

SUMMARY
Genome wide association studies (GWAS) have increased our knowledge of loci associated with a range of human diseases. However, applying such findings to elucidate pathophysiology and promote drug discovery remains challenging. Here, we created isogenic human embryonic stem cells (hESCs) with mutations in GWAS-identified susceptibility genes for type 2 diabetes. In pancreatic betalike cells differentiated from these lines, we found that mutations in CDKAL1, KCNQ1 and KCNJ11 led to impaired glucose secretion in vitro and in vivo, coinciding with defective glucose homeostasis. CDKAL1 mutant insulin+ cells were also hypersensitive to glucolipotoxicity. A high-content chemical screen identified a candidate drug that rescued CDKAL1 specific defects in vitro and in vivo by inhibiting the FOS/JUN pathway. Our approach of a proof-of-principle platform, which uses isogenic hESCs for functional evaluation of GWAS-identified loci and identification of a drug candidate that rescues gene-specific defects, paves the way for precision therapy of metabolic diseases.
eTOC Blurb
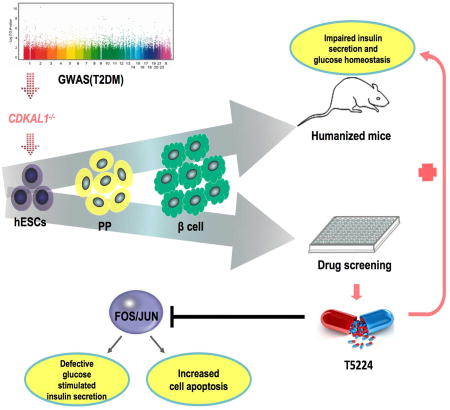
Zeng et al. report functional evaluation of GWAS identified candidate diabetes genes in an isogenic hESC-based platform. The authors find that biallelic mutations in CDKAL1, KCNQ1, and KCNJ11 caused impaired insulin secretion both in vitro and in vivo, and identified the compound T5224 which rescued mutant CDKAL1 associated pancreatic beta cell defects by inhibiting the FOS/JUN pathway.
INTRODUCTION
Multiple genome wide association studies (GWAS) have correlated Type 2 Diabetes Mellitus (T2DM) with genetic variants, yielding a large number of loci and associated gene products that are linked to the disease phenotype – often with little or no insight into the mechanism underlying that link (Hivert et al., 2014). The current challenge is to establish robust systems to systematically evaluate the role of these loci using disease relevant cells. Previous studies have used patient samples, cell lines, or animal models to seek mechanistic insight, but with significant limitations. Large variation is observed in primary patient samples, perhaps due to genetic heterogeneity, while animal models present major physiological and metabolic differences that hamper understanding of the precise function of human genes in T2DM. Therefore, a robust system to systematically evaluate the role of T2DM-associated genes using disease-relevant human cells will provide an important tool for diabetes research and spur the development of precision (allele-specific) therapies, exemplified by the use of sulfonylurea drugs to treat patients carrying certain KCNJ11 mutations (Gloyn et al., 2004).
Human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs) provide platforms to recapitulate cellular pathology of human diseases. While two iPSC models have been used to mimic pancreatic beta cell defects in neonatal and inherited forms of diabetes: Maturity Onset Diabetes of Young 2 (Hua et al., 2013) and Wolfram Syndrome patients (Shang et al., 2014), there is no robust model reported for T2DM-associated loci in the literature. Here, we focused on CDKAL1, KCNQ1, and KCNJ11 loci that were identified and confirmed through the first wave of T2DM GWAS. Risk alleles of the genetic variants at these loci are associated with aspects of beta cell function (HOMA-B) rather than insulin resistance (HOMA-IR) (Saxena et al., 2007; Scott et al., 2007; Steinthorsdottir et al., 2007; Unoki et al., 2008; Yasuda et al., 2008). Some studies suggested potential roles of these genes in pancreatic beta cell function or survival. For example, knockdown of Cdkal1 enhanced endoplasmic reticulum (ER) stress in insulinoma cells (Brambillasca et al., 2012), while Cdkal1−/− mice show reduced first phase insulin exocytosis (Ohara-Imaizumi et al., 2010) and are hypersensitive to high fat diet-induced ER stress (Wei et al., 2011) and defects in glucose-stimulated insulin secretion (Okamura et al., 2012).
For other T2DM-linked genes, population and rodent studies reported mixed or even conflicting results. The risk allele of lead SNP at KCNQ1 is associated with impaired insulin secretion (Unoki et al., 2008; Yasuda et al., 2008) and reduced insulin exocytosis in patients (Rosengren et al., 2012) and Kcnq1−/− mice have impaired glucose stimulated insulin secretion (GSIS)(Boini et al., 2009). However, forced expression of Kcnq1 in an insulinoma cell line resulted in impairment of insulin secretion (Yamagata et al., 2011) and islets isolated from Kcnq1−/− mice revealed no difference in insulin secretion compared to wildtype islets (Asahara et al., 2015). Several activating mutations in KCNJ11 result in permanent neonatal DM (Massa et al., 2005; Proks et al., 2004; Shimomura et al., 2006) and a polymorphism E23K is consistently linked with T2DM (Gloyn et al., 2003; Nielsen et al., 2003). A number of heterozygous mutations result in congenital hyperinsulinism (Bitner-Glindzicz et al., 2000). Heterozygous loss of murine Kcnj11 causes a hyperinsulinaemic phenotype, whereas complete loss underlies eventual secretory failure (Remedi et al., 2006). These mixed results suggest that GWAS-identified genes may play a context dependent role in human pancreatic beta cells. Furthermore, using mouse models, it can be challenging to differentiate whether the GWAS-associated alteration causes cell autonomous defects or acts indirectly through extra-pancreatic tissues.
We built on recent work deriving glucose-responsive pancreatic beta-like cells from hESCs/iPSCs (Pagliuca et al., 2014; Rezania et al., 2014) and used isogenic hESC-derived glucose-responding cells to systematically examine the role of several GWAS-identified genes in pancreatic beta cell function and survival. While the mutations do not affect the generation of insulin+ cells, they impaired insulin secretion both in vitro and in vivo, coinciding with defective glucose homeostasis. CDKAL1−/− insulin+ cells also displayed hypersensitivity to glucolipotoxicity. A high-content chemical screen identified a candidate drug that rescued CDKAL1−/−-specific defects by inhibiting the FOS/JUN pathway. These studies represent a proof-of-principle for the use of isogenic hESC-derived cells to define the precise role of genes associated with disease though GWAS in human pancreatic beta cells, as well as the lead-compound identification for pharmacological intervention of T2DM.
RESULTS
Generation of Biallelic Mutant hESC Lines by CRISPR-Cas9 Gene Targeting
We targeted indel mutations to CDKAL1, KCNQ1, or KCNJ11 in INSGFP/W HES3 cells, since this reporter line allows for the purification of insulin producing (insulin+) cells (Micallef et al., 2012). First, qRT-PCR was used to monitor the expression of the targeted genes in insulin-GFP+ cells derived from INSGFP/W HES3 cells. The transcript levels of CDKAL1, KCNQ1, and KCNJ11 were detected at levels comparable to those observed in primary human adult beta cells (Figure 1A), suggesting that these genes are likely to function in the insulin+ cells.
Figure 1. Biallelic mutation of CDKAL1, KCNQ1 or KCNJ11 does not affect differentiation or the expression of mature pancreatic beta cell markers.

