clone BM8 | eBioscience | Cat#17-4801-82 |
| Anti-mouse/human GL7, Biotin, clone GL7 | eBioscience | Cat#13-5902-82 |
| Anti-mouse/human GL7, Alexa647, clone GL7 | BD Biosciences | Cat#561529 |
| Anti-mouse I-A/I-E, BV421, clone ME/114.15.2 | BioLegend | Cat#107632 |
| Anti-mouse I-A/I-E, PE, clone ME/114.15.3 | eBioscience | Cat#12-5321-82 |
| Anti-mouse IgD, APC-Cy7, clone 11-26c2a | BioLegend | Cat#405716 |
| Anti-mouse IgG (1+2a+2b+3), Alexa488 | Jackson ImmunoResearch | Cat#115-545-164 |
| Anti-mouse IgG (1+2a+2b+3), PerCP | Jackson ImmunoResearch | Cat#115-125-164 |
| Anti-mouse IgG (1+2a+2b+3) | Jackson ImmunoResearch | Cat#115-005-164 |
| Anti-Goat IgG (H+L), DyLight350 | Immunoreagent, INC | RbxGt-003-E35NHSX |
| Anti-mouse IgM, PE-Cy7, clone R6-60.2 | BD Biosciences | Cat#552867 |
| Anti-mouse IL-1β, FITC, clone NJTEN3 | eBioscience | Cat#11-7114-82 |
| Anti-mouse Ly6C, FITC, clone AL-21 | BD Biosciences | Cat#553104 |
| Anti-mouse T-bet, eFluor660, clone eBio4B10 | eBioscience | Cat#50-5825-80 |
| Anti-mouse V⍺2, APC, clone B20.1 | BioLegend | Cat#127810 |
| Anti-mouse Vβ5, eFluor450, clone MR9-4 | eBioscience | Cat#48-5796-80 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Collagenase D | Roche | Cat#11088882001 |
| DNase I | Roche | Cat#04716728001 |
| Live/Dead fixable Aqua Dead cell stain kit | Invitrogen | Cat#L34957 |
| 7-AAD | BD Biosciences | Cat#559925 |
| Streptavidin, PE | eBioscience | Cat#12-4317-82 |
| Streptavidin, APC-Cy7 | eBioscience | Cat#47-4317-82 |
| Streptavidin, PerCP-Cy5.5 | BD Biosciences | Cat#551419 |
| Recombinant mouse IL-1b | Peprotech | Cat#211-11B |
| Recombinant mouse IFN-b | R&Dsystems | Cat#12401-1 |
| Diphteria Toxin | emdmillipore | Cat#322326-1MG |
| Red blood cell lysis buffer | Sigma-Aldrich | Cat#R7757 |
| Thymidine | Sigma-Aldrich | Cat#T9250 |
| Trimethoprim | Sigma-Aldrich | Cat#T7883 |
| Critical Commercial Assays | ||
| CD4+ T Cell Isolation Kit, Mouse | Miltenyi Biotec | Cat#130-048-454 |
| Foxp3/Transcription Factor Staining Buffer Set | eBioscience | Cat#00-5523-00 |
| RNeasy Midi Kit | Qiagen | Cat#74144 |
| SuperScript III First-Strand Synthesis System | Invitrogen | Cat#18080-051 |
| Trizol LS Reagen | Thermo Fisher | Cat#10296028 |
| Brefeldin A | Sigma | Cat#B7651-5MG |
| SBA Clonotyping System C57BL/6-HRP | SouthernBiotech | Cat#5300-05B |
| Maxima SYBR Green/ROX qPCR Master Mix (2X) | Thermo Fisher | Cat#K0222 |
| Deposited Data | ||
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | Jackson Laboratory | Cat#000664 |
| Mouse: muMT−/− | Jackson Laboratory | Cat#002249 |
| Mouse: Zbtb46-DTR | Jackson Laboratory | Cat#019506 |
| Mouse: Il1r1−/− | Jackson Laboratory | Cat#003245 |
| Mouse: CD45.1 | Jackson Laboratory | Cat#002014 |
| Mouse: CD11c-DTR | Jackson Laboratory | Cat#004509 |
| Mouse: MyD88−/− | From Drs. Akira and Medzhitov | |
| Mouse: Trif−/− | From Drs. Akira and Medzhitov | |
| Mouse: OT-II | From Drs. Akira and Medzhitov | |
| Mouse: Casp1−/− Casp11129mt/129mt | From Dr. Flavell | |
