

Abstract
Focal adhesions (FAs) are micron-sized protein assemblies that coordinate cell adhesion, migration, and mechanotransduction. How the many proteins within FAs are organized into force sensing and transmitting structures is poorly understood. We combined fluorescent molecular tension sensors with super-resolution light microscopy to visualize traction forces within FAs with <100 nm spatial resolution. We find that αvβ3 integrin selectively localizes to high force regions. Paxillin, which is not generally considered to play a direct role in force transmission, shows a higher degree of spatial correlation with force than vinculin, talin, or α-actinin, proteins with hypothesized roles as force transducers. These observations suggest that αvβ3 integrin and paxillin may play important roles in mechanotransduction.
Keywords: Mechanobiology, integrin, focal adhesion, tension, molecular tension sensor, super-resolution, traction force microscopy
Mechanical forces play an important role in a variety of biological settings, including cell adhesion, migration, stem-cell differentiation, and disease progression1–4. A prominent means by which cells sense the physical properties of their surroundings is via integrins, heterodimeric transmembrane proteins that anchor cells to the surrounding extracellular matrix (ECM)5,6. Distinct integrin heterodimers are expressed in different cell types, and specific integrins are implicated in immunological function, angiogenesis, stem cell differentiation, and cancer metastasis7–10. Integrins cluster into micron-sized assemblies termed focal adhesions (FAs) that link the actin cytoskeleton to the ECM. FAs contain hundreds of different proteins, many of which transmit and respond to mechanical tension11–14. How exactly these proteins assemble into force producing and force sensing structures within FAs is thus the subject of intense interest.
Conventional optical microscopy lacks the spatial resolution necessary to observe structure within individual FAs. Recent measurements using super-resolution light microscopy revealed that the proteins within FAs are arranged in distinct layers in the vertical dimension relative to the cell membrane15. Experiments that track the diffusion of single, fluorescently-labeled proteins showed that integrins and other membrane proteins can diffuse freely within FAs, suggesting that integrins within FAs may be arranged in distinct clusters16,17. This view is supported by data showing that local clusters of 4 or more integrins within 60 nm of each other are required to support adhesion18–20. In addition, FA components, for example paxillin and focal adhesion kinase (FAK), are likely recruited to adhesions as pre-assembled clusters21,22. Finally, several studies show that integrins and integrin-associated proteins are differentially recruited to FAs in response to mechanical tension23–26.
Collectively, these and other studies support the general idea that proteins within FAs assemble into higher order structures on the ~100 nm length scale27. How exactly these structures orchestrate force production and force sensing remains poorly understood, due principally to a lack of techniques that can measure cellular traction forces on this length scale. This gap in measurement capabilities represents a central limitation in understanding the molecular mechanisms that underlie cell adhesion, motility, and mechanotransduction in a wide range of physiological and disease settings.
In this study we address this challenge by combining FRET-based molecular tension sensors28–38 and super-resolution light microscopy to directly visualize integrin-generated mechanical tension within individual FAs with sub-second and <100 nm spatial resolution (Supplementary Information). The improved spatial resolution of our system relative to traction force microscopy or micropost arrays allows us to observe distinct patterns of localization for α5β1 integrin, αvβ3 integrin, paxillin, vinculin, talin, α-actinin-1, actin, and non-muscle myosin II regulatory light chain (MRLC) with respect to local tension generation. These data reveal that αvβ3 integrin recruitment is highly correlated with local force production, while α5β1 integrin localizes to both force-producing regions as well as areas of the cell’s basal surface where local traction forces are similar to background. In addition, we found that recruitment of paxillin, a protein with no known direct role in force transmission, is highly correlated with local force production, suggesting a possible role in mechanotransduction.
Results
The molecular tension sensors (MTSs) used here are shown in Figure 1 and SI Materials and Methods. Integrins bind to the MTSs via an arginine-glycine-aspartate (RGD) sequence derived from fibronectin. Previous versions of MTSs relied on biotin-avidin conjugation for attachment to a glass coverslip. However, we and others have observed that cell-generated forces applied over 1–2 hours can break the biotin-avidin connection39. To address this challenge, we re-engineered the MTSs to covalently attach to a glass coverslip via a modified, cysteine-free HaloTag domain (Figure 1; SI). The covalent attachment of the MTSs prevents cell traction-mediated MTS detachment from the coverslip surface and thus facilitates time-lapse imaging. Mechanical stretch is detected via changes in FRET between the FRET donor Alexa 546 and the FRET acceptor ATTO 647N, which are connected via an extensible (GPGGA)8 peptide linker (Figure 1 and SI Materials and Methods)31. Covalently modifying the glass coverslip with a high fraction of fluorescently labeled MTS molecules provides a continuous FRET map to visualize localized tension35. In addition, this FRET pair uses the red portion of the visible spectrum, allowing us to simultaneously visualize GFP-labeled proteins inside the cell. The FRET donor and acceptor are site-specifically attached to the MTSs via a unique cysteine residue and an unnatural amino acid (4-azido-L-phenylalanine) at positions that flank the (GPGGA)8 spring (SI Materials and Methods, Figure S1). This site-specific labeling strategy allows for a short resting distance for the FRET pair, resulting in a higher no-load FRET efficiency compared to first generation MTSs (71% vs. 49% resting FRET, Figure S2)35. As a result, the dynamic range of the sensor is expanded (Figure S3, S4). It should be noted that the physiochemical properties of the acceptor dye, ATTO 647N, made it difficult to obtain 100% purity of donor and acceptor-labeled sensors in our hands, which would maximize the signal-to-background ratio (Figure S1, S4). However, ATTO 647N is far more photostable than any of the other far-red FRET acceptors we have tested, which is essential to obtaining the super-resolution measurements reported here.
Figure 1.
