

Abstract
Key points
Na+ current inactivation is biphasic in insulin‐secreting cells, proceeding with two voltage dependences that are half‐maximal at ∼−100 mV and −60 mV.
Inactivation of voltage‐gated Na+ (NaV) channels occurs at ∼30 mV more negative voltages in insulin‐secreting Ins1 and primary β‐cells than in HEK, CHO or glucagon‐secreting αTC1‐6 cells.
The difference in inactivation between Ins1 and non‐β‐cells persists in the inside‐out patch configuration, discounting an involvement of a diffusible factor.
In Ins1 cells and primary β‐cells, but not in HEK cells, inactivation of a single NaV subtype is biphasic and follows two voltage dependences separated by 30–40 mV.
We propose that NaV channels adopt different inactivation behaviours depending on the local membrane environment.
Abstract
Pancreatic β‐cells are equipped with voltage‐gated Na+ channels that undergo biphasic voltage‐dependent steady‐state inactivation. A small Na+ current component (10–15%) inactivates over physiological membrane potentials and contributes to action potential firing. However, the major Na+ channel component is completely inactivated at −90 to −80 mV and is therefore inactive in the β‐cell. It has been proposed that the biphasic inactivation reflects the contribution of different NaV α‐subunits. We tested this possibility by expression of TTX‐resistant variants of the NaV subunits found in β‐cells (NaV1.3, NaV1.6 and NaV1.7) in insulin‐secreting Ins1 cells and in non‐β‐cells (including HEK and CHO cells). We found that all NaV subunits inactivated at 20–30 mV more negative membrane potentials in Ins1 cells than in HEK or CHO cells. The more negative inactivation in Ins1 cells does not involve a diffusible intracellular factor because the difference between Ins1 and CHO persisted after excision of the membrane. NaV1.7 inactivated at 15‐20 mV more negative membrane potentials than NaV1.3 and NaV1.6 in Ins1 cells but this small difference is insufficient to solely explain the biphasic inactivation in Ins1 cells. In Ins1 cells, but never in the other cell types, widely different components of NaV inactivation (separated by 30 mV) were also observed following expression of a single type of NaV α‐subunit. The more positive component exhibited a voltage dependence of inactivation similar to that found in HEK and CHO cells. We propose that biphasic NaV inactivation in insulin‐secreting cells reflects insertion of channels in membrane domains that differ with regard to lipid and/or membrane protein composition.
Keywords: voltage‐gated sodium channels, pancreatic beta‐cell, voltage‐dependent inactivation, insulin secretion, electrical activity
Key points
Na+ current inactivation is biphasic in insulin‐secreting cells, proceeding with two voltage dependences that are half‐maximal at ∼−100 mV and −60 mV.
Inactivation of voltage‐gated Na+ (NaV) channels occurs at ∼30 mV more negative voltages in insulin‐secreting Ins1 and primary β‐cells than in HEK, CHO or glucagon‐secreting αTC1‐6 cells.
The difference in inactivation between Ins1 and non‐β‐cells persists in the inside‐out patch configuration, discounting an involvement of a diffusible factor.
In Ins1 cells and primary β‐cells, but not in HEK cells, inactivation of a single NaV subtype is biphasic and follows two voltage dependences separated by 30–40 mV.
We propose that NaV channels adopt different inactivation behaviours depending on the local membrane environment.
Introduction
Voltage‐gated Na+ (NaV) channels are expressed in nearly all electrically excitable cells where they play a key role in action potential initiation and generation (Hille, 2001). NaV channels exhibit a dual dependence on voltage: depolarization results in both rapid activation and a slower time‐dependent inactivation. During inactivation, the NaV channels enter a non‐conducting state. Reversal of the ‘inactivated’ state requires hyperpolarization of the membrane, the extent of which can vary according to cell type and NaV subtype (Catterall et al. 2017). The balance between activation and inactivation results in a ‘sodium window current’ that determines cellular excitability.
