

Abstract
Mitotic chromosomes fold as compact arrays of chromatin loops. To identify the pathway of mitotic chromosome formation, we combined imaging and Hi-C of synchronous DT40 cell cultures with polymer simulations. We show that in prophase, the interphase organization is rapidly lost in a condensin-dependent manner and arrays of consecutive 60 kb loops are formed. During prometaphase ~80 kb inner loops are nested within ~400 kb outer loops. The loop array acquires a helical arrangement with consecutive loops emanating from a central spiral-staircase condensin scaffold. The size of helical turns progressively increases during prometaphase to ~12 Mb. Acute depletion of condensin I or II shows that nested loops form by differential action of the two condensins while condensin II is required for helical winding.
Summary figure
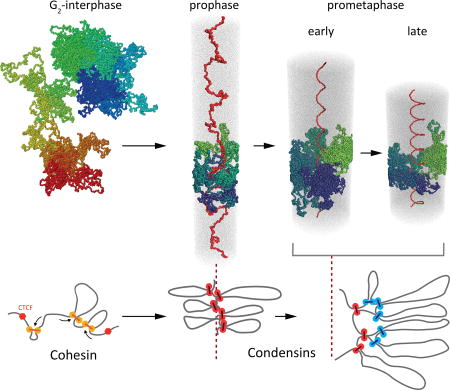
In prophase, condensins mediate the loss of interphase conformation and loop arrays are formed. In prometaphase, combined action of condensin I and II forms helically arranged nested loop arrays.
Introduction
Chromosomes dramatically change their conformation as cells progress through the cell cycle. Throughout most of interphase, chromosomes of vertebrates display two layers of organization: topologically associating domains (TADs) (1, 2) and A- and B-compartments (3). At a finer scale, chromatin looping between promoters, enhancers and CTCF-bound sites (4, 5) facilitates gene regulation. During mitosis, these features disappear and chromosomes are compacted into dense arrays of randomly positioned consecutive chromatin loops (6–9).
Although the organization of these two states is now increasingly understood, much less is known about how cells convert from one state into the other. Previous microscopy observations revealed that chromosomes become recognizable during prophase and form linearly organized structures where sister chromatids are initially mixed (10–13). By late prophase, sister chromatid arms separate and each chromatid is thought to be organized as an array of loops that emanate from an axial core containing condensin complexes and topoisomerase II alpha (14–18). During prometaphase, the chromatids shorten and become thicker (11), ultimately forming fully condensed metaphase chromosomes (19). How compaction of loop arrays occurs during prometaphase is not known.
Here we employ a chemical-genetic system for highly synchronous entry of DT40 cells into prophase. DT40 cells are karyotypically stable, near diploid (Fig. S1) and have been extensively used for analysis of mitotic chromosome organization (20). Use of chemical genetics (21) in this cell system allowed us to apply Hi-C with high temporal resolution and to determine how chromosome conformation changes as cells disassemble the interphase nucleus and form mitotic chromosomes (22, 23). These data, combined with polymer simulations (24, 25) and direct imaging reveal a mitotic chromosome morphogenesis pathway with distinct transitions, including compartment and TAD loss, loop array formation by late prophase and chromosome shortening during prometaphase through growing and winding of loops around a central helical scaffold. Using an auxin-inducible degron approach (26, 27) we then identify distinct key roles for condensin I and II in this pathway.
Results
Synchronous progression into mitosis
To obtain cultures of cells that synchronously enter mitosis we arrested cells in G2 by selectively inhibiting CDK1. We stably expressed a variant of Xenopus laevis CDK1 cDNA (CDK1as) harboring a F80G mutation in DT40 cells (22, 28). This mutation renders CDK1as sensitive to inhibition by the ATP analog 1NM-PP1 (22). We then disrupted the endogenous CDK1 gene using CRISPR/Cas9. Growing cells for 10 hours in the presence of 1NM-PP1 efficiently arrested >90% of cells in G2 as indicated by FACS (Table S1, Fig. S2) and by microscopy analysis of chromosome and nuclear morphology (Fig. 1A). Washing out 1NM-PP1 led to rapid release of cells from the G2 arrest and synchronous entry into prophase.
Fig. 1. Chromosome morphogenesis during synchronous mitosis.

(A) Representative DAPI images of nuclei and chromosomes in CDK1as DT40 cells taken at indicated time points (in minutes) after release from 1NM-PP1-induced G2 arrest show mitotic chromosome formation. Bar indicates 5 micron. (B) Hi-C interaction maps of chromosome 7 (binned at 100 kb) from cells collected indicated time points in prophase and prometaphase show large-scale changes in contact frequencies as cell progress through mitosis (C) The average interaction maps center around G2 TAD boundaries. TAD boundaries disappear. (D) Compartmentalization saddle plots: average distance-normalized interaction frequencies between cis-pairs of 100-kb bins arranged by their G2 eigenvector value. Compartments disappear.
This system allowed us to study chromosome morphogenesis by harvesting cells at sequential time points for imaging and Hi-C analysis as they synchronously progress through mitosis. For some cultures collected at later time points (30 – 60 minutes), we added nocodazole 30 minutes prior to their release from the 1NM-PP1 arrest, to block the metaphase-anaphase transition (see Methods). All time courses described here were performed in duplicate and results were highly concordant. DAPI staining showed the expected chromosome condensation and individualization in prophase (Fig. 1A). Nuclear envelope breakdown (NEBD) occurred around t = ~7 – 10 minutes as evidenced by staining for Lamin B1, which diffuses into the cytoplasm upon NEBD (Fig. S3) (29) and by measuring the association of previously cytoplasmic condensin I subunits with the chromosomes (CAPD2, CAPG1, CAPH and increased levels of SMC2 and SMC4; Fig. S4A,B). Previous studies, and our proteomic analysis (Fig. S4B) show that by late prophase, cohesin (SMC1 and SMC3) has dissociated from the arms of sister chromatids, which separate, but remain aligned (11, 12, 30, 31). Chromosome shortening subsequently occurred during prometaphase and at the late time points fully condensed chromosomes were observed (Fig. 1A).
Loss of compartments and TADs in Prophase
Hi-C analysis showed that G2-arrested cells display all features characteristic of vertebrate interphase cells (8). First, chromosomes form territories as indicated by relatively high levels of intra-chromosomal interactions (3). Second, chromosomes display the characteristic pattern of active A- and inactive B- compartments as revealed by the plaid pattern of Hi-C interactions (3) (Fig. 1B). The locations of these compartments in G2 resembled those detected in exponentially growing cells, though the compartment signal strength was stronger and the pattern sharper in the synchronous cells likely as a result of uniformity in cell cycle stage. Third, TADs were readily visible in the Hi-C interaction maps as squares of relatively high interaction frequencies along the diagonal (Fig. 1C). TAD boundaries were similar in position and strength to those in non-synchronous cells as determined using an insulation score calculated from a 250 kb sliding window (32) (Fig. S5). Finally, we analyzed how contact frequency (P) of contacts between locus pairs depends on their genomic distance (s). P(s) decays with genomic distance and this relationship changes with cell cycle stages (8). For G2 cells, we found P(s) to be highly similar to that observed previously in non-synchronous cells (Fig. S6, Fig. S7). Together, these analyses show that G2 chromosomes, which are composed of two closely aligned and likely catenated sister chromatids, are organized similarly to G1 chromosomes (8).
This interphase chromosomal organization was rapidly lost upon release of cells into prophase. As soon as 5 minutes after release we detected a marked reduction in the typical plaid pattern of long-range interactions, indicating a loss of compartments (Fig. 1B). By 10 minutes (late prophase) compartments were mostly gone. At the same time, TADs were also lost (Fig. 1C, Fig. S8).
We used eigenvector decomposition to quantify the disappearance of compartments (33). The first eigenvector readily captured compartments at t = 0 and 2.5 minutes, but starting at t = 5 minutes it explained progressively less of the variance in the Hi-C interaction maps, indicating weakening of the compartment structure. By t = 7.5 minutes the strength of the first eigenvector fell to 17% (from 80% at t = 0 minutes) and by t = 10 minutes, it no longer captured compartments. Loss of compartments was also quantified by calculating the ratio of A-to-A or B-to-B interactions over A-to-B interactions over the time course. From t = 0 to t = 2.5 minutes and onward this fraction decreased steadily, indicating that preferential interactions within compartments are lost (Fig. 1D, Fig. S9).
The strength of TADs can be quantified using the insulation score, which indicates the amount of contacts formed across a locus up to a certain distance (32). TAD boundaries have a low score (indicative of high insulation), whereas loci inside TADs show a high score value (no insulation). The genome-wide variance of insulation scores provides a quantitative measure of the presence of TADs (8). Starting at t = 2.5 minutes the variance of the insulation profiles progressively decreased, indicating loss of TADs (Fig. 1C, Fig. S5B). By t = 7.5 minutes the variance was reduced more than 2-fold and by t = 10 minutes no TADs were detected. This conclusion was confirmed by plotting the average Hi-C interaction pattern at and around TAD boundaries identified in G2 at different time points during mitosis (Fig. 1C). Insulation is strongest in G2 and by late prophase insulation values are near background levels (quantified in Fig. S10). We conclude that compartments and TADs disappear rapidly during early prophase.