| Mouse: Ifnar1−/− | From Dr. Lopez | |
| Mouse: Irf3−/− | From Dr. Lopez | |
| Mouse: CCR2-CFP-DTR | From Dr. Pamer Lab (Hohl et al., 2009) | |
| Mouse: CX3CR1-STOP-DTR/CD11c-CRE | From Dr. Iliev Lab (Diehl et al., 2013) | |
| Oligonucleotides | ||
| Mouse Actb forward primer for qRT-PCR: GAAGTCCCTCACCCTCCCAA | This paper | N/A |
| Mouse Actb reverse primer for qRT-PCR: GGCATGGACGCGACCA | This paper | N/A |
| Mouse Il21forward primer for qRT-PCR: GCTCCACAAGATGTAAAGGGGC | This paper | N/A |
| Mouse Il21 reverse primer for qRT-PCR: CCACGAGGTCAATGATGAATGTC | This paper | N/A |
| Mouse Bcl6 promoter forward primer for qRT-PCR: CCTGTGAAATCTGTGGCACTCG | This paper | N/A |
| Mouse Bcl6 promoter forward primer for qRT-PCR: CGCAGTTGGCTTTTGTGACG | This paper | N/A |
| Mouse Tbx21 promoter forward primer for qRT-PCR: ACCAACAACAAGGGGGCTTC | This paper | N/A |
| Mouse Tbx21 promoter forward primer for qRT-PCR: CTCTGGCTCTCCATCATTCACC | This paper | N/A |
| Recombinant DNA | ||
| Software and Algorithms | ||
| GraphPad Prism 5 | GraphPad Software | N/A |
| FlowJo 8.7 | Tree Star | https://www.flowjo.com/solutions/flowjo/downloads |
| Image J 2.0.0-rc-43/1.51s | NIH Image | https://imagej.net/ |
| Other | ||
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, J. Magarian Blander (jmblander@med.cornell.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6J and μMT, Il1r1−/−, and Zbtb46-DTR mice were purchased from the Jackson Laboratories. Trif−/− mice were originally provided by S. Akira and obtained from Ruslan Medzhitov at Yale University. Casp1−/−Casp11129mt/129mt were provided by Richard Flavell at Yale University. Ifnar1−/− and Irf3−/− were provided by Carolina Lopez at the University of Pennsylvania. Trif−/−, Irf3−/− or Casp1−/−Casp11129mt/129mt mice were interbred with μMT mice to generate μMTxTrif−/−, μMTx Irf3−/− and μMTx Casp1−/−Casp11129mt/129mt mice. CCR2-CFP-DTR mice were provided by Eric Pamer at Memorial Sloan Kettering (Hohl et al., 2009), and CX3CR1-STOP-DTR and CD11c-CRE mice were provided by Iliyan Iliev at Weill Cornell Medicine (Diehl et al., 2013). We used 6-10 week-old mice (male and female) for all experiments. All experiments were approved by the Institutional Animal Care and Use Committee (IACUC) at the Icahn School of Medicine at Mount Sinai as well as Weill Cornell Medicine (New York, NY, USA), and carried out in accordance with the ‘Guide for the Care and Use of Laboratory Animals’ (NIH publication 86-23, revised 1985).
Bone marrow-derived dendritic cells
Bone marrow-derived dendritic cell (BMDC) cultures were grown for 6 days in RPMI 1640 supplemented with granulocyte-macrophage colony-stimulating factor (GM-CSF) and 5% fetal bovine serum (FBS), plus 100 U/ml penicillin, 100 μg/ml, 2 mM L-glutamine, 10 mM HEPES, 1 mM sodium pyruvate, 1% MEM non-essential amino acids, and 55 μM β -mercaptoethanol (all Sigma-Aldrich). BMDCs were harvested and re-plated 12-16 hours before stimulation with bacteria in appropriate medium at a concentration of 125,000 cells per cm2.