NaV channels consist of a pore‐forming αsubunit, which can form heterodimers or heterotrimers with auxiliary βsubunits that modify their gating properties (Calhoun & Isom, 2014; Kruger & Isom, 2016). Insulin‐secreting β‐cells are equipped with NaV channels and express NaV1.3, NaV1.6 and NaV1.7 α‐subunits that are encoded by Scn3a, Scn8a and Scn9a genes, respectively. Moreover, they principally express Scn1b, which encodes the β1subunit (Benner et al. 2014; Adriaenssens et al. 2016; DiGruccio et al. 2016).
In mouse and rat (but not in human) β‐cells, NaV channels exhibit an unusual voltage dependence of inactivation (Hiriart & Matteson, 1988; Lou et al. 2003; Braun et al. 2008; Zhang et al. 2014). In most mouse β‐cells, inactivation proceeds at unphysiologically negative membrane potentials such that no Na+ voltage‐gated currents remain activatable at membrane potentials above −70 mV. In β‐cells, full NaV reactivation requires membrane potentials as negative as ∼−120 mV. This is 40–50 mV more negative than the most repolarized membrane potential of the βcell. As a result, most NaV channels are ‘locked’ in the non‐conducting inactivated state (Plant, 1988; Gopel et al. 1999). However, more recently it was reported that inactivation in β‐cells is biphasic and consists of an additional small Na+ current component (10–15% of the total Na+ current) that persists at physiologically relevant membrane potentials in one‐third of the β‐cells (Vignali et al. 2006; Zhang et al. 2014).
It has been proposed that the two components of inactivation reflect different Na+ channel subtypes (Vignali et al. 2006; Zhang et al. 2014). Indeed, we have shown that whereas NaV1.7 gives rise to the component inactivating at hyperpolarized voltages, NaV1.3 accounts for the component inactivating over more physiological membrane potentials (Zhang et al. 2014). This would suggest that NaV1.7 channels in β‐cells inactivate at 40 mV more negative membrane potentials than in other cells and that β‐cells contain a factor modulating NaV1.7 channels in a subtype‐specific fashion.
Here we have compared the inactivation properties of different NaV channel subtypes when expressed in insulin‐secreting cells and in HEK, CHO and glucagon‐secreting αTC1‐6 cells. To isolate the expressed current, we generated a tetrodotoxin (TTX)‐resistant form of the channels and blocked endogenous channels by inclusion of TTX in the bath medium. Our data confirm that NaV1.7 currents do indeed inactivate at more hyperpolarized membrane potentials than NaV1.3 and NaV1.6 in Ins1 cells but that the difference is small and insufficient to explain the biphasic inactivation observed in pancreatic β‐cells. Intriguingly, expression of a single NaV subtype gives rise to currents that undergo biphasic inactivation over distinct and widely separated membrane potential ranges in individual Ins1 cells but never in the other cell types. We propose a model that accounts for the αsubunit‐independent biphasic inactivation of NaV channels in β‐cells.
Methods
Ethical approval
With the exception of Fig. 13, all measurements were made in cell lines. Experiments in Fig. 13 were conducted in accordance with the UK Animals (Scientific Procedures) Act 1986 and University of Oxford ethical guidelines.
Figure 13. Biphasic inactivation in mouse β‐cells.
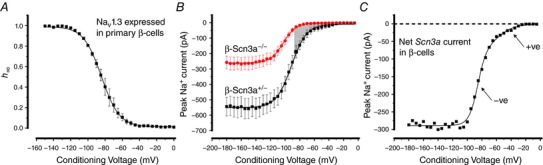
A, Na+ current inactivation in primary mouse β‐cells transfected with TTXresistant NaV1.3 (n = 3). B, voltage dependence of Na+ current inactivation in Scn3a +/– (n = 7) and Scn3a –/– β‐cells (n = 8). Same data as in Zhang et al. (2014) but current amplitudes are presented in absolute rather than relative terms. C, net NaV1.3 current isolated by subtracting NaV currents in Scn3a –/– β‐cells from those in Scn3a+ /– β‐cells. The dashed line represents the zero‐current level. Note biphasic inactivation. A double Boltzmann function was fitted to the data points, yielding V h values of −86 and −57 mV. [Color figure can be viewed at http://wileyonlinelibrary.com]
Mice and islet isolation
The procedures for islet isolation, primary cell culture and generation of the Scn3a knockout mice were as described previously (Zhang et al. 2014).
