

SUMMARY
Telomeric repeat binding factor 1 (TRF1) is essential to the maintenance of telomere chromatin structure and integrity. However, how telomere integrity is maintained, especially in response to damage, remains poorly understood. Here, we identify Nek7, a member of the Never in Mitosis Gene A (NIMA) kinase family, as a regulator of telomere integrity. Nek7 is recruited to telomeres and stabilizes TRF1 at telomeres after damage in an ATM activation-dependent manner. Nek7 deficiency leads to telomere aberrations, long-lasting γH2AX and 53BP1 foci, and augmented cell death upon oxidative telomeric DNA damage. Mechanistically, Nek7 interacts with and phosphorylates TRF1 on Ser114, which prevents TRF1 from binding to Fbx4, an Skp1-Cul1-F box E3 ligase subunit, thereby alleviating proteasomal degradation of TRF1, leading to a stable association of TRF1 with Tin2 to form a shelterin complex. Our data reveal a mechanism of efficient protection of telomeres from damage through Nek7-dependent stabilization of TRF1.
Graphical abstract
In Brief: Tan et al. discover a mechanism of efficient protection of telomeres from damage by Nek7-dependent stabilization of TRF1. S114 phosphorylated TRF1 by the NIMA kinase Nek7 favors the binding of shelterin protein TIN2 and disfavors E3 ligase Fbx4 interaction, thus preventing TRF1 ubiquitination and proteasome degradation to maintain telomere integrity.

INTRODUCTION
Telomeres play important roles in preventing premature aging and/or cancer development by maintaining genome stability. Telomeric DNA damage accumulated from chronic stresses leads to reorganization and relaxation of the compacted telomere structure (Cesare et al., 2009; O’Sullivan et al., 2010). Moreover, cancer cells must address abnormal oxidative metabolism and consequent telomere DNA damage for survival. However, the regulation of telomeric structure in the face of stresses, such as oxidation-induced DNA damage, is not fully understood.
Telomeric DNA repeat sequences are coated by shelterin proteins whose binding to telomeres is essential to prevent damaged telomeres from activating unwanted DNA damage response (DDR) and harmful chromosomal fusion (de Lange, 2005; Palm and de Lange, 2008). Insufficient protection of telomeres by shelterin proteins exposes telomeres to detrimental stresses, leading to compromised telomere and genome integrity. Genetic deletion of TRF1, an essential component of the shelterin complex, triggers extensive de-condensation of telomeric chromatin, which can activate the ATM and Rad3-related (ATR) pathway to initiate the DDR at telomeres and replication fork stalling, leading to telomere association and fragile telomeres (Martínez et al., 2009; Sfeir et al., 2009). TRF1 has been shown to periodically and rapidly switch between the free- and the telomere-associated forms (Mattern et al., 2004). The high-order association of TRF1 with telomeres closely correlates with telomere length and telomere replication and has been implicated in the regulation of the DDR at telomeres (Martínez et al., 2009; Sfeir et al., 2009; van Steensel and de Lange, 1997). TRF1 contains an N-terminal acidic domain, a middle TRF homology (TRFH) domain, and a C-terminal Myb/SANT telomeric DNA-binding domain (Broccoli et al., 1997; Fairall et al., 2001; Nishikawa et al., 2001). The TRFH domain of TRF1 is essential for the dimerization of TRF1, as well as interaction with Tin2, and also serves as a versatile docking site for specific subsets of regulatory proteins that are involved in the cell cycle or DNA damage signal pathway (Chen et al., 2008). The association of TRF1 with Tin2 is important to maintain a highly condensed shelterin structure at telomeres, thereby inhibiting an inappropriate DDR (Bandaria et al., 2016). Therefore, stringent regulation of TRF1 is critical for maintaining the appropriate shelterin complex and protection of chromosome ends upon oxidative DNA damage.
Recently, members of the NIMA family of kinases have emerged as important regulators of the DDR as well as the cell cycle (Lee et al., 2008; Meirelles et al., 2011, 2014). However, little is known about the role of Nek7, the smallest member of the NIMA family, in the DDR and in telomere integrity. Cells lacking Nek7 show microtubule instability and abnormal spindle formation, indicating a role of Nek7 in mitosis (Kim et al., 2007). Nek7 is known to associate with the centrosome microtubule-organizing center (MTOC), the spindle mid-zone, and the mid-body during mitosis, suggesting a regulatory role in G2/M for cell-cycle control (Kim et al., 2007; O’Regan and Fry, 2009; Yissachar et al., 2006). Interestingly, Nek7 is also found both in the nucleus and cytoplasm during interphase (O’Regan and Fry, 2009). High Nek7 mRNA and protein levels are commonly found in breast, colorectal, and lung cancers (Capra et al., 2006). The protein level of Nek7 appears to be relatively constant throughout the cell cycle (O’Regan and Fry, 2009), and the constitutive expression of Nek7 implies that it may be involved in other cellular processes beyond mitotic regulation.
In this study, we have elucidated a molecular mechanism of Nek7-mediated telomere structure maintenance via phosphorylation of TRF1. Using a technique developed recently to induce localized oxidative DNA damage at telomeres (Sun et al., 2015), we discovered that Nek7 is recruited to telomeres and acts as a regulator of TRF1 in response to oxidative telomeric DNA damage. Mechanistically, Nek7 binds and phosphorylates TRF1 at Ser114, which in turn inhibits the interaction of TRF1 with the E3 ligase subunit, Fbx4, and leads to a stable association of TRF1 with Tin2 to form a shelterin complex in response to oxidative telomeric DNA damage. Importantly, Nek7 deficiency was shown to lead to telomere aberrations in a phenotype recapitulating that of TRF1-depleted cells. Taken together, our data reveal how TRF1 and a shelterin structure are maintained and regulated to protect the telomere in countering oxidative damage.
RESULTS
Nek7 Is Preferentially Recruited to Damaged Telomeres and Protects Telomeres from Oxidative DNA Damage
To understand how telomeres maintain integrity in response to oxidative telomeric DNA damage, we used a KillerRed (KR)-induced telomere damage system (Figure 1A) (Sun et al., 2015). Upon visible light exposure, the KR-TRF1 fusion enables KR protein to target telomeres and allows dose-dependent introduction of oxidative DNA damage confined to the telomeres. RFP-TRF1- and KR-TRF1-expressing cells were cultured in complete darkness (to shield the cells from oxidative damage accumulation) or exposed to light for 1 hr. We screened factors that may potentially be involved in the DNA damage response and found that Nek7 is specifically recruited to the telomeres in response to oxidative telomeric DNA damage. Before light exposure, a basal 20% of the cell population showed colocalization of Nek7 at RFP-TRF1- and KR-TRF1-labeled telomere sites. However, light exposure induced a strong Nek7 accumulation in nearly 80% of the cells at the telomeres in the KR-TRF1-expressing telomerase-negative (U2OS) cells (Figure 1B) and telomerase-positive (HeLa 1.3) cells (Figure 1C) but had little effect on the control, RFP-TRF1-expressing cells (Figures 1B and 1C).
Figure 1. Nek7 Is Recruited to Telomeres and Protects Cells from Telomeric DNA Damage.

(A) Schematic illustration of the KillerRed-TRF1-induced oxidative DNA damage at sites of telomeres.
(B) Recruitment of Nek7 to telomeres in the presence or absence of telomeric DNA damage in U2OS cells. The percentages of cells with positive Nek7 foci (≥5) at telomeres are shown. The scale bars represent 2 μm. Error bars represent ± SEM (n = 3, ***p < 0.001).
(C) Recruitment of Nek7 to telomeres in the presence or absence of telomeric DNA damage in HeLa 1.3 cells.
(D) Telomere aberrations (arrows) in control (shCtrl) and Nek7 knockdown (shNek7) HeLa cells. Telomeric DNA was stained by Tel C-Cy3 (red) and total DNA by DAPI (blue). The percentage of sister telomere loss (a) and sister telomere association (b) are shown. Error bars represent ± SEM (n = 3, 4,500 chromosomes, **p < 0.01).
(E) Telomere and chromosomal aberrations in shNek7-treated HeLa cells after KR-TRF1-induced oxidative telomeric DNA damage (shNek7+KR-TRF1). (a) Fragile telomeres; (b1 and b2) intrachromosomal telomeric insertions. The table shows quantification of telomere aberrations in cells.
(F) Colony formation assays for shCtrl- or shNek7-U2OS cells transiently expressing KR-TRF1. Nek7 expression levels are shown (insert).
(G) Recruitment of Nek7 under treatment of ATM inhibitor. U2OS cells were either untreated (control) or treated with ATM inhibitor KU55933 (10 μM) for 1 hr before being exposed to light for 1 hr. Error bars represent ± SEM (n = 3).
See also Figures S1 and S2.
Nek7 is important for mitosis, but its role in controlling genome stability has not been fully explored. We induced global DNA damage by treating the cells with hydrogen peroxide (H2O2) and ionizing radiation (IR). We did not observe obvious Nek7 foci under either H2O2 treatment or IR treatment at sites of telomere or genomic DNA damage, which was indicated by γH2AX staining (Figures S1A and S1B). Telomeric DNA is less than 0.5% of total genomic DNA; therefore, the percentage of its damage induced by global treatment should be less than 0.5% of the total genomic DNA damage, which may not be easily detected because of the large background of non-telomeric DNA damage. Telomeric and subtelomeric regions are enriched with epigenetic markers similar to heterochromatin (Gonzalo et al., 2006). Therefore, we also introduced KR to a non-telomeric genomic site within a heterochromatin structure via the tetracycline repressor (TetR) fused-KR (TetR-KR), which is capable of binding to the integrated TetO repeats in the genome (Figure S1C). We found that DNA glycosylase and other repair enzymes are recruited to the TetR-KR-induced damage (Lan et al., 2014). However, Nek7 was not recruited at sites of TetR-KR-induced damage, indicating that Nek7 is specifically recruited to oxidatively damaged telomeric DNA sites (Figure S1C).
To further evaluate the role of Nek7 at telomeres, we first disrupted telomeric structure by knocking down (KD) the shelterin protein TRF2 (Celli and de Lange, 2005; Denchi and de Lange, 2007), but we did not observe Nek7 recruitment at telomeres under this condition (Figure S1D). Second, we introduced telomeric DNA injury by using Flag-TRF1-Fok1 endonuclease to cut telomeres specifically (Cho et al., 2014) or by treating cells with low and high levels of the DNA polymerase inhibitor aphidicolin, which could induce fragile telomeres, an indicator of replication folk stall. Interestingly, under both conditions, Nek7 was not recruited to the damaged telomere sites (Figures S1E and S1F). Thus, the recruitment of Nek7 is likely restricted to oxidative telomeric DNA damage.