Bacterial strains
Escherichia coli K12, strain DH5α was purchased from Invitrogen. Naturally occurring thymidine auxotrophs (thyA−) were selected as previously described (Sander et al., 2011), and were grown in Luria-Bertani (LB) supplemented with 500 μg/ml Thymidine and 50 μg /ml Trimethoprim (Sigma).
METHOD DETAILS
Bone marrow chimeric mice
For bone marrow chimeric mice, C57BL/6J female mice were lethally γ-irradiated twice with 2 doses of 600 Rads at a 4-hour interval between irradiations. Irradiated mice were reconstituted with 5×106 bone marrow cells from donor mice. Reconstituted mice were used for experiments 8 weeks after. For mixed bone marrow chimera, lethally irradiated mice were reconstituted with a mixture of 20% bone marrow cells from B cell-sufficient mice (either WT or Trif−/−) and 80% of bone marrow cells from B cell-deficient μMT (Kitamura et al., 1991) mice on either a Trif−/−, Irf3−/− or Casp1−/−Casp11129mt/129mt background (Neves et al., 2010).
Specific cell depletion by injection of Diphtheria toxin
Diphtheria toxin (DT) was injected 2 days prior injection at 4ng/g in CD11c-DTR bone marrow chimera mice or at 200ng in CCR2-CFP-DTR, CX3CR1-STOP-DTR/CD11c-CRE or Zbtb46-DTR/Trif−/− (1:1) bone chimera mice. DT injection was repeated every other day until mice were euthanized.
Injection of bacteria and cytokines
ThyA− E. coli grew only in the presence of Thymidine and were resistant to Trimethoprim. For experiments, thyA− E. coli were grown to mid-log phase, washed in phosphate buffered saline (PBS) before injection into mice or addition to cells. For heat killing, ThyA− E. coli were grown to log phase and subsequently incubated at 60 °C for 60 min with frequent mixing. 5×107 ThyA− E. coli (live or heat killed) were injected intraperitoneally. When indicated the injection of HKEC was supplemented with 30μg of total bacterial RNA. Cytokines were injected intravenously 20 hours after injection of 5×107 HKEC. 50 U of IFN-β and 1 μg of IL-1β were injected in PBS.
Anti-E. coli antibody ELISA
Mice were injected with 5×107 live or heat-killed ThyA− E. coli at day 1, day 10 and day 20, as previously described (Sander et al., 2011). Sera were collected at day 25. 96-well microtiter plates were coated overnight with bacterial lysates (3 μg/ml) generated from log-phase cultures of thyA− E. coli. Serum samples from immunized mice were serially diluted (12 dilutions) in PBS and incubated in the pre-coated plates for 12 hours at 4 °C followed by washing and incubation with rabbit anti-mouse isotype-specific Ig–HRP (Southern Biotech) for 1 hour. Bound rabbit anti-mouse Ig–HRP was visualized by the addition of O-phenylenediamine dihydrochloride from tablets (Sigma) and the anti-E. coli antibody titers for each mouse were determined by the dilution of the last detectable absorbance readings at 490 nm using a Molecular Devices spectrophotometer.
Immunofluorescence staining and microscopy
Seven days post vaccination, spleens were isolated and frozen in OCT medium. Frozen spleens were sectioned at 8 μm, fixed in cold acetone, blocked with PBS 5% fetal calf serum and stained with a goat anti-mouse total IgG (Jackson Immunoresearch), Alexa647-labeled anti-mouse B220, biotinylated anti-mouse GL-7 (eBioscience) and FITC-labeled anti-CD3 followed by Alexa564- conjugated streptavidin (Life Technologies) and DyLight350®-labelled anti-Goat antibody (Fisher Scientific). Fluorescent images were captured using the ×4 objective on a Nikon fluorescent microscope. All images were analyzed on Image J software. For germinal center counts, at least 5 fields of view were counted per mouse. Number of mice ranged from 3 to 7 mice. For the extra-follicular IgG MFI measurement, the number of mice investigated ranged from 3 to 7 mice with a minimum of 7 extra-follicular regions measured per mouse.