By late prophase, when sister arms have resolved (11, 34), and around the time of nuclear envelope breakdown (t ~ 7.5–10 minutes), the Hi-C maps are characterized by a general decay of contact frequency P with genomic distance s (Fig. 2A). The shape of the P(s) curve changes as prophase progresses. In G2 cells, it is shallow (P(s) ~ s−0.5) up to a distance of several hundred kb, reflecting compaction within TADs (35, 36), but for larger distances the decay becomes steeper. During prophase, the initial shallow decay extends for longer-range interactions, with a steeper drop at 2 Mb at t = 10 minutes, suggesting a higher degree of compaction. As we demonstrate below, this decay and shape are consistent with formation of a linearly arranged, layered organization of the chromosome (8), where the size of each layer corresponds to the position of the steep drop in the P(s) curve.
Fig. 2. Prophase chromosomes fold as axially compressed loop arrays.

(A) Genome-wide curves of contact frequency P(s) vs genomic distance s, normalized to unity at s=100 kb. The curves are derived from prophase Hi-C data at the indicated time points after release from G2 arrest. The dotted line indicates P(s) = s−0.5 observed for mitotic chromosomes (8). (B) Overview of the coarse grained model of prophase chromosomes. The chromosome is compacted into a series of consecutive loops and compressed into a cylindrical shape. The loop bases form a scaffold at the chromosomal axis, each loop occupies a cylindrical sector of height h and angular size φ, oriented at angle ϴi. The coarse-grained model predicts the P(s) curve to have three distinct regions: an intra loop (I), intra layer (II) and inter layer (III) regions. (C) The best fitting P(s) predictions by the coarse grained model for late prophase (t = 7.5 minutes) under two different assumptions on loop orientations: (top panels) uncorrelated and (bottom panels) correlated orientations of consecutive loops. Uncorrelated angular loop orientations lead to a plateau in P(s) in the intra-layer, whereas correlated angles lead to the experimentally observed P(s) = s−0.5 (right panels). (D) Polymer models of prophase chromosomes. Chromatin fibers are modeled as chains of particles (dark grey circles), compacted into arrays of consecutive loops (loop bases indicated in orange). Chromosomes are compacted into a cylinder with a density of one nucleosome per 11×11×11nm cube (lower right). (E) Goodness of fit for simulated vs experimental P(s). Polymer simulations were performed for a range of loop densities and loop lengths, and for each simulation P(s) was calculated. The heatmap shows the quality of a match between the predicted and experimental P(s) curves at late prophase (t = 7.5 minutes). (F) P(s) derived from late prophase Hi-C experiments (green line) and the best fitting polymer models (grayscale lines). Average loop size and linear density of loops along the chromosome axis are listed.. (G) Top and side view of the best fitting polymer model of late prophase chromosomes. Loops bases are shown in red and several loops rendered in different colors. (H) The average loop size and linear density of the 3 best-fitting models of prophase chromosomes at different time points.
Appearance of a second diagonal band in Hi-C maps from prometaphase cells
At t = 15 minutes, when cells have entered prometaphase, the Hi-C maps produce a P(s) curve with a drop at 2 Mb. Strikingly, a distinct second diagonal band appears, running in parallel with the primary diagonal for all loci and chromosomes (Fig. 1B, Fig. S11). This second diagonal represents increased interaction frequencies between any pair of loci separated by around 3 Mb. At 15 min, this feature is clearly observed in P(s) plots as a local peak at ~3 Mb (Fig. 3A, Fig. S6, Fig. S7). As cells progress through prometaphase, the position of the drop in P(s) and the position of the second diagonal migrate to larger genomic distances (Fig. 3A; Fig. S6, S7). By t = 60 minutes, when compact metaphase chromosomes have formed, the second diagonal is positioned at ~12 Mb and appears more diffuse. The second diagonal appears in all chromosomal maps and its position is independent of chromosome size over two orders of magnitude (Fig. S11). The appearance and movement of the second diagonal is not dependent or affected by nocodazole: no nocodazole was added to the t = 15 minute sample, and a replicate Hi-C dataset obtained from a culture collected at t = 30 minutes in the absence of nocodazole was nearly identical to the data obtained in the presence of nocodazole (Table S2, Fig. S7, Fig. S8). Together, these P(s) curves reveal a periodicity of interactions that reflects chromosome structure at the scale of megabases.
Fig. 3. Helical organization of prometaphase chromosomes.

(A) Genome-wide curves of contact frequency P(s) vs genomic distance (separation, s), normalized to unity at s=100 kb. The curves are derived from Hi-C data obtained from prometaphase cells (t = 10–60 minutes after release from G2 arrest). The dashed line indicates P(s) = s−0.5. Arrows indicate positions of a local peak in P(s) representing the second diagonal band observed in Hi-C interaction maps. (B) The coarse grained model of prometaphase chromosomes with staircase loop arrangement. Left, top: the staircase loop arrangement implies that loops rotate in genomic order around a central scaffold (see Supplemental Materials). Left, bottom: angles of adjacent loops are correlated and steadily increasing, reflecting helical arrangement of loops. Right: this helical arrangement can be observed as gyres by DNA staining and a helical scaffold can be observed in cells expressing GFP-tagged condensins. (C) The best fitting P(s) predictions by the staircase coarse grained model for late prometaphase t = 30 minutes (30min; left panel) and t = 60 minutes (60min, right panel) after release from G2 arrest (Hi-C data: colored lines; model; gray lines). (D) Polymer model of prometaphase chromosomes. Chromosomes are modeled as arrays of consecutive nested loops with a helical scaffold (outer loops in red, inner loops in blue, also indicated diagrammatically bottom right). (E) Goodness of fit for simulated vs experimental P(s). Polymer simulations were performed varying the helix height (nm), the size of a helical turn (Mb), and the sizes of inner and outer loops. For each simulation P(s) was calculated. The heatmaps show the quality of the best match between the predicted and experimental P(s) at prometaphase (t = 30 minutes), when two out of four parameters were fixed to the specified values. (F) P(s) derived from prometaphase Hi-C experiments (colored lines) and the best fitting polymer models (gray lines). Left panel: t = 30 minutes, right panel t = 60 minutes after release from G2 arrest. Average size of outer and inner loops, the length of a helix turn and the helical pitch are indicated. (G) Parameters of the helical scaffolds from the best fitting polymer models. X-axis: ratio of the radius of the helical scaffold to that of the whole chromatid; Y-axis: ratio of the pitch to the helix radius. The dashed lines show the corresponding values (0.46 and 2.5122) for the optimal space-filling helix (84). Classical solenoid configurations are predicted to be in sector III, while the “spiraling staircase” configurations are in I and II. On the right, three examples of models of type I, II and III are shown with loops bases in red and several individual loops rendered in different colors. Also shown is a schematic of a prometaphase chromosome with the helical winding of loops indicated by arrow around the loop array. (H) Parameters of the best 3 models of prometaphase chromosomes at different time points.
The only known regular periodic structural feature of chromosomes is helical coiling, which was first described in 1880 (37) and can be observed in certain chromosome preparations (10, 38–40). Experimentally induced banding of the chromosome arms is much more irregular (41). In helical chromosomes with a pitch (the length of a complete turn) of ~3 Mb, each locus is in relatively close proximity to loci located one turn up or down the chromosome, i.e. 3 Mb up or downstream along the DNA. The progressive movement of the second diagonal band to larger distances during prometaphase would reflect an increased “winding up” and shortening of the helix.
Although this is the first Hi-C data revealing the helical coiling of chromosomes, the chicken DT40 late prometaphase (t = 60 minutes) Hi-C contact maps strongly resemble those for mitotic human HeLa S3 chromosomes that we had reported earlier (8). In fact, re-analysis of mitotic HeLa S3 Hi-C data in more detail by deeper sequencing revealed a weak second diagonal band at ~10 Mb distance (Fig. S12), suggesting this periodic folding is a conserved feature of vertebrate mitotic chromosomes.
Testing models of chromosomes
Previous studies suggested that mitotic chromosomes are organized as arrays of consecutive loops emanating from a condensin-rich scaffold, forming a polymer bottlebrush (42, 43), with a layered organization of loops (6, 19, 44). To understand chromosome organization at different stages of compaction we built coarse-grained models of chromosomes as arrays of loops aiming to reproduce P(s) curves of Hi-C data, separately for each time point. In these models, a chromosome is represented by a cylinder with an axial scaffold; loop bases are arranged consecutively along the scaffold, and each chromatin loop, emanating from the scaffold in a particular direction, is represented by a blob of loci (Fig. 2B, see Supplemental Material). Loops are regularly placed along the axis, with angular positions determined by a stochastic model; loop sizes are exponentially distributed and bases of loops are not positioned at defined genomic sequences or loci (8). Analysis of condensin ChIP data for DT40 cells (45) supports sequence-independent positioning for >95% of loops (Supplemental Material, Fig. S13; see discussion). For specific models of loop arrangements, presented below, the P(s) curve can be found analytically as the return probability of a stochastic process describing angular positions of loops (see Supplemental Material, section “Coarse-grained model of contact probability decay in mitotic chromosomes”). The resulting P(s) always has three regions (Fig. 2B): (i) the intra-loop region at short separations, where two loci are likely to be within the same loop and P(s) reflects the internal organization of loops; (ii) the “intra-layer” region at larger genomic separations, where loci are located on different loops within the same axial layer of the cylindrical chromosome and P(s) reflects the specific arrangement of loops relative to each other; (iii) the “inter-layer” that appears as a steep drop in contact frequency at large genomic distances, where loci are located in loops that are so distant along the scaffold that their blobs can no longer overlap. In the P(s) plot of experimental Hi-C data throughout mitosis, the intra-layer region and the drop-off can be readily discerned (Fig. 2A, 2C).