Plasmid constructs
The constructs coding for human brain SCN9A isoform, for human SCN1B and SCN2B expressed in tandem and for human SCN3B were kindly provided by Frank Reimann (University of Cambridge, UK) (Cox et al. 2006). The constructs for βsubunit expression also coded for green fluorescent protein (GFP), thus allowing tracing of transfected cells.
The constructs coding for human SCN3A (NM_006922), SCN5A (NM_198056) and SCN8A (NM_014191) bearing a Myc‐DDK‐tag at the C‐terminus were purchased from OriGene Technologies, Inc. (Rockville, MD, USA).
The α‐subunits of NaV1.3, NaV1.6 and NaV1.7 were rendered TTX‐resistant by replacing the amino acid tyrosine with serine at positions 384, 371 and 362, respectively (Cummins et al. 2001). Site‐directed mutagenesis was achieved using the Q5 Site‐Directed Mutagenesis Kit (New England Biolabs, Ipswich, MA, USA) with back‐to‐back primers according to the manufacturer's instructions. To generate NaV1.7–NaV1.3 hybrid constructs, Nterminal cytoplasmic domain, loop 1 (L1) and loop 2 (L2), loop 3 (L3) and the C‐terminal cytoplasmic domain of NaV1.7 (see Fig. 1) were replaced with the corresponding sequences of NaV1.3 using overlap PCR and Gibson assembly techniques. All mutations and the integrity of the open reading frames (ORFs) were verified by Sanger sequencing.
Figure 1. Generation of NaV1.7 and NaV1.3 chimera constructs.
CLUSTAL format alignment of NaV1.7 (NP_002968) and NaV1.3 (NP_008853) amino acid sequences was performed using MAFFT software. Corresponding residue numbers are labelled on the left. Sequences of the generated chimeric αsubunits are shown at the bottom. Only partial alignments of the regions where changes were introduced are depicted. NaV1.3 sequences introduced into NaV1.7 backbone are highlighted in yellow. [Color figure can be viewed at http://wileyonlinelibrary.com]



Cell culture
The rat insulinoma cell line Ins1 832/13 (referred to as Ins1) was provided by J. Lang (Université de Bordeaux, France) and cultured in RPMI supplemented with 11 mm glucose (also 5 mm and 25 mm glucose in the indicated experiments), 10% fetal calf serum, 10 mm HEPES, 1 mm sodium pyruvate, 50 μm β‐mercaptoethanol and 100 U ml−1 penicillin and 100 μg ml−1 streptomycin. In specified experiments Ins1 cells were cultured for 48 h with 10 nm phorbol 12‐myristate 13‐acetate (PMA) (Sigma‐Aldrich, St Louis, MO, USA) or 100 μm diazoxide (Sigma‐Aldrich); or for 24 h with the insulin receptor antagonist S961 (Sigma‐Aldrich). The human embryonic kidney cell line (AD‐293; referred to as HEK cells from here on) was obtained from Agilent Technologies (Santa Clara, CA, USA), the mouse pancreatic α‐cell line (αTC1‐6) was obtained from ATCC (Manassas, VA, USA), the Chinese hamster ovary cell line (CHO) was obtained from European Collection of Authenticated Cell Cultures (Salisbury, UK). All cell lines were cultured according to the providers’ protocols.
Transfection of plasmids and small interfering RNA (siRNA) duplexes was performed using Lipofectamine® 2000 (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer's guidelines.