Next, we analyzed chromosome abnormalities in Nek7-deficient cells using telomere fluorescence in situ hybridization (FISH) in HeLa cells with a short hairpin RNA (shRNA)-mediated Nek7 knockdown to investigate whether Nek7 has a direct role in regulating chromosome function and telomere integrity. We observed more telomere aberrations in shNek7-KD HeLa cells than in control shRNA (shCtrl)-treated cells (15% in shNek7 versus 5% in shCtrl, respectively) (Figure 1D). Almost all of the telomere aberrations involved telomere loss and sister telomere association in shNek7-KD cells (Figure 1D, bottom), suggesting that Nek7 may have a telomere protection-related function. Dysfunctional telomeres can be marked by telomere-induced foci (TIF), in which 53BP1 foci label the strand breaks at telomeres (d’Adda di Fagagna et al., 2003; Takai et al., 2003). We also examined 53BP1 TIF formation in a telomerase-negative U2OS cell line and observed more 53BP1 TIF in shNek7-KD U2OS cells compared with shCtrl-treated cells (Figure S1G), suggesting that the Nek7-mediated telomere protection is not dependent on its regulation of telomerase (Cerone et al., 2011).
To further determine the physiological significance of the Nek7-mediated telomere protection from telomeric oxidative damage, we expressed KR-TRF1 in both telomerase-positive (HeLa and 293) and negative (U2OS) cells. Knocking down Nek7 in HeLa or 293 cells with shNek7 treatment did not cause immediate cell-cycle arrest following the KR-induced telomeric DNA damage (Figures S2A and S2B). Interestingly, Nek7-KD HeLa cells with telomeric DNA damage exhibited multiple chromosomal aberrations, including fragile telomeres, doublet telomeric signals, and broken chromosomes (Figure 1E), that were rarely found in either Nek7-KD cells in the absence of induced telomere DNA damage (no KR-TRF1) or in normal Nek7 expressing cells with KR-TRF1-induced DNA damage (light induced). Furthermore, we found that shNek7-KD U2OS cells also had increased sensitivity to telomeric DNA damage, as demonstrated by a significant reduction in cell survival (Figure 1F). Consistently, the shNek7-KD HeLa cells had reduced clonogenicity compared with shCtrl-treated HeLa cells (Figure S2C). The high incidence of telomere loss, sister telomere association, chromosomal instability, and increased cell death in Nek7-depleted cells following telomeric oxidative DNA damage suggests a crucial role of Nek7 in maintaining telomere integrity and in protecting cells from telomeric DNA damage.
Both ATM- and ADP-ribosylation pathways are important for the upstream regulation of the DNA damage response. ATM is a kinase that amplifies subsequent DNA damage signals via phosphorylation of multiple target proteins (Huang et al., 2004; Kishi et al., 2001; Shiloh and Ziv, 2013). ADP-ribosylation is a modification necessary for single-strand damage repair (SSDR). PARP1 is also reported to be involved in the alt-NHEJ pathway at telomeres (Sfeir and de Lange, 2012). To investigate if Nek7 telomeric recruitment is regulated by these upstream signals, we inhibited ATM with KU55933 and found diminished Nek7 telomeric recruitment in response to telomeric DNA damage (Figure 1G). In contrast, inhibition of either tankyrase 1 with G007-LK, or PARP with PJ34, or by tankyrase knockdown, had no effect on the recruitment of Nek7 under the same conditions (Figures S2D and S2E). Consistently, light exposure-induced telomeric DNA damage led to ATM recruitment to telomeres in cells expressing KR-TRF1 but not RFP-TRF1 (Figure S2F).
Nek7 Stabilizes TRF1 upon Oxidative DNA Damage
Given a critical role of Nek7 in protecting cells from telomeric DNA damage and its recruitment to telomeres following oxidative telomeric DNA damage, we next asked if knockdown of Nek7 may impair the DNA repair process at telomeres. H2AX is phosphorylated rapidly at Ser132 to form γH2AX after DNA damage or at the collapsed DNA replication forks; this serves as a platform to recruit and retain DNA damage repair proteins but is dissolved after the completion of DNA repair (Bonner et al., 2008; Macůrek et al., 2010; Nakada et al., 2008). Interestingly, we found that although the initial formation of TIF (γH2AX and 53BP1 foci) was similar in the Nek7-KD and control cells (Figure S2G), the resolution of TIF, the disappearance of γH2AX, and dissociation of 53BP1 from the telomeres after telomeric DNA damage took a much longer time in Nek7-KD cells. Under these conditions, more than 70% of telomeres maintained γH2AX foci in Nek7-KD cells compared with 30% in control cells 24 hr after DNA damage induction (Figure 2A; Figure S2H). Up to 90% of telomeres exhibited 53BP1 foci 48 hr after DNA damage in Nek7 knockdown cells, whereas only 40% were found in control cells (Figure 2B; Figure S2H). Consistent with these results, significantly more γH2AX protein was detected in Nek7-KD cells than in control cells 24 hr after DNA damage (Figure 2C). Surprisingly, the recruitment of the key proteins known to play critical roles in the oxidative DNA damage repair process, including XRCC1, Fen1, Polb, Ligase IV, and PCNA, was not affected in Nek7-KD cells in response to telomeric DNA damage (Figure S3), indicating that Nek7 loss does not affect the recruitment of DNA repair factors to telomeres directly.
Figure 2. Nek7 Stabilizes TRF1 upon Oxidative Telomeric DNA Damage.

(A) Percentage of γH2AX TIF in siCtrl- or siNek7-U2OS cells expressing KR-TRF1. Cells were exposed to light for 10 min and then recovered in the dark for indicated time periods. Error bars represent ± SEM (n = 3).
(B) Percentage of 53BP1 TIF in siCtrl- or siNek7-U2OS cells co-expressing KR-TRF1 and GFP-53BP1 were analyzed.
(C) Immunoblotting to detect γH2AX expression in shCtrl- or shNek7-HeLa cells stably expressing KR-TRF1. Relative expression of γH2AX was normalized to KR-TRF1.
(D) Expression of TRF1, TRF2, Pot1, Tin2 in shCtrl, shNek7 knockdown Flp-in T-REX KR-TRF1 293 cells and shNek7 knockdown cells rescued by Flag-Nek7. Cells had light-induced telomere damage.
(E) Expression of TRF1 and KR-TRF1 in shCtrl and two Nek7 shRNA knockdown Flp-in T-REX KR-TRF1 293 cell lines was determined.
(F) The Myc-TRF1 expression level in HeLa cells with increasing Flag-Nek7 expression was determined. Tubulin expression was used as a loading control.
(G) The design for Cas9-CRISPR KO cells (top). Genotyping results and colony formation assays are shown (bottom).
(H) Expression of TRF1, TRF2, Pot1, and Tin2 in WT and two Flp-in T-REX 293 KR-TRF1 Nek7 KO cell lines.
See also Figures S2 and S3.
Telomeres are shielded by six shelterin proteins. DNA damage repair proteins are recruited at the telomere when shelterin proteins are insufficient. We then tested the expression level of TRF1, TRF2, Pot1, and Tin2 in KR-TRF1-expressing Flp-in-control and Nek7-KD 293 cells. Nek7 knockdown did not change TRF2, Tin2 and Pot1 expression (Figure 2D; Figure S4A). Whereas the expression of the TRF1 protein was relatively stable under unperturbed conditions (Figure S4B), interestingly it was greatly reduced in response to telomeric DNA damage in Nek7-deficient cells but not in control cells (Figure 2D; Figure S4A). KR-released superoxide might lead to protein oxidation; however, we did not observe alterations of measurable protein functions after light-induced KR activation (Sun et al., 2015). More important, the reduced expression of TRF1 could be rescued by re-expression of Nek7 both in 293 cells and HeLa cells (Figure 2D; Figure S4C). Similar results were observed in the two independent Nek7 shRNA-KD, KR-TRF1-expressing Flp-in 293 cell lines (Figure 2E). Furthermore, we found that Nek7 overexpression could enhance TRF1 protein expression (Figure 2F), suggesting that Nek7 may regulate TRF1 expression either at the transcription or posttranscriptional levels. We also created CRISPR knockout (KO) cells by designing two single guide RNAs (sgRNAs) targeted at the start code region and the first intro region of Nek7 in U2OS and 293 cells, respectively. The Nek7 KO was confirmed by PCR genotyping and WB (Figure 2G). Consistent with results in shRNA knockdown cells, Nek7 CRISPR KO U2OS cells showed increased sensitivity to telomeric damage (Figure 2G). Nek7 KO in Flp-in KR-TRF1 293 cells leads to the decreased expression of TRF1 (Figure 2H), showing the same effect obtained with shNek7 cells.
To determine if the Nek7-mediated TRF1 expression is at the transcriptional level, we measured the trf1 mRNA level and found that Nek7 expression had no effect on trf1 transcription (Figure 3A), indicating that Nek7 may regulate TRF1 expression at the protein level. Indeed, we found that both exogenous and endogenous TRF1 protein was degraded faster in Nek7-depleted Flp-in KR-TRF1 293 cells (Figure 3B) or HeLa cells (Figure S4D) than in the shCtrl-treated cells after damage. Both cell lines stably expressed KR-TRF1, which allowed us to assess TRF1 stability upon specific telomeric DNA damage. Following light-induced DNA damage, cells were treated with cycloheximide, and the TRF1 levels were determined at various time points by immunoblotting. TRF1 protein degradation was prevented by treatment of cells with the proteasome inhibitor MG132 (Figure 3B), indicating that TRF1 is subjected to proteasome-mediated degradation. We further confirmed this finding in Nek7-overexpressing cells (Figure 3C). Again, the turnover rate of TRF1 was slower in Nek7-overexpressing cells compared with that in control cells (Figure 3C).
Figure 3. Stabilization of TRF1 by Nek7 Occurs at the Post-translational Level.
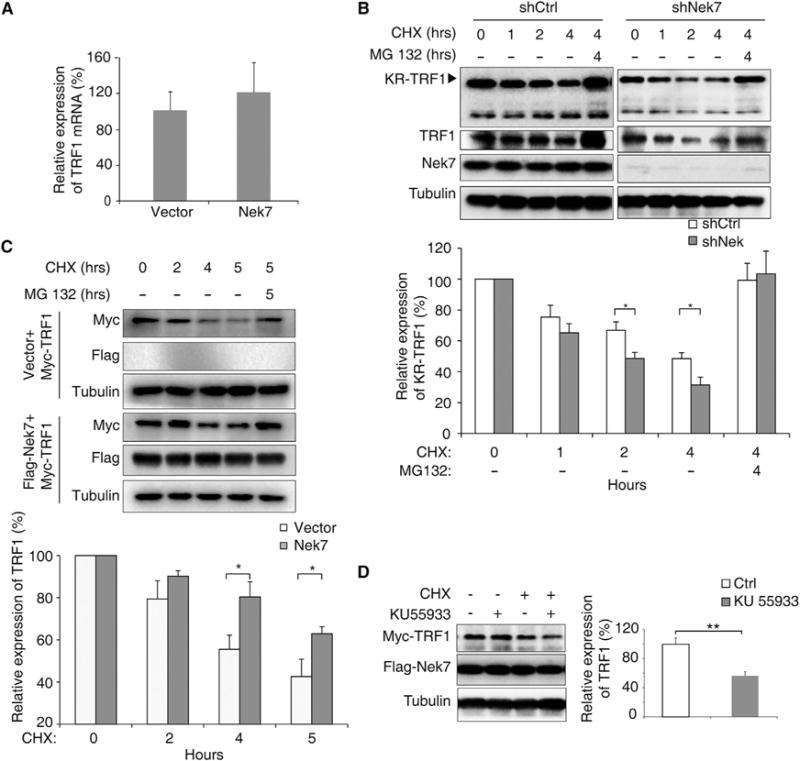
(A) TRF1 mRNA level in control or Flag-Nek7 transfected HeLa cells was measured by real-time PCR. Error bars represent ± SEM (n = 3, p = 0.23).