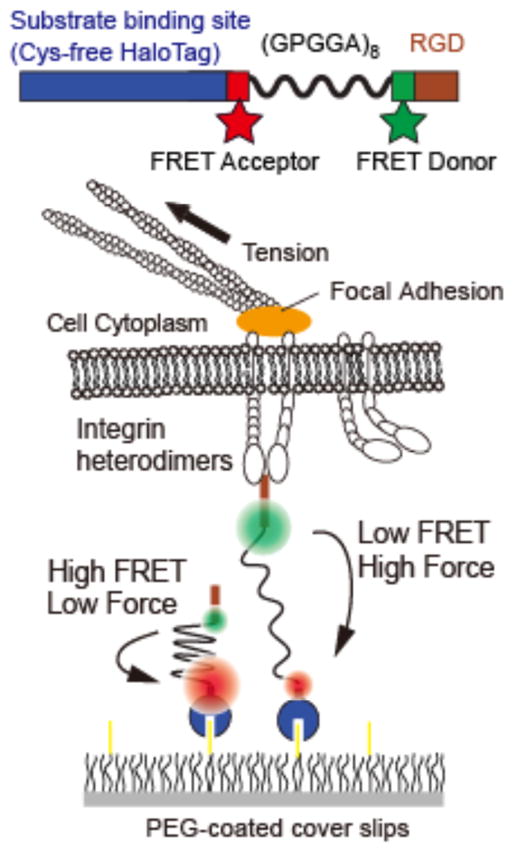
Experimental schematic. Sensors are attached to a PEG-coated coverslip using a cysteine-free HaloTag domain. The FRET pair flanks an elastic protein spring consisting of 8 repeats of the GPGGA amino acid sequence. The FRET acceptor ATTO 647N is attached using alkyne-azide click chemistry at the unnatural amino acid 4-azido-L-phenylalanine, and the FRET donor Alexa 546 is specifically attached at a unique cysteine (SI Materials and Methods). Cells are plated on a sensor-coated surface. Integrins engage the RGD domain and apply tension that pulls the FRET pair apart. High tension leads to low FRET.
We used total internal reflection fluorescence (TIRF) microscopy to measure MTS FRET in the presence of human foreskin fibroblasts (HFFs). HFFs are an advantageous cell type for imaging due to their large size (~50 μm diameter), large adhesions, and relatively slow migration. We employ an MTS density of ~120 molecules/μm2 (~90 nm spacing between MTSs), which in our hands is sufficient to ensure robust cell spreading (Figure S5). At these densities individual fluorophores are not resolved, resulting in a continuous fluorescent field. HFFs plated on the MTSs exhibit low FRET (high traction) at the cell periphery, consistent with previous results (Video S1)35. The low FRET regions near the cell periphery adopt FA-like structures, and are coincident with FA protein recruitment. For visualization, the FRET index (defined as the ratio of acceptor intensity to summed intensity of donor and acceptor) is inverted such that a bright pixel intensity corresponds to low FRET and thus high traction (Figure S6). FRET data can equivalently be analyzed to yield spatial maps of FRET efficiency (Figure S7), or, using a previously determined calibration curve (Figure S3)31, the average force per MTS (Figure S7) or traction stress maps (Figure S8).
We recorded analogous FRET maps for mouse embryonic fibroblasts and human microvascular endothelial cells, demonstrating the general applicability of the technique (Figure S9). Control measurements with donor-only labeled sensors indicate that changes in fluorescence intensity are not due to cell or integrin binding (Figure S10). Additional control measurements show minimal spectral bleed-through between the GFP, FRET-donor, and FRET-acceptor channels, such that the presence of GFP-labeled proteins does not affect FRET measurements, and vice versa (SI Materials and Methods, see also ref. 35).
In addition to simultaneous imaging of GFP-tagged proteins and FRET maps in live cells, we developed a procedure that combines the MTS force measurement with immunofluorescence. This allows us to image endogenous and phosphorylated proteins with tension. We measured MTS FRET in the presence of living HFFs, fixed the cells in situ (on the microscope), and re-measured FRET in the same area (Figure S11). We tested for possible fixation artifacts by tracking MTS-measured force and GFP-paxillin during fixation (Figure S12). Though we observe subtle shifts for both force and GFP-paxillin in small, peripheral adhesions, shifts are much less pronounced in larger, more central adhesions and the regions of low FRET and paxillin remain coincident. Interestingly, we observe that the low-FRET force signal persists for ~2 hrs immediately following fixation. Cells grown on micropost arrays also continue to exert tension on their substrate following fixation40. The preservation of the patterns of force production following fixation provides a simple and potentially powerful means of combining MTS traction force measurements with standard immunocytochemistry.
Previous studies suggest that αvβ3 integrins are immobilized in large and static FAs, whereas α5β1 integrin exhibits a more widespread localization across the cell’s basal surface23. In addition, the integrin β3 subunit was reported to localize to areas inferred to experience high traction forces, while β1 recruitment remained low in these sites23. However, other experiments show that activation of β1 but not β3 increases cell traction in mouse embryonic fibroblasts (MEFs)41. Which class of integrin is responsible for traction force generation in specific cellular systems is thus unclear.
We used complementary assays to assess the localization of α5β1 and αvβ3 integrins with respect to tension generation. First, we simultaneously measured local traction generation using MTS-functionalized coverslips and either α5β1 or αvβ3 integrin localization using standard immunocytochemical protocols (Figure 2A, B, SI Materials and Methods). We observed that αvβ3 integrin is concentrated in force-producing FA-like structures, whereas α5β1 integrin localizes both to force-producing FAs and other regions on the basal side of the cell membrane (Figure 2A, 2B, S13, S14). Next, we imaged MTS FRET for HFFs expressing either mEmerald-tagged αv integrin or eGFP-tagged α5 integrin in an expression construct incorporating a crippled CMV promoter42,43 (Figure 2C, D). As in the fixed cells, we observe a strong coincidence of mEmerald-αv with regions of high force, but limited localization of GFP-α5 integrin with respect to traction generation (Figure 2D, S15). In summary, under our experimental conditions we find that low FRET (high force) is highly correlated with αvβ3 recruitment, while α5β1 integrin is more broadly distributed and only partially present at low FRET regions.
Figure 2.