Cells plated on 35 mm dishes were co‐transfected with 1.5 μg of DNA encoding NaV1.3, NaV1.6 and NaV1.7 αsubunits and 50 ng of DNA encoding βsubunits (as specified) and assayed 24–48 h after transfection. GFP fluorescence was used to select for transfected cells, which subsequently were tested for channel expression by whole‐cell patch‐clamp recording techniques.
siRNA‐mediated knockdown experiments were performed in Ins1 cells. siRNA duplexes against rat Scn3b and scrambled negative control (OriGene Technologies, Inc.) were applied at a final concentration of 60 nm. For efficient knockdown, the cells were transfected on day 1 and day 3 and used for experiments on day 4. The efficiency of knockdown was assessed by qPCR.
RNA isolation and quantitative RT‐PCR
RNA was isolated using a combination of TRI reagent and Ambion PureLink RNA Mini Kit (Thermo Fisher Scientific). On‐column DNase treatment was performed to eliminate genomic DNA contamination. cDNA was synthesized using the High Capacity RNA‐to‐cDNA Kit (Thermo Fisher Scientific). Real‐time qPCR was performed using SYBR Green detection and gene specific QuantiTect Primer Assays (Qiagen, Hileden, Germany). Relative expression was calculated using the ΔC t method. Glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) and peptidylprolyl isomerase A (PPIA) were used as reference genes.
Electrophysiological recordings
Whole‐cell Na+ currents were recorded from untreated or transfected Ins1, HEK, αTC1‐6 and CHO cells using the standard whole‐cell configuration as previously described (Zhang et al. 2014). Voltage‐clamp experiments were performed using an EPC‐9 amplifier and Pulse (Version 8.80) software (HEKA Electronik, Lambrecht/Pfalz, Germany). A DMZ‐Zeitz‐Puller (Zeitz, Martinsreid, Germany) was used for fabrication of polished patch‐clamp electrodes (Harvard Apparatus, Cambridge, MA, USA) that had a resistance of 2–4.5 MΩ when filled with the electrode‐filling solution. Capacitive transients were compensated for using a computer‐controlled algorithm. The remaining capacitive transients as well as leak subtraction were removed using a –P/4 protocol. Series resistance compensation between 50 and 80% was used every 2 μs. The recorded currents were filtered by 2.9 kHz and digitized at >10 kHz. Recordings of cells with a seal resistance of less than 1GΩ, a voltage error greater than 5 mV and currents less than 50 pA (except in Fig. 7) were not used. Since Na+ current activation and inactivation undergo a time‐dependent shift towards more negative membrane potentials in the whole‐cell configuration (Fernandez et al. 1984), all measurements reported here were obtained within ∼1 min of achieving the whole‐cell configuration.