(B) Relative TRF1 expression in shCtrl and shNek7 knockdown Flp-in T-REX KR-TRF1 293 cells was analyzed. Cells were treated with CHX (100 mg/ml) for the indicated time periods, in the absence or presence of 15 mM MG132, and expression was determined. Error bars represent ± SEM (n = 3, *p < 0.05).
(C) Myc-TRF1 expression in control (Flag-empty vector) or Flag-Nek7 transfected HeLa cells was analyzed.
(D) shNek7 knockdown HeLa cells co-transfected with KR-TRF2, Myc-TRF1, and Flag-Nek7 were either untreated, or treated with CHX or an ATM inhibitor alone, or together as indicated. Cells were then exposed to light for an additional 1 hr before being analyzed for Myc-TRF1 expression. Error bars represent ± SEM (n = 3).
See also Figure S4.
Because TRF1 becomes unstable in the absence of Nek7 upon telomeric DNA damage, we also examined the stability of TRF1 in the presence or absence of an ATM inhibitor. KR-TRF2 induces oxidative DNA damage at telomeres and leads to γH2AX expression and the recruitment of base excision repair factors, including NTH1 and XRCC1, to telomeres in a manner similar to KR-TRF1 (Figure S5A). We also observed the recruitment of Nek7 to telomeres after the KR-TRF2-induced damage observed in U2OS cells (Figure S5B). Using KR-TRF2 to induce telomeric DNA damage, we found that TRF1 degradation was also exacerbated when ATM was inhibited after damage induction (55.2% of mock-treated cells) (Figure 3D), indicating that the ATM signal pathway is required for Nek7-mediated TRF1 stabilization.
Nek7 Interacts with TRF1 upon Oxidative DNA Damage
The above results suggest that Nek7 may regulate TRF1 through a close interaction. To determine whether Nek7 could directly interact with TRF1, we used a bimolecular fluorescence complementation (BiFC) assay (Figure 4A). In this assay, N-terminal (residues 1–155, Yn) and C-terminal (residues 156–239, Yc) fragments of the Venus variant of yellow fluorescent protein (YFP) are fused to interacting target protein pairs. If the Venus fragment fusion proteins interact upon co-expression in the same cell, then the Yn and Yc fragments are juxtaposed, allowing structural complementation of the Venus fluorophore and a bright fluorescent signal (Hu and Kerppola, 2003; Lee et al., 2010). We fused Nek7 and TRF1 with Yc and Yn, respectively (Nek7-Yc and TRF1-Yn), and co-expressed them in HeLa 1.3 cells (Figure 4B) and U2OS cells (Figure 4C). We observed bright fluorescent signals in the nuclei that co-localized with telomeres, indicative of a direct TRF1-Nek7 interaction (Figures 4B and 4C). No fluorescence was observed when either fusion protein was expressed alone. Similar interaction of Nek7 and TRF1 at telomeres was observed in HeLa cells (Figure S5C). We further performed in vitro binding assay to consolidate that TRF1 and Nek7 could interact directly (Figure S5D). Consistently, reciprocal co-immunoprecipitation studies confirmed that telomeric DNA damage enhanced Nek7-TRF1 interaction and telomeric recruitment of Nek7 (Figures 4D and 4E; Figures S5E and S5F). Furthermore, ATM inhibition abolished the DNA damage-induced Nek7 and TRF1 interaction (Figure 4F), demonstrating that ATM activation is required for the telomeric recruitment and further function of Nek7 upon DNA damage.
Figure 4. Nek7 Is Recruited and Interacts with TRF1 at Telomeres in Response to Oxidative Telomeric DNA Damage.

(A) Schematic illustration of a bimolecular fluorescence complementation (BiFC) assay.
(B) Interaction of TRF1 and Nek7 using a BiFC assay in HeLa 1.3 cells. The nuclei were stained with DAPI (blue).
(C) The interaction of TRF1 and Nek7 in U2OS cells at telomeres. Telomeres were stained by PNA Tel C-Cy3 (red).
(D) Co-immunoprecipitation of Myc-TRF1 and Flag-Nek7 in HeLa cells after telomeric DNA damage.
(E) TRF1 binds to endogenous Nek7 in HeLa cells.
(F) Inhibition of ATM prevents TRF1 and Nek7 interaction in Flp-in TREX KR-TRF1 293 cells.
See also Figure S5.
TRF1 Stabilization Requires Nek7 Catalytic Activity
To determine whether the catalytic activity of Nek7 is necessary for TRF1 stability, we first examined the telomeric recruitment of a Nek7 kinase-dead mutant (KM), in which lysines 63/64 at the ATP binding site are replaced with methionines (O’Regan and Fry, 2009), and a constitutively active Nek7 mutant (Y97A) in which a Tyr residue at the auto-inhibitory domain was mutated to an Ala, thus lacking auto-inhibitory function (Richards et al., 2009). We transfected cells in parallel with equal amounts of the plasmids. However, telomere recruitment of either mutant was indistinguishable from that of the wild-type (WT) Nek7 in response to telomeric DNA damage (Figure 5A). Consistently, Nek7(Y97A) and Nek7(KM) showed a similar interaction with TRF1 in response to light-induced DNA damage (Figure 5B). Interestingly, only the Nek7 WT and mutant (Y97A) but not Nek7(KM) had the ability to restore TRF1 expression in Nek7-KD HeLa cells (Figure 5C; Figure S5G). Therefore, the kinase activity of Nek7, although not essential for its telomere recruitment and TRF1 binding, is necessary for TRF1 stabilization.
Figure 5. Nek7 Catalytic Activity Is Required to Phosphorylate and Stabilize TRF1.
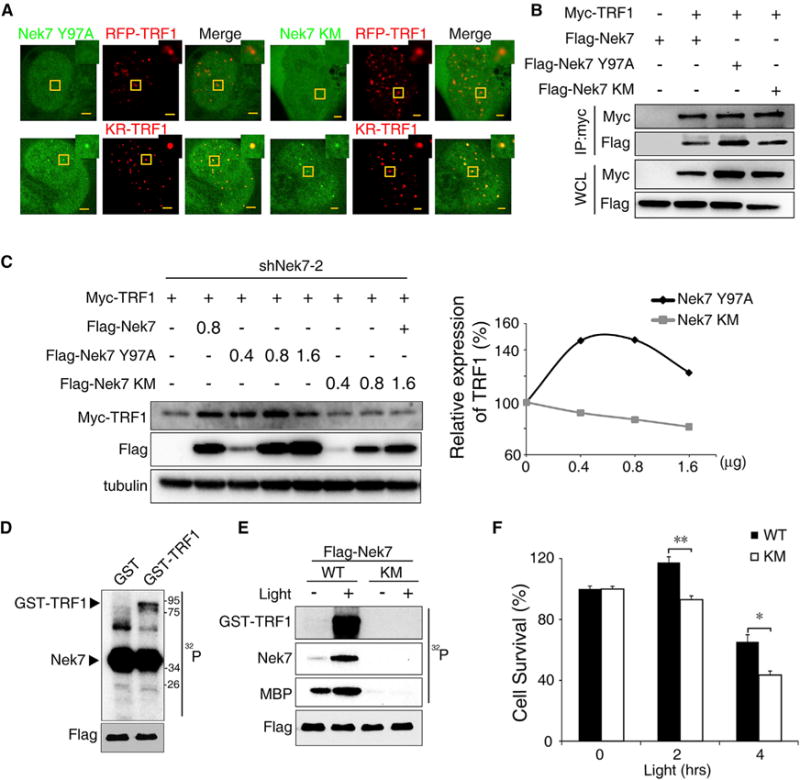
(A) The recruitment of Nek7 mutants to telomere damage sites.
(B) Co-immunoprecipitation of TRF1 with Nek7 mutants.
(C) Myc-TRF1 was co-transfected with Nek7 mutants (Y97A or KM) in Nek7-KD HeLa cells, and its expression determined by IB. The relative Myc-TRF1 expression is shown.
(D) In vitro kinase assay for Nek7 activity. Recombinant GST and GST-TRF1 were purified and used as substrates. Flag-Nek7 was immunoprecipitated from shNek7 Flp-in T-REX KR-TRF1 293 cells and used as the enzyme for the kinase assay.
(E) In vitro kinase assay for Nek7 on TRF1 with or without telomeric DNA damage. GST-TRF1 and MBP were used as substrates for the Flag-Nek7 or Flag-Nek7 KM mutant.
(F) Colony formation assays for shNek7 U2OS cells co-expressing KR-TRF1 and an shRNA-resistant Flag-Nek7 or an Flag-Nek7 KM mutant. Error bars represent ± SEM (n = 3).
Nek7 Is a Regulatory Kinase for TRF1
The inability of the kinase-dead Nek7 mutant to rescue TRF1 expression suggests that Nek7 may directly phosphorylate TRF1 to regulate its stability. We thus purified a WT Flag-tagged Nek7 and a mutant Flag-tagged Nek7(KM) protein from 293 cells and used them to phosphorylate recombinant TRF1 in vitro. WT Nek7 did not phosphorylate recombinant GST but phosphorylated GST-TRF1, indicating that phosphorylation sites are located in TRF1 (Figure 5D). In addition, only WT Nek7 but not the kinase-dead Nek7 mutant could phosphorylate GST-TRF1 (Figure 5E). Significantly, we found that the Nek7 activity was enhanced following light-induced telomeric DNA damage, as evidenced by the increased phosphorylation of the pseudo-substrate myelin basic protein and Nek7 autophosphorylation (Figure 5E). Concomitantly, this telomeric DNA damage-induced Nek7 activation also led to increased phosphorylation of GST-TRF1.
To further determine if the kinase activity of Nek7 is functionally important, we performed a clonogenic assay using either a WT Nek7 or a kinase-dead Nek7(KM) mutant to rescue Nek7-KD cells from oxidative damage-induced cell death (Figure 5F). As expected, we found that clonogenic survival was consistently lower for the Nek7(KM)-rescued cells as compared with WT Nek7 in a light dose-dependent manner. This result indicates that Nek7 kinase activity is important to maintain cell viability when encountering oxidative telomeric damage.