(A) Top: immunofluorescence staining for αvβ3 integrin. Bottom: normalized, inverted FRET signal calculated from TIRF images of the FRET donor and acceptor (SI, Figure S6). Bright red indicates high tension. (B) Immunofluorescence staining for α5β1 integrin (top) and inverted FRET signal (bottom). (C) Live cell images of mEmerald-tagged αv integrin (top) and inverted FRET signal (bottom). (D) Live cell images of GFP-tagged α5 integrin (top) and inverted FRET signal (bottom). (E) GFP actin (top), inverted FRET (middle), and overlay (bottom). (F) GFP-MRLC (green) overlaid with inverted FRET (red). The GFP-TIRF image shows both myosin-containing stress fibers and a more diffuse contractile band in the lamellum. Scale bar is 10 μm.
We further investigated the difference in colocalization of αv and α5β1 integrins with tension using blocking antibodies against αv (L230) and β1 integrin (P5D2) (Figure S16). We found that treatment with either antibody resulted in partial cell detachment after 60 minutes, and that the addition of both antibodies yielded a synergistic effect in which 70% of the HFFs detached over this time period. These data indicate that both integrins contribute to the formation of stable adhesions, as has been previously suggested23. We noted that treatment with L230 resulted in a rapid decrease in cellular traction force (Figure S16), suggesting that αv-containing integrins transmit a large fraction of the cell-generated forces to the MTS-coated surface.
Actin polymerization and actomyosin contractility are the primary means by which cells exert traction forces on their surroundings and are required for surface stiffness sensing and adhesion maturation44–46. To study the relationship between force and actomyosin contractility, we observed tension maps simultaneously with GFP-actin and GFP-MRLC localization (Figure 2E, F). In TIRF images, GFP-actin localizes closely with high tension (Figure 2E). GFP-MRLC localizes to stress fibers that terminate in a fraction of the low FRET regions (Figure 2F). Inhibition of myosin light chain kinase using ML-7 results in a reduction in MTS-measured tension prior to the disassembly of GFP-MRLC containing stress fibers (Video S2). Thus, although myosin regulatory light chain does not localize to sites of integrin-mediated tension, myosin contractility is required for traction force generation in our system.
We next sought to determine which FA proteins participated in localized tension transmission. We simultaneously imaged MTS FRET and GFP fusions of paxillin, talin (eGFP at the N-terminus), vinculin, and α-actinin in living cells (Figure 3A; Video S3, S4). Paxillin is a canonical FA protein, but is not commonly thought to have a direct role in force transmission47,48. In contrast, talin and α-actinin have been proposed to form direct links between integrins and actin, while vinculin is thought to play a prominent role in both tension transmission and tension sensing24,31,45,49,50. We note that some caution is required in interpreting data derived using GFP-tagged proteins, as these attributes could in principle be influenced by the fusion protein’s expression levels relative to the endogenous protein, or by the presence of the GFP tag. However, all of the GFP-fusions used here have either been used previously to study FA dynamics or are functionally equivalent to previously described constructs (see SI Materials and Methods). These prior data support the use of the particular GFP fusions used here as reasonable proxies for the localization and dynamics of the endogenous proteins.
Figure 3.

Internal architecture of tension generation within focal adhesions. (A) GFP-tagged paxillin, talin, vinculin, and α-actinin (green) were transfected into HFFs and visualized simultaneously with FRET. Here the FRET index map is inverted and normalized such that bright red indicates low FRET and hence high force. Scale bar in all images is 10 μm. (B) Schematic showing segmented GFP (green oval) and low FRET (red oval) regions. Solid dots represent their respective centers of mass, and the black arrow represents the vector connecting the two (see Supplemental Information). (C) 2D correlation coefficient for mEmerald-tagged αv integrin and GFP-tagged paxillin, talin, vinculin, and α-actinin with inverted FRET within segmented FAs. A correlation coefficient approaching 1 indicates a high degree of coincidence between protein localization and local tension generation. (D) Distance shift between the FA protein and inverted FRET centers of mass. Positive distances indicate that the protein center of mass is shifted toward the cell center. The control represents a GFP image taken before and after the FRET image to verify that FA movement during the FRET acquisition does not account for the observed correlation and shift differences. αv Integrin: 95 FAs over 9 cells, Paxillin: 114 FAs over 17 cells, Vinculin: 92 FAs over 10 cells, α-Actinin: 86 FAs over 12 cells, Talin: 75 FAs over 11 cells, Control: 27 FAs over 3 cells. Note that only isolated adhesions were selected for the analysis (see SI). Statistical two-sample Kolmogorov-Smirnov test: * = p < 2.5 × 10−5. Statistical two-sample t-test: * = p < 2.5 × 10−5.
We segmented FA regions in both GFP and FRET images and manually selected adhesions that were well isolated for analysis (SI Methods, Figure S17). For these segmented regions, we calculated the 2-D correlation coefficient between GFP intensity and inverted FRET for each of the proteins (Figure 3B, C, S17). We find that paxillin and αv-integrin have the highest correlation with low FRET (and presumably localized tension generation), while talin and vinculin exhibit markedly lower correlation values. GFP-α-actinin shows a relatively low correlation with low FRET, and was even anti-correlated in some regions (Figure 3C). We also find that phosphorylated paxillin has a distinct localization pattern relative to total paxillin in certain adhesions on MTS-coated surfaces (Figure S18). These data suggest that regions of tension generation are not completely synonymous with the recruitment of talin, vinculin, or α-actinin, but are well correlated with paxillin and αv integrin on our surfaces.
We next sought complementary means to determine how different proteins participated in tension transmission (Figure 3D). Following the methodology from a previous report51, we quantified the center of mass positions for both GFP intensity and inverted FRET index of the segmented FA regions (SI Methods, Figure S17). We observed that the centers of mass for αv-integrin, paxillin, talin, and vinculin are modestly shifted toward the cell center relative to FRET (~100 nm for αv integrin and paxillin, ~200 nm for talin and vinculin), whereas for α-actinin we see a much larger, ~400 nm shift toward the cell center with a large spread in the distribution. In addition, we find that the angle between the vector connecting the two centers of mass and the major axis of the FA is generally close to zero, implying that the positional shift is along the major axis of the FA (presumably along the length of the attached actin bundle or bundles). These data are in agreement with the 2D correlation calculations, with paxillin and αv integrin being most closely associated with localized tension.