(A) qRT-PCR experiments confirmed the expression of CDKAL1, KCNQ1, and KCNJ11 in insulin-GFP+ (INS-GFP+) cells derived from INSGFP/W HES3 cells (n=4 independent experiments, error bars indicate S.D.) The expression level of CDKAL1, KCNQ1, and KCNJ11 transcripts in primary human beta cells was calculated by dividing the expression level in primary human islets by the percentage of insulin+ cells. (B) Western blotting analysis of wildtype (wt) and isogenic mutant hESC-derived D30 cells. (C) Representative flow cytometry analysis and quantification of wt and isogenic mutant hESC-derived cells at day 30, n=3. (D) Immunocytochemistry analysis of wt and isogenic mutant hESC-derived D30 cells. The insulin+ cells express mature beta cell markers, including PDX1, NKX6.1 and NKX2.2. Scale bar = 100 µm. (E) Intracellular FACS analysis of D30 cells. (F) Total c-peptide content per 1 k insulin-GFP+ cells as measured by ELISA, n=3. Total c-peptide content in primary human beta cells was calculated by dividing the total c-peptide in primary human islets by the percentage of insulin+ cells. Clone #1 and #2 are two independent isogenic hESC clones carrying different frameshift mutations. hESCs were differentiated using protocol 2. The data is presented as mean±S.D. p values calculated by unpaired two-tailed Student’s t-test were *p<0.05, **p<0.01, ***p<0.001. See also Figure S1.
To mutate each gene, INSGFP/W HES3 cells were electroporated with a vector expressing Cas9 and a specific sgRNA targeted to the first or second exon of each gene (Table S1). After sub-cloning, an efficiency of 11–15% (Table S2) was observed for the creation of biallelic mutant lines. Multiple independent clones for each mutation were expanded. All established clones have typical hESC colony morphology and express pluripotency markers, including OCT3/4, NANOG, TRA-1-60 and TRA-1-81 (Figure S1A). To account for possible variation between different clones, two clones (#1 and #2) were chosen of each mutant line for further analysis. Biallelic indel mutations for each of the targeted genes were verified by genomic DNA sequencing (Figure S1B). Each indel mutation creates an early frame-shift that is predicted to generate null alleles. Western blotting experiments further validated the knockout of the target genes in day 30 (D30) differentiated cells derived from each mutant hESC line (Figure 1B).
Biallelic Mutation of CDKAL1, KCNQ1, or KCNJ11 Does Not Affect the Stepwise Differentiation towards Insulin+ Cells or the Expression of More Mature Beta Cell Markers
The isogenic lines were differentiated using a strategy modified slightly from a previously reported protocol (Rezania et al., 2014), which is summarized in Table S3. Immunocytochemistry analysis with antibodies against stage-specific markers was used to quantify differentiation efficiency. No significant difference was detected between wildtype and any of the isogenic mutant lines with respect to their capacity to differentiate toward definitive endoderm (SOX17+/FOXA2+, DE) (Figures S1C and S1D) or pancreatic progenitors (PDX1+/NKX6.1+/SOX9+, PP) (Figures S1E–S1H). Flow cytometry analysis showed the percentage of insulin-GFP+ cells in D30 populations to be indistinguishable between wildtype and the isogenic mutant lines (Figure 1C; Figure S1I). Together, these data suggest that biallelic mutation of CDKAL1, KCNQ1 or KCNJ11 does not affect the stepwise differentiation of insulin+ cells.
The expression of pancreatic beta cell makers in D30 hESC-derived insulin+ cells was analyzed by immunocytochemistry, and all cells, regardless of genotype, were found to express markers indicative of mature pancreatic beta cells, including PDX1, NKX6.1 and NKX2.2 (Figure 1D). Intracellular FACS analysis showed that most hESC-derived insulin+ cells express the mature beta cell marker NKX6.1, but not the alpha cell marker glucagon (Figure 1E; Figure S1J). Wildtype and isogenic mutant cell lines did not differ with respect to the NKX6.1+/insulin+ cell or insulin+/glucagon− cell fractions (Figure 1E; Figure S1J). Next, insulin-GFP+ cells were purified by cell sorting and analyzed for transcript expression levels with qRT-PCR (Figure S1K). Undifferentiated hESCs served as a negative control and primary human islets as a positive control. Transcripts encoding mature pancreatic beta cells markers, including NKX6.1, NKX2.2, PDX1, ISLET1, PAX6, NEUROD1, GCK, G6PC2, UCN3 and MAFA are highly expressed at levels comparable to human islets in hESC-derived insulin-GFP+ cells. No significant difference was observed between wildtype and isogenic mutant insulin-GFP+ cells (Figure S1K). The total c-peptide level of wildtype and mutant hESC-derived insulin-GFP+ cells, as measured by ELISA, was comparable to levels in primary human islets (Figure 1F, Table S4). Thus, mutation of CDKAL1, KCNQ1, or KCNJ11 does not significantly affect the generation of mature beta-like cells or insulin production.
Mutation of CDKAL1, KCNQ1 or KCNJ11 Differentially Impairs Insulin Secretion in Response to Multiple Secretagogues
The major function of pancreatic beta cells is to secret insulin/c-peptide upon induction by secretagogues. D30 differentiated wildtype or mutant cells were stimulated with 30 mM KCl, and secreted human c-peptide was measured by ELISA. Wildtype cells respond with a 4.5±1.6 fold induction of c-peptide secretion (Figures 2A and 2B; Figure S2A). CDKAL1−/− cells showed a small but insignificant decreased response, while KCNQ1−/− and KCNJ11−/− cells were severely and significantly impaired in their response to KCl stimulation (Figures 2A and 2B). The cells were further queried for their response to 10 mM arginine. Again, both wildtype and CDKAL1−/− D30 cells responded well, while KCNQ1−/− and KCNJ11−/− cells failed to respond (Figures 2C and 2D; Figure S2B). D30 cells were also stimulated with 20 µM forskolin or 50 µM IBMX to measure cAMP induced insulin secretion. Wildtype cells responded well to both, yielding 7.2±1.9 and 6.2±1.8 fold induction of c-peptide secretion, respectively (Figures 2E and 2F; Figure S2C). Cells carrying the three mutant alleles were able to respond to both forskolin and IBMX stimulation (Figure 2E), but compared to wildtype cells the fold induction was significantly decreased (Figure 2F). Finally, wildtype cells stimulated with 2 mM (low) or 20 mM (high) D-glucose responded to high glucose with a 2.3±0.8 fold induction of c-peptide secretion, while all three mutant genotypes failed to respond (Figures 2G and 2H, Figure S2D). Thus, loss of KCNQ1 or KCNJ11 affects insulin secretion. Since CDKAL1−/− cells respond to KCl and arginine (Figures 2A and 2C), but not cAMP or glucose stimulation (Figures 2E and 2G), CDKAL1 may be involved in cAMP and glucose sensing rather than exocytosis of insulin granules.
Figure 2. Biallelic mutation of CDKAL1, KCNQ1 or KCNJ11 impairs insulin secretion upon various stimulations.

(A and B) Human c-peptide (% of content) (A) and fold change (B) of wildtype (wt) and isogenic mutant cells at day 30 with or without 30 mM KCl stimulation in the presence of 2 mM D-glucose, n=3. (C and D) Human c-peptide (% of content) (C) and fold change (D) of wildtype and isogenic mutant cells at day 30 with and without 10 mM arginine stimulation in the presence of 2 mM D-glucose, n=3. (E and F) Human c-peptide (% of content) (E) and fold change (F) of wildtype and isogenic mutant cells at day 30 with or without 20 µM forskolin and 50 µM IBMX stimulation in the presence of 2 mM D-glucose, n=3. (G and H) Human c-peptide (% of content) (G) and fold change (H) of wildtype and isogenic mutant cells at day 30 with 2 mM or 20 mM D-glucose, n=3. Arg: arginine; forsk: forskolin; LG: 2 mM D-glucose; HG: 20 mM D-glucose. Human c-peptide secretion was calculated by dividing the secreted c-peptide by the total c-peptide of insulin-GFP+ cells or primary human beta cells. Clones #1 and #2 are two independent isogenic hESC clones carrying different frameshift mutations. hESCs were differentiated using protocol 2. The data is presented as mean±S.D. n.s. indicates a non-significant difference. p values calculated by unpaired two-tailed Student’s t-test were *p<0.05, **p<0.01, ***p <0.001, ****p <0.0001. See also Figure S2.