Figure 7. Current inactivation of NaV1.7 and NaV1.3 in cell‐attached and inside‐out configuration.
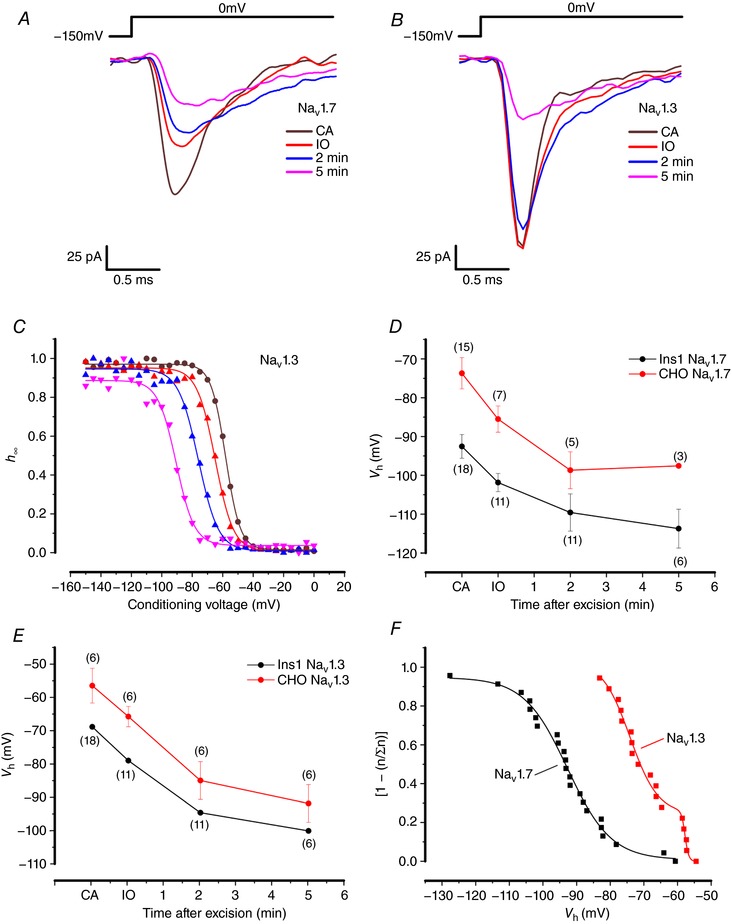
A, NaV1.7 currents when the α‐subunit is co‐expressed with β1 and β2 in Ins1 cells in response to a step depolarization from −150 to 0 mV in cell‐attached (CA), inside‐out (IO) and 2 and 5 min after excision. B, same as in A, but with NaV1.3 currents in Ins1 cells. C, voltage dependence of inactivation of NaV1.3 currents shown in B (same colour code). D, summarized voltage dependence of inactivation of NaV1.7 currents in Ins1 (black) and CHO cells (red) of indicated number of patches/cells (n). E, same as in D but for NaV1.3. F, cumulative distribution of NaV1.3 (red) and NaV1.7 (black) V h recorded in cell‐attached patches on Ins1 cells. Data have been normalized (n/∑n) to the total number of patches (n = 18 patches for NaV1.3 and n = 23 patches for NaV1.7). For display and to facilitate comparison with inactivation curves, data are shown as (1 − [n/∑n]). The cumulative distributions have been fitted to a double Boltzmann function for NaV1.3 and a single Boltzmann function for NaV1.7. [Color figure can be viewed at http://wileyonlinelibrary.com]
The standard extracellular medium for the electrophysiological measurements consisted of (mm): 118 NaCl, 20 tetraethylammonium‐Cl (TEA‐Cl), 5.6 KCl, 1.2 MgCl2, 5 HEPES, 5 d‐glucose and 2 CoCl2 (to block Ca2+ channels; also used at 0.2 and 10 mm final concentrations in the indicated experiments), adjusted to pH 7.4 using NaOH. TTX (Alomone Labs, Jerusalem, Israel) was used at a final concentration of 0.1 μg ml−1, to block endogenous Na+ currents. In the specified experiments BIM23056 (Tocris Bioscience, Bristol, UK) or PMA was present in the extracellular solution at a 100 nm and 10 nm final concentration, respectively. The pipette solution contained (mm): 120 CsCl, 1 MgCl2.6 H2O, 1 CaCl2, 10 EGTA, 10 HEPES) and 3 Mg‐ATP, adjusted to pH 7.15 with CsOH. In the indicated experiments, phosphatidylinositol 4,5‐bisphosphate (PIP2) diC8 (Echelon Bioscience Inc, Salt Lake City, UT, USA) or neomycin (Sigma‐Aldrich) was present in the pipette solution at 50 μm final concentration.
The effect of acute changes in the extracellular glucose concentration were measured in the perforated‐patch configuration, as previously described (De Marinis et al. 2010). Perforation was achieved using amphotericin B (Sigma‐Aldrich), present in the intracellular solution at 0.4 mg ml−1 final concentration. The intracellular pipette solution was composed of (mm): 76 Cs2SO4, 10 NaCL, 10 KCl, 1 MgCl2 and 5 Hepes, adjusted to pH 7.15 with CsOH. The extracellular solution used in the whole‐cell configuration was also used for perforated patch clamp measurements at a final glucose concentration of 1 or 20 mm.