The photostability of the ATTO 647N dye and the covalent attachment of MTSs to the glass coverslip allow for both long-term, time-lapse imaging, and, as will be discussed, super-resolved traction force maps. To illustrate this point, we took time-lapse movies of both FRET and GFP fusions of paxillin and α-actinin to verify that the FRET signal is robust over an hour timescale when imaged at 1 frame per minute (Figure 4A, 4B, Video S3, S4). We found that paxillin and low FRET were closely related in space and time in both assembling and disassembling adhesions (Figure S18, Video S4). Interestingly, time-lapse movies of GFP α-actinin plus MTS FRET showed that the relation of α-actinin with local traction force production is more complicated (Figure S18, Video S3), which may reflect α-actinin’s multiple roles in both FA formation and stress fiber reinforcement24,52,53.
Figure 4.

Time-lapse imaging of GFP-fusions of (A) paxillin and (B) α-actinin and inverted, normalized FRET. White arrows indicate example adhesions that assemble or disassemble. Scale bar is 10 μm.
To further examine the close association of paxillin and tension localization, we employed a super-resolution imaging technique, Bayesian localization microscopy (3B)54. We applied the Bayesian analysis method to the FRET donor channel to generate super-resolved tension maps: high donor intensity corresponds to low FRET, and hence localized tension (Figure 5, SI Methods, Video S5). 3B analysis reveals a strong correspondence between paxillin, αv-integrin, and low-FRET regions, consistent with their high 2D correlation coefficients (Figure 3C). Consistent with prior reports55–57, we observed that many though not all FAs consist of a single, discontinuous line in GFP-paxillin (Figure 5D). Additional images reveal a similar correspondence of GFP-paxillin and Alexa 555-stained actin (Supplemental Figure S20). Vinculin also localizes to similar linear structures, though with what appears to be qualitatively reduced co-localization with low FRET (Figure 5J), in agreement with the moderate correlation of vinculin with local tension (Figure 3C).
Figure 5.

Super-resolution imaging reveals paxillin recruitment and force generation within FAs. Bayesian localization microscopy (3B)54 was used to resolve the location of GFP-paxillin (A) and the FRET donor (B) to generate super-resolved paxillin and tension maps (D, E). Overlay of GFP-paxillin and the FRET donor (C, F) show the close association of paxillin and low FRET. Scale bar is 10 μm. Insets (i, ii, iii) focus on specific adhesions and show a strong correspondence between paxillin (green) and low FRET (red). Linescans through insets Gi and Gii (Hi, Hii) show improved resolution of Bayesian localization microscopy over widefield images (green: paxillin, red: low FRET, solid lines: 3B results, dotted lines: widefield). Scale bar is 1.0 μm. (I) Raw images of mEmerald tagged αv integrin (green) and the FRET donor (red) (i) and super-resolved images of mEmerald-αv integrin and the FRET donor (ii). Scale bar is 5 μm. (J) Raw images of GFP-vinculin (green) and the FRET donor (red) (i) and super-resolved images of GFP-vinculin and the FRET donor (ii). Scale bar is 5 μm.
We also used structured illumination microscopy (SIM) to record combined αv integrin, paxillin, and vinculin localization and force maps with ~100 nm resolution (Figure S21). These results corroborate the reliability of 3B imaging and provide a complementary means of visualizing tension via MTSs at high spatial resolutions. We note in the 3B images, the super-resolved FRET donor and GFP images sometimes show small lateral offsets. These offsets may reflect intricacies of the FA architecture, chromatic aberrations across a field of view, or alternatively may stem from the small time delay between the acquisition of the FRET and GFP images (~30 s), during which time the cell is expected to move a small amount.
Discussion
Here we describe technical advances that allow us to visualize the internal architecture of tension generation within FAs. These improvements include covalent attachment of MTSs to the glass coverslip, which allows measurements over long periods of time, and site-specific labeling of FRET pairs immediately proximal to the (GPGGA)8 spring, which increases the resting FRET efficiency. Coupling these improvements with Bayesian localization microscopy or structured illumination microscopy generates super-resolved FA protein localization and tension maps. These advances provide a potentially powerful means to directly observe the cell’s force-generating and force-sensing protein machinery, both in the context of integrins and other adhesion complexes32,58.
We find that much of the force exerted at FAs is generated remotely by myosin and transmitted to FAs via the actin cytoskeleton. Specifically, we observe that addition of ML-7, a myosin light chain kinase inhibitor, rapidly reduces force production. This finding is consistent with previous reports showing that nonmuscle myosin II is the major contributor to cellular traction stress generation59. We find that MRLC is localized to stress fibers that extend to some, but not all, adhesions. How force is generated at the adhesions that are not obviously attached to myosin-containing filaments is not firmly established. It is plausible that dorsal stress fibers that extend out of the TIRF illumination exert force on some of these adhesions60. Alternatively, force transmission may occur via the dendritic actin meshwork within the lamellum, where actin retrograde flow leads to transient application of tension on FA components59.
Integrins with overlapping ECM binding preferences, for example α5β1 and αvβ3 integrin studied here, are thought to play both distinct and redundant roles in mechanosignaling. However, how this differential function is achieved is incompletely understood. In our system, we find that αvβ3 integrin localization invariably coincides with areas of high traction, while α5β1 integrin localizes to both high traction regions as well as adhesions that display MTS FRET similar to background (Figure 2A, B, Figure S13). These data support previous observations that β3 integrins localize to regions inferred from prior traction force microscopy measurements to experience high traction, a technique that has an order of magnitude lower resolution than MTS-measured traction23. Our data thus reinforce and extend the previously proposed model in which αvβ3 integrins support the formation of large, stable adhesions that require myosin II–generated tension stabilization, whereas the stability of α5β1-based adhesions is less contractility-dependent23. Our observations also support the notion that αvβ3 integrin plays a direct role in cellular force sensing, a model supported by the observation that αvβ3 integrin (but not α5β1 integrin) was required for the stiffening response resulting from oscillatory force application to fibronectin-coated beads attached to cells61.