Patch-Clamp experiments were used to determine KATP channel activity in KCNJ11−/− cells. To perform KATP current recordings, wildtype insulin-GFP+ cells were held at 0 mV to inactivate any voltage gated ion channels, and KATP currents were elicited by depolarization from HP=0 mV to +80 mV. KATP channels were activated by the KATP channel-specific activator diazoxide (Pasyk et al., 2004) (Figure S2E), and inhibited by KATP channel-specific blocker glybenclamide. The effect of diazoxide was reversible. After washout of diazoxide, glybenclamide further reduced current amplitude from 400 pA to ~ 200 pA (Figure S2F), suggesting that in the absence of diazoxide, there were basal KATP channel activities, which was likely induced by the pipette solution. Whereas KATP currents were recorded in wildtype insulin-GFP+ cells, diazoxide and glybenclamide did not produce any effects in the recordings from insulin-GFP+ KCNJ11−/− mutant cells (Figure S2G), suggesting the absence of KATP channel activity.
CDKAL1−/− Insulin-GFP+ Cells are Hypersensitive to Glucolipotoxicity
Hyperglycemia and hyperlipidemia are two major risk factors associated with pancreatic beta cell death in diabetic patients. Wildtype and isogenic CDKAL1−/−, KCNQ1−/−, and KCNJ11−/− D30 insulin-GFP+ cells were cultured in the presence of 35 mM D-Glucose for 96 hours, or 1 mM palmitate for 48 hours. Cells were stained with propidium iodide (PI) to determine the cell death rate (Figure 3A). No significant difference was detected between wildtype and mutant insulin+ cells under control conditions. However, the percentage of PI+/insulin+ cells in CDKAL1−/− insulin+ cells was significantly higher compared to wildtype insulin+ cells exposed to 35 mM D-Glucose or 1 mM palmitate, indicating that CDKAL1−/− insulin+ cells are hypersensitive to glucotoxicity and lipotoxicity (Figure 3B). In contrast, neither KCNQ1−/− nor KCNJ11−/− insulin+ cells showed increased sensitivity to glucotoxicity or lipotoxicity (Figure S3A). Treated cells were stained with the apoptosis marker Annexin V, as well as the cell death marker 7AAD and evaluated by flow cytometry to measure apoptosis in insulin-GFP+ cells (Figure 3C and S3B). Consistent with the PI staining results, the percentage of Annexin V+/7AAD− cells in CDKAL1−/− insulin-GFP+ cells was significantly higher than wildtype (Figure 3D), KCNQ1−/− or KCNJ11−/− insulin-GFP+ cells when cultured in the presence of 35 mM D-Glucose or 1 mM palmitate (Figure S3C and S3D). We also measured the proliferation rate of wildtype and CDKAL1−/− insulin-GFP+ cells (Figure S3E), which showed no significant difference (Figure S3F). RNA-seq was used to compare the gene expression profiles in wildtype and CDKAL1−/− cells cultured in the presence or absence of palmitate. ER stress-related genes were found significantly upregulated in CDKAL1−/− cells cultured under palmitate conditions (Figures 3E and 3F). This suggests, consistent with the literature (Brambillasca et al., 2012; Wei et al., 2011), that loss of CDKAL1 induces elevated ER stress under exposure to high levels of fatty acids.
Figure 3. CDKAL1−/− insulin-GFP+ cells are hypersensitive to glucotoxicity and lipotoxicity.

(A and B) Immunocytochemistry analysis (A) and quantification of the percentage (B) of PI+/insulin+ cells in wildtype (wt) or CDKAL1−/− insulin+ cells cultured in the presence of 2 mM D-Glucose (ctrl-g), 35 mM D-Glucose (glu), no palmitate (ctrl-p) or 1 mM palmitate (palm). PI+/insulin+ cells are highlighted by arrows. (C and D) Flow cytometry analysis (C) and quantification of the percentage (D) of Annexin V+ cells in wildtype and CDKAL1−/− insulin-GFP+ cells cultured as in (A). (E) Heat map representing the expression profiles of ER-stress related genes comparing wildtype and CDKAL1−/− insulin+ cells cultured in the absence or presence of 1 mM palmitate. (F) Ingenuity Pathway Analysis of genes that are >2 fold upregulated in CDKAL1−/− insulin+ cells cultured in the presence of 1 mM palmitate. INS: insulin; PI: propidium iodide. n=3 independent biological replicates. n.s. indicates a non-significant difference. Clone #1 and #2 are two independent isogenic hESC clones carrying different frameshift mutations. hESCs were differentiated using protocol 2. p values calculated by unpaired two-tailed Student’s t-test were *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Scale bar = 100 µm. See also Figure S3.
CDKAL1−/−, KCNQ1−/−, and KCNJ11−/− hESC-Derived Beta-Like Cells Show Defective GSIS and Impaired Capacity to Maintain Glucose Homeostasis in vivo
To determine the survival and functional capacities of CDKAL1−/−, KCNQ1−/−, and KCNJ11−/− hESC-derived beta-like cells in vivo, wildtype and isogenic mutant glucose-responding cells were transplanted under the kidney capsule of immuno-deficient SCID-Beige mice. Two days after transplantation, the mice were treated with 200 mg/kg streptozotocin (STZ) to chemically ablate endogenous murine pancreatic beta cells (Figure S4A). After STZ treatment, the levels of mouse insulin are below the detection limit of the ELISA kit (Figure S4B). Two weeks post-transplantation, SCID-beige mice carrying human cells were fasted overnight and monitored for GSIS, measuring by ELISA human insulin in serum at fasting and 30 min after stimulation with 3 g/kg glucose (Figure 4A and S4C). SCID-beige mice transplanted with wildtype or mutant cells displayed indistinguishable concentrations of human insulin (Figure 4A). By six weeks post transplantation, SCID-beige mice carrying wildtype cells showed significantly increased insulin secretion after glucose stimulation (Figure 4B; Figure S4D), while SCID-beige mice carrying CDKAL1−/−, KCNQ1−/−, or KCNJ11−/− cells continued to fail to respond to glucose stimulation (Figure 4B). Since SCID-beige mice transplanted with wildtype or mutant cells displayed indistinguishable concentrations of human insulin at two weeks after transplantation (Figure 4A), the failed GSIS of mice carrying mutant cells at 6 weeks after transplantation is due to the impaired function of the transplanted cells, rather than unsuccessful transplantation. These results validate in vivo the impaired glucose response measured in mutant cells in vitro (Figure 2G).
Figure 4. CDKAL1−/−, KCNQ1−/− and KCNJ11−/− cells show defective glucose stimulated insulin secretion and impaired ability to maintain glucose homeostasis after transplantation into streptozotocin-treated immunodeficient mice.

(A) human insulin GSIS at 2 weeks after transplantation of the mutant cells compared to wildtype cells (wt). (B) GSIS secretion of SCID-beige mice carrying human cells at 6 weeks after transplantation. p values calculated by one-way repeated measures ANOVA. (C and D) Intraperitoneal glucose tolerance test (IPGTT) (C) and area under the curve (AUC) (D) of STZ treated mice 6 weeks after transplantation. p values calculated by two-way repeated measures ANOVA with a Bonferroni test for multiple comparisons between wt and mutant cells. n=8 mice for each condition. hESCs were differentiated using protocol 2. n.s. indicates a non-significant difference. p values were *p<0.05 and ** p<0.01, *** p<0.001, **** p<0.0001. See also Figure S4.