Flow cytometry
At day 5 or day 7 after injection of bacteria, splenocyte suspensions were obtained by mechanical dissociation in PBS supplemented with 2% FBS and a 3 mM EDTA solution in PBS. Cells were stained with fluorochrome-conjugated monoclonal antibodies to mouse CD25 (PC61.5), GL7 (GL7), CD4 (GK1.5), CD3 (2C11) CD44 (IM7), CD19 (MB19-1), MHC-II (M5/114.15.2), CD21 (ebio8D9), CD23 (B3B4), CD5 (537.3) (all from eB ioscience); PD-1 (29F1.A12), ICOS (398.4A), IgD (1-26c2a), B220 (RA3-6B2) (all from Biolegend); CD138 (281-2), CXCR5 (2G8), IgM (R6-60.2), CD43 (S7) (all from BD Bioscience), and IgG (anti-IgG1, IgG2a, IgG2b and IgG3) from Jackson Immunoresearch. For every antibody mixture, cells were stained for their viability with either the live/dead marker Aqua (Life Technologies) or 7AAD (both from Life Technologies). CXCR5 and IgG staining were performed for 45 minutes at 4 °C prior to the addition of other antibodies. Thewo distinct distinct populations of CD4+ T cells defined in Figure 3B were based on the expression of CD25 and CXCR5 as described previously (Choi et al., 2011). Data were acquired with a LSR Fortessa (Becton Dickinson) and analyzed with FlowJo 8.3.3 software (TreeStar). For intracellular staining, cells were fixed and permeabilized after extracellular staining with transcription factor staining kit (eBioscience) according to the manufacturer’s recommendation. Antibodies used for intracellular staining were anti-IgG (anti-IgG1, IgG2a, IgG2b and IgG3) from Jackson Immunoresearch or anti-Bcl6 (mGI191E, eBioscience).
Intracellular staining of IL-1β producing cells
1 hour after mouse vaccination with live EC, Brefeldin A (Sigma) was injected intravenously. Mice were euthanized 4 hours after Brefeldin A injection and the spleens were digested in Collagenase (1mg/mL, Roche) and DNase (0.5mg/mL, Roche) for 15 min at 37°C. Extracellular staining was then performed before fixation and intracellular IL-1β staining (anti-IL-1β, clone NJTEN3, eBioscience).
Determination of bacterial burdens in the spleens of vaccinated mice
Vaccinated mice were re-challenged 6 months after the first inoculation with 5×108 E. coli (DH5α, K12 strain). 24 hours after re-challenge, the spleens were homogenized in PBS and spread on LB agar plates and incubated overnight. Colony forming unit (CFU) were counted.
Fluorescence activated cell sorting
Total T cells and B cells were purified by magnetic bead isolation using specific anti-CD4 coated beads or anti-CD19 coated beads, respectively, according to the manufacturer’s recommendations (Miltenyi Biotec). For specific follicular T helper cell isolation, we used a FACS Aria II (BD bioscience) with the antibody mixture used for Tfh monitoring. This mixture consisted of anti-ICOS (398.4A), anti-CD3 (2C11), anti-CD4 (GK1.5), anti-CD44 (IM7) (all from eBioscience), CXCR5 (2G8, BD bioscience), and the Aqua live/dead marker (Life Technologies).
Quantitative Real Time Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
Total RNA was isolated from sorted T cells using the RNeasy kit (Qiagen). Contaminating genomic DNA was removed by DNase digestion (DNase I, Roche). Reverse transcription was performed using Superscript III (Invitrogen) and cDNA was used for subsequent qRT-PCR reactions, which were conducted on an ABI Prism 7900 instrument using the Maxima SYBR green qPCR Master Mix (Fermentas) with the following primer pairs.
All reactions were performed in duplicates and the samples were normalized to β-actin.
Adoptive cell transfer
T cell transfer
CD4+ splenocytes from either OT-II, Ifnar1−/− or Il1r1−/− mice were purified by magnetic separation according to the manufacturer’s recommendations (Miltenyi biotech). Recipient mice (either C57BL/6J or C57BL/6 CD45.1) were injected intravenously with 5×106 CD4+ T cells, 2 days prior to vaccination.