TRF1 Ser114 Is the Target Phosphorylation Site by Nek7
Given that Nek7 binds to TRF1 in response to telomeric DNA damage and is able to phosphorylate TRF1 in vitro, we next performed MS analyses based on the in vitro kinase assay to identify the Nek7 targeted phosphorylation sites in TRF1 (Figure 6A). We identified a single phosphorylation site at Ser114 in TRF1, which exhibited an approximate 20-fold increase in phosphopeptide counts between the WT Nek7 and the Nek7(KM) mediated kinase reaction. To evaluate the phosphorylation site in vivo, we undertook mass spectrometry for TRF1 in cells. We also detected the phosphorylation of Serine 114 on TRF1 in cells in which we induced oxidative telomere damage (Figure S6A). Ser114 is localized at the LSSL motif of the TRFH domain in TRF1, a region known to be critical for binding to Fbx4, a subunit of the E3 ligase complex SCFFBX4 that plays a key role in the regulation of TRF1 stability (Lee et al., 2006). This result strongly suggests that Nek7 may regulate TRF1 stability through phosphorylating its Ser114 site. Indeed, molecular modeling based on the crystal structure of the TRF1 and Fbx4 interaction surface revealed that negative charges introduced by Nek7-mediated Ser114 phosphorylation perturb the interaction of TRF1 with Fbx4 through the LSSL motif (Figure 6B).
Figure 6. Nek7-Mediated TRF1 S114 Phosphorylation Prevents TRF1 Interaction with Fbx4, Leading to an Increased Interaction with Tin2.

(A) TRF1 phosphor peptides identified by a liquid chromatography-tandem mass spectrometry (LC/MS) analysis. The ratio of S114 phosphorylation between the Nek7 and Nek7 KM samples are shown. Error bars represent ± SEM (n = 5).
(B) Surface representation of S114 phosphorylated TRF1 with Fbx4 (left), or non-phosphorylated TRF1 with Fbx4 (right). Green ribbons represent TRF1 structure. Blue indicates positive charges, and red indicates negative charges. Enlarged images from the interacting surface are shown.
(C) Phosphor-mimicry TRF1 S114D mutation disrupts the TRF1 interaction with Fbx4.
(D) The Fbx4 interaction with TRF1 or TRF1 S114A mutant in the presence of Nek7 or Nek7(KM) expression after DNA damage. shTRF1 and shNek7 Flp-in TREX 293 cells were co-transfected KR-TRF2, Flag-Fbx4 with plasmids as indicated. The relative Fbx4 binding to TRF1 was quantified (n = 4). Error bars represent ± SEM (n = 4).
(E) The interaction of Flag-Tin2 with Myc-TRF1 or Myc-TRF1 mutants (S114A or S114D). shTRF1 stably expressing Flp-in TREX 293 cells was co-transfected with Flag-Tin2, Myc-TRF1, or Myc-TRF S114A or S114D mutants as indicated.
(F) Interaction of TRF1 with Tin2 in HeLa cells with or without Nek7 knockdown in the presence of telomeric DNA damage. Flag-control vector or an shRNA-resistant Flag-Nek7 plasmid was co-transfected with Myc-TRF1 and HA-Tin2 in shNek7 HeLa cells as indicated. The relative TRF1 binding to Tin2 was quantified (n = 3). Error bars represent ± SEM (n = 3).
(G) A model illustrating Nek7 regulation of TRF1 and Fbx4 interaction.
See also Figure S6.
TRF1 Ser114 Phosphorylation Disrupts the Interaction between TRF1 and Fbx4
To further determine if indeed the Nek7-mediated TRF1 Ser114 phosphorylation plays a key role in regulating the TRF1-Fbx4 protein-protein interaction, we substituted TRF1 Ser114 with an aspartate as a phosphomimetic (S114D mutant) or alanine (S114A) (non-phosphorylated). We first verified that both of the TRF1(S114A) and TRF1(S114D) mutants can localize to telomeres in the same way as WT TRF1 (Figure S6B). Next, we examined the interaction of these mutants with Fbx4. We found that Fbx4 interacted with WT TRF1 but not the phosphomimetic TRF1(S114D) (Figure 6C; Figure S6C). To further confirm that TRF1 Ser114 phosphorylation specifically mediated by Nek7 regulates TRF1 interaction with Fbx4, we co-expressed shRNA-resistant WT TRF1 or TRF1(S114A) mutant with either WT Nek7 or Nek7(KM) mutant in TRF1/Nek7 double-KD cells. Following light-induced telomeric DNA damage, we found that TRF1 interaction with Fbx4 was greatly reduced in WT Nek7-expressing cells compared with that in Nek7(KM)-expressing cells (Figure 6D). However, under the same conditions, TRF1(S114A) binding to Fbx4 was similar in WT Nek7 and Nek7(KM)-expressing cells (Figure 6D), indicating strongly that Nek7 was the specific kinase for TRF1 Ser114 phosphorylation, and this phosphorylation event prevents TRF1 interaction with Fbx4.
The shelterin protein Tin2 serves as a linker protein between TRF1 and TRF2 dimers. More important, Tin2 acts to shield TRF1 from degradation by preventing TRF1 from binding to SCFFBX4. Given that the Nek7-mediated phosphorylation of TRF1 at Ser114 prevents the interaction of TRF1 with Fbx4, we reasoned that TRF1 Ser114 phosphorylation may increase its association with Tin2. Indeed, we found that the phosphomimetic mutant TRF1(S114D) displayed an increased association with Tin2 compared with that of WT TRF1 or the TRF1(S114A) mutant in a pull-down assay (Figure 6E). Consistently, re-expression of Nek7 in Nek7-KD cells led to increased TRF1-Tin2 interaction in response to telomeric DNA damage (Figure 6F). The high level of Tin2 could stabilize TRF1 expression in Nek7-KD cells to some extent (Figure S6D), which also supports the idea that Tin2 serves as the major factor for stabilizing TRF1 in counteracting E3 ligase binding (Zeng et al., 2010). However, the efficiency of stabilization was much lower compared with Nek7 (Figure 2F; Figure S6E), indicating Nek7 is a key factor to protect TRF1 stability after damage.
Nek7-Mediated TRF1 Ser114 Phosphorylation Prevents TRF1 from Proteasomal Degradation
Phosphorylation of TRF1 at Ser114 by Nek7 prevents its interaction with Fbx4 E3 ligase, suggesting that phosphorylated TRF1 might be resistant to ubiquitin E3 ligase-mediated proteasomal degradation. We thus measured the stability of TRF1(S114A) and TRF1(S114D) in HeLa cells treated with the protein synthesis inhibitor cycloheximide. As expected, the phosphomimetic TRF1(S114D) mutant exhibited a slower degradation rate than the non-phosphorylated TRF1(S114A) mutant (Figure 7A). Furthermore, we found that Nek7 expression had little effect on TRF1(S114A) expression (Figure 7B, bottom).
Figure 7. Nek7-Mediated TRF1 Phosphorylation Prevents TRF1 Ubiquitination and Proteasome-Targeted Protein Degradation.

(A) Relative expression of Myc-TRF1mutants (S114A or S114D) in HeLa cells were analyzed. See also Figure 3B.
(B) Wild-type Myc-TRF1 and Myc-TRF1 S114A expression levels in HeLa cells with increased Flag-Nek7 expression were determined by IB.
(C) Ubiquitination of TRF1 in the absence or presence of telomeric DNA damage. Myc-ubiquitin (Myc-Ub) and KR-TRF1 plasmids were co-transfected into shCtrl-treated or shNek7-2-treated HeLa cells.
(D). Colony formation assays for shTRF1 and shNek7 double-knockdown U2OS cells co-expressing KR-TRF2 and an shRNA-resistant Myc-TRF1 or an Myc-TRF1 S114D. Error bars represent ± SEM (n = 3).
(E) Model of how Nek7 protects telomere integrity after DNA damage.
Finally, we knocked down Nek7 and determined if TRF1 could be degraded through the ubiquitin-mediated proteasome degradation pathway. Using Myc-tagged ubiquitin, we detected substantially increased poly-ubiquitinated TRF1 in Nek7 knockdown cells in response to light-induced DNA damage compared with that in control cells (Figure 7C), indicating that non-phosphorylated TRF1 was more susceptible to polyubiquitin-dependent degradation.
Nek7-Mediated TRF1 Ser114 Phosphorylation Protects Cells from DNA Damage-Induced Cell Death
Because the kinase-dead Nek7(KM) mutant had reduced ability to rescue cells from oxidative telomeric DNA damage-induced cell death compared with WT Nek7 in a clonogenic assay (Figure 5F), we reasoned that Nek7 may protect cells from oxidative DNA damage through its phosphorylation of TRF1 at Ser114. To test this possibility, we introduced WT TRF1 or the phosphomimetic TRF1(S114D) mutant into Nek7/TRF1 double-knockdown cells (Figure 7D). We found that the phosphomimetic TRF1(S114D) mutant was more capable of supporting cell survival than WT TRF1 following light-induced telomeric DNA damage (Figure 7D). This result suggests that the telomere protection function of Nek7 is delivered primarily through phosphorylation and stabilization of TRF1.
DISCUSSION
The intrinsic G-rich sequence of telomeres renders it a predisposed genomic region for oxidative damage (Hewitt et al., 2012; Petersen et al., 1998). Telomeric oxidative damage triggers telomere shortening and dysfunction. Shelterin proteins play central roles to prevent telomeres from being recognized as strand breaks and thus inhibit an inappropriate DDR at telomeres (Sfeir and de Lange, 2012). In this study, we identify Nek7 as an upstream regulator for TRF1 to protect telomeres in the face of oxidative telomeric DNA damage. Nek7-deficient cells manifest cell death and chromosome instability with accumulated oxidative damage, implicating unstable telomeres under inadequate TRF1 protection. We demonstrate that Nek7 is a critical factor to maintain telomere integrity by phosphorylating TRF1 and preserving TRF1 in a functional shelterin complex at telomeres.