To monitor the capacity of the transplanted cells to maintain glucose homeostasis in STZ-treated mice beyond an acute glucose response, an intraperitoneal glucose tolerance test (IPGTT) with 2 g/kg glucose was used. In contrast to SCID-beige mice carrying wildtype cells, those transplanted with CDKAL1−/−, KCNQ1−/−, or KCNJ11−/− cells show glucose intolerance (Figure 4C; Figure S4E). The area under the curve (AUC) for the glucose tolerance test in SCID-beige mice carrying mutant cells was significantly higher compared to that of SCID-beige mice carrying wildtype cells (Figure 4D; Figure S4F). Immunohistochemistry was used to document the persistence of human beta-like cells in transplanted human grafts. Mature pancreatic beta cells markers, including PDX1, NKX6.1, NKX2.2 and insulin were detected in the grafts regardless of genotype (Figure S4G). Taken together, beta-like cells derived from CDKAL1−/−, KCNQ1−/−, or KCNJ11−/− hESCs present with impaired glucose induced insulin secretion as well as glucose tolerance in SCID-beige mice carrying glucose-responding cells.
A High Content Chemical Screen Identifies A Candidate Drug That Rescues CDKAL1−/− Specific Glucolipotoxicity and Impaired GSIS
A high content chemical screen was performed to identify drug candidates capable of rescuing CDKAL1−/− specific glucolipotoxicity. D30 differentiated CDKAL1−/− cells were replated in 384-well plates and treated for 48 hours with chemicals from a collection of FDA-approved drugs and drug candidates in clinical trials at 10 µM in the presence of 1 mM palmitate. We screened 2000 compounds for the capacity to decrease cell death by at least 80% in CDKAL1−/− derived beta-like cells exposed to glucolipotoxicity, while also increasing the number of insulin+ cells at least two-fold (Figure S5A). Of six initial lead hits, one compound, T5224 (Figure 5A) was validated to protect CDKAL1−/− insulin+ cells from glucolipotoxicity in follow up experiments. Using the same platform as for the primary screening (1 mM palmitate), addition of T5224 caused increased numbers of insulin+ cells (Figure 5B) and a decreased percentage of PI+/INS+ cells in CDKAL1−/− insulin+ cells (Figures 5C) in a dose-dependent manner with an EC50 of 16.2 µM. In addition, T5224 rescued the increased the cell death rate in CDKAL1−/− insulin+ cells when cultured with high glucose or high palmitate (Figure 5D and 5E). As measured using the Annexin V assay for apoptosis, T5224 also rescued the increased apoptotic rate in CDKAL1−/− insulin+ cells under conditions of high fatty acid concentration, without affecting the rate in wildtype insulin+ cells (Figures 5F and 5G; Figure S5B), thus blunting hypersensitivity to glucolipotoxicity.
Figure 5. A high content chemical screen identifies a drug candidate that rescues glucolipotoxicity caused specifically by mutations in CDKAL1.

(A) Chemical structure of T5224. (B and C) Efficacy curve of T5224 on the number of insulin+ cells (B) and the percentage of PI+INS+ cells (C). PI: propidium iodide. (D and E) Immunocytochemistry analysis (D) and quantification of the percentage (E) of PI+/insulin+ cells in wildtype (wt) and CDKAL1−/−, insulin+ cells treated with 30 µM T5224 when cultured in the presence of 2 mM D-Glucose (ctrl-g), 35 mM D-Glucose (glu), no palmitate (ctrl-p) or 1 mM palmitate (palm). PI+/insulin+ cells are highlighted by arrows. (F and G) Flow cytometry analysis (F) and quantification (G) of apoptotic rate for wt or CDKAL1−/− insulin-GFP+ cells treated with DMSO or T5224. (H and I) T5224 also rescues the impaired forskolin-induced (H) and glucose-induced insulin secretion (I). Experiments in Figure 5A–C were performed using cells derived from protocol 1. Experiments in Figure 5D–I were performed using cells derived from protocol 2. n=3 independent biological replicates for each condition. n.s. indicates a non-significant difference. p values calculated by unpaired two-tailed Student’s t-test were *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Scale bar = 50 µm. See also Figure S5.
The CDKAL1−/− cells were treated with 30 µM T5224 for 48 hours and examined for impaired response to forskolin or glucose stimulated insulin secretion (FSIS and GSIS). Remarkably, the mutant cells treated with T5224 showed increased insulin secretion in response to forskolin treatment (Figure 5H and Figure S5C), significantly elevated compared to cells treated with DMSO and at a level of insulin secretion comparable to wildtype cells (Figure 5H). Similarly, T5224 treatment also rescued the impaired GSIS of CDKAL1−/− cells (Figure 5I and Figure S5D). Notably, T5224 treatment did not significantly affect FSIS or GSIS in wildtype cells.
T5224 Rescues CDKAL1−/− Induced Beta Cell Defects Through Inhibition of The FOS/JUN Pathway
T5224 was reported to be an inhibitor of FOS/JUN activator protein-1 (AP-1) (Aikawa et al., 2008). To explore this potential mechanism of action, RNA-seq was used to compare the global gene expression profiles in CDKAL1−/− and wildtype insulin-GFP+ cells. Pathway enrichment analysis highlighted the FOS/JUN and Focal Adhesion pathways as highly changed in CDKAL1−/− insulin-GFP+ cells (Figure 6A). Genes associated with the Focal Adhesion GO term were consistently downregulated (Figure 6B and Figure S6A) while the FOS/JUN pathway (Figure 6C) was consistently upregulated in CDKAL1−/− insulin-GFP+ cells. Among the top 20 genes showing relatively increased expression in CDKAL1−/− insulin-GFP+ cells are FOSB (6.3 fold), FOS (3.5 fold), and JUNB (2.4 fold) (Figure 6D), which was confirmed by qRT-PCR (Figure 6E). Finally, western blotting experiments validated the relatively increased expression of FOS protein in mutant cells (Figure 6F).
Figure 6. T5224 rescues beta cell defects caused by CDKAL1 mutation through inhibiting the FOS/JUN pathway.

(A) Pathway enrichment analysis on up/down-regulated genes in CDKAL1−/− insulin-GFP+ cells using the DAVID function annotation tool. (B) Heat map of Focal Adhesion pathway associated genes comparing wildtype (wt) and CDKAL1−/− insulin-GFP+ cells. (C) Heat map of FOS/JUN pathway associated genes comparing wt and CDKAL1−/− insulin-GFP+ cells. (D) Top 20 upregulated genes in CDKAL1−/− insulin-GFP+ cells as compared to wildtype cells. (E) qRT-PCR analysis of JUNB, FOS, FOSB expression in wildtype and CDKAL1−/− insulin-GFP+ cells. (F) Western blotting analysis of FOS protein in wildtype and CDKAL1−/− cells at D30 of differentiation. (G) Targeted mutation of FOS rescues the high death rate in CDKAL1−/− insulin-GFP+ cells in the presence of 35 mM D-glucose or 1 mM palmitate. (H and I) Flow cytometry analysis (H) and quantification of apoptotic rate (I) of CDKAL1−/− insulin-GFP+ cells expressing Cas9 and either scrambled sgRNA or sgFOS. (J and K) Mutation of FOS rescues the impaired forskolin-induced (J) and glucose-induced (K) insulin secretion that is caused by mutation of CDKAL1. SgFOS 1# and 2# represent two independent sgRNAs targeting different locations of exon1 of c-FOS. Scramble sgRNA #1 and Scramble #2 “target” controls were designed to have low homology to the human genome and are used as non-targeting controls. hESCs were differentiated using protocol 2. The data is presented as mean±S.D. n.s. indicates a non-significant difference. p values calculated by unpaired two-tailed Student’s t-test were *p<0.05, **p<0.01, ***p<0.001. See also Figure S6.