Leukocyte transfer
Zbtb46-DTR mice were bled and blood was treated with Red Blood Cell lysis buffer according the manufacturer’s recommendations (Sigma). The equivalent of 200μl of initial blood volume or 0.5×106 leukocytes were injected intravenously into Trif−/− mice. 2 hours later, mice were injected intraperitoneally with 200ng of DT 24 hours prior to vaccination.
Zbtb46-DTR leukocyte and OT-II T cell co-culture
Zbtb46-DTR mice were bled and blood was treated with Red Blood Cell lysis buffer according to the manufacturer’s recommendations (Sigma). The equivalent of 50μl of initial blood volume or 1.25×105 Zbtb46-DTR leukocytes were plated with DT at 100ng/ml for 2 hours prior to in vitro infection with live OVA-expressing EC (MOI 20). 2×105 CD4+ OT-II T cells sorted by magnetic column (Miltenyi) were added 5 hours later in the presence of anti-IFN-γ (40μg/ml), anti-IL-4 (40μg/ml) and anti-TGF-β (40μg/ml). Upregulation of ICOS and CXCR5 was assessed after 4 days of co-culture.
QUANTIFICATION AND STATISTICAL ANALYSES
Statistical significance was determined by a two-sided unpaired Student’s t-test (GraphPad Prism). P < 0.05 was considered significant. NS, not significant *, P<0.05; **, P≤0.01; ***, P≤0.001. Variances were similar among groups in all experiments. No specific randomization method was used for the animal experiments. Outlier values were excluded on the basis of the Grubbs’ test. The sample sizes were chosen to ensure adequate power to detect percent change for each parameter according to recommendations by a consulted statistician. The investigators were not blinded to the group allocation during experiments and analyses.
Supplementary Material
HIGHLIGHTS.
Innate detection of bacterial RNA directs Tfh differentiation and antibody responses
TRIF-dependent IRF3 and inflammasome pathways orchestrate the Tfh response
CX3CR1+ CCR2–monocytes secrete IL-1β in response to live bacteria and drive Tfh cells
IFN-β-dependent secretion of IL-1β engages T cell IL-1R1 to mediate Tfh differentiation
Acknowledgments
This work was supported by the National Institutes of Health (NIH) grants AI095245 and AI127658 to J.M.B. and DK113136 to I.I. J.M.B. and her laboratory were supported by NIH grants AI123284, AI073899, DK072201, and DK111862. A.C. and his laboratory were supported by NIH grants AI57653, AI95613, AI61093, and U19 096187. We thank the Crohn’s and Colitis Foundation (to G.B.), European Advanced Grant (ERC-2011-ADG-20110310 to A.C.), Ministerio de Ciencia e Innovacio´ n (SAF2011-25241 to A.C.), Marie Curie reintegration grant (PIRG-08-GA-2010-276928 to A.C.), the German Research Council (DFG SA1940-2/1 and SFB-TR84 TP C8 to L.E.S.), the Burroughs Wellcome Fund and the Leukemia and Lymphoma Society (to J.M.B.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
G.B. and J.M.B. designed and directed the study; G.B. conducted most experiments; M.G. assisted with serum antibody ELISAs; A.C. provided experimental guidance and discussed data; L.E.S. initiated and performed experiments related to the antibody response; I.I. and I.L. provided help and reagents pertaining to CX3CR1 phagocytes. G.B., and J.M.B. wrote the manuscript and J.M.B. conceived of the study.
COMPETING FINANCIAL INTERESTS
J.M.B. and L.E.S. have a patent related to this work: PCT/US2012/047087 “Use of Bacterial RNA or Structural Motifs thereof as adjuvants for vaccines”.