Prophase chromosomes
The coarse-grained models show that the relative orientation of consecutive loops strongly affects the shape of the P(s) curve in the intra-layer region. If the orientations of consecutive loops are independent of each other, the contact frequency P(s) does not decay with genomic distance in the intra-layer region, as any pair of loops within a layer are equally likely to interact (Fig. 2C). In contrast, introducing correlations between orientations of consecutive loops, i.e. forcing neighboring loops to project in similar directions, makes them follow an angular random walk. The angular random walk is a 1D random walk on a circle and has a return probability of P(s)~s−0.5 until the full turn is made by the walk. The P(s)~s−0.5 decay followed by a drop is in good agreement with the late prophase Hi-C data (t = 7–10 minutes – Fig. 2C). Taken together, these results suggest that by late prophase chromosomes are already organized into arrays of consecutive loops with correlated angular orientations.
We developed detailed polymer models to test whether specific classes of conformations can reproduce experimental Hi-C data, though they do not prove mechanisms by which these structures form. Further, all our simulations produce equilibrium models and do not reflect kinetics of chromosome folding. In these models, chromatin is represented as a 10 nm fiber (46, 47), where one monomer corresponds to one nucleosome (Fig. 2D), allowing us to simulate up to 40 Mb of chromatin. Prophase chromosomes are modeled as arrays of consecutive loops of exponentially distributed length and random genomic locations, emanating from a flexible scaffold, as would result from a loop extrusion process (48). The loop array is further condensed by imposing poor solvent conditions to the density observed in electron microscopy (one nucleosome per 11×11×11nm cube, i.e. ~40% volume fraction) (49), while preserving the overall cylindrical shape of the chromosome (Fig. 2D). We systematically varied two parameters: the average loop size and the linear loop density along the chromosomal scaffold (Fig. 2E). For all combinations, we generated equilibrium conformations, simulated a Hi-C experiment, and evaluated its ability to reproduce P(s) curves from Hi-C data for different time points during prophase (Fig. 2E–H).
These polymer models can accurately reproduce P(s) (20 kb<s<4 Mb) for all prophase time points, in agreement with the prediction of the coarse-grained model (Fig. 2C). The best matching models for later prophase time points, when sister chromatids are separate and lie side-by-side, have gradually increasing average loop size: from 40–50 kb at t = 5 minutes to ~60–70 kb at t = 10 minutes (Fig. 2H), reproducing the gradually shifting position of the drop-off from 2 to 3.5Mb (i.e. increase of the layer size), while maintaining about the same ~50 loops per layer and ~250 loops per µm. These results are consistent with a model where loop arrays are formed early in prophase, and loop sizes grow gradually, e.g. by merging smaller adjacent loops (25). Thus, both coarse grained modes and polymer simulations indicate that by late prophase, chromosomes fold as dense arrays of loops, with consecutive loops positioned with correlated radial orientations.
Prometaphase spirals
A striking feature of prometaphase Hi-C data is the appearance of the second diagonal band, which appears as a distinct peak on the P(s) curves (Fig. 3A). This feature cannot be explained by interactions between sister chromatids, as these become minimal in prometaphase, and simulations show that no amount of overlap between sisters gives rise to such periodic pattern in interactions (Fig. S14). As argued above, periodic interactions seen by Hi-C are most readily explained by a helical organization of mitotic loop arrays, which has been observed microscopically (6, 10, 40, 50). Two classes of chromosome architecture can give rise to periodicity in contact frequencies: an “external” helix when the whole chromosome is folded into a solenoid (50) (the solenoid model), and a “staircase” model in which consecutive loops wind in a helical order around a centrally located scaffold (“internal” helix). We note that by “scaffold” we do not necessarily imply a solid integrated structure stretching from one end of the chromosome to the other. The “scaffold” could equally be a dynamic association of smaller complexes linked by DNA that pack with helical symmetry. By modeling we examined these classes of architectures and the continuum of models in between them.
To explore whether an internal helix can arise through reorganization of loop orientations, while preserving the cylindrical morphology of the whole chromosome, we extended our coarse-grained prophase model (Fig. 2B) by adding a preferred angular orientation for each loop: (1) as in prophase, the orientation of each loop is correlated with its neighbors; (2) these loops have preferred, but not fixed, orientations that follow a helical path, thus winding around the chromosomal scaffold (Fig. 3B). Loops in this spiral staircase model follow an angular Ornstein-Uhlenbeck random walk with bias toward preferred positions, and P(s) can be found analytically (51) (Supplemental Material, section “Coarse-grained model of contact probability decay in mitotic chromosomes – loops with spiral staircase orientation”). This coarse-grained model yields a P(s) curve that closely follows the experimental prometaphase P(s) and displays both the P(s)~s−0.5 decay and the narrow peak corresponding to the second diagonal band (Fig. 3C). These results indicate that, (i) the emergent second diagonal band in Hi-C data can result from a spiral organization, and (ii) such organization can arise from preferred orientations of loops around the central scaffold. Further support for a centrally located spiraling scaffold is provided by analysis of chromosome shape and SMC2-mAID-GFP, CAP-H-mAID-GFP or CAP-H2-mAID-GFP localization along mitotic chromosomes from colchicine arrested DT40 cells (Fig. 3B, Fig. S15, Fig. S16). We observe a pattern of condensin localization that is consistent with a helical path of the scaffold.
Detailed polymer modeling allowed us to explore a broader range of architectures, with both external and internal helices, and to obtain quantitative estimates of loop sizes and other aspects of organization. Two aspects of the prometaphase organization must be captured by any model: (i) a higher linear density of chromatin of up to 50–70 Mb/µm, necessitating an evolution of the loop architecture; and (ii) spiraling of the scaffold. The higher density of loops can be achieved by a nested loop organization where several smaller (inner) loops are organized consecutively within each larger (outer) loop whose bases form the central axis (Fig. 3D). The presence of nested loops is an essential feature for prometaphase models, as models with a single layer of loops could not reproduce Hi-C P(s) curves even when other parameters were varied (Fig. S14B). To model helical architecture we made the scaffold follow a helical path in 3D, while allowing loops to adopt their equilibrium conformations within an otherwise cylindrical chromosome (Fig. 3D).
We systematically varied the model parameters, such as geometry of the spiral scaffold and loop sizes (Fig. 3E). This also probed different lengths and widths of chromosomes as the volume density was kept constant. For t = 30 minutes the best agreement was achieved for a relatively narrow internal spiral staircase-like scaffold (R=30–60nm) (Fig. 3F–G). This spiral is much more narrow than the ~300nm diameter of the chromatid, and has a small pitch (the height of one turn, 100–200nm) (Fig. 3H). Interestingly, this spiral arrangement of loop bases can achieve helical winding of loops that reproduces the second diagonal in the interaction maps and the peak on the P(s) curves for t = 15, 30 and 60 minutes (Fig. 3F–H). Wider spiraling of the scaffold (Fig. 3G–III) approximating external helix architectures (50) failed to accurately reproduce P(s) (Fig. S14C). Taken together, coarse-grained and polymer models that agree with Hi-C data overwhelmingly support the spiral staircase (“internal” helix) architecture of the scaffold (I and II in Fig. 3G), and subsequent helical winding of loops.
Fitting consecutive time points probed by Hi-C, we found that the linear chromatin density of the best-matching models continued to grow throughout prometaphase, in agreement with the observed steadily shortening of mitotic chromosomes (Fig. 1A, 3H, Fig. S18). Simulations show that shifting the peak in P(s) to larger genomic distances representing the second diagonal in consecutive time points (Fig. 3A) can be achieved by increasing the radius of the helical scaffold from 30 to 100 nm, the radius of the chromatid from 300 to 360 nm, and increasing the pitch from 100 to 250 nm, while maintaining a constant outer and inner loop size (~400 kb and ~80 kb respectively) (Fig. 3H). These changes lead to the increase in amount of DNA (Mb) per turn of the spiral from ~3 Mb in early prometaphase up to ~12 Mb by late prometaphase.
Comparison of the dimensions of our best models with direct microscopic measurements of prometaphase chromosomes prepared according to the Hi-C protocol reveals good agreement between experiment and predictions (Fig. S17).