To determine whether the mutation of CDKAL1 induces pancreatic beta cell defects through activation of the FOS/JUN pathway, two sgRNAs and two scrambled sgRNAs were designed to knockout human FOS (Table S6). Wildtype and CDKAL1−/− hESC-derived day 10 PPs were infected with lentivirus expressing either sgFOS, or a scrambled sgRNA and following 4–6 days selection with puromycin, the cells were differentiated to beta-like cells for an additional 16–20 days. In cells expressing sgFOS, the expression of FOS was decreased by more than 99% based on western blotting experiments, validating the targeting efficiency (Figure S6B). The cells were cultured in the absence or presence of 35 mM D-glucose or 1 mM palmitate and analyzed with respect to the rates of cell death and apoptosis by PI staining and Annexin V, respectively. Mutation of FOS using sgRNA rescues the increased cell death rate of CDKAL1−/− insulin-GFP+ cells (Figure 6G), and cell apoptotic rate of CDKAL1−/− insulin-GFP+ cells (Figures 6H and 6I; Figure S6C). In contrast, mutation of FOS does not affect cell death (Figure 6G) or apoptosis in wildtype insulin-GFP+ cells (Figures 6H and 6I). In addition to sgRNA, two shRNA against FOS were cloned into a lentivirial vector and used to knockdown FOS. The knockdown efficiency in day 10 PPs is more than 50% based on western blotting experiments (Figure S6D). Consistent with the knockout using sgFOS, knockdown of FOS using shRNAs rescued the increased cell apoptotic rate of CDKAL1−/− insulin-GFP+ cells when cultured in high fatty acid condition (Figures S6E and S6F).
Likewise, wildtype and CDKAL1−/− hESC-derived PPs infected with lentivirus expressing sgFOS or a scrambled sgRNA were differentiated for 20 days and measured for FSIS and GSIS. CDKAL1−/− cells infected with lentivirus containing scrambled sgRNA showed impaired FSIS and GSIS compared to wildtype cells. Transfection with lentivirus expressing sgFOS rescued those phenotypes (Figures 6J and 6K; Figures S6G and S6H). However, knockout of FOS did not affect FSIS (Figure 6J) or GSIS (Figure 6K) in wildtype cells. Consistently, knockdown of FOS using shRNAs recued the impaired FSIS (Figures S6I and S6K) and GSIS (Figures S6J and S6L) in CDKAL1−/− cells without affecting wildtype cells. Together, this suggests that loss of CDKAL1 causes hypersensitivity to glucolipotoxicity and impairs FSIS and GSIS through the FOS/JUN pathway.
T5224 and Loss of FOS Rescues the Function of CDKAL1−/− Cells in vivo
To examine the effect of T5224 on CDKAL1−/− cells in vivo, mice transplanted with wildtype and CDKAL1−/− cells were examined for GSIS at 10 weeks after transplantation. Consistent with the 6 week results reported above, mice transplanted with wildtype cells respond well to glucose stimulation. In contrast, mice transplanted with CDKAL1−/− cells showed impaired GSIS (Figure 7A and S7A). After glucose stimulation, the insulin level of mice transplanted with CDKAL1−/− cells was significantly lower than for mice transplanted with wildtype cells.
Figure 7. T5224 or loss of FOS rescues the function of CDKAL1−/− cells in SCID-beige mice carrying human cells.

(A) Human insulin GSIS at 10 weeks after transplantation of mutant cells compared to wildtype cells (wt). (B) GSIS secretion of SCID-beige mice carrying human cells after glucose stimulation 48 hours after treatment with 300 mg/kg T5224 or vehicle. (C and D) IPGTT (C) and AUC (D) of mice transplanted with CDKAL1−/− cells treated with 300 mg/kg T5224 or vehicle. (E) GSIS secretion of SCID-beige mice carrying human cells after glucose stimulation after treatment with T5224 or vehicle twice a week for four weeks. (F and G) IPGTT (F) and AUC (G) of mice transplanted with CDKAL1−/− cells treated with 300 mg/kg T5224 or vehicle twice a week for 4 weeks. (H) GSIS secretion of SCID-beige mice transplanted with CDKAL1−/− cells carrying scramble sgRNA or CDKAL1−/− cells carrying sgFOS. (I and J) IPGTT (I) and AUC (J) of mice transplanted with CDKAL1−/− cells carrying scramble sgRNA or CDKAL1−/− cells carrying sgFOS at 6 weeks after transplantation. n=8 mice for each condition. hESCs were differentiated using protocol 2. In GSIS assay, p values were calculated by one-way repeated measures ANOVA. In IPGTT assay, p values were calculated by two-way repeated measures ANOVA with a Bonferroni test for multiple comparisons between DMSO and T5224 treated conditions. p values were *p<0.05, ** p<0.01, ***p<0.001. See also Figure S7.
Subsequently, mice were treated with 300 mg/kg T5224 orally and measured for GSIS 48 hours after treatment. Mice treated with T5224 restored the capacity to respond to glucose stimulation (Figure 7B and S7B). T5224 treatment significantly increased the level of insulin secretion after glucose stimulation. In addition, the mice carrying CDKAL1−/− cells showed glucose intolerance. T5224 treatment restored the capacity of the SCID-beige mice carrying human cells to maintain glucose homeostasis (Figure 7C and S7C). The AUC for mice after T5224 treatment was significantly lower than for mice treated with control vehicle (Figure 7D and S7D). T5224 treatment was also examined using mice carrying wildtype cells. Consistent with the in vitro results (Figure 5), T5224 treatment affects neither GSIS (Figure S7E and S7F) nor glucose tolerance of mice carrying wildtype cells (Figure S7G–S7J). To determine the long-term effect of T5224, mice carrying CDKAL1−/− cells were treated with 300 mg/kg T5224 orally twice a week and measured for GSIS and glucose tolerance 4 weeks after treatment. The long-term treatment of T5224 restored both GSIS (Figure 7E and S7K) and glucose tolerance (Figure 7F, 7G and S7L, S7M) for mice carrying CDKAL1−/− cells. Finally, D30 CDKAL1−/− cells carrying scrambled sgRNA, and D30 CDKAL1−/− cells carrying sgFOS were transplanted into mice that were than measured for function in vivo 6 weeks after transplantation. Consistent with in vitro results (Figure 6), mice with CDKAL1−/− cells carrying sgFOS showed improved GSIS (Figure 7H and S7N) and a stronger ability to maintain glucose homeostasis (Figure 7I, 7J and S7O and S7P) than mice transplated with CDKAL1−/− cells carrying scrambled sgRNA. Together, these data suggest that T5224 or loss of FOS rescues the function of CDKAL1−/− cells in vivo.
DISCUSSION
With more than 80 loci associated with T2DM identified by GWAS, a robust platform to evaluate the role of these loci using disease relevant cells is urgently needed. Here we report proof-of-principle for using isogenic hESC-derived glucose-responding cells to evaluate the role of these loci in the function and survival of human pancreatic beta cells under conditions mimicking both health and disease. The derived glucose-responding cells share the same genetic background, providing a unique resource to determine the precise role of genes or loci in human pancreatic beta cells independent of complications from genetic heterogeneity implied by other approaches such as patient-derived iPSCs.