References
- Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, Geissmann F. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science. 2007;317:666–670. doi: 10.1126/science.1142883. [DOI] [PubMed] [Google Scholar]
- Baumgarth N. The double life of a B-1 cell: self-reactivity selects for protective effector functions. Nat Rev Immunol. 2011;11:34–46. doi: 10.1038/nri2901. [DOI] [PubMed] [Google Scholar]
- Blander JM. A long-awaited merger of the pathways mediating host defence and programmed cell death. Nat Rev Immunol. 2014;14:601–618. doi: 10.1038/nri3720. [DOI] [PubMed] [Google Scholar]
- Blander JM, Sander LE. Beyond pattern recognition: five immune checkpoints for scaling the microbial threat. Nat Rev Immunol. 2012;12:215–225. doi: 10.1038/nri3167. [DOI] [PubMed] [Google Scholar]
- Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407–420. doi: 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- Cerutti A, Cols M, Puga I. Marginal zone B cells: virtues of innate-like antibody-producing lymphocytes. Nat Rev Immunol. 2013;13:118–132. doi: 10.1038/nri3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crotty S. T follicular helper cell differentiation, function, and roles in disease. Immunity. 2014;41:529–542. doi: 10.1016/j.immuni.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cucak H, Yrlid U, Reizis B, Kalinke U, Johansson-Lindbom B. Type I interferon signaling in dendritic cells stimulates the development of lymph-node-resident T follicular helper cells. Immunity. 2009;31:491–501. doi: 10.1016/j.immuni.2009.07.005. [DOI] [PubMed] [Google Scholar]
- Detmer A, Glenting J. Live bacterial vaccines–a review and identification of potential hazards. Microbial cell factories. 2006;5:23. doi: 10.1186/1475-2859-5-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson LD, Foy TM, Waldschmidt TJ. Murine B1 B cells require IL-5 for optimal T cell-dependent activation. J Immunol. 2001;166:1531–1539. doi: 10.4049/jimmunol.166.3.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eto D, Lao C, DiToro D, Barnett B, Escobar TC, Kageyama R, Yusuf I, Crotty S. IL-21 and IL-6 are critical for different aspects of B cell immunity and redundantly induce optimal follicular helper CD4 T cell (Tfh) differentiation. PLoS One. 2011;6:e17739. doi: 10.1371/journal.pone.0017739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fazilleau N, McHeyzer-Williams LJ, Rosen H, McHeyzer-Williams MG. The function of follicular helper T cells is regulated by the strength of T cell antigen receptor binding. Nat Immunol. 2009;10:375–384. doi: 10.1038/ni.1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finco O, Rappuoli R. Designing vaccines for the twenty-first century society. Frontiers in immunology. 2014;5:12. doi: 10.3389/fimmu.2014.00012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissmann F, Jung S, Littman DR. Blood monocytes consist of two principal subsets with distinct migratory properties. Immunity. 2003;19:71–82. doi: 10.1016/s1074-7613(03)00174-2. [DOI] [PubMed] [Google Scholar]
- Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol. 2017;17:349–362. doi: 10.1038/nri.2017.28. [DOI] [PubMed] [Google Scholar]
- Kanagavelu S, Flores C, Hagiwara S, Ruiz J, Hyun J, Cho EE, Sun F, Romero L, Shih DQ, Fukata M. TIR-Domain-Containing Adapter-Inducing Interferon-beta (TRIF) Regulates CXCR5+ T helper Cells in the Intestine. J Clin Cell Immunol. 2016;7 doi: 10.4172/2155-9899.1000458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnaswamy JK, Gowthaman U, Zhang B, Mattsson J, Szeponik L, Liu D, Wu R, White T, Calabro S, Xu L, et al. Migratory CD11b(+) conventional dendritic cells induce T follicular helper cell-dependent antibody responses. Sci Immunol. 2017;2 doi: 10.1126/sciimmunol.aam9169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubinak JL, Petersen C, Stephens WZ, Soto R, Bake E, O’Connell RM, Round JL. MyD88 signaling in T cells directs IgA-mediated control of the microbiota to promote health. Cell Host Microbe. 2015;17:153–163. doi: 10.1016/j.chom.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022. doi: 10.1016/j.cell.2014.04.007. [DOI] [PubMed] [Google Scholar]