Condensins are critical for prophase chromosome morphogenesis
To determine the role of condensin complexes in chromosome morphogenesis, we fused a minimal auxin inducible degron domain (mAID) to SMC2 (Supplemental Materials, Table S3). In the presence of the plant F-box protein osTIR1, addition of auxin induces rapid proteasome-dependent degradation of the SMC2-mAID protein, thus disrupting both condensin I and II complexes (18, 26, 27). Incubation of cells for 3 hours in the presence of auxin during the 1NM-PP1-induced G2–arrest (Supplementary Materials) reduced SMC2 levels to <5% (Fig. S19). Remarkably, this did not affect global chromosome organization as compartments and TADs were comparable to those in WT G2 arrested cells (Fig. 4A, Fig. S20). Cells entered prophase rapidly after washout of 1NM-PP1, and the onset of NEBD, as indicated by DAPI staining, occurred as in wild type at ~7–10 minutes (Fig. S21).
Fig. 4. Defects in chromosome morphogenesis in condensin depleted cells.

(A–C). Hi-C interaction frequency maps (binned at 100 kb) for chromosome 7 at indicated time points (top right in each heatmap) after release from G2 arrest. The first plot below each Hi-C interaction map displays the compartment signal (Eigenvector 1). The bottom graph shows the insulation score (TADs; binned at 50 kb). (A) SMC2-mAID cells were treated with auxin for three hours prior to release from G2 arrest to deplete SMC2. SMC2+: Hi-C interaction map for G2-arrested cells prior to auxin treatment. SMC2-: Hi-C interaction map for G2-arrested cells after three hours of auxin treatment. (B) Hi-C data for CAP-H2-mAID cells treated for three hours with auxin prior to release from G2 arrest to deplete CAP-H2. (C). Hi-C data for CAP-H-mAID cells treated for three hours with auxin prior to release from G2 arrest to deplete CAP-H.
Chromosomes in SMC2-depleted cells did not form well-resolved chromatids as cells progressed to prometaphase, confirming previous observations (Fig. S21) (18, 52–54). Chromatin in such cells lacks functional condensin (55, 56), but nonetheless achieves a normal degree of chromatin compaction despite the absence of individualized chromosomes (57). FACS analysis confirmed that these cells are incapable of normal mitotic exit. They ultimately undergo mitotic slippage, forming tetraploid interphase cells (Fig. S2).
Hi-C analysis revealed that in the absence of SMC2, interphase compartments and TADs were still present and largely unaffected by late prophase, at a time when they were completely disassembled in WT (t = 10 minutes, Fig. 4A, Fig. S20A). NEBD and spindle assembly did occur; indicating cells progressed to physiological prometaphase. In prometaphase (t = 45 minutes and t = 75 minutes), compartments and TADs progressively weakened, but remained detectable (Fig. 4A, Fig. S22–23). No second diagonal, characteristic for WT prometaphase ever appeared in Hi-C maps (Fig. 4A), instead P(s) curves show little change from G2 (Figs. S20A, S24–26). Preferential A-to-A interactions and B-to-B interactions became progressively weaker (Fig. S20A, S22A). Analysis of the variation of the insulation score along chromosomes indicated that TAD boundaries were reduced in strength but not eliminated (Fig. 4A, Fig. S23). Further, removal of cohesin (SMC1/3) and CTCF from chromatin, as assessed by chromatin enrichment for proteomics (ChEP) (58) was delayed and reduced compared to WT (Fig. S4D). This may explain the incomplete loss of TAD boundaries. Combined, these data reveal that condensin is not required for TAD and compartment architecture during interphase. In its absence, mitotic chromatin is compacted but chromosomes do not become individualized or acquire the normal mitotic morphology, while partially preserving elements of interphase architecture. This indicates (i) that compaction and formation of rod shaped mitotic chromosomes are two separate processes, as assumed by our model; and (ii) a critical role for condensin is in the formation of proper morphology and internal organization of mitotic chromosomes, and in disassembly of the interphase architecture (59).
Condensin I and II play distinct roles in chromosome morphogenesis
Next, we determined the roles of condensin I and II separately. Condensin I and II bind chromatin independently (52, 56, 60, 61), and recent in vitro mitotic chromosome assembly experiments show that they can act independently (62). Therefore, depletion of one condensin complex is unlikely to affect the other, though we cannot rule out more subtle interplay between the complexes. We fused auxin inducible degron domains to the condensin II-specific kleisin CAP-H2 (CAP-H2-mAID) or the condensin I-specific kleisin CAP-H (CAP-H-mAID) in CDK1as DT40 cells (Supplemental Methods). Addition of auxin lead to >95% protein depletion in G2–arrested CAP-H-mAID or CAP-H2-mAID cells (Fig. S19). Cells were then released from the G2 block and chromosome conformation was determined by microscopy and Hi-C as cells progressed through mitosis.
Depleting either condensin I or II alone led to less severe phenotypes than depleting both together (Fig. S21). In contrast to cells lacking both condensin I and II (SMC2-mAID), these cells exited mitosis within 3 hours after entry into prophase (Fig. S2).
Comparison of Hi-C interaction matrices (Fig. 4BC, Fig. S20BC) and P(s) curves (Fig. 5A, 5B, Fig. S24BC, S25–26) for CAP-H and CAP-H2 depleted cells in late prometaphase (t = 30 minutes and 60 minutes) shows that they capture different aspects of the WT architecture. The P(s) curve for CAP-H2-depleted cells, where only condensin I remains active, matches that of the intra-layer organization of WT up to ~6 Mb, and lacks the second diagonal band (Fig. 5B). The P(s) curve for CAP-H depleted cells (active condensin II), matches that of WT only for the long-range organization (6–20 Mb), including the second diagonal band (Fig. 5B). CAP-H depleted cells have a much lower contact frequency between loci separated less than 6 Mb than do WT and CAP-H2 depleted cells. Thus, condensin I and II play distinct roles at different structural levels, in mitotic chromosome morphogenesis, providing a mechanistic explanation for earlier microscopic studies (60, 61, 63, 64).
Fig. 5. Distinct roles for condensin I and II in mitotic chromosome formation.

(A) Genome-wide curves of contact frequency P(s) vs genomic distance s, normalized to unity at s=100 kb. The curves are P(s) derived from Hi-C data obtained from CAP-H2-depleted (left panel) and CAP-H-depleted cells (right panel), at t = 7–60 minutes after release from G2 arrest. Dashed line indicates P(s) = s−0.5. (B) Overlayed P(s) curves of WT, CAP-H-and CAP-H2-depleted chromosomes show independent contributions of two condensin complexes to short- and long-distance contacts. (C) Polymer models of CAP-H2 (top) and CAP-H (bottom) depleted chromosomes. Top: depletion of CAP-H2 is modeled via removal of outer loops and relaxation of the helix.. Bottom: depletion of CAP-H is modeled via removal of the inner loops, while preserving the helical arrangement of the scaffold. Condensin II loop anchors are shown in red, condensin I loop anchors are shown in blue. (D) P(s) derived from late prometaphase CAP-H2 depletion Hi-C experiments (red line) and the three best fitting polymer models (grayscale lines). The average loop size and linear density of loops along the chromosome axis are indicated. (E) The average loop size and linear DNA density of the 3 best-fitting models of CAP-H2-depleted chromosomes at different time points. (F) P(s) derived from late prometaphase CAP-H depletion Hi-C experiments (blue line) and the best fitting polymer models with and without nested inner loops (grayscale lines). The average size of outer and inner loops, the length of a helix turn in Mb and the helical pitch are indicated. (G) The best fitting P(s) predictions by the staircase coarse grained model for late prometaphase CAP-H depletion Hi-C experiments at t = 30 minutes after release of G2 arrest (gray lines; experimental P(s): red lines. Top: loop size is 200 kb, bottom: loop size is 400 kb.
Helical winding during prometaphase requires condensin II
In condensin II-depleted cells, both A- and B-compartments and TADs were lost starting around the prophase-prometaphase transition (t = 10 – 15 minutes; Fig. 4B, Fig. S20, Fig. S22). In late prometaphase (t = 30 – 60 minutes), chromosomes in these cells were longer and narrower than WT chromosomes, as previously observed (60, 63, 64) (Fig. S21). P(s) curves for t = 10 and 15 minutes (early prometaphase) resembled those in WT for late prophase (t = 10 minutes; compare Fig. 5A with Fig. 2A), displaying a mild decay followed by a steep drop that is characteristic for a densely packed loop array (Fig. 2B). Most strikingly, CAP-H2 depletion prevented emergence of the second diagonal band in prometaphase in Hi-C contact frequency maps and P(s) plots (Fig. 4B, 5B; Fig. S20B, S24B).
The close similarity between CAP-H2 prometaphase and WT prophase Hi-C, and lack of the second diagonal, allowed us to model CAP-H2 chromosomes as a prophase-like array of a single layer of loops emanating from a flexible, non-helical scaffold. By systematically varying the loop size and the degree of linear compaction, we obtained excellent agreement with experimental P(s) curves for ~40–60 kb loops, and a linear density of 15 Mb/µm for all prometaphase time points (Fig. 5D, 5E). This linear density is 3–4 times smaller than that of WT prometaphase chromosomes (50–70 Mb/µm). These simulations indicate that in the absence of condensin II, prometaphase chromosomes form extended prophase-like loop arrays and do not progress to further longitudinal shortening and helical winding.