We found that mutation of KCNJ11 resulted in impaired insulin secretion upon KCl, arginine, forskolin, IBMX, and glucose stimulation, suggesting that KCNJ11 plays an essential role in insulin secretion, which is consistent with results in homozygous Kcnj11−/− KO mice, as well as in homozygous Kcnj11−/− null mice (Remedi et al., 2006) (Boini et al., 2009). In the context of reports that forced expression of KCNQ1 in a mouse beta cell line results in impairment of insulin secretion (Yamagata et al., 2011) and islets isolated from Kcnq1−/− mice reveal no difference in the extent of basal or stimulated insulin secretion compared to islet from wildtype mice (Asahara et al., 2015), we were surprised to find impaired insulin secretion in KCNQ1−/− insulin-secreting cells. This apparent discrepancy may suggest dose and/or species-specific roles in pancreatic beta cell function, highlighting the importance of using human relevant cell types.
An ultimate goal of exploring loci or genetic variants associated with disease through GWAS is to identify locus/variant-specific treatments. Risk alleles of SNPs at the CDKAL1 locus associated with diabetes are thought to be loss-of-function alleles, which we modeled generating null mutations. We found that CDKAL1−/− insulin+ cells showed impaired FSIS and GSIS, which is consistent with Cdkal1−/− mice showing reduced first phase insulin exocytosis (Ohara-Imaizumi et al., 2010). CDKAL1−/− insulin+ cells also show increased ER stress, cell apoptosis and death when cultured in high glucose and high fatty acid conditions. Although there are papers describing the potential contribution of lipotoxicity in T2DM, direct evidence that lipotoxicity affect pancreatic beta cell death in vivo under normal physiological and pathological conditions needs to be further explored. Here, we found that CDKAL1−/− insulin+ cells are hypersensitive to both high glucose and high fatty acid induced pancreatic beta-like cell death. Moreover, CDKAL1−/− insulin+ cells display defective GSIS and impaired ability to maintain glucose homeostasis following transplantation into STZ-treated mice. This is consistent with the in vitro functional defects of CDKAL1−/− insulin+ cells. Since the mice are hyperglycemic after STZ treatment, the observed glucotoxicity may further worsen the defects of CDKAL1−/− insulin+ cells. From a high content chemical screen, T5224 was found to rescue the CDKAL1 mutation-mediated pancreatic beta cell defects. T5224 has been investigated in clinical trials for patients with rheumatoid arthritis (Adis, 2014) and may have the potential to be repurposed for CDKAL1-specific treatment of T2DM. T5224 is able to strikingly rescue CDKAL1 mutation-mediated pancreatic beta cell dysfunction in vivo, which is a proof-of-concept for a T2DM drug candidate rescuing a gene-specific defect in vivo.
By combining high content chemical screening and RNA-seq, we found the FOS/JUN pathway to be significantly upregulated in CDKAL1−/− insulin+ cells, and that reducing FOS/JUN pathway activity either chemically or genetically rescued CDKAL1 mutation-induced defects. Previous studies have shown that FOS/JUN activation is involved in cytokine and mechanical stress-induced beta cell death (Abdelli et al., 2007; Hughes et al., 1990) and amylin-induced apoptosis (Zhang et al., 2002). Here, we found that CDKAL1−/− mediated activation of the FOS/JUN pathway through fatty acids may be a further effector of FOS/JUN regulated beta cell survival, providing mechanistic insight into how CDKAL1 locus may contribute to diabetes progression.
In summary, we established an isogenic hESC platform to systematically evaluate the role of disease-associated loci in the survival and function of human pancreatic beta-like cells in vitro and in vivo. The platform can be used to study other disease-associated loci/variants with respect to beta-like cell function. It is worth noting that the glucose-responding cells derived using the current reported protocols are not equivalent to primary human beta cells. Ca2+ flux assays suggested that approximately 30–40% of the insulin-GFP+ cells show increased cytosolic Ca2+ concentrations in response to glucose stimulation (Figure. S7Q), whereas robust glucose-induced signaling was observed in more than 70% of human beta cells, based on the previous report (Rezania et al., 2014). The restricted functionality of pancreatic beta-like cells derived using current protocols might limit their application for evaluating subtle contributions of genes to glucose metabolism and Ca2+ signaling. Thus, additional work is needed to further improve the protocol to derive mature pancreatic beta-like cells. In addition, the platform established here can also be applied to study the role of disease-associated loci/variants in other diabetes-related cell types, such as hepatocytes, adipocytes, muscles and/or intestinal neuroendocrine cells. Finally, the system may be used as a high throughput/content chemical screening platform to identify candidate drugs correcting allele-specific defects, for precision therapy of metabolic diseases.
EXPERIMENTAL PROCEDURES
Cell Culture and Chemicals
All experiments were performed using INSGFPW HES3 cells. hESCs were grown on Matrigel-coated 6-well plates in mTeSR1 medium (STEM CELL Technologies). Cells were maintained at 37°C with 5% CO2. T5224 was purchased from MedChem Express (HY-12270). Human islets were provided by IIDP (Integated Islet Distribution Program).
Creation of Isogenic Mutant hESC Lines
To mutate the target genes, two sgRNAs targeting the first two exons of the target gene were designed, cloned into a vector carrying a CRISPR-Cas9 gene, and validated using the surveyor assay in 293T cells. After validation, INSGFP/W HES3 cells were dissociated using Accutase (STEM CELL Technologies) and transfected (8 ×105 cells per sample) in suspension using Human Stem Cell Nucleofector™ solution (Lonza) using electroporation and following the manufacturer’s instructions. Cells were co-transfected with the vector expressing Cas9/sgRNA at 10 nM final concentration and a vector expressing puromycin. After replating, the transfected cells were treated with 500 ng/ml puromycin. After two days of puromycin selection, hESCs were dissociated into single cells by Accutase and re-plated at low density. 10 µM Y-27632 was added. After approximately 10 days, individual colonies were picked, mechanically disaggregated and re-plated into two individual wells of 96-well plates. A portion of the cells was analyzed by genomic DNA sequencing. For biallelic frameshift mutants, we chose both homozygous mutants and compound heterozygous mutants. Wild-type clonal lines from the corresponding targeting experiments were included as wild-type controls to account for potential nonspecific effects associated with the gene targeting process.
Stepwise Differentiation
Wildtype and isogenic mutant hESCs were differentiated using either of two slightly modified protocols from what was previously reported (Rezania et al., 2014). The details of protocol 1 and 2 are listed as supplemental Figure 1C and described in detail in supplemental methods.
In vivo Transplantation, GSIS and IPGTT
Wildtype and isogenic mutant hESCs at day 30 of differentiation were resuspended in 40 µl DMEM+B27 and transplanted under the kidney capsule of 6–8 week old male SCID-beige mice. Two days after transplantation, the mice were treated with 200 mg/kg STZ. To perform GSIS, mice were starved for about 20 hours. Mouse blood was collected under fasting conditions and at 15 min after intraperitoneal injection with 3 g/kg glucose solution. The mouse sera were analyzed using the ultrasensitive human insulin ELISA kit (ALPCO, 80-INSHUU-E01.1). To perform IPGTT analysis, the mice were fasted overnight and treated with 2 g/kg glucose. Blood glucose level (mg/dl) in each animal was measured before and every 15 minutes in the first hour and every 30 minutes in the second hour after glucose injection. The mice transplanted with wildtype or CDKAL1−/− cells were orally treated with 300 mg/kg T5224 dissolved in polyvinylpyrrolidone K 60 solution (Sigma). After 48 hours treatment, the mice were examined for GSIS and IPGTT. The mice treated with polyvinylpyrrolidone K 60 solution (vehicle) were used as the controls. For long-term treatment, the mice were orally treated with 300 mg/kg T5224 twice a week for four weeks. GSIS and IPGTT were measured 48 hours after the last treatment.