- Lee SK, Rigby RJ, Zotos D, Tsai LM, Kawamoto S, Marshall JL, Ramiscal RR, Chan TD, Gatto D, Brink R, et al. B cell priming for extrafollicular antibody responses requires Bcl-6 expression by T cells. J Exp Med. 2011;208:1377–1388. doi: 10.1084/jem.20102065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonardi I, Li X, Semon A, Li D, Doron I, Putzel G, Bar A, Prieto D, Rescigno M, McGovern DPB, et al. CX3CR1(+) mononuclear phagocytes control immunity to intestinal fungi. Science. 2018;359:232–236. doi: 10.1126/science.aao1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Lu E, Yi T, Cyster JG. EBI2 augments Tfh cell fate by promoting interaction with IL-2-quenching dendritic cells. Nature. 2016;533:110–114. doi: 10.1038/nature17947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin IY, Van TT, Smooker PM. Live-Attenuated Bacterial Vectors: Tools for Vaccine and Therapeutic Agent Delivery. Vaccines (Basel) 2015;3:940–972. doi: 10.3390/vaccines3040940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locci M, Wu JE, Arumemi F, Mikulski Z, Dahlberg C, Miller AT, Crotty S. Activin A programs the differentiation of human TFH cells. Nat Immunol. 2016;17:976–984. doi: 10.1038/ni.3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith MM, Liu K, Darrasse-Jeze G, Kamphorst AO, Schreiber HA, Guermonprez P, Idoyaga J, Cheong C, Yao KH, Niec RE, Nussenzweig MC. Expression of the zinc finger transcription factor zDC (Zbtb46, Btbd4) defines the classical dendritic cell lineage. J Exp Med. 2012;209:1153–1165. doi: 10.1084/jem.20112675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mond JJ, Lees A, Snapper CM. T cell-independent antigens type 2. Annual review of immunology. 1995;13:655–692. doi: 10.1146/annurev.iy.13.040195.003255. [DOI] [PubMed] [Google Scholar]
- Moretti J, Roy S, Bozec D, Martinez J, Chapman JR, Ueberheide B, Lamming DW, Chen ZJ, Horng T, Yeretssian G, et al. STING Senses Microbial Viability to Orchestrate Stress-Mediated Autophagy of the Endoplasmic Reticulum. Cell. 2017;171:809–823 e813. doi: 10.1016/j.cell.2017.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayamada S, Poholek AC, Lu KT, Takahashi H, Kato M, Iwata S, Hirahara K, Cannons JL, Schwartzberg PL, Vahedi G, et al. Type I IFN induces binding of STAT1 to Bcl6: divergent roles of STAT family transcription factors in the T follicular helper cell genetic program. J Immunol. 2014;192:2156–2166. doi: 10.4049/jimmunol.1300675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nutt SL, Hodgkin PD, Tarlinton DM, Corcoran LM. The generation of antibody-secreting plasma cells. Nat Rev Immunol. 2015;15:160–171. doi: 10.1038/nri3795. [DOI] [PubMed] [Google Scholar]
- Plotkin SA, Plotkin SL. The development of vaccines: how the past led to the future. Nat Rev Microbiol. 2011;9:889–893. doi: 10.1038/nrmicro2668. [DOI] [PubMed] [Google Scholar]
- Riteau N, Radtke AJ, Shenderov K, Mittereder L, Oland SD, Hieny S, Jankovic D, Sher A. Water-in-Oil-Only Adjuvants Selectively Promote T Follicular Helper Cell Polarization through a Type I IFN and IL-6-Dependent Pathway. J Immunol. 2016;197:3884–3893. doi: 10.4049/jimmunol.1600883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritvo PG, Churlaud G, Quiniou V, Florez L, Brimaud F, Fourcade G, Mariotti-Ferrandiz E, Klatzmann D. Tfr cells lack IL-2Ralpha but express decoy IL-1R2 and IL-1Ra and suppress the IL-1-dependent activation of Tfh cells. Sci Immunol. 2017;2 doi: 10.1126/sciimmunol.aan0368. [DOI] [PubMed] [Google Scholar]