TRF1 plays a major role in the regulation of telomere replication (Sfeir et al., 2009). However, we did not observe the recruitment of Nek7 to telomeres under replication stress or throughout the S phase (Figure S1F), indicating that the function of Nek7 in stabilizing TRF1 may not result from the telomere replication problems and may not be necessary for facilitating telomere replication. During mitosis, the association of TRF1 with telomeres displays a cell cycle-dependent oscillation, exhibiting decreased telomeric binding during the S phase and increased binding afterward (Verdun et al., 2005; Verdun and Karlseder, 2006). The release of TRF1 prompts telomeric chromatin changes and creates a temporarily open status to facilitate end processing (Poulet et al., 2012). It is possible that a repair process for oxidative telomeric DNA damage might also be accompanied by temporary or partial TRF1 release, leading to a transient capping and uncapping intermediate status at the damaged telomere sites. The released TRF1 is subsequently targeted by the Fbx4-E3 ligase complex for proteasomal degradation. However, because the intermediate configuration is inevitable and necessary for both mitosis and DNA repair progression, if the intermediate status is persistent, telomeres will be unstable because of insufficient shelterin proteins for telomere protection. This intermediate telomere configuration is recognized by DDR proteins but is still resistant to DNA repair-mediated telomere fusions or cell-cycle arrest at the initial stage (Cesare et al., 2009). Knockdown of Nek7 in cells did not affect immediate cell-cycle progression (Figure S2), indicating that Nek7 deficiency did not cause acute activation of a cell-cycle checkpoint. However, the light-induced reduction of cell survival suggests that the high level and constant accumulation of damage will eventually cause telomere catastrophe and cell death in Nek7-deficient cells. In this regard, Nek7 is more likely to be involved in telomere end protection at the stage of intermediate configuration. Interestingly, we also found that Nek7 is preferentially recruited to telomeres during the cell cycle in the later G2/M phase without exogenous DNA damage (Figure S7), indicating that there is some similarity in the biology of this cell-cycle stage to that of oxidative DNA damage at the telomeres. Other cell-cycle kinases, such as CDK1, are reported to negatively regulate telomeric binding of TRF1 during the cell cycle (McKerlie et al., 2012; McKerlie and Zhu, 2011). How Nek7 cooperates with other pathways to elastically regulate the abundance of telomeric TRF1 during the cell cycle needs further investigation. Together, Nek7 likely acts as a feedback loop to prevent excessive loss of TRF1 and to balance TRF1 expression upon oxidative damage and during the cell cycle, thereby maintaining telomere physiological function.
Stabilization of the shelterin complex at telomeres plays a decisive role in controlling the compacted telomere configuration and protecting the telomere from the DDR. Within the complex, TRF1 binds telomeric DNA and works with Tin2 to control the compacted telomeric chromatin, thereby contributing to the suppression of telomeric DNA damage (Bandaria et al., 2016). We identified phosphorylation of the TRF1 S114 site both in an in vitro kinase assay and in vivo using mass-spectrometry analysis (Figure 6A; Figure S6A). We show that Nek7 phosphorylates TRF1 at Ser114 and in turn maintains stability of the shelterin complex at telomeres. It is known that Tin2 stabilizes the whole shelterin complex by bridging several key telomeric binding proteins, TRF1, TRF2, and Pot1. Loss of any telomeric DNA-binding proteins or disruption of their interaction with Tin2 enlarges the telomere volume and leads to the accumulation of damage foci. TRF1 Ser114 is located at the interface region LSSL of TRF1, which is responsible for association with Tin2 and Fbx4. Previous studies have shown that mutation of Leu115 or Leu120 to arginine in LSSL motif abrogates the interaction of TRF1 with Fbx4, confirming the critical importance of this motif for Fbx4 recognition (Zeng et al., 2010). Hence, in terms of the mutually exclusive interaction of TRF1 with Fbx4 and Tin2, Nek7 phosphorylation of TRF1 at Ser114 safeguards TRF1 from degradation by disrupting interaction with its cognate E3 ligase, thus allowing it to be retained at telomeres and increasing the association with Tin2 to maintain the shelterin complex (Figure 7E, left). Nek7-mediated TRF1 Ser114 phosphorylation is likely to be the key regulatory mechanism that governs the dynamic stability of TRF1 through binding to Tin2 and Fbx4 under oxidative stress (Figure 6G). The increase in Tin2-bound TRF1 may be an indirect effect of inhibiting the Fbx4-dependent TRF1 degradation. This suggests that the Nek7-mediated Ser114 phosphorylation, but not the Tin2 binding to TRF1, is important for TRF1 stabilization after damage. The shelterin complex stabilized by Nek7 may also prevent exposure of the telomeric ends and protect cells from excessive telomeric DNA damage.
The efficient recruitment of Nek7 to telomeres is dependent on ATM but not tankyrase 1. It is known that ATM phosphorylates DNA damage sensors required for the accumulation of regulatory proteins involved in the DDR and chromosome end processing (Chowdhury et al., 2005; Dantuma and van Attikum, 2015). It is unclear how ATM regulates or recruits Nek7 to telomeric oxidative damage. Both the constitutively active and the kinase-dead Nek7 mutants could be recruited to telomeres, indicating that the Nek7 recruitment and Nek7 activation are independent processes. The Nek7 recruitment may not be directly associated with its activation by ATM or a yet to be determined kinase. However, the dependency on ATM also points to the possibility that ATM may phosphorylate histones or other proteins after DNA damage to facilitate recruitment of Nek7 to the damaged telomeres. Interestingly, it has been reported that the telomeric association of ATM is reduced in S phase but gradually increased during the G2/M phase (Verdun et al., 2005) in a manner very similar to that of Nek7 (Figure S7). Future studies will be necessary to understand the role of the ATM-Nek7-TRF1 pathway at the molecular level in this process.
With all of our data taken together, we propose that Nek7 stabilizes TRF1 via phosphorylating TFR1 at Ser114, which protects TRF1 from Fbx4-mediated proteasomal degradation, thereby protecting telomeres from oxidative DNA damage and possibly during mitosis (Figure 7E). When Nek7 is deficient, TRF1 Ser114 is not phosphorylated; thus it is degraded by the Fbx4-mediated proteasome pathway, resulting in a sustained DDR at telomeres and unstable telomeres. Oxidative and end-replicative stress then accelerates the telomere shortening-coupled shelterin loss, which eventually leads to telomere crisis and cell death (Hayashi et al., 2015; Maciejowski et al., 2015). The gradual accumulation of DNA damage at telomeres due to insufficient TRF1 signals telomere dysfunction, which is considered as a pre-crisis telomere (Figure 7E, right). Such a protection mechanism involving Nek7-mediated phosphorylation and stabilization of TRF1 strikes a balance between accessibility of DNA repair proteins and the detrimental impact of over-exposing telomere DNA.
STAR★METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-γH2AX | EMD Millipore | Cat# 05-636, clone JBW301; RRID: AB_309864 |
| Anti-53BP1 | Novus | Cat# NB100-304; RRID: AB_10003037 |
| anti-Nek7 | Cell Signaling Technology | Cat# 3057; RRID: AB_2150676 |
| TelC-Cy3 | PNA Bio | Cat# F1002 |
| anti-TRF1 | Sigma-Aldrich | Cat# T1948; RRID: AB_477562 |
| anti-TRF1 | Abcam | Cat# ab10579; RRID: AB_2201461 |
| anti-Myc (Rabbit) | Santa Cruz | Cat# SC-789; RRID: AB_631274 |
| anti-Myc (Mouse) | Sigma-Aldrich | Cat# M4439, clone 9E10; RRID: AB_439694 |
| anti-TRF2 | Santa Cruz | Cat# SC-9143; RRID: AB_2201333 |
| anti-Pot1 | Abcam | Cat# ab124784; RRID: AB_10975313 |
| anti-Tin2 | Abcam | Cat# ab197894 |
| anti-Flag | Sigma-Aldrich | Cat# F1804, clone M2; RRID: AB_262044 |
| anti-Tubulin | Homemade | Ascite |
| Alexa Fluor goat anti-Mouse 488 | Thermo Fisher Scientific | Cat# A28175; RRID: AB_2536161 |
| Alexa Fluor goat anti-Rabbit 488 | Thermo Fisher Scientific | Cat# A27034; RRID: AB_2536097 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Ku-55933 | Sigma-Aldrich | Cat# SML1109 |
| PJ34 | Sigma-Aldrich | Cat# P4365 |
| G007-LK | EMD Millipore | Cat# 504907 |
| KaryoMAX Colcemid | Thermo Fisher Scientific | Cat# 15212012 |
| Yeast tRNA | Thermo Fisher Scientific | Cat# 15401-011 |
| Crystal violet | Sigma-Aldrich | Cat# C6158 |
| 20 × SSC | Thermo Fisher Scientific | Cat# 15557036 |
| Aphidicolin | Sigma-Aldrich | Cat# 89458 |
| Cycloheximide | Sigma-Aldrich | C7698 |
| MG132 | Sigma-Aldrich | C2211 |
| Formamide (Deionized) | Ambion | Cat# AM9342 |
| IPTG | Sigma-Aldrich | Cat# I6758 |
| ANTI-FLAG M2 Affinity Gel | Sigma-Aldrich | A2220 |
| Dynabeads M-280 Streptavidin | Thermo Fisher Scientific | 11205D |
| Anti-c-Myc Agarose | Thermo Fisher Scientific | 20168 |
| glutathione Sepharose 4B | GE Healthcare Life Sciences | 17-0756-01 |
| Pierce Protein G Agarose | Thermo Fisher Scientific | 20398 |
| L-glutathione | Sigma-Aldrich | G4251 |
| GST-TRF1 | Home-made | N/A |
| Critical Commercial Assays | ||
| QuikChange site-directed mutagenesis kit | Agilent Technologies | Cat# 200524 |
| KOD PCR kits | EMD Millipore | Cat# 71086 |
| Deposited Data | ||
| TRF1-Fbx4 structure | Zeng et al., 2010 | PDB 3L82 |
| Experimental Models: Cell Lines | ||
| U2OS | ATCC | HTB-96 |
| Flp-in KR-TRF1 293 cells | Sun et al., 2015 | N/A |
| HeLa 1.3 | Dr. Robert Tsai Lab | N/A |
| HeLa | This paper | N/A |
| 293FT Cell Line | Thermo Fisher Scientific | R70007 |
| Recombinant DNA | ||
| EGFP-C1-Nek7/Nek7 Y97A (Y97)/Nek7 K63, 64M (KM) | This paper | N/A |
| pLVX-KR-TRF1 | Sun et al., 2015 | N/A |
| pLVX -KR-TRF2 | This paper | N/A |
| pCMV-RFP-TRF1 | Sun et al., 2015 | N/A |
| pRK5-Myc-TRF1/TRF1 S114D/TRF1 S114A | This paper | N/A |
| pRK5-Flag-Nek7/Nek7 Y97A (Y97)/Nek7 K63, 64M (KM) | This paper | N/A |
| pRK5-HA-Tin2 | This paper | N/A |
| pRK5-Flag-Fbx4 | This paper | N/A |
| Myc-Ubiquitin | This paper | N/A |
| pDest-Nek7-cYc | This paper | N/A |
| pDest-TRF1-Yn | Lee et al., 2010 | N/A |
| 3×Flag-TRF1-FokI | Cho et al., 2014 | N/A |
| GFP-53BP1 | Lan et al., 2014 | N/A |
| pcDNA-flag-ATM | Lan et al., 2014 | N/A |
| tetR-KR | Lan et al., 2014 | N/A |
| EGFP-XRCC1 | Lan et al., 2014 | N/A |
| EGFP-FEN1 | Lan et al., 2014 | N/A |
| EGFP-PCNA | Lan et al., 2014 | N/A |
| EGFP-NTH1 | Lan et al., 2014 | N/A |
| EGFP-Ligase IV | Lan et al., 2014 | N/A |
| EGFP-Polymerase β(Polβ) | Lan et al., 2014 | N/A |
| GST-TRF1 | Dr. Kunping Lu Lab | N/A |
| pET22-6 × His-Nek7 | This paper | N/A |
| pACT-TRF1/TRF1 S114D | This paper | N/A |
| pBTM116-Fbx4 | This paper | N/A |
| CRISPR p300 | Dr. Feng Zhang Lab | N/A |
| Sequence-Based Reagents | ||
| shControl sequence: aattctccgaacgtgtcacgt | This paper | N/A |
| shRNA-2 Nek7-1 targeted Nek7 sequence: ccggatatgggctataataca | This paper | N/A |
| shRNA-2 targeted Nek7 sequence: ctccgacagttagttaatatg | This paper | N/A |
| shRNA targeted TRF1 sequence: aacgtattctgtaaagcttaa | This paper | N/A |
| Nek7 siRNA | Thermo Fisher Scientific | Cat# M-003795-02-0005 |
| siRNA Targeted Tankyrase sequence: AACAAUUCACCGUCGUCCUCUU | This paper | N/A |
| sgRNA targeted Nek7 sequence: CATCCATTGTCTGAAGCAAC | This paper | N/A |
| sgRNA targeted Nek7 sequence: ACAATGGATGAGCAATCACA | This paper | N/A |
| sgRNA targeted Nek7 sequence: TACTAACGCTCAGAGCTTAC | This paper | N/A |
| sgRNA targeted Nek7 sequence: GGGGCAGGTACAAAATGTAC | This paper | N/A |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Olympus FV1000 confocal software | Olympus | N/A |
| PyMOL (For TRF1-Fbx4 crystal structure) | Warren Lyford DeLano | https://www.pymol.org/ |
| MASCOT search engine (For Mass Spectrometry) | Matrix Science Ltd | Version 2.4.0 |
| Scaffold (For Mass Spectrometry) | Proteome Software | http://www.proteomesoftware.com/products/scaffold/ |
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to and will be fulfilled by the corresponding author Li Lan (lil64@pitt.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
shRNA stable cell lines
HeLa cells stably expressing KR-TRF1 or RFP-TRF1 with either shNek7, or scrambled shRNA or 293 cells stably expressing shTRF1 were generated by lentiviral infection and selected with puromycin (1 mg/ml, Hyclone). Overexpressing KR-TRF1 in shNek7 HeLa cells was achieved by first infecting HeLa cells with shNek7-expressing lentivirus followed by puromycin selection, then sequentially infecting with KR-TRF1-expressing lentivirus twice at 24 hr intervals. All cells were cultured in DMEM with 5% CO2.