High Content Chemical Screening
To perform the high content chemical screening, CDKAL1−/− D30 cells were plated on 804G-coated 384 well plates at 5000 cells/40 µl medium/well. After overnight incubation, cells were treated at 10 µM with compounds from a chemical collection containing the Prestwick FDA approved drug library and drugs in clinical trials. DMSO treatment was used as a negative control. After 48 hours incubation, cells were first stained with 100 µg/ml PI and then fixed and stained using an insulin antibody (DAKO). Plates were analyzed using a Molecular Devices ImageXpress High-Content Analysis System. Two dimensional analysis was used. Compounds decreasing the cell death rate in excess of 80% and increasing the number of insulin+ cells by 2-fold were picked as primary hits.
Statistical Analysis
n=3 independent biological replicates if not otherwise specifically indicated. n.s. indicates non-significant difference. p values were calculated by unpaired two-tailed Student’s t-test if not otherwise specifically indicated. n=8 mice for in vivo experiments if not otherwise specifically indicated. p values were calculated by one-way repeated measures ANOVA or two-way repeated measures ANOVA with a Bonferroni test for multiple comparisons between wildtype and knockout cells. *p<0.05, **p<0.01, ***p<0.001, and ****p<0.0001.
Supplementary Material
Highlights.
An isogenic hESC-based platform to define precise roles of T2D associated genes.
CDKAL1−/−, KCNJ11−/−, KCNQ1−/− beta-like cells show defective insulin secretion.
CDKAL1−/− beta-like cells are hypersensitive to glucotoxcity and lipotoxicity;
T5224 rescues CDKAL1−/− induced beta cell defects by inhibiting the FOS/JUN pathway.
Acknowledgments
S.C. is funded by The New York Stem Cell Foundation (R-103), American Diabetes Association (1-12-JF-06), Tri-institutional Starr Stem Cell Grant (2014-030). S.C. is New York Stem Cell Foundation-Robertson Investigator. T.Z. is funded by a NYSTEM postdoctoral fellowship. A.Y. and L. Y. are supported by NIH HL078960 (to L.Y.) and AHA grant 16GRNT26430113 (to L.Y.). J.G. was supported by “Biomedical Research Program” funds at Weill Cornell Medical College in Qatar, a program funded by Qatar Foundation. Q. Q. is supported by a Scientist Development Award (K01HL129892) from the NHLBI. This study was also supported by a Shared Facility contract to T.E. and S.C. from the New York State Department of Health (NYSTEM C029156). NKX6.1 and NKX2.2 antibodies were provided by U of lowa Hybridoma bank. Human islets were provided by The Integrated Islet Distribution Program. The plentiCRISPR v2 vector and Puro 2.0 were purchased from Addgene (plasmid#52961 and #24970). We are also very grateful for technical support and advice provided by Harold S. Ralph in the Cell Screening Core Facility and Jason McCormick in the Flow Cytometry Facility at Weill Cornell Medical College, NY. The authors have filled a patent “AP-1 inhibitors for precison therapy of diabetic pateints”
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
AUTHOR CONTRIBUTIONS
S.C., H.Z., J.G., T.E. designed the project, H. Z. and M. G. performed most experiments, T. Z., L. T., C. N. C., X. D., A. Y. and L. Y. performed other necessary experiments, T. Z. and J. Z. X. performed the bioinformatics analysis, H. Z., M. G., and S.C. analyzed data, H. Z., S.C. J.G., Q.Q. and T.E. wrote the manuscript.
References
- Abdelli S, Abderrahmani A, Hering BJ, Beckmann JS, Bonny C. The c-Jun N-terminal kinase JNK participates in cytokine- and isolation stress-induced rat pancreatic islet apoptosis. Diabetologia. 2007;50:1660–1669. doi: 10.1007/s00125-007-0704-2. [DOI] [PubMed] [Google Scholar]
- Adis. Drugs in Clinical Development for Rheumatoid Arthritis Summary and Table. Pharmaceutical Medicine. 2014;28:195–213. [Google Scholar]
- Aikawa Y, Morimoto K, Yamamoto T, Chaki H, Hashiramoto A, Narita H, Hirono S, Shiozawa S. Treatment of arthritis with a selective inhibitor of c-Fos/activator protein-1. Nature biotechnology. 2008;26:817–823. doi: 10.1038/nbt1412. [DOI] [PubMed] [Google Scholar]
- Asahara S, Etoh H, Inoue H, Teruyama K, Shibutani Y, Ihara Y, Kawada Y, Bartolome A, Hashimoto N, Matsuda T, et al. Paternal allelic mutation at the Kcnq1 locus reduces pancreatic beta-cell mass by epigenetic modification of Cdkn1c. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:8332–8337. doi: 10.1073/pnas.1422104112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitner-Glindzicz M, Lindley KJ, Rutland P, Blaydon D, Smith VV, Milla PJ, Hussain K, Furth-Lavi J, Cosgrove KE, Shepherd RM, et al. A recessive contiguous gene deletion causing infantile hyperinsulinism, enteropathy and deafness identifies the Usher type 1C gene. Nature genetics. 2000;26:56–60. doi: 10.1038/79178. [DOI] [PubMed] [Google Scholar]
- Boini KM, Graf D, Hennige AM, Koka S, Kempe DS, Wang K, Ackermann TF, Foller M, Vallon V, Pfeifer K, et al. Enhanced insulin sensitivity of gene-targeted mice lacking functional KCNQ1. American journal of physiology Regulatory, integrative and comparative physiology. 2009;296:R1695–1701. doi: 10.1152/ajpregu.90839.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brambillasca S, Altkrueger A, Colombo SF, Friederich A, Eickelmann P, Mark M, Borgese N, Solimena M. CDK5 regulatory subunit-associated protein 1-like 1 (CDKAL1) is a tail-anchored protein in the endoplasmic reticulum (ER) of insulinoma cells. The Journal of biological chemistry. 2012;287:41808–41819. doi: 10.1074/jbc.M112.376558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gloyn AL, Pearson ER, Antcliff JF, Proks P, Bruining GJ, Slingerland AS, Howard N, Srinivasan S, Silva JM, Molnes J, et al. Activating mutations in the gene encoding the ATP-sensitive potassium-channel subunit Kir6.2 and permanent neonatal diabetes. The New England journal of medicine. 2004;350:1838–1849. doi: 10.1056/NEJMoa032922. [DOI] [PubMed] [Google Scholar]
- Gloyn AL, Weedon MN, Owen KR, Turner MJ, Knight BA, Hitman G, Walker M, Levy JC, Sampson M, Halford S, et al. Large-scale association studies of variants in genes encoding the pancreatic beta-cell KATP channel subunits Kir6.2 (KCNJ11) and SUR1 (ABCC8) confirm that the KCNJ11 E23K variant is associated with type 2 diabetes. Diabetes. 2003;52:568–572. doi: 10.2337/diabetes.52.2.568. [DOI] [PubMed] [Google Scholar]
- Hivert MF, Vassy JL, Meigs JB. Susceptibility to type 2 diabetes mellitus--from genes to prevention. Nature reviews Endocrinology. 2014;10:198–205. doi: 10.1038/nrendo.2014.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua H, Shang L, Martinez H, Freeby M, Gallagher MP, Ludwig T, Deng L, Greenberg E, Leduc C, Chung WK, et al. iPSC-derived beta cells model diabetes due to glucokinase deficiency. The Journal of clinical investigation. 2013;123:3146–3153. doi: 10.1172/JCI67638. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Hughes JH, Watson MA, Easom RA, Turk J, McDaniel ML. Interleukin-1 induces rapid and transient expression of the c-fos proto-oncogene in isolated pancreatic islets and in purified beta-cells. FEBS letters. 1990;266:33–36. doi: 10.1016/0014-5793(90)81499-e. [DOI] [PubMed] [Google Scholar]