Condensin I modulates the internal organization of prometaphase helical layers
Cells depleted for CAP-H (Fig. S19) seemed to progress through prophase normally: Hi-C data show a rapid loss of compartments and TADs (Fig. 4C, S20C, S22, S23) and by late prophase individual chromosomes were discerned by DAPI staining (Fig. S21). Deviation from the WT morphogenesis pathway was observed during prometaphase, i.e. after NEBD, when the bulk of condensin I normally loads in WT (Fig. S4B, C). A second diagonal was observed at 30 minutes indicating helical winding of the chromatids (Fig. 4C, S20C) but this was located at a genomic distance of ~12 Mb, which in WT cells was only observed at t = 60 minutes. Therefore, the progression to larger helical turns during prometaphase is accelerated in cells lacking CAP-H.
Despite a spiral organization, loss of condensin I leads to a different loop arrangement and folding, as seen from differences in the P(s) curves: the intra-layer arrangement of loops shows a characteristic P(s)~s−0.5 from 400 kb to ~3 Mb, with P(s) for the s<400 kb region having a different slope, possibly reflecting a different intra-loop organization. These features are captured well by the coarse-grained model with 200–400 kb loops emanating with correlated angular orientations from a spiral scaffold (Fig. 5G). This loop size agrees well with the sizes of outer loops in the best models for WT chromosomes at t = 60 minutes (Fig. 3H).
Strikingly, when we matched the t = 30 minutes P(s) curve with the simulations of prometaphase chromosomes with helical scaffolds and nested loops, the best match was achieved with either a single layer of 200 kb loops, or a nested system of loops, with 400 kb outer loops and 200 kb inner loops (Fig. 5F). Together these results suggest that CAP-H (condensin I) is essential for formation of short (60–80 kb) inner loops but is dispensable for ~200–400 kb outer loops emanating from a helical staircase scaffold. The helical arrangement appears weaker in condensin I depleted chromosomes, as illustrated by the reduced strength of the second diagonal and reduced peaks in the P(s) plots. One possible reason for this could be the much larger loop sizes in condensin I depleted chromosomes that may allow larger disorder in their angular arrangement. Taken together, our data obtained with CAP-H and CAP-H2 depleted cells support the formation of nested loops during prometaphase.
Discussion
We delineate a folding pathway from interphase to metaphase at minute time resolution. Hi-C data reveal a periodic pattern of interactions that we show to be consistent with a helical staircase model of mitotic chromosome folding. This model unifies many disparate observations made over the last several decades. We demonstrate that mitotic chromosomes have nested loops that are formed by differential action of condensin I and II, with condensin II being required for helical coiling of mitotic chromosomes. Finally, we find that condensins are required for the timely loss of the interphase nuclear architecture.
A mitotic chromosome morphogenesis pathway
The data and modeling presented here suggest a chromosome morphogenesis pathway by which cells convert interphase chromosome organization into compacted mitotic chromosomes (Fig. 6). Together, our imaging and Hi-C data, coarse-grained models and polymer simulations, and previous observations (11) reveal that upon entry into prophase, interphase features such as compartments and TADs are lost within minutes in a condensin-dependent process and by late prophase, chromosomes are organized as radial loop arrays. The mechanism by which TADs and compartments are lost is not known. Our data show that condensin is required. Additional contributing factors could include loss of CTCF and cohesin binding (Fig. S4B, S4C) and increased levels of loop extrusion that can erase boundaries even when CTCF is still bound (Supplemental Materials, Fig. S27). Importantly, activation of the mitotic kinase cascade is not sufficient to disassemble interphase chromatin organization without the action of condensin.
Fig. 6. A mitotic chromosome morphogenesis pathway.

In prophase, condensin II compacts chromosomes into arrays of consecutive loops and sister chromatids split along their length. The scaffold of condensin II-mediated loop bases is indicated in red. Upon nuclear envelope breakdown and entry into prometaphase, condensin II-mediated loops become increasingly large as they split into smaller ~80 kb loops by condensin I. Chromosomes are shown as arrays of loops (only inner loops can be observed microscopically; top: cross-section, bottom: side view. For clarity, loops are indicated as separate entities pointing in one direction, while in reality loops are unstructured and can mix). The nested arrangements of centrally located condensin II-mediated loop bases and more peripherally located condensin I-mediated loop bases are indicated in red and blue respectively. During prometaphase central scaffold acquires a helical arrangement with loops rotating around the scaffold as steps in a “spiral staircase” (helical path of loops is indicated by arrows). As prometaphase progresses outer loops grow and the number of loops per turn increases and chromosomes shorten to form the mature mitotic chromosome.
Our models that achieve best agreement with Hi-C data show that during prophase, condensin II-dependent loops grow from 30–40 kb to 60 kb in size, leading to a ~2-fold increase in linear chromatin density from ~7 Mb/µm to 15 Mb/µm. Condensins at loop bases form a chromosomal scaffold (19, 62), which may be a dynamic, rather than static structure, and loops are arranged consecutively along it (one loop every ~5 nm of the axis). Interestingly, the radial arrangement of loops around the central flexible scaffold is not random, with consecutive loops projecting in similar directions i.e. with an angularly correlated arrangement.
Chromosomes shorten along their longitudinal axis and become wider during prometaphase. Our simulations show that condensin II loops continue to grow to 200–400 kb by 30 min and 400–700 kb by 60 min, accompanied by an increase in the linear chromatin density, which reaches 60 Mb/µm. However, two important reorganizations take place during prometaphase. First, large condensin II-mediated loops are subdivided into smaller 80 kb loops in a condensin I-dependent process, thus producing a nested loop arrangement with ~400 kb outer loops and ~80 kb inner loops. Second, the loop array acquires a helical arrangement as evidenced by the appearance of a second diagonal band in Hi-C maps for all loci and chromosomes. Models show that this helical arrangement of loops can be achieved if the scaffold forms a narrow helical “spiral staircase” inside an otherwise homogeneous cylindrical chromosome. Interestingly, the period, radius and pitch of this helix continue to grow through prometaphase, and this growth is to some extent restrained by condensin I. An emerging model of the prometaphase chromosome thus has a central helical scaffold formed by condensin II (62) that organizes 200–400 kb outer loops, that are further subdivided into 80 kb condensin Imediated inner loops in order to achieve a high volume density.
Comparison to previous and classical studies
While specific details of this model emerge from an unbiased fitting of models to the data, the emerging organization and its quantitative characteristics agree with earlier studies. First, the 60–80 kb sizes of the inner loops are remarkably similar to values suggested by an extensive survey of the literature (44), measurements from electron microscopy (6, 19) and Hi-C analysis of mitotic HeLa cells (8). Similarly, changes of linear density from prophase to prometaphase in the best models (from 15 Mb/µm to 50 Mb/µm) are consistent with prophase chromosomes being at least two-fold longer than metaphase chromosomes (11, 61).
Second, helical prometaphase chromosomes have long been observed in certain chromosome preparations (10, 37, 38, 40), and this has led to diverse models for how mitotic chromosomes are folded. Our analysis of Hi-C data indicates that the prometaphase chromosome is organized around a helical central region or scaffold: loops emanate with helical packing from a centrally located “spiral staircase” scaffold. Modeling shows that other helical arrangements of loop arrays, e.g. coiling of the entire loop array itself (40, 50, 65, 66), are not consistent with our Hi-C data.
Our helical scaffold/loop model unifies a range of models and observations made over the years. It explains how a helical chromatin packing arrangement can be achieved while scaffold proteins such as condensins and topoisomerase II are localized centrally (15–17), within a cylindrical chromatid that is not obviously helical when visualized with a DNA dye such as DAPI (67). Interestingly, by late prometaphase we estimate the height of one helical turn to be around 200 nm, which is also the size of the layer (12 Mb layer at linear density 60 Mb/um), and is consistent with microscope observations suggesting that consecutive genomic loci follow a helical gyre with a pitch of ~250 nm within the cylindrical shape of chromatids (68).
Possible mechanisms
Such loop arrangements can naturally emerge due to a process of loop extrusion. Loop extrusion has been hypothesized as a mechanism of chromosome compaction (69, 70) and most recently examined by simulations (25, 48, 71) and supported by single-molecule studies (72). In this process, each condensin starts forming a progressively larger loop until it dissociates or stops being blocked by neighboring condensins or other DNA-binding proteins. A recent study demonstrated that this process can form an array of consecutive loops (8) with condensins forming a central scaffold in the middle of a cylindrical chromosome (48), essential features of mitotic chromosomes. We note that sister chromatids are resolved by late prophase (11–13) indicating that the formation of loop arrays occurs as sister chromatid arms become separated. Another aspect of loop extrusion is that loop sizes are established by a dynamic process of condensin exchange, without a need for barrier elements or specific loading sites (25). This is consistent with our Hi-C data that suggests that loop bases are not positioned at specific reproducible positions (e.g. scaffold or matrix attachment regions - (73, 74)) in a population of cells. Analysis of published chromatin immunoprecipitation data for SMC2 in mitotic DT40 cells (45) shows a low level of condensin binding throughout the genome and only very few loci enriched in condensin binding: only 289 sites show more than a 5-fold enrichment compared to DNA input and 4,617 sites show over 2-fold enrichment. These numbers are much lower than the 16,000 inner loops our data and models predict. Interestingly, the condensin-enriched sites do show a Hi-C interaction pattern consistent with them being at the bases of loops slightly more frequently than other loci (Supplemental Materials, Fig. S13). Based on these analyses, we estimate that over 95% of mitotic loops are not positioned at specific loci.