CRISPR-Cas9 design and analysis
The plasmids for CRISPR-Cas9 were obtained from Feng Zhang’s Lab. All sgRNAs were designed using the website (http://crispr.mit.edu). The target sequences were indicated in Figure 2. For sgRNA construction, a pair of synthesized oligos was annealed and digested by Bpi I, and then ligated into the linearized vector. For CRISPR knockout cells, U2OS and Flp-in TREX KR-TRF1 293 cells were co-transfected with plasmids containing the targeting sequences. The cells were passaged on 96 well plates to form single cell colonies. All the single cell colonies were primarily identified by PCR using KOD polymerase and then western blotting with Nek7 antibody.
METHOD DETAILS
Colony formation assay
HeLa cells stably expressing shCtrl/shNek7 and KR-TRF1 shCtrl/shNek7+KR-TRF1 or shNek7 stable U2OS cells transiently expressing KR-TRF1 were seeded into 60 mm petri dishes (350 cells/dish). Cells were illuminated with or without 15 W white light for the indicated time period 8 hr later until cells were attached. After 10 days, cells were fixed and stained with 0.3% crystal violet in methanol.
Immunofluorescence and microscopy and CO-FISH
For endogenous γH2AX and 53BP1 staining, cells were seeded on 3.5 cm glass bottom dishes (MatTekCo.) and were activated after 48 hr of siRNA transfection and 24 hr of KR-TRF1 transfection. After the indicated recovery time, cells were quickly washed with PBS three times and fixed in 3.7% formaldehyde solution for 15 min at room temperature in the dark, then washed three times with PBS and permeabilized with 0.2% Triton X-100 in PBS for 10 min. Cells were then blocked for 1 hr at room temperature in a blocking buffer containing 5% BSA (Sigma) in PBS. Cells were then incubated with primary antibody dilutions (1:1000) at 4°C overnight. After washing three times with PBST (0.05% Tween-20 in PBS), cells were incubated with the secondary antibody dilution for 1 hr at room temperature in the dark. Following three 5 min washes with PBST, cells were stained with DAPI and imaged using an Olympus FV1000 confocal microscopy system (Cat. F10PRDMYR-1, Olympus). FV1000 software was used to acquire images. ImageJ was applied for the calculation of the percentage of co-localization at the site of KR.
For CO-FISH, cells were fixed, permeabilized and blocked as described. After being blocked, telomere FISH was performed by adding hybridizing solution (70% deionized formamide [Ambion, AM9342], 0.1% BSA, 0.5 ng/ml tRNA [Invitrogen], 0.06 3 SSC [Roche], and 125 nM TelC-Cy3 [PNA Bio F1002]) and heating for 5 min at 80°C on a heat block. After incubation for 2 hr, cells were washed twice with wash buffer A (70% deionized formamide, 10 mM Tris-HCl). Cells were then immunostained with primary antibody, followed by an Alexa Fluor-488-conjugated secondary antibody (Thermo Fisher Scientific).
Immunoprecipitation and immunoblotting
HeLa or Flp-in TREX KR-TRF1 293 cells were transfected with indicated plasmids by Lipofectamine 2000 (Invitrogen). KR-TRF1 expression was induced in Flp-in TREX KR-TRF1 293 cells by tetracycline, 2 μg/ml, for 24 hr. For damage induced interaction, cells were light exposed for 1 hr before immunoprecipitation. For immunoprecipitation, cells were washed with PBS twice and then lysed in buffer A(20 mM Tris-HCl, pH 7.9; 1 mM EDTA; 0.2% NP-40; 20 mM NaF; 10 mM β-glycerophosphate; 10 mM pyrophosphate; 0.5mM Na3VO4; and 1 mM PMSF proteasome inhibitor cocktail). After incubation on ice for 10 min with occasional mixing, the cell lysates were centrifuged at 5,000 rpm for 1 min at 4°C to pellet the nuclei. Then buffer B (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 1 mM EDTA, 0.2% NP-40, 20 mM NaF, 10 mM β-glycerophosphate, 10 mM pyrophosphate, 0.5 mM NaVO3, and 1 mM PMSF cocktail) was added with 3 × pellet volume. The nuclear extracts were homogenized by passing through 25-gauge needles 5 to 7 times and then kept on ice for another 20 min. The lysates were centrifuged at 12,000 rpm for 10 min at 4°C and supernatants were collected for immunoprecipitation. Anti-myc antibody (1 μg) was added for 5 hr and 30 μl of a 50% slurry of protein G-Sepharose beads (Thermo Fisher Scientific) was added for another 1 hr. Fortelomeic DNA IP, a biotin conjugated double strand telomeric sequence (TTAGGG7) was incubated with streptavidin dynbeads (Dynabeads® M-280 Streptavidin, Thermo Scientific) at room temperature for 2 hr. Then the dynabeads were added into the lysate and incubated for 4 hr. The precipitated proteins were washed four times with buffer B and then subjected to immunoblot analysis. For ubiquitination of TRF1, interaction of TRF1 with Tin2 or Fbx4, cells were lysed as described before (Chen et al., 2008; Lee et al., 2006). For immunoblotting, cells were lysed directly in lysis buffer (LB) (50 mM Tris-HCl, pH 7.5; 150 mM NaCl; 1 mM EDTA; 1% Triton X-100; 20 mM NaF; 10 mM β-glycerophosphate; 10 mM pyrophosphate; 0.5 mM Na3VO4; and 1 mM PMSF proteasome inhibitor cocktail). In the protein stability assay, cells were treated with cycloheximide (100 μg/ml, Sigma) and collected at indicated time points. Relative protein levels in the immunoblots were quantified using ImageJ image analysis software. The protein levels were normalized to tubulin and the relative levels at time 0 defined as 100%.
Telomere fluorescence in situ hybridization (FISH) on metaphase spreads
HeLa cells stably expressing KR-TRF1 were light-activated for 1 hr, then allowed to recover for 12 hr before being treated with colcemid (Invitrogen). For synchronization, 0.1 μg/ml colcemid was added to cells for 3 hr. Following treatment, cells were collected, swollen in 75 mM KCl, fixed four times with methanol/acetic acid (3:1), and dropped on to glass slides with 50% humidity. After incubation overnight, slides were washed with PBS for 5 min, fixed with 3.7% formaldehyde for 10 min, washed twice with PBS, and then incubated with RNase 100 μg/ml in PBS at 37°C for 10 min. After being washed with PBS, slides were consecutively immersed in a cold ethanol series (75%, 85%, and 100%) for 2 min each. Slides were left in a vertical position or a cold air stream was used to dry the slides. Hybridizing solution (70% deionized formamide [Ambion, AM9342], 0.1% BSA, 0.5 ng/μl tRNA [Invitrogen], 0.06 × SSC [Roche], and 125 nM TelC-Cy3 [PNA Bio F1002]) was added to the slides and covered with a coverslip. The spreads were denatured for 3 min at 80°C on a heat block and hybridized at 37°C for 2 hr, then washed twice for 15 min each with 37°C pre-warmed wash buffer A (70% deionized formamide, 10 mM Tris-HCl). Slides were then washed twice for 5 min each with buffer B (100 mM Tris-HCl, pH 7.0; 150 mM NaCl; 0.08% Tween-20). The chromosomal DNA was stained with DAPI, which was added to the second wash. Slides were then mounted and images were captured with the Olympus FV1000 confocal microscopy system.