- Sander LE, Davis MJ, Boekschoten MV, Amsen D, Dascher CC, Ryffel B, Swanson JA, Muller M, Blander JM. Detection of prokaryotic mRNA signifies microbial viability and promotes immunity. Nature. 2011;474:385–389. doi: 10.1038/nature10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy AT, Kc W, Albring JC, Edelson BT, Kretzer NM, Bhattacharya D, Murphy TL, Murphy KM. Zbtb46 expression distinguishes classical dendritic cells and their committed progenitors from other immune lineages. J Exp Med. 2012;209:1135–1152. doi: 10.1084/jem.20120030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt N, Liu Y, Bentebibel SE, Munagala I, Bourdery L, Venuprasad K, Banchereau J, Ueno H. The cytokine TGF-beta co-opts signaling via STAT3-STAT4 to promote the differentiation of human TFH cells. Nat Immunol. 2014;15:856–865. doi: 10.1038/ni.2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt N, Ueno H. Regulation of human helper T cell subset differentiation by cytokines. Curr Opin Immunol. 2015;34:130–136. doi: 10.1016/j.coi.2015.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprokholt JK, Kaptein TM, van Hamme JL, Overmars RJ, Gringhuis SI, Geijtenbeek TBH. RIG-I-like receptor activation by dengue virus drives follicular T helper cell formation and antibody production. PLoS pathogens. 2017;13:e1006738. doi: 10.1371/journal.ppat.1006738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strengell M, Julkunen I, Matikainen S. IFN-alpha regulates IL-21 and IL-21R expression in human NK and T cells. J Leukoc Biol. 2004;76:416–422. doi: 10.1189/jlb.1003488. [DOI] [PubMed] [Google Scholar]
- Sunderkotter C, Nikolic T, Dillon MJ, Van Rooijen N, Stehling M, Drevets DA, Leenen PJ. Subpopulations of mouse blood monocytes differ in maturation stage and inflammatory response. J Immunol. 2004;172:4410–4417. doi: 10.4049/jimmunol.172.7.4410. [DOI] [PubMed] [Google Scholar]
- Swanson CL, Pelanda R, Torres RM. Division of labor during primary humoral immunity. Immunologic research. 2013;55:277–286. doi: 10.1007/s12026-012-8372-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueno H, Banchereau J, Vinuesa CG. Pathophysiology of T follicular helper cells in humans and mice. Nat Immunol. 2015;16:142–152. doi: 10.1038/ni.3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong MT, Ye JJ, Alonso MN, Landrigan A, Cheung RK, Engleman E, Utz PJ. Regulation of human Th9 differentiation by type I interferons and IL-21. Immunol Cell Biol. 2010;88:624–631. doi: 10.1038/icb.2010.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YS, Kageyama R, Eto D, Escobar TC, Johnston RJ, Monticelli L, Lao C, Crotty S. ICOS receptor instructs T follicular helper cell versus effector cell differentiation via induction of the transcriptional repressor Bcl6. Immunity. 2011;34:932–946. doi: 10.1016/j.immuni.2011.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diehl GE, Longman RS, Zhang JX, Breart B, Galan C, Cuesta A, Schwab SR, Littman DR. Microbiota restricts trafficking of bacteria to mesenteric lymph nodes by CX(3)CR1(hi) cells. Nature. 2013;494:116–120. doi: 10.1038/nature11809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohl TM, Rivera A, Lipuma L, Gallegos A, Shi C, Mack M, Pamer EG. Inflammatory monocytes facilitate adaptive CD4 T cell responses during respiratory fungal infection. Cell Host Microbe. 2009;6:470–481. doi: 10.1016/j.chom.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura D, Roes J, Kuhn R, Rajewsky K. A B cell-deficient mouse by targeted disruption of the membrane exon of the immunoglobulin mu chain gene. Nature. 1991;350:423–426. doi: 10.1038/350423a0. [DOI] [PubMed] [Google Scholar]
- Neves P, Lampropoulou V, Calderon-Gomez E, Roch T, Stervbo U, Shen P, Kuhl AA, Loddenkemper C, Haury M, Nedospasov SA, et al. Signaling via the MyD88 adaptor protein in B cells suppresses protective immunity during Salmonella typhimurium infection. Immunity. 2010;33:777–790. doi: 10.1016/j.immuni.2010.10.016. [DOI] [PubMed] [Google Scholar]
- Sander LE, Davis MJ, Boekschoten MV, Amsen D, Dascher CC, Ryffel B, Swanson JA, Muller M, Blander JM. Detection of prokaryotic mRNA signifies microbial viability and promotes immunity. Nature. 2011;474:385–389. doi: 10.1038/nature10072. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.