The MTSs used here contain the RGD-containing peptide sequence derived from the 10th type III domain of fibronectin. A secondary integrin-binding site located on the 9th type III domain, termed the synergy site, has been identified as playing a role in mediating adhesion by α5β1 but not αvβ3 integrin62. Previous work demonstrates that mechanical force can switch α5β1 integrins into a tension-dependent conformation that engages the synergy site and increases bond strength via a catch-bond mechanism62,63. Despite the absence of the synergy site, HFFs plated on MTS-functionalized surfaces form peripheral paxillin-rich adhesions that are similar to those formed when HFFs are plated on full-length fibronectin (Figure S20, S22). Importantly, these results suggest that the synergy site is dispensable for αvβ3 integrin localization to areas of traction force production, and by extension αvβ3-based mechanotransduction. Both α5β1 and αvβ3 integrins have approximately micromolar affinities for linear RGD peptides such as the ones used here64,65; furthermore, membrane diffusivities of GFP-tagged β1 and β3 integrins are similar both inside and outside adhesions17. These considerations suggest that the differences in integration localization that we observe may arise from a more complex mechanism than a simple difference in affinities for the MTS RGD sequence. Measurements that compare the localization of specific integrin states, for example their activated conformation(s), with local traction force generation may thus be useful in clarifying the molecular mechanisms that underlie integrin-based mechanotransduction. More broadly, MTSs that incorporate the fibronectin synergy site and other integrin subtype-specific motifs may provide a uniquely effective means of probing mechanotransduction at specific integrin complexes, in the context of stem cell66, cancer4, and endothelial biology67.
The cytoplasmic FA components paxillin, talin, vinculin and α-actinin are shifted toward the center of the cell relative to force production (Figure 3C). The magnitudes of these shifts are in reasonable agreement with previously-measured heights of these proteins relative to the plasma membrane15. We observe a ~100 nm shift between low FRET and paxillin along the length of an adhesion, and a ~400 nm shift between low FRET and α-actinin; we thus deduce that the shift between α-actinin and paxillin is ~300 nm along the length of a FA. Previous results indicate that α-actinin on average lies ~50 nm above paxillin in the vertical direction above the membrane surface15. Combined, these values predict that the actin filaments extend from the FA at an angle of 10°, a physically reasonable picture given the thinness of the cell’s lamellum. In a separate study, traction force measurements using bead-embedded polyacrylamide gels showed that the peak traction stress in mature FAs was shifted from the peak paxillin intensity by an average distance of ~1 μm68. We observe a smaller shift between traction and paxillin in our experiments, which may reflect the difference in substrate stiffness used in these two studies.
While some earlier studies are consistent with the idea that paxillin is recruited very early in adhesion formation either with or by FAK21,69, other data support models in which paxillin is instead recruited by, and after, talin70. Additional reports suggest that integrin recruitment to adhesions can occur independently of talin and instead may involve α-actinin24,52. It is thus striking that, in our data, paxillin is more, not less, correlated with local force production than either talin or α-actinin. It is plausible that integrins may connect to actin via talin, FAK, or α-actinin at varying points in the adhesion lifetime and turnover cycle, or in a cell-type or matrix-dependent manner. The relatively modest correlation of both talin and α-actinin with local tension generation may thus reflect multiple load-bearing assemblies within the FAs studied here49,53. It is also possible that talin, α-actinin, and vinculin (see below) may be maximally recruited in response to optimal, intermediate tensions, rather than showing a monotonic correlation or anticorrelation with force.
A large body of work implicates vinculin as a mechanosensor at FAs31,49,71,72. Both cell biological71 and biophysical73 data indicate that mechanical tension unfolds domains in talin, which in turn recruits and activates vinculin such that it forms additional, reinforcing connections to actin. Separate work likewise suggests that a direct interaction between FAK, paxillin, and vinculin contributes to matrix stiffness sensing, though the underlying molecular mechanism remains unclear49,68. Though indirect evidence, mostly co-immunoprecipitation, suggests that paxillin may bind α- or β-integrins74–76, to our knowledge a direct binding interaction between paxillin and any integrin remains to be firmly established. Further, cells lacking paxillin can still form robust adhesions to fibronectin substrates, arguing against an essential role for paxillin in force transmission47. For these reasons, paxillin is thus not generally thought to play a direct role in force transmission between integrins and the cytoskeleton. We nevertheless observe that paxillin colocalizes with tension to a higher degree than vinculin as measured by 2D correlation coefficients, relative centers of mass, and superresolution imaging. We therefore speculate that αvβ3 integrin and paxillin may participate in a complex, perhaps with additional FA proteins, that functions upstream or in parallel to vinculin in tension sensing, at least under the circumstances studied here.
Super-resolution tension maps allowed us to directly visualize the structure of force-generating structures within individual adhesions. We find two types of structures in our data: thin, linear adhesions and larger plaques. Unlike native fibronectin, which assembles into fibrils, the integrin-binding sites provided by the MTS-functioned coverslips are spatially homogenous. The super-resolved structures we observe may thus reflect a default physical structure adopted by FAs in the absence of spatial constraints provided by the ECM. The linear patterns we observe for actin, paxillin, vinculin, αv integrin, and tension suggest that many, though not all, of the adhesions in our system are templated by actin bundles. Conversely, we and others also see roughly linear actin and paxillin localization patterns for cells adhering to full-length fibronectin, arguing that these structures are not a consequence of adhesion specifically to the MTSs (Figure S22)55–57. We therefore speculate that the fundamental unit of FA assembly may be an actin bundle that is templated by VASP77,78, formins6,79, or other actin nucleators, and connected to integrins via a variety of integrin- and actin-binding complexes. The approaches described here provide a potentially powerful means to determine how FA components are arranged in space and time relative to traction forces, and how they function together to sense and transduce mechanical signals.