Simulations show that loop extrusion slowly approaches steady state by exchanging condensins and gradually increasing loop sizes during this process (25). This is consistent with gradual growth of loops up to 500 kb by slowly-exchanging condensin II, and relatively rapid formation of 60–80 kb inner loops by the more rapidly exchanging condensin I (75).
Formation of nested loops was critical for our polymer simulations to reproduce prometaphase Hi-C data because only this allowed a higher linear chromatin density. In this architecture, the outer loop bases are located at the central scaffold, while the inner, nested loop bases are radially displaced. Our analysis of condensin I or II depletion reveals that condensin II generates outer loops and condensin I generates inner loops. Our simulations reveal that this nested loop arrangement can be explained by the longer half-life of condensin II and shorter half life of condensin I on chromatin as measured by FRAP (75) (Supplemental Material, Supplemental Movie 1). Indeed, nested loops only form in prometaphase, when condensin I gains access to chromatin. Thus, loop extrusion models can explain the nested loop arrangement of condensed mitotic chromosomes.
Why condensin II-based scaffolds only acquire helicity in prometaphase, and not in prophase is not known, but this could involve interactions with other proteins, such as DNA topoisomerase IIalpha or KIF4A. Our estimates of the radius of the prometaphase scaffold of 30–100 nm is consistent with a 50 nm length of SMC coiled coils that can interact with each other through HEAT repeats (76) which are known for ability to self-assemble into a helical “spiral staircase” (77). Gradual formation of such a HEAT-mediated staircase and binding of other factors can explain how the pitch and the radius of the helix increase in time.
We note that mitotic chromatin still condenses in the absence of both condensin I and II, although individualized rod-shaped chromosomes are not formed and cells cannot progress into anaphase. This indicates that there are other mechanisms by which chromatin fibers become condensed during mitosis. Our simulations also show that to achieve agreement with Hi-C data, chromatin should also be condensed (computationally analogous to poor solvent conditions) forming densely packed chromatin loops within mitotic chromosomes analogous to the dense packing of chromatin observed in mitotic chromosomes by electron microscopy (46, 78, 79). The molecular basis for this condensation is not known but may involve mitosis-specific chromatin modifications (80, 81) or active motor proteins such as KIF4A (82, 83).
The chromosome morphogenesis pathway described here, and the identification of distinct architectural roles for condensin I and II in organizing chromosomes as nested loop arrays winding around a helical “spiral staircase” within a cylindrical chromatid can guide future experiments to uncover the molecular mechanisms by which these complexes, and other key components such as topoisomerase IIalpha and KIF4A, act in generating, (re-)arranging and condensing chromatin loops to build the mitotic chromosome.
Methods Summary
DT40 Cell cultures synchronously entering mitosis were analyzed by Hi-C, imaging and proteomics to determine the structure of chromosomes. Hi-C data were used to quantify chromosome compartmentalization and to derive relationships between contact frequency P and genomic distance s. Coarse grained models and equilibrium polymer simulations were performed to test models of prophase and prometaphase chromosome organization against Hi-C data, and to identify best fitting parameters for size of loops, helical turn and pitch, linear density (Mb/micron chromosome length). Imaging of chromosome dimensions and condensin localization were performed to validate model predictions. Cell lines expressing condensin subunits fused to auxin-inducible degron domains were used to efficiently deplete these subunits prior to cells entering mitosis. Hi-C and imaging analysis were then performed to assess the effects of depletion of condensins on mitotic chromosome formation. Detailed procedures for all methods are described in the Supplementary Materials.
Supplementary Material
Acknowledgments
Supported by grants from the National Human Genome Research Institute (HG003143) and the National Institutes of Health Common Fund (DK107980) to J. Dekker and L. Mirny. J. Dekker is an investigator of the Howard Hughes Medical Institute. Work in the Mirny lab was also supported by the National Science Foundation (Physics of Living Systems, 1504942), and the National Institute of General Medical Sciences (GM114190). Work in the Earnshaw lab was funded by Wellcome, of which W.C.E. is a Principal Research Fellow (Grant 107022). The Wellcome Centre for Cell Biology is supported by core funding from the Wellcome Trust [203149]. M.T.K. was supported by JSPS KAKENHI Grant (16K5095), and research grants from Mochida Memorial Foundation for Medical and Pharmaceutical Research, SGH Foundation, The Sumitomo Foundation, and The Canon Foundation. Sorting of GFP positive and negative cells was performed in the Flow Cytometry facility, Institute of Immunology & Infection Research, Edinburgh with assistance of Dr. Martin Waterfall. We thank Dr. Hakan Ozadam for assistance with Hi-C data processing. All Hi-C data has been submitted to GEO and will be publicly available upon publication (accession number GSE102740). This paper is dedicated to the memory of Donald S. Coffey, who pioneered the study of chromatin loops in the nucleus.
Footnotes
Author contributions
KS generated cell lines, established cell synchronization protocols, performed synchronized timecourses and performed imaging analyses. JHG and NN performed Hi-C analyses. AG and JN performed polymer simulations. LAM and AG designed and analyzed coarse grained models. AG analyzed condensin chromatin immunoprecipitation data. LX and JRP prepared 1NM-PP1. IS performed chromatin enriched proteomics analyses. MK provided constructs for cell line engineering. AG, JHG, KS, JN, WCE, LAM and JD designed the project and analyzed data. All contributed to writing the manuscript.
References (85–99)
References
- 1.Dixon JR, et al. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012;485:376–380. doi: 10.1038/nature11082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nora EP, et al. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012;485:381–385. doi: 10.1038/nature11049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lieberman-Aiden E, et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science (80-.) 2009;326:289–293. doi: 10.1126/science.1181369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rao SS, et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014;159:1665–1680. doi: 10.1016/j.cell.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sanyal A, Lajoie BR, Jain G, Dekker J. The long-range interaction landscape of gene promoters. Nature. 2012;489:109–113. doi: 10.1038/nature11279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Earnshaw WC, Laemmli UK. Architecture of metaphase chromosomes and chromosome scaffolds. J. Cell Biol. 1983;96 doi: 10.1083/jcb.96.1.84. (available at http://jcb.rupress.org/content/96/1/84.long) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marsden MP, Laemmli UK. Metaphase chromosome structure: evidence for a radial loop model. Cell. 1979;17:849–58. doi: 10.1016/0092-8674(79)90325-8. [DOI] [PubMed] [Google Scholar]
- 8.Naumova N, et al. Organization of the mitotic chromosome. Science (80-.) 2013;342:948–953. doi: 10.1126/science.1236083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nagano T, et al. Cell-cycle dynamics of chromosomal organization at single-cell resolution. Nature. 2017;547:61–67. doi: 10.1038/nature23001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ohnuki Y. Structure of chromosomes. I. Morphological studies of the spiral structure of human somatic chromosomes. Chromosoma. 1968;25:402–28. doi: 10.1007/BF02327721. [DOI] [PubMed] [Google Scholar]
- 11.Liang Z, et al. Chromosomes Progress to Metaphase in Multiple Discrete Steps via Global Compaction/Expansion Cycles. Cell. 2015;161:1124–1137. doi: 10.1016/j.cell.2015.04.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nagasaka K, Hossain MJ, Roberti MJ, Ellenberg J, Hirota T. Sister chromatid resolution is an intrinsic part of chromosome organization in prophase. Nat. Cell Biol. 2016 doi: 10.1038/ncb3353. [DOI] [PubMed] [Google Scholar]