Purification of GST-TRF1, His-Nek7 and in vitro kinase assay
GST-TRF1 was a gift from the Kuping Lu lab. Nek7 was constructed into a pET22 6 × His tag vector using EcoRI and HindIII. GST-TRF1 and His-Nek7 were expressed in Rosetta bacteria or BL21. One-liter cultures of E. coli transformed with the GST-TRF1/His-Nek7 expression vector were incubated overnight in the presence of 1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG, Sigma) at 25°C. The cell pellet was re-suspended in LB (50 mM Tris-HCl, pH 7.9; 500 mM NaCl; 1 mM EDTA; 5% glycerol; 1 mM DTT; 1% Triton) and then lysed by sonication. Following centrifugation, the supernatant of His-Nek7 was purified with a His column. The supernatant of GST-TRF1 was collected and incubated with 500 μl of a 50% slurry of glutathione Sepharose 4B (GE Healthcare Life Sciences, 17-0756-01) for 30 min at room temperature. Then the beads were washed three times with LB and three times with elution buffer (50 mM Tris-HCl, pH 7.9; 150 mM NaCl). The bound proteins were eluted with LB containing 30 mM L-glutathione (Sigma). Flag-Nek7 was isolated from Flp-in TREX 293 KR-TRF1 and shNek7 stable cells that were transiently transfected with Flag-Nek7 or a mutant. For light induction, the cells were light exposed for 1 hr. The immunoprecipitation of Flag-Nek7 was as described above. After incubation with 30 μl of a 50% slurry of Flag M2 agarose beads (Sigma, A2222) for 2 hr, immunoprecipitates were washed twice with the lysis buffer B and three times with the kinase buffer (50 mM HEPES, pH 7.5; 5 mM MgCl2; 0.2% NP-40; 2 mM pyrophosphate). For the Nek7 kinase assay, immunoprecipitates were incubated in a final volume of 50 μl of kinase buffer containing 500 μM ATP, 1.0 mCi of [γ32P]-ATP, and 2 μg GST-TRF1 at 30°C for 20 min. The reaction was terminated by the addition of 12.5 μl of 5 × SDS sample buffer and incubated at 95°C for 5 min. Kinase reactions were resolved by SDS-PAGE, and radiolabeled bands were visualized by autoradiography.
Mass spectrometry analysis for phosphorylation sites of TRF1
Samples were prepared for mass spectrometry by incubating Nek7 immunoprecipitates in a final volume of 50 μl of kinase buffer containing 1 mM ATP and 2 μg GST-TRF1 at 30°C for 20 min. The reactions were resolved by loading on SDS-PAGE and stained with Coomassie blue. The GST-TRF1 bands were cut from the gels, and each gel slice was destained with 50% acetonitrile/25 mM ammonium bicarbonate. Reduction and alkylation was conducted by incubating the gel bands in the presence of 10 mM DTT at 56°C for 1 hr followed by 1 hr of incubation in the dark with the addition of 55 mM iodoacetamide. Enzymes (trypsin, chymotrypsin, or endopeptidases GluC or LysC) were added to dried gel pieces and digestion was allowed to proceed overnight at 37°C. The resultant proteolytic peptides were extracted with 70% acetonitrile/5% formic acid, vacuum dried, and re-constituted in 0.1% formic acid for LC-MS/MS analysis. For LC-MS/MS analysis, proteolytic peptides were analyzed by nano LC-MS/MS with a Dionex HPLC system (Dionex Ultimate 3000, ThermoFisher Scientific, San Jose, CA) interfaced to a linear ion trap MS (LTQ-XL, ThermoFisher Scientific). The Dionex HPLC system was operated with a double-split system to provide an in-column nano-flow rate (~300 nl/min). Peptides were separated on a C18 column (PicoChip column packed with 10.5 cm Reprosil C18 3 μm 120 A chromatography media with a 75 μm ID column and a 15 μm tip, New Objective, Inc., Woburn, MA). The mass spectrometer was operated in a data-dependent MS/MS mode in which each full MS spectrum was followed by MS/MS scans of the 8 most abundant molecular ions. Dynamic exclusion was enabled to minimize redundant selection of peptides previously selected for CID. MS/MS spectra were searched against a human protein sequence database (modified to include the GST-TRF1 sequence) using the MASCOT search engine (Version 2.4.0, Matrix Science Ltd). The mass tolerance was set at 1.4 Da for the precursor ions and 0.8 Da for fragment ions. Enzyme specificity was set according to the corresponding enzyme used for each digestion, with two missed cleavages allowed. Carboxyaminomethylation of cysteine residues was set as a static modification and oxidation of methionine residues was set as a variable modification. Identification results were further filtered with Scaffold (Proteome Software, Portland, Oregon, USA). Extracted ion chromatograms from full scan spectral data (MS1) were extracted through Skyline software and used as a surrogate metric for peptide abundance. The extracted ion chromatogram peaks were manually inspected to make sure that the proper peaks were selected. The observed full MS peaks for GST-TRF1 peptides were all within 5 parts per million (ppm).
Yeast two-hybrid assay
Yeast two-hybrid assays were performed as previously described (Moretti et al., 1994; Zeng et al., 2010). Colonies containing the L40 strain harboring pBTM116 and PACT2 (Clontech) fusion plasmids were selected on Leu-Trp plates. β-Galactosidase activities were measured by a liquid assay.
QUANTIFICATION AND STATISTICAL ANALYSIS
Image and immunoblotting Analysis
Image and immunoblotting analysis was performed using ImageJ software (NIH). Mean intensity of immunoblotting was measured within an defined area containing gel band. Foci analysis of Nek7, γH2AX and 53BP1 was performed by analysis of foci colocalization with KR-TRF1 using ImageJ.
Statistics
Statistical analysis was done using GraphPad Prism version 6.0 (GraphPad Software Inc). Statistical parameters and tests including the exact value of n, the definition of center, dispersion and precision measures (mean ± SEM) and statistical significance are reported in the Figures and Figure Legends. Data are judged to be statistically significant when p < 0.05 by two-tailed Student’s t test. In figures, asterisks denote statistical significance as calculated by Student’s t test (* p < 0.05; **p < 0.01; ***p < 0.001).
Supplementary Material
Highlights.
Nek7 is an important regulator for telomere stability
Nek7 interacts and stabilizes TRF1 at telomeres upon oxidative damage
Nek7 phosphorylates TRF1 at S114 and prevents Fbx4-mediated degradation
TRF1 stabilization by Nek7 is critical for cell survival after oxidative damage
Acknowledgments
This work was supported by grants from the NIH (GM118833 and AG045545) to L.L., the National Natural Science Foundation of China (31470845 and 81430033) to B.S., and the Shanghai Science and Technology Commission (13JC1404700) to B.S. Support for the UPCI Imaging Facility and UPCI Cytometry Facility was provided by the Cancer Center Support Grant from the National Institutes of Health (P30 CA047904). Funding for open-access charge was provided by the National Institutes of Health (GM118833). This project used the UPCI Cancer Proteomics Facility/Biomedical Mass Spectrometry Center, which is supported in part by award P30CA047904. We thank Dr. Yinsheng Wang at the University of California, Riverside, for the mass spectrometry analysis. We acknowledge the Peptide & Peptoid Synthesis Center at the University of Pittsburgh Health Sciences Core Research Facilities for synthesis, characterization, and purification of the specialized peptide used in this work. We thank Dr. Yuheng Han at Shanghai JiaoTong University School of Medicine for in vitro binding assays. We thank Dr. Kunping Lu for the gifts of GST-TRF1 and GFP-TRF1, Dr. Robert Tsai for Fbx4 cDNA, and Dr. Roger A. Greenberg for Flag-TRF1-Fok1. We thank Dr. Songyang Zou at Baylor College of Medicine for the TRF1-Yc and TRF1-Yn constructs and the BiFC system.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and can be found with this article online at http://dx.doi.org/10.1016/j.molcel.2017.01.015.
AUTHOR CONTRIBUTIONS
R.T. designed and performed the major experiments. Q.W., H.S., S.H., J.X., J.W., X.Z., N.A.Y., and Y.J. assisted with experiments. Y.J., T.E.S., S.N., M.L., A.S.L., B.S., and L.L. helped design the experiments and discussed and aided in planning the entire research theme. All authors were involved in manuscript preparation.
References
- Bandaria JN, Qin P, Berk V, Chu S, Yildiz A. Shelterin protects chromosome ends by compacting telomeric chromatin. Cell. 2016;164:735–746. doi: 10.1016/j.cell.2016.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. GammaH2AX and cancer. Nat Rev Cancer. 2008;8:957–967. doi: 10.1038/nrc2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broccoli D, Smogorzewska A, Chong L, de Lange T. Human telomeres contain two distinct Myb-related proteins, TRF1 and TRF2. Nat Genet. 1997;17:231–235. doi: 10.1038/ng1097-231. [DOI] [PubMed] [Google Scholar]
- Capra M, Nuciforo PG, Confalonieri S, Quarto M, Bianchi M, Nebuloni M, Boldorini R, Pallotti F, Viale G, Gishizky ML, et al. Frequent alterations in the expression of serine/threonine kinases in human cancers. Cancer Res. 2006;66:8147–8154. doi: 10.1158/0008-5472.CAN-05-3489. [DOI] [PubMed] [Google Scholar]
- Celli GB, de Lange T. DNA processing is not required for ATM-mediated telomere damage response after TRF2 deletion. Nat Cell Biol. 2005;7:712–718. doi: 10.1038/ncb1275. [DOI] [PubMed] [Google Scholar]
- Cerone MA, Burgess DJ, Naceur-Lombardelli C, Lord CJ, Ashworth A. High-throughput RNAi screening reveals novel regulators of telomerase. Cancer Res. 2011;71:3328–3340. doi: 10.1158/0008-5472.CAN-10-2734. [DOI] [PubMed] [Google Scholar]
- Cesare AJ, Kaul Z, Cohen SB, Napier CE, Pickett HA, Neumann AA, Reddel RR. Spontaneous occurrence of telomeric DNA damage response in the absence of chromosome fusions. Nat Struct Mol Biol. 2009;16:1244–1251. doi: 10.1038/nsmb.1725. [DOI] [PubMed] [Google Scholar]
- Chen Y, Yang Y, van Overbeek M, Donigian JR, Baciu P, de Lange T, Lei M. A shared docking motif in TRF1 and TRF2 used for differential recruitment of telomeric proteins. Science. 2008;319:1092–1096. doi: 10.1126/science.1151804. [DOI] [PubMed] [Google Scholar]
- Cho NW, Dilley RL, Lampson MA, Greenberg RA. Interchromosomal homology searches drive directional ALT telomere movement and synapsis. Cell. 2014;159:108–121. doi: 10.1016/j.cell.2014.08.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhury D, Keogh MC, Ishii H, Peterson CL, Buratowski S, Lieberman J. gamma-H2AX dephosphorylation by protein phosphatase 2A facilitates DNA double-strand break repair. Mol Cell. 2005;20:801–809. doi: 10.1016/j.molcel.2005.10.003. [DOI] [PubMed] [Google Scholar]