Supplementary Material
Acknowledgments
Funding Sources
This work is supported by a Stanford Bio-X IIP award (ARD), the National Science Foundation (NSF) under Emerging Frontiers in Research and Innovation (EFRI) Grant 1136790 (ARD), National Institute of Health (NIH) New Innovator Award 1DP2OD007078 (ARD), Stanford Cardiovascular Institute Seed Grant (ARD), Burroughs-Wellcome Career Award at the Scientific Interface (ARD), and NSF Graduate Research Fellowships 1000121811 (AHM) and 1000125096 (ACC).
We thank the members of the A.R.D. laboratory for insightful discussions, specifically Jack Chai for extensive help with data analysis. This work is supported by a Stanford Bio-X IIP award (ARD), the National Science Foundation (NSF) under Emerging Frontiers in Research and Innovation (EFRI) Grant 1136790 (ARD), National Institute of Health (NIH) New Innovator Award 1DP2OD007078 (ARD), Stanford Cardiovascular Institute Seed Grant (ARD), Burroughs-Wellcome Career Award at the Scientific Interface (ARD), NSF Graduate Research Fellowships 1000121811 (AHM) and 1000125096 (ACC), and a Stanford Graduate Fellowship (SJT). In addition, we thank Jon Mulholland and the Cell Sciences Imaging Facility for imaging support. The project described was supported, in part, by Award Number 1S10OD01277601 (HEIG to CSIF for OMX-BLAZE) from the National Center for Research Resources (NCRR). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NCRR or the National Institutes of Health. We thank Theresa Mary McLaughlin and Stanford University Mass Spectrometry for mass spectrometry support.
Abbreviations
- FA
focal adhesion
- ECM
extracellular matrix
- FRET
Förster resonance energy transfer
- MTS
molecular tension sensor
- HFF
human foreskin fibroblast
- RGD
arginine-glycine-aspartic acid
- PEG
poly(ethylene glycol)
- eGFP
enhanced green fluorescent protein
Footnotes
Author Contributions
The manuscript was written through contributions from all of the authors. All authors have given approval to the final version of the manuscript.
Competing financial interests
The author declares no competing financial interests.
Detailed materials and methods, supplementary figures, and supplementary videos.
References
- 1.Jaalouk DE, Lammerding J. Nat Rev Mol Cell Biol. 2009;10:63–73. doi: 10.1038/nrm2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hoffman BD, Grashoff C, Schwartz MA. Nature. 2011;475:316–323. doi: 10.1038/nature10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Engler AJ, Sen S, Sweeney HL, Discher DE. Cell. 2006;126:677–689. doi: 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 4.Butcher DT, Alliston T, Weaver VM. Nat Rev Cancer. 2009;9:108–122. doi: 10.1038/nrc2544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwarz US, Gardel ML. J Cell Sci. 2012;125:3051–3060. doi: 10.1242/jcs.093716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Geiger B, Spatz JP, Bershadsky AD. Nat Rev Mol Cell Biol. 2009;10:21–33. doi: 10.1038/nrm2593. [DOI] [PubMed] [Google Scholar]
- 7.Ramsay AG, Marshall JF, Hart IR. Cancer Metastasis Rev. 2007;26:567–578. doi: 10.1007/s10555-007-9078-7. [DOI] [PubMed] [Google Scholar]
- 8.Hynes RO. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- 9.Felding-Habermann B, O’Toole TE, Smith JW, Fransvea E, Ruggeri ZM, Ginsberg MH, Hughes PE, Pampori N, Shattil SJ, Saven A, Mueller BM. Proc Natl Acad Sci U S A. 2001;98:1853–1858. doi: 10.1073/pnas.98.4.1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Barczyk M, Carracedo S, Gullberg D. Cell Tissue Res. 2010;339:269–280. doi: 10.1007/s00441-009-0834-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geiger T, Zaidel-Bar R. Curr Opin Cell Biol. 2012;24:562–568. doi: 10.1016/j.ceb.2012.05.004. [DOI] [PubMed] [Google Scholar]
- 12.Kuo JC, Han X, Hsiao CT, Yates JR, Waterman CM. Nat Cell Biol. 2011;13:383–393. doi: 10.1038/ncb2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Humphries JD, Byron A, Bass MD, Craig SE, Pinney JW, Knight D, Humphries MJ. Sci Signal. 2009;2:ra51. doi: 10.1126/scisignal.2000396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schiller HB, Friedel CC, Boulegue C, Fässler R. EMBO Rep. 2011;12:259–266. doi: 10.1038/embor.2011.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanchanawong P, Shtengel G, Pasapera AM, Ramko EB, Davidson MW, Hess HF, Waterman CM. Nature. 2010;468:580–584. doi: 10.1038/nature09621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shibata ACE, Fujiwara TK, Chen L, Suzuki KGN, Ishikawa Y, Nemoto YL, Miwa Y, Kalay Z, Chadda R, Naruse K, Kusumi A. Cytoskeleton (Hoboken) 2012;69:380–392. doi: 10.1002/cm.21032. [DOI] [PubMed] [Google Scholar]
- 17.Rossier O, Octeau V, Sibarita JB, Leduc C, Tessier B, Nair D, Gatterdam V, Destaing O, Albigès-Rizo C, Tampé R, Cognet L, Choquet D, Lounis B, Giannone G. Nat Cell Biol. 2012;14:1057–1067. doi: 10.1038/ncb2588. [DOI] [PubMed] [Google Scholar]