- 13.Moore LL, Roth MB. HCP-4, a CENP-C-like protein in Caenorhabditis elegans, is required for resolution of sister centromeres. J. Cell Biol. 2001;153:1199–208. doi: 10.1083/jcb.153.6.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adolph KW, Cheng SM, Paulson JR, Laemmli UK. Isolation of a protein scaffold from mitotic HeLa cell chromosomes. Proc. Natl. Acad. Sci. U. S. A. 1977;74:4937–41. doi: 10.1073/pnas.74.11.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Earnshaw WC, Heck MM. Localization of topoisomerase II in mitotic chromosomes. J. Cell Biol. 1985;100:1716–25. doi: 10.1083/jcb.100.5.1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gasser SM, Laemmli UK. The organisation of chromatin loops: characterization of a scaffold attachment site. EMBO J. 1986;5:511–518. doi: 10.1002/j.1460-2075.1986.tb04240.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Saitoh Y, Laemmli UK. Metaphase chromosome structure: Bands arise from a differential folding path of the highly AT-rich scaffold. Cell. 1994;76:609–622. doi: 10.1016/0092-8674(94)90502-9. [DOI] [PubMed] [Google Scholar]
- 18.Samejima K, et al. Mitotic chromosomes are compacted laterally by KIF4 and condensin and axially by topoisomerase IIα. J. Cell Biol. 2012;199:755–70. doi: 10.1083/jcb.201202155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paulson JR, Laemmli UK. The structure of histone-depleted metaphase chromosomes. Cell. 1977;12:817–28. doi: 10.1016/0092-8674(77)90280-x. [DOI] [PubMed] [Google Scholar]
- 20.Okada M, Hori T, Fukagawa T. The DT40 system as a tool for analyzing kinetochore assembly. Subcell. Biochem. 2006;40:91–106. doi: 10.1007/978-1-4020-4896-8_7. [DOI] [PubMed] [Google Scholar]
- 21.Bishop AC, et al. Design of allele-specific inhibitors to probe protein kinase signaling. Curr. Biol. 1998;8:257–66. doi: 10.1016/s0960-9822(98)70198-8. [DOI] [PubMed] [Google Scholar]
- 22.Hochegger H, et al. An essential role for Cdk1 in S phase control is revealed via chemical genetics in vertebrate cells. J Cell Biol. 2007;178:257–268. doi: 10.1083/jcb.200702034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Samejima K, et al. A promoter-hijack strategy for conditional shutdown of multiply spliced essential cell cycle genes. Proc. Natl. Acad. Sci. 2008;105:2457–2462. doi: 10.1073/pnas.0712083105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fudenberg G, et al. Formation of Chromosomal Domains by Loop Extrusion. Cell Rep. 2016;15:2038–49. doi: 10.1016/j.celrep.2016.04.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goloborodko A, Marko JF, Mirny LA. Chromosome Compaction by Active Loop Extrusion. Biophys. J. 2016;110:2162–2168. doi: 10.1016/j.bpj.2016.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Holland AJ, Fachinetti D, Han JS, Cleveland DW. Inducible, reversible system for the rapid and complete degradation of proteins in mammalian cells. Proc Natl Acad Sci U S A. 2012;109:E3350–7. doi: 10.1073/pnas.1216880109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nishimura K, Fukagawa T, Takisawa H, Kakimoto T, Kanemaki M. An auxin-based degron system for the rapid depletion of proteins in nonplant cells. Nat. Methods. 2009;6:917–22. doi: 10.1038/nmeth.1401. [DOI] [PubMed] [Google Scholar]
- 28.Sonoda E, Takata M, Yamashita YM, Morrison C, Takeda S. Homologous DNA recombination in vertebrate cells. Proc. Natl. Acad. Sci. U. S. A. 2001;98:8388–94. doi: 10.1073/pnas.111006398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Georgatos SD, Pyrpasopoulou A, Theodoropoulos PA. Nuclear envelope breakdown in mammalian cells involves stepwise lamina disassembly and microtubule-drive deformation of the nuclear membrane. J. Cell Sci. 1997:2129–40. doi: 10.1242/jcs.110.17.2129. [DOI] [PubMed] [Google Scholar]
- 30.Waizenegger IC, Hauf S, Meinke A, Peters JM. Two distinct pathways remove mammalian cohesin from chromosome arms in prophase and from centromeres in anaphase. Cell. 2000;103:399–410. doi: 10.1016/s0092-8674(00)00132-x. [DOI] [PubMed] [Google Scholar]
- 31.Losada A, Hirano M, Hirano T. Cohesin release is required for sister chromatid resolution, but not for condensin-mediated compaction, at the onset of mitosis. Genes Dev. 2002;16:3004–16. doi: 10.1101/gad.249202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Crane E, et al. Condensin-driven remodelling of X chromosome topology during dosage compensation. Nature. 2015;523:240–244. doi: 10.1038/nature14450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Imakaev M, et al. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat Methods. 2012;9:999–1003. doi: 10.1038/nmeth.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abe S, et al. The initial phase of chromosome condensation requires Cdk1-mediated phosphorylation of the CAP-D3 subunit of condensin II. Genes Dev. 2011;25:863–74. doi: 10.1101/gad.2016411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwarzer W, et al. Two independent modes of chromosome organization are revealed by cohesin removal. bioRxiv. 2016 doi: 10.1038/nature24281. (available at http://biorxiv.org/content/early/2016/12/15/094185.abstract) [DOI] [PMC free article] [PubMed]
- 36.Nora EP, et al. Targeted Degradation of CTCF Decouples Local Insulation of Chromosome Domains from Genomic Compartmentalization. Cell. 2017;169:930–944. e22. doi: 10.1016/j.cell.2017.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baranetzky J. Die Kerntheilung in den Pollenmutterzellen einiger Tradescantien. Bot. Zeitung. 1880;38:281–296. [Google Scholar]
- 38.Kuwada Y. Chromosome Structure A critical review. Cytologia (Tokyo) 1939;10:213–256. [Google Scholar]
- 39.Woodcock CL, Frado LL, Rattner JB. The higher-order structure of chromatin: evidence for a helical ribbon arrangement. J. Cell Biol. 1984;99:42–52. doi: 10.1083/jcb.99.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boy de la Tour E, Laemmli UK. The metaphase scaffold is helically folded: sister chromatids have predominantly opposite helical handedness. Cell. 1988;55:937–44. doi: 10.1016/0092-8674(88)90239-5. [DOI] [PubMed] [Google Scholar]
- 41.Craig JM, Bickmore WA. Chromosome bands--flavours to savour. Bioessays. 1993;15:349–54. doi: 10.1002/bies.950150510. [DOI] [PubMed] [Google Scholar]
- 42.Paturej J, Sheiko SS, Panyukov S, Rubinstein M. Molecular structure of bottlebrush polymers in melts. Sci. Adv. 2016;2 doi: 10.1126/sciadv.1601478. doi: https://doi.org/10.1126/sciadv.1601478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marko JF, Siggia ED. Polymer models of meiotic and mitotic chromosomes. Mol. Biol. Cell. 1997;8:2217–31. doi: 10.1091/mbc.8.11.2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pienta KJ, Coffey DS. A structural analysis of the role of the nuclear matrix and DNA loops in the organization of the nucleus and chromosome. J. Cell Sci. Suppl. 1984;1:123–35. doi: 10.1242/jcs.1984.supplement_1.9. [DOI] [PubMed] [Google Scholar]
- 45.Kim JH, et al. Condensin I associates with structural and gene regulatory regions in vertebrate chromosomes. Nat. Commun. 2013;4:2537. doi: 10.1038/ncomms3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ou HD, et al. Science (80-.) doi: 10.1126/science.aag0025. in press. [DOI] [Google Scholar]
- 47.Nishino Y, et al. Human mitotic chromosomes consist predominantly of irregularly folded nucleosome fibres without a 30-nm chromatin structure. EMBO J. 2012;31:1644–53. doi: 10.1038/emboj.2012.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goloborodko A, V Imakaev M, Marko JF, Mirny L. Compaction and segregation of sister chromatids via active loop extrusion. Elife. 2016;5 doi: 10.7554/eLife.14864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Booth DG, et al. 3D-CLEM Reveals that a Major Portion of Mitotic Chromosomes Is Not Chromatin. Mol. Cell. 2016 doi: 10.1016/j.molcel.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rattner JB, Lin CC. Radial loops and helical coils coexist in metaphase chromosomes. Cell. 1985;42:291–6. doi: 10.1016/s0092-8674(85)80124-0. [DOI] [PubMed] [Google Scholar]
- 51.Barry BD, Hughes D. Random walks and random environments. Clarendon Press; 1995. https://global.oup.com/academic/product/random-walks-and-random-environments-9780198537885?cc=us&lang=en&) [Google Scholar]
- 52.Hirota T, Gerlich D, Koch B, Ellenberg J, Peters J-M. Distinct functions of condensin I and II in mitotic chromosome assembly. J. Cell Sci. 2004;117:6435–45. doi: 10.1242/jcs.01604. [DOI] [PubMed] [Google Scholar]
- 53.Ono T, Fang Y, Spector DL, Hirano T. Spatial and temporal regulation of Condensins I and II in mitotic chromosome assembly in human cells. Mol. Biol. Cell. 2004;15:3296–308. doi: 10.1091/mbc.E04-03-0242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hudson DF, Vagnarelli P, Gassmann R, Earnshaw WC. Condensin is required for nonhistone protein assembly and structural integrity of vertebrate mitotic chromosomes. Dev Cell. 2003;5:323–336. doi: 10.1016/s1534-5807(03)00199-0. [DOI] [PubMed] [Google Scholar]
- 55.Ohta S, et al. The protein composition of mitotic chromosomes determined using multiclassifier combinatorial proteomics. Cell. 2010;142:810–21. doi: 10.1016/j.cell.2010.07.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ohta S, et al. Proteomics Analysis with a Nano Random Forest Approach Reveals Novel Functional Interactions Regulated by SMC Complexes on Mitotic Chromosomes. Mol. Cell. Proteomics. 2016;15:2802–18. doi: 10.1074/mcp.M116.057885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Samejima K, et al. Rapid degradation and 3D CLEM of condensin reveal chromatin compaction uncoupled from chromosome architecture in mitosis. bioRxiv. 2017 doi: 10.1242/jcs.210187. (available at http://biorxiv.org/content/early/2017/08/09/173633.abstract) [DOI] [PMC free article] [PubMed]