- Massa O, Iafusco D, D’Amato E, Gloyn AL, Hattersley AT, Pasquino B, Tonini G, Dammacco F, Zanette G, Meschi F, et al. KCNJ11 activating mutations in Italian patients with permanent neonatal diabetes. Human mutation. 2005;25:22–27. doi: 10.1002/humu.20124. [DOI] [PubMed] [Google Scholar]
- Micallef SJ, Li X, Schiesser JV, Hirst CE, Yu QC, Lim SM, Nostro MC, Elliott DA, Sarangi F, Harrison LC, et al. INS(GFP/w) human embryonic stem cells facilitate isolation of in vitro derived insulin-producing cells. Diabetologia. 2012;55:694–706. doi: 10.1007/s00125-011-2379-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen EM, Hansen L, Carstensen B, Echwald SM, Drivsholm T, Glumer C, Thorsteinsson B, Borch-Johnsen K, Hansen T, Pedersen O. The E23K variant of Kir6.2 associates with impaired post-OGTT serum insulin response and increased risk of type 2 diabetes. Diabetes. 2003;52:573–577. doi: 10.2337/diabetes.52.2.573. [DOI] [PubMed] [Google Scholar]
- Ohara-Imaizumi M, Yoshida M, Aoyagi K, Saito T, Okamura T, Takenaka H, Akimoto Y, Nakamichi Y, Takanashi-Yanobu R, Nishiwaki C, et al. Deletion of CDKAL1 affects mitochondrial ATP generation and first-phase insulin exocytosis. PloS one. 2010;5:e15553. doi: 10.1371/journal.pone.0015553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura T, Yanobu-Takanashi R, Takeuchi F, Isono M, Akiyama K, Shimizu Y, Goto M, Liang YQ, Yamamoto K, Katsuya T, et al. Deletion of CDKAL1 affects high-fat diet-induced fat accumulation and glucose-stimulated insulin secretion in mice, indicating relevance to diabetes. PloS one. 2012;7:e49055. doi: 10.1371/journal.pone.0049055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagliuca FW, Millman JR, Gurtler M, Segel M, Van Dervort A, Ryu JH, Peterson QP, Greiner D, Melton DA. Generation of functional human pancreatic beta cells in vitro. Cell. 2014;159:428–439. doi: 10.1016/j.cell.2014.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasyk EA, Kang Y, Huang X, Cui N, Sheu L, Gaisano HY. Syntaxin-1A binds the nucleotide-binding folds of sulphonylurea receptor 1 to regulate the KATP channel. The Journal of biological chemistry. 2004;279:4234–4240. doi: 10.1074/jbc.M309667200. [DOI] [PubMed] [Google Scholar]
- Proks P, Antcliff JF, Lippiat J, Gloyn AL, Hattersley AT, Ashcroft FM. Molecular basis of Kir6.2 mutations associated with neonatal diabetes or neonatal diabetes plus neurological features. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:17539–17544. doi: 10.1073/pnas.0404756101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remedi MS, Rocheleau JV, Tong A, Patton BL, McDaniel ML, Piston DW, Koster JC, Nichols CG. Hyperinsulinism in mice with heterozygous loss of K(ATP) channels. Diabetologia. 2006;49:2368–2378. doi: 10.1007/s00125-006-0367-4. [DOI] [PubMed] [Google Scholar]
- Rezania A, Bruin JE, Arora P, Rubin A, Batushansky I, Asadi A, O’Dwyer S, Quiskamp N, Mojibian M, Albrecht T, et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nature biotechnology. 2014;32:1121–1133. doi: 10.1038/nbt.3033. [DOI] [PubMed] [Google Scholar]
- Rosengren AH, Braun M, Mahdi T, Andersson SA, Travers ME, Shigeto M, Zhang E, Almgren P, Ladenvall C, Axelsson AS, et al. Reduced insulin exocytosis in human pancreatic beta-cells with gene variants linked to type 2 diabetes. Diabetes. 2012;61:1726–1733. doi: 10.2337/db11-1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxena R, Voight BF, Lyssenko V, Burtt NP, de Bakker PI, Chen H, Roix JJ, Kathiresan S, Hirschhorn JN, Daly MJ, et al. Genome-wide association analysis identifies loci for type 2 diabetes and triglyceride levels. Science. 2007;316:1331–1336. doi: 10.1126/science.1142358. [DOI] [PubMed] [Google Scholar]
- Scott LJ, Mohlke KL, Bonnycastle LL, Willer CJ, Li Y, Duren WL, Erdos MR, Stringham HM, Chines PS, Jackson AU, et al. A genome-wide association study of type 2 diabetes in Finns detects multiple susceptibility variants. Science. 2007;316:1341–1345. doi: 10.1126/science.1142382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang L, Hua H, Foo K, Martinez H, Watanabe K, Zimmer M, Kahler DJ, Freeby M, Chung W, LeDuc C, et al. beta-cell dysfunction due to increased ER stress in a stem cell model of Wolfram syndrome. Diabetes. 2014;63:923–933. doi: 10.2337/db13-0717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimomura K, Girard CA, Proks P, Nazim J, Lippiat JD, Cerutti F, Lorini R, Ellard S, Hattersley AT, Barbetti F, et al. Mutations at the same residue (R50) of Kir6.2 (KCNJ11) that cause neonatal diabetes produce different functional effects. Diabetes. 2006;55:1705–1712. doi: 10.2337/db05-1640. [DOI] [PubMed] [Google Scholar]
- Steinthorsdottir V, Thorleifsson G, Reynisdottir I, Benediktsson R, Jonsdottir T, Walters GB, Styrkarsdottir U, Gretarsdottir S, Emilsson V, Ghosh S, et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nature genetics. 2007;39:770–775. doi: 10.1038/ng2043. [DOI] [PubMed] [Google Scholar]
- Unoki H, Takahashi A, Kawaguchi T, Hara K, Horikoshi M, Andersen G, Ng DP, Holmkvist J, Borch-Johnsen K, Jorgensen T, et al. SNPs in KCNQ1 are associated with susceptibility to type 2 diabetes in East Asian and European populations. Nature genetics. 2008;40:1098–1102. doi: 10.1038/ng.208. [DOI] [PubMed] [Google Scholar]
- Wei FY, Suzuki T, Watanabe S, Kimura S, Kaitsuka T, Fujimura A, Matsui H, Atta M, Michiue H, Fontecave M, et al. Deficit of tRNA(Lys) modification by Cdkal1 causes the development of type 2 diabetes in mice. The Journal of clinical investigation. 2011;121:3598–3608. doi: 10.1172/JCI58056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagata K, Senokuchi T, Lu M, Takemoto M, Fazlul Karim M, Go C, Sato Y, Hatta M, Yoshizawa T, Araki E, et al. Voltage-gated K+ channel KCNQ1 regulates insulin secretion in MIN6 beta-cell line. Biochemical and biophysical research communications. 2011;407:620–625. doi: 10.1016/j.bbrc.2011.03.083. [DOI] [PubMed] [Google Scholar]
- Yasuda K, Miyake K, Horikawa Y, Hara K, Osawa H, Furuta H, Hirota Y, Mori H, Jonsson A, Sato Y, et al. Variants in KCNQ1 are associated with susceptibility to type 2 diabetes mellitus. Nature genetics. 2008;40:1092–1097. doi: 10.1038/ng.207. [DOI] [PubMed] [Google Scholar]
- Zhang S, Liu J, MacGibbon G, Dragunow M, Cooper GJ. Increased expression and activation of c-Jun contributes to human amylin-induced apoptosis in pancreatic islet beta-cells. Journal of molecular biology. 2002;324:271–285. doi: 10.1016/s0022-2836(02)01044-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.