- 18.Yu C, Law JBK, Suryana M, Low HY, Sheetz MP. Proc Natl Acad Sci U S A. 2011;108:20585–20590. doi: 10.1073/pnas.1109485108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schvartzman M, Palma M, Sable J, Abramson J, Hu X, Sheetz MP, Wind SJ. Nano Lett. 2011;11:1306–1312. doi: 10.1021/nl104378f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang J, Grater SV, Corbellini F, Rinck S, Bock E, Kemkemer R, Kessler H, Ding J, Spatz JP. Nano Lett. 2009;9:1111–1116. doi: 10.1021/nl803548b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bachir AI, Zareno J, Moissoglu K, Plow EF, Gratton E, Horwitz AR. Curr Biol. 2014;24:1845–1853. doi: 10.1016/j.cub.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoffmann J-E, Fermin Y, Stricker R, Lo Ickstadt K, Zamir E. Elife. 2014;3:e02257. doi: 10.7554/eLife.02257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schiller HB, Hermann MR, Polleux J, Vignaud T, Zanivan S, Friedel CC, Sun Z, Raducanu A, Gottschalk KE, Théry M, Mann M, Fässler R. Nat Cell Biol. 2013;15:625–636. doi: 10.1038/ncb2747. [DOI] [PubMed] [Google Scholar]
- 24.Roca-Cusachs P, del Rio A, Puklin-Faucher E, Gauthier NC, Biais N, Sheetz MP. Proc Natl Acad Sci U S A. 2013;110:E1361–E1370. doi: 10.1073/pnas.1220723110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gardel ML, Schneider IC, Aratyn-Schaus Y, Waterman CM. Annu Rev Cell Dev Biol. 2010;26:315–333. doi: 10.1146/annurev.cellbio.011209.122036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Oakes PW, Beckham Y, Stricker J, Gardel ML. J Cell Biol. 2012;196:363–374. doi: 10.1083/jcb.201107042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Byron A, Morgan MR, Humphries MJ. Curr Biol. 2010;20:R1063–R1067. doi: 10.1016/j.cub.2010.10.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Blakely BL, Dumelin CE, Trappmann B, McGregor LM, Choi CK, Anthony PC, Duesterberg VK, Baker BM, Block SM, Liu DR, Chen CS. Nat Methods. 2014;11:1229–1232. doi: 10.1038/nmeth.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu Y, Medda R, Liu Z, Galior K, Yehl K, Spatz JP, Cavalcanti-Adam EA, Salaita K. Nano Lett. 2014;14:5539–5546. doi: 10.1021/nl501912g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Ge C, Zhu C, Salaita K. Nat Commun. 2014;5:5167. doi: 10.1038/ncomms6167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grashoff C, Hoffman BD, Brenner MD, Zhou R, Parsons M, Yang MT, McLean MA, Sligar SG, Chen CS, Ha T, Schwartz MA. Nature. 2010;466:263–266. doi: 10.1038/nature09198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Borghi N, Sorokina M, Shcherbakova OG, Weis WI, Pruitt BL, Nelson WJ, Dunn AR. Proc Natl Acad Sci U S A. 2012;109:12568–12573. doi: 10.1073/pnas.1204390109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Meng F, Suchyna TM, Sachs F. FEBS J. 2008;275:3072–3087. doi: 10.1111/j.1742-4658.2008.06461.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krieg M, Dunn AR, Goodman MB. Nat Cell Biol. 2014;16:224–233. doi: 10.1038/ncb2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Morimatsu M, Mekhdjian AH, Adhikari AS, Dunn AR. Nano Lett. 2013;13:3985–3989. doi: 10.1021/nl4005145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y, Yehl K, Narui Y, Salaita K. J Am Chem Soc. 2013;135:5320–5323. doi: 10.1021/ja401494e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang X, Ha T. Science. 2013;340:991–994. doi: 10.1126/science.1231041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stabley DR, Jurchenko C, Marshall SS, Salaita KS. Nat Methods. 2012;9:64–67. doi: 10.1038/nmeth.1747. [DOI] [PubMed] [Google Scholar]
- 39.Jurchenko C, Chang Y, Narui Y, Zhang Y, Salaita KS. Biophys J. 2014;106:1436–1446. doi: 10.1016/j.bpj.2014.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tan JL, Tien J, Pirone DM, Gray DS, Bhadriraju K, Chen CS. Proc Natl Acad Sci U S A. 2003;100:1484–1489. doi: 10.1073/pnas.0235407100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lin GL, Cohen DM, Desai RA, Breckenridge MT, Gao L, Humphries MJ, Chen CS. FEBS Lett. 2013;587:763–769. doi: 10.1016/j.febslet.2013.01.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen L, Vicente-Manzanares M, Potvin-Trottier L, Wiseman PW, Horwitz AR. PLoS One. 2012;7 doi: 10.1371/journal.pone.0040202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Laukaitis CM, Webb DJ, Donais K, Horwitz AF. J Cell Biol. 2001;153:1427–1440. doi: 10.1083/jcb.153.7.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lavelin I, Wolfenson H, Patla I, Henis YI, Medalia O, Volberg T, Livne A, Kam Z, Geiger B. PLoS One. 2013;8:e73549. doi: 10.1371/journal.pone.0073549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Parsons JT, Horwitz AR, Schwartz MA. Nat Rev Mol Cell Biol. 2010;11:633–643. doi: 10.1038/nrm2957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wolfenson H, Bershadsky A, Henis YI, Geiger B. J Cell Sci. 2011;124:1425–1432. doi: 10.1242/jcs.077388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hagel M, George EL, Kim A, Tamimi R, Opitz SL, Turner CE, Imamoto A, Thomas SM. Mol Cell Biol. 2002;22:901–915. doi: 10.1128/MCB.22.3.901-915.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brown MC, Turner CE. Physiol Rev. 2004;84:1315–1339. doi: 10.1152/physrev.00002.2004. [DOI] [PubMed] [Google Scholar]
- 49.Pasapera AM, Schneider IC, Rericha E, Schlaepfer DD, Waterman CM. J Cell Biol. 2010;188:877–890. doi: 10.1083/jcb.200906012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lawson C, Lim ST, Uryu S, Chen XL, Calderwood DA, Schlaepfer DD. J Cell Biol. 2012;196:223–232. doi: 10.1083/jcb.201108078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zamir E, Katz BZ, Aota S, Yamada KM, Geiger B, Kam Z. J Cell Sci. 1999;112:1655–1669. doi: 10.1242/jcs.112.11.1655. [DOI] [PubMed] [Google Scholar]