- 58.Kustatscher G, Wills KLH, Furlan C, Rappsilber J. Chromatin enrichment for proteomics. Nat. Protoc. 2014;9:2090–9. doi: 10.1038/nprot.2014.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yanagida M. Clearing the way for mitosis: is cohesin a target? Nat. Rev. Mol. Cell Biol. 2009;10:489–96. doi: 10.1038/nrm2712. [DOI] [PubMed] [Google Scholar]
- 60.Ono T, et al. Differential Contributions of Condensin I and Condensin II to Mitotic Chromosome Architecture in Vertebrate Cells. Cell. 2003;115:109–121. doi: 10.1016/s0092-8674(03)00724-4. [DOI] [PubMed] [Google Scholar]
- 61.Green LC, et al. Contrasting roles of condensin I and condensin II in mitotic chromosome formation. J. Cell Sci. 2012;125:1591–604. doi: 10.1242/jcs.097790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shintomi K, et al. Mitotic chromosome assembly despite nucleosome depletion in Xenopus egg extracts. Science (80-.) 2017;356:1284–1287. doi: 10.1126/science.aam9702. [DOI] [PubMed] [Google Scholar]
- 63.Shintomi K, Hirano T. The relative ratio of condensin I to II determines chromosome shapes. Genes Dev. 2011;25:1464–9. doi: 10.1101/gad.2060311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhang T, et al. Condensin I and II behaviour in interphase nuclei and cells undergoing premature chromosome condensation. Chromosome Res. 2016;24:243–69. doi: 10.1007/s10577-016-9519-7. [DOI] [PubMed] [Google Scholar]
- 65.Belmont AS, Sedat JW, Agard DA. A three-dimensional approach to mitotic chromosome structure: evidence for a complex hierarchical organization. J. Cell Biol. 1987;105:77–92. doi: 10.1083/jcb.105.1.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Belmont AS, Dietzel S, Nye AC, Strukov YG, Tumbar T. Large-scale chromatin structure and function. Curr. Opin. Cell Biol. 1999;11:307–11. doi: 10.1016/S0955-0674(99)80041-6. [DOI] [PubMed] [Google Scholar]
- 67.Maeshima K, Laemmli UK. A Two-Step Scaffolding Model for Mitotic Chromosome Assembly. Dev. Cell. 2003;4:467–480. doi: 10.1016/s1534-5807(03)00092-3. [DOI] [PubMed] [Google Scholar]
- 68.Strukov YG, Wang Y, Belmont AS. Engineered chromosome regions with altered sequence composition demonstrate hierarchical large-scale folding within metaphase chromosomes. J. Cell Biol. 2003;162:23–35. doi: 10.1083/jcb.200303098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nasmyth K. Disseminating the Genome: Joining, Resolving, and Separating Sister Chromatids During Mitosis and Meiosis. Annu. Rev. Genet. 2001;35:673–745. doi: 10.1146/annurev.genet.35.102401.091334. [DOI] [PubMed] [Google Scholar]
- 70.Riggs AD. DNA methylation and late replication probably aid cell memory, and type I DNA reeling could aid chromosome folding and enhancer function. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 1990;326:285–97. doi: 10.1098/rstb.1990.0012. [DOI] [PubMed] [Google Scholar]
- 71.Alipour E, Marko JF. Self-organization of domain structures by DNA-loop-extruding enzymes. Nucleic Acids Res. 2012;40:11202–11212. doi: 10.1093/nar/gks925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Terekawa T, et al. The Condensin Complex Is A Mechanochemical Motor That Translocates Along DNA. bioRxiv. 2017 doi: 10.1126/science.aan6516. (available at http://www.biorxiv.org/content/early/2017/05/13/137711) [DOI] [PMC free article] [PubMed]
- 73.Mirkovitch J, Gasser SM, Laemmli UK. Scaffold attachment of DNA loops in metaphase chromosomes. J. Mol. Biol. 1988;200:101–9. doi: 10.1016/0022-2836(88)90336-1. [DOI] [PubMed] [Google Scholar]
- 74.Cockerill PN, Garrard WT. Chromosomal loop anchorage of the kappa immunoglobulin gene occurs next to the enhancer in a region containing topoisomerase II sites. Cell. 1986;44:273–82. doi: 10.1016/0092-8674(86)90761-0. [DOI] [PubMed] [Google Scholar]
- 75.Gerlich D, et al. Condensin I stabilizes chromosomes mechanically through a dynamic interaction in live cells. Curr. Biol. 2006;16:333–44. doi: 10.1016/j.cub.2005.12.040. [DOI] [PubMed] [Google Scholar]
- 76.Yoshimura SH, Hirano T. HEAT repeats - versatile arrays of amphiphilic helices working in crowded environments? J. Cell Sci. 2016;129:3963–3970. doi: 10.1242/jcs.185710. [DOI] [PubMed] [Google Scholar]
- 77.Kobe B, et al. Turn up the HEAT. Structure. 1999;7:R91–7. doi: 10.1016/s0969-2126(99)80060-4. [DOI] [PubMed] [Google Scholar]
- 78.Robbins E, Gonatas NK. THE ULTRASTRUCTURE OF A MAMMALIAN CELL DURING THE MITOTIC CYCLE. J. Cell Biol. 1964;21:429–63. doi: 10.1083/jcb.21.3.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Eltsov M, Maclellan KM, Maeshima K, Frangakis AS, Dubochet J. Analysis of cryo-electron microscopy images does not support the existence of 30-nm chromatin fibers in mitotic chromosomes in situ. Proc. Natl. Acad. Sci. U. S. A. 2008;105:19732–7. doi: 10.1073/pnas.0810057105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wilkins BJ, et al. A cascade of histone modifications induces chromatin condensation in mitosis. Science. 2014;343:77–80. doi: 10.1126/science.1244508. [DOI] [PubMed] [Google Scholar]
- 81.Zhiteneva A, et al. Mitotic post-translational modifications of histones promote chromatin compaction in vitro. Open Biol. 2017 doi: 10.1098/rsob.170076. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mazumdar M, Sundareshan S, Misteli T. Human chromokinesin KIF4A functions in chromosome condensation and segregation. J. Cell Biol. 2004;166:613–20. doi: 10.1083/jcb.200401142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Takahashi M, Wakai T, Hirota T. Condensin I-mediated mitotic chromosome assembly requires association with chromokinesin KIF4A. Genes Dev. 2016 doi: 10.1101/gad.282855.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Maritan A, Micheletti C, Trovato A, Banavar JR. Optimal shapes of compact strings. Nature. 2000;406:287–90. doi: 10.1038/35018538. [DOI] [PubMed] [Google Scholar]
- 85.Cox J, Mann M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008;26:1367–72. doi: 10.1038/nbt.1511. [DOI] [PubMed] [Google Scholar]
- 86.Schwanhäusser B, et al. Global quantification of mammalian gene expression control. Nature. 2011;473:337–42. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- 87.Wiśniewski JR, Hein MY, Cox J, Mann M. A "proteomic ruler" for protein copy number and concentration estimation without spike-in standards. Mol. Cell. Proteomics. 2014;13:3497–506. doi: 10.1074/mcp.M113.037309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Landt SG, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22:1813–31. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Belton JM, et al. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods. 2012;58:268–276. doi: 10.1016/j.ymeth.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lajoie BR, Dekker J, Kaplan N. The Hitchhiker’s guide to Hi-C analysis: Practical guidelines. Methods. 2015;72:65–75. doi: 10.1016/j.ymeth.2014.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Imakaev M, et al. Iterative correction of Hi-C data reveals hallmarks of chromosome organization. Nat Methods. 2012 doi: 10.1038/nmeth.2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Warren WC, et al. A New Chicken Genome Assembly Provides Insight into Avian Genome Structure. G3. 2017;7:109–117. doi: 10.1534/g3.116.035923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang Y, et al. Spatial organization of the mouse genome and its role in recurrent chromosomal translocations. Cell. 2012;148:908–921. doi: 10.1016/j.cell.2012.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Flyamer IM, et al. Single-nucleus Hi-C reveals unique chromatin reorganization at oocyte-to-zygote transition. Nature. 2017;544:110–114. doi: 10.1038/nature21711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kihoon J, Hong K, Satchell S. Defining Single Asset Price Momentum in Terms of a Stochastic Process. 2012;2 [Google Scholar]
- 96.Eastman P, et al. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput. Biol. 2017;13:e1005659. doi: 10.1371/journal.pcbi.1005659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Eastman P, Pande VS. Efficient nonbonded interactions for molecular dynamics on a graphics processing unit. J. Comput. Chem. 2010;31:1268–72. doi: 10.1002/jcc.21413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Nuebler J, Fudenberg G, Imakaev M, Abdennur N, Mirny L. Chromatin Organization by an Interplay of Loop Extrusion and Compartmental Segregation. bioRxiv. 2017 doi: 10.1073/pnas.1717730115. (available at http://biorxiv.org/content/early/2017/10/03/196261.abstract) [DOI] [PMC free article] [PubMed]
- 99.Molnár J, et al. The Genome of the Chicken DT40 Bursal Lymphoma Cell Line. G3. 2014;4:2231–2240. doi: 10.1534/g3.114.013482. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.