- d’Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–198. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]
- Dantuma NP, van Attikum H. Spatiotemporal regulation of post-translational modifications in the DNA damage response. EMBO J. 2015;35:6–23. doi: 10.15252/embj.201592595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lange T. Shelterin: the protein complex that shapes and safeguards human telomeres. Genes Dev. 2005;19:2100–2110. doi: 10.1101/gad.1346005. [DOI] [PubMed] [Google Scholar]
- Denchi EL, de Lange T. Protection of telomeres through independent control of ATM and ATR by TRF2 and POT1. Nature. 2007;448:1068–1071. doi: 10.1038/nature06065. [DOI] [PubMed] [Google Scholar]
- Fairall L, Chapman L, Moss H, de Lange T, Rhodes D. Structure of the TRFH dimerization domain of the human telomeric proteins TRF1 and TRF2. Mol Cell. 2001;8:351–361. doi: 10.1016/s1097-2765(01)00321-5. [DOI] [PubMed] [Google Scholar]
- Gonzalo S, Jaco I, Fraga MF, Chen T, Li E, Esteller M, Blasco MA. DNA methyltransferases control telomere length and telomere recombination in mammalian cells. Nat Cell Biol. 2006;8:416–424. doi: 10.1038/ncb1386. [DOI] [PubMed] [Google Scholar]
- Hayashi MT, Cesare AJ, Rivera T, Karlseder J. Cell death during crisis is mediated by mitotic telomere deprotection. Nature. 2015;522:492–496. doi: 10.1038/nature14513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt G, Jurk D, Marques FD, Correia-Melo C, Hardy T, Gackowska A, Anderson R, Taschuk M, Mann J, Passos JF. Telomeres are favoured targets of a persistent DNA damage response in ageing and stress-induced senescence. Nat Commun. 2012;3:708. doi: 10.1038/ncomms1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu CD, Kerppola TK. Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat Biotechnol. 2003;21:539–545. doi: 10.1038/nbt816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Halicka HD, Darzynkiewicz Z. Detection of histone H2AX phosphorylation on Ser-139 as an indicator of DNA damage (DNA double-strand breaks) Curr Protoc Cytom. 2004 doi: 10.1002/0471142956.cy0727s30. Chapter 7. Unit 7.27. [DOI] [PubMed] [Google Scholar]
- Kim S, Lee K, Rhee K. NEK7 is a centrosomal kinase critical for microtubule nucleation. Biochem Biophys Res Commun. 2007;360:56–62. doi: 10.1016/j.bbrc.2007.05.206. [DOI] [PubMed] [Google Scholar]
- Kishi S, Zhou XZ, Ziv Y, Khoo C, Hill DE, Shiloh Y, Lu KP. Telomeric protein Pin2/TRF1 as an important ATM target in response to double strand DNA breaks. J Biol Chem. 2001;276:29282–29291. doi: 10.1074/jbc.M011534200. [DOI] [PubMed] [Google Scholar]
- Lan L, Nakajima S, Wei L, Sun L, Hsieh CL, Sobol RW, Bruchez M, Van Houten B, Yasui A, Levine AS. Novel method for site-specific induction of oxidative DNA damage reveals differences in recruitment of repair proteins to heterochromatin and euchromatin. Nucleic Acids Res. 2014;42:2330–2345. doi: 10.1093/nar/gkt1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee TH, Perrem K, Harper JW, Lu KP, Zhou XZ. The F-box protein FBX4 targets PIN2/TRF1 for ubiquitin-mediated degradation and regulates telomere maintenance. J Biol Chem. 2006;281:759–768. doi: 10.1074/jbc.M509855200. [DOI] [PubMed] [Google Scholar]
- Lee MY, Kim HJ, Kim MA, Jee HJ, Kim AJ, Bae YS, Park JI, Chung JH, Yun J. Nek6 is involved in G2/M phase cell cycle arrest through DNA damage-induced phosphorylation. Cell Cycle. 2008;7:2705–2709. doi: 10.4161/cc.7.17.6551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee OH, Kim H, He Q, Baek HJ, Yang D, Chen LY, Liang J, Chae HK, Safari A, Liu D, et al. Genome-wide YFP fluorescence complementation screen identifies new regulators for telomere signaling in human cells. Mol Cell Proteomics. 2010;10:M110.001628. doi: 10.1074/mcp.M110.001628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maciejowski J, Li Y, Bosco N, Campbell PJ, de Lange T. Chromothripsis and kataegis induced by telomere crisis. Cell. 2015;163:1641–1654. doi: 10.1016/j.cell.2015.11.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macůrek L, Lindqvist A, Voets O, Kool J, Vos HR, Medema RH. Wip1 phosphatase is associated with chromatin and dephosphorylates gammaH2AX to promote checkpoint inhibition. Oncogene. 2010;29:2281–2291. doi: 10.1038/onc.2009.501. [DOI] [PubMed] [Google Scholar]
- Martínez P, Thanasoula M, Muñoz P, Liao C, Tejera A, McNees C, Flores JM, Fernández-Capetillo O, Tarsounas M, Blasco MA. Increased telomere fragility and fusions resulting from TRF1 deficiency lead to degenerative pathologies and increased cancer in mice. Genes Dev. 2009;23:2060–2075. doi: 10.1101/gad.543509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattern KA, Swiggers SJ, Nigg AL, Löwenberg B, Houtsmuller AB, Zijlmans JM. Dynamics of protein binding to telomeres in living cells: implications for telomere structure and function. Mol Cell Biol. 2004;24:5587–5594. doi: 10.1128/MCB.24.12.5587-5594.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKerlie M, Zhu XD. Cyclin B-dependent kinase 1 regulates human TRF1 to modulate the resolution of sister telomeres. Nat Commun. 2011;2:371. doi: 10.1038/ncomms1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKerlie M, Lin S, Zhu XD. ATM regulates proteasome-dependent subnuclear localization of TRF1, which is important for telomere maintenance. Nucleic Acids Res. 2012;40:3975–3989. doi: 10.1093/nar/gks035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meirelles GV, Silva JC, Mendonça YdeA, Ramos CH, Torriani IL, Kobarg J. Human Nek6 is a monomeric mostly globular kinase with an unfolded short N-terminal domain. BMC Struct Biol. 2011;11:12. doi: 10.1186/1472-6807-11-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meirelles GV, Perez AM, de Souza EE, Basei FL, Papa PF, Melo Hanchuk TD, Cardoso VB, Kobarg J. ‘‘Stop Ne(c)king around’’: how interactomics contributes to functionally characterize Nek family kinases. World J Biol Chem. 2014;5:141–160. doi: 10.4331/wjbc.v5.i2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti P, Freeman K, Coodly L, Shore D. Evidence that a complex of SIR proteins interacts with the silencer and telomere-binding protein RAP1. Genes Dev. 1994;8:2257–2269. doi: 10.1101/gad.8.19.2257. [DOI] [PubMed] [Google Scholar]
- Nakada S, Chen GI, Gingras AC, Durocher D. PP4 is a gamma H2AX phosphatase required for recovery from the DNA damage checkpoint. EMBO Rep. 2008;9:1019–1026. doi: 10.1038/embor.2008.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikawa T, Okamura H, Nagadoi A, König P, Rhodes D, Nishimura Y. Solution structure of a telomeric DNA complex of human TRF1. Structure. 2001;9:1237–1251. doi: 10.1016/s0969-2126(01)00688-8. [DOI] [PubMed] [Google Scholar]
- O’Regan L, Fry AM. The Nek6 and Nek7 protein kinases are required for robust mitotic spindle formation and cytokinesis. Mol Cell Biol. 2009;29:3975–3990. doi: 10.1128/MCB.01867-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan RJ, Kubicek S, Schreiber SL, Karlseder J. Reduced histone biosynthesis and chromatin changes arising from a damage signal at telomeres. Nat Struct Mol Biol. 2010;17:1218–1225. doi: 10.1038/nsmb.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–334. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- Petersen S, Saretzki G, von Zglinicki T. Preferential accumulation of single-stranded regions in telomeres of human fibroblasts. Exp Cell Res. 1998;239:152–160. doi: 10.1006/excr.1997.3893. [DOI] [PubMed] [Google Scholar]
- Poulet A, Pisano S, Faivre-Moskalenko C, Pei B, Tauran Y, Haftek-Terreau Z, Brunet F, Le Bihan YV, Ledu MH, Montel F, et al. The N-terminal domains of TRF1 and TRF2 regulate their ability to condense telomeric DNA. Nucleic Acids Res. 2012;40:2566–2576. doi: 10.1093/nar/gkr1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards MW, O’Regan L, Mas-Droux C, Blot JM, Cheung J, Hoelder S, Fry AM, Bayliss R. An autoinhibitory tyrosine motif in the cell-cycle-regulated Nek7 kinase is released through binding of Nek9. Mol Cell. 2009;36:560–570. doi: 10.1016/j.molcel.2009.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A, de Lange T. Removal of shelterin reveals the telomere end-protection problem. Science. 2012;336:593–597. doi: 10.1126/science.1218498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sfeir A, Kosiyatrakul ST, Hockemeyer D, MacRae SL, Karlseder J, Schildkraut CL, de Lange T. Mammalian telomeres resemble fragile sites and require TRF1 for efficient replication. Cell. 2009;138:90–103. doi: 10.1016/j.cell.2009.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiloh Y, Ziv Y. The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat Rev Mol Cell Biol. 2013;14:197–210. [PubMed] [Google Scholar]
- Sun L, Tan R, Xu J, LaFace J, Gao Y, Xiao Y, Attar M, Neumann C, Li GM, Su B, et al. Targeted DNA damage at individual telomeres disrupts their integrity and triggers cell death. Nucleic Acids Res. 2015;43:6334–6347. doi: 10.1093/nar/gkv598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takai H, Smogorzewska A, de Lange T. DNA damage foci at dysfunctional telomeres. Curr Biol. 2003;13:1549–1556. doi: 10.1016/s0960-9822(03)00542-6. [DOI] [PubMed] [Google Scholar]
- van Steensel B, de Lange T. Control of telomere length by the human telomeric protein TRF1. Nature. 1997;385:740–743. doi: 10.1038/385740a0. [DOI] [PubMed] [Google Scholar]
- Verdun RE, Karlseder J. The DNA damage machinery and homologous recombination pathway act consecutively to protect human telomeres. Cell. 2006;127:709–720. doi: 10.1016/j.cell.2006.09.034. [DOI] [PubMed] [Google Scholar]
- Verdun RE, Crabbe L, Haggblom C, Karlseder J. Functional human telomeres are recognized as DNA damage in G2 of the cell cycle. Mol Cell. 2005;20:551–561. doi: 10.1016/j.molcel.2005.09.024. [DOI] [PubMed] [Google Scholar]
- Yissachar N, Salem H, Tennenbaum T, Motro B. Nek7 kinase is enriched at the centrosome, and is required for proper spindle assembly and mitotic progression. FEBS Lett. 2006;580:6489–6495. doi: 10.1016/j.febslet.2006.10.069. [DOI] [PubMed] [Google Scholar]
- Zeng Z, Wang W, Yang Y, Chen Y, Yang X, Diehl JA, Liu X, Lei M. Structural basis of selective ubiquitination of TRF1 by SCFFbx4. Dev Cell. 2010;18:214–225. doi: 10.1016/j.devcel.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.