- 52.Choi CK, Vicente-Manzanares M, Zareno J, Whitmore LA, Mogilner A, Horwitz AR. Nat Cell Biol. 2008;10:1039–1050. doi: 10.1038/ncb1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jockusch BM, Isenberg G. Proc Natl Acad Sci U S A. 1981;78:3005–3009. doi: 10.1073/pnas.78.5.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cox S, Rosten E, Monypenny J, Jovanovic-Talisman T, Burnette DT, Lippincott-Schwartz J, Jones GE, Heintzmann R. Nat Methods. 2012;9:195–200. doi: 10.1038/nmeth.1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shroff H, Galbraith CG, Galbraith JA, Betzig E. Nat Methods. 2008;5:417–423. doi: 10.1038/nmeth.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shroff H, Galbraith CG, Galbraith JA, White H, Gillette J, Olenych S, Davidson MW, Betzig E. Proc Natl Acad Sci U S A. 2007;104:20308–20313. doi: 10.1073/pnas.0710517105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Van Hoorn H, Harkes R, Spiesz EM, Storm C, van Noort D, Ladoux B, Schmidt T. Nano Lett. 2014;14:4257–4262. doi: 10.1021/nl5008773. [DOI] [PubMed] [Google Scholar]
- 58.Paszek MJ, DuFort CC, Rossier O, Bainer R, Mouw JK, Godula K, Hudak JE, Lakins JN, Wijekoon AC, Cassereau L, Rubashkin MG, Magbanua MJ, Thorn KS, Davidson MW, Rugo HS, Park JW, Hammer Da, Giannone G, Bertozzi CR, Weaver VM. Nature. 2014;511:319–325. doi: 10.1038/nature13535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gardel ML, Sabass B, Ji L, Danuser G, Schwarz US, Waterman CM. J Cell Biol. 2008;183:999–1005. doi: 10.1083/jcb.200810060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Burnette DT, Shao L, Ott C, Pasapera AM, Fischer RS, Baird MA, Der Loughian C, Delanoe-Ayari H, Paszek MJ, Davidson MW, Betzig E, Lippincott-Schwartz J. J Cell Biol. 2014;205:83–96. doi: 10.1083/jcb.201311104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roca-Cusachs P, Gauthier NC, Del Rio A, Sheetz MP. Proc Natl Acad Sci U S A. 2009;106:16245–16250. doi: 10.1073/pnas.0902818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Friedland JC, Lee MH, Boettiger D. Science. 2009;323:642–644. doi: 10.1126/science.1168441. [DOI] [PubMed] [Google Scholar]
- 63.Kong F, García AJ, Mould AP, Humphries MJ, Zhu C. J Cell Biol. 2009;185:1275–1284. doi: 10.1083/jcb.200810002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Orlando RA, Cheresh DA. J Biol Chem. 1991;266:19543–19550. [PubMed] [Google Scholar]
- 65.Pfaff M, Tangemann K, Müller B, Gurrath M, Müller G, Kessler H, Timpl R, Engel J. J Biol Chem. 1994;269:20233–20238. [PubMed] [Google Scholar]
- 66.Goessler UR, Bugert P, Bieback K, Stern-Straeter J, Bran G, Hörmann K, Riedel F. Int J Mol Med. 2008;21:271–279. [PubMed] [Google Scholar]
- 67.Su G, Hodnett M, Wu N, Atakilit A, Kosinski C, Godzich M, Huang XZ, Kim JK, Frank JA, Matthay MA, Sheppard D, Pittet JF. Am J Respir Cell Mol Biol. 2007;36:377–386. doi: 10.1165/rcmb.2006-0238OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Plotnikov SV, Pasapera AM, Sabass B, Waterman CM. Cell. 2012;151:1513–1527. doi: 10.1016/j.cell.2012.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Choi CK, Zareno J, Digman MA, Gratton E, Horwitz AR. Biophys J. 2011;100:583–592. doi: 10.1016/j.bpj.2010.12.3719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pinon P, Pärssinen J, Vazquez P, Bachmann M, Rahikainen R, Jacquier MC, Azizi L, Määttä JA, Bastmeyer M, Hytönen VP, Wehrle-Haller B. J Cell Biol. 2014;205:265–281. doi: 10.1083/jcb.201308136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dumbauld DW, Lee TT, Singh A, Scrimgeour J, Gersbach CA, Zamir EA, Fu J, Chen CS, Curtis JE, Craig SW, García AJ. Proc Natl Acad Sci U S A. 2013;110:9788–9793. doi: 10.1073/pnas.1216209110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cohen DM, Kutscher B, Chen H, Murphy DB, Craig SW. J Biol Chem. 2006;281:16006–16015. doi: 10.1074/jbc.M600738200. [DOI] [PubMed] [Google Scholar]
- 73.Del Rio A, Perez-Jimenez R, Liu R, Roca-Cusachs P, Fernandez JM, Sheetz MP. Science. 2009;323:638–641. doi: 10.1126/science.1162912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Legate KR, Fässler R. J Cell Sci. 2009;122:187–198. doi: 10.1242/jcs.041624. [DOI] [PubMed] [Google Scholar]
- 75.Schaller MD, Otey CA, Hildebrand JD, Parsons JT. J Cell Biol. 1995;130:1181–1187. doi: 10.1083/jcb.130.5.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu S, Thomas SM, Woodside DG, Rose DM, Kiosses WB, Pfaff M, Ginsberg MH. Nature. 1999;402:676–681. doi: 10.1038/45264. [DOI] [PubMed] [Google Scholar]
- 77.Brindle NP, Holt MR, Davies JE, Price CJ, Critchley DR. Biochem J. 1996;318(Pt 3):753–757. doi: 10.1042/bj3180753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Reinhard M, Halbrügge M, Scheer U, Wiegand C, Jockusch BM, Walter U. EMBO J. 1992;11:2063–2070. doi: 10.1002/j.1460-2075.1992.tb05264.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gupton SL, Eisenmann K, Alberts AS, Waterman-Storer CM. J Cell Sci. 2007;120:3475–3487. doi: 10.1242/jcs.006049. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.