For cell‐attached experiments, the pipette solution had an extracellular composition of (mm): 118 NaCl, 20 tetraethylammonium‐Cl, 5.6 KCl, 1.2 MgCl2, 5 HEPES, 2 CoCl2 and 5 d‐glucose, adjusted to pH 7.4 with NaOH. During seal formation, the cells were immersed in standard extracellular medium containing (mm): 137 NaCl, 5.6 KCl, 10 Hepes (pH 7.4 using NaOH), 1.1 MgCl2 and 2.6 CaCl2. Once the seal was formed, the high K+ extracellular solution was perfused, consisting of (mm): 125 KCl, 1 MgCl2, 1 CaCl2, 10 EGTA, 10 Hepes and 5 d‐glucose, adjusted to pH 7.4 with KOH to depolarize the cell to ∼0 mV (Nernst potential) to allow accurate control of the membrane potential. After recordings were made in the cell‐attached configuration, an extracellular solution with an intracellular ion composition was perfused, ready for patch excision into an inside‐out configuration. The extracellular medium used for the inside‐out patch experiments was identical to the high‐K+ solution specified above except that pH was adjusted to 7.2 and 3 mm MgATP was added. All electrophysiological experiments were performed at 34°C.
For ‘patch cramming’ experiments, Ins1 and HEK cells were cultured in droplets of their respective culture media in the same 35 mm dish. The same solutions that were used for cell‐attached and inside‐out configurations were then applied. However, upon excision of the membrane, the electrode was ‘crammed’ into a neighbouring HEK cell followed by subsequent measurements of voltage‐dependent activation and inactivation (Kramer, 1990).
A standard two‐pulse protocol was used to assess inactivation. The pulse protocol consisted of a 50 ms conditioning pulse of potentials varying from −150 to 0 mV (in increments of 5 mV) followed by a 5 ms test pulse to 0 mV. The peak inward current (I) produced after each depolarization to 0 mV was measured and normalized to the maximum Na+ current amplitude (I max). The data are presented as h ∞ (= I/I max), which was plotted against the conditioning voltage. For each cell, the data points were approximated to a single or a double Boltzmann function to determine the half‐maximal inactivation (V h) and the slope factor (k). The most appropriate fit to the data (single or double Boltzmann) was determined using the Akaike information criterion using Origin software (OriginLab Corp., Northampton, MA, USA). Inactivation curves were fitted to a single Boltzmann function when the fit to a double Boltzmann function resulted in either two V h values with a difference of less than 15 mV or a V h composed of less than 15% of the total current.
Steady‐state activation was assessed by applying a depolarizing pulse, between −70 and +60 mV (in increments of 10 mV) for 20 ms, from a holding potential of −150 mV. The peak inward current (I) produced after depolarization to each voltage step was measured and used to calculate the Na+ conductance (G) by the relation G = I/(V − V r) where V is the membrane potential and V r is the estimated reversal potential, calculated using the Nernst equation from the extra‐ and intracellular Na+ concentrations in the media (Gonoi & Hille, 1987). The sigmoidal curve produced was fitted to a Boltzmann function, which was used to determine the half‐maximal activation (V h) and the slope factor (k).
Data analysis
All data are given as mean values ± SEM of the indicated number of experiments (n). Statistical significances were calculated using Student's t test or ANOVA (for multiple comparisons, as appropriate).
Results
Characterization of TTX‐resistant Na+ channels
To further explore the role of the different Na+ channel αsubunits and their contribution to voltage dependence of inactivation, it was important to isolate the current from individual NaV channel αsubunits. As there are currently no reliable αsubunit‐specific Na+ blockers, we generated TTX‐resistant αsubunits by sitedirected mutagenesis (see Methods) and expressed them in clonal β‐cells and HEK cells. Figure 2 A and B shows Na+ currents recorded from non‐transfected Ins1 and HEK cells during a voltage‐clamp depolarization to 0 mV. All untransfected Ins1 cells contained TTX‐sensitive voltage‐gated Na+ currents (NaV currents; n = 21). By contrast, none of the HEK cells contained any NaV currents. After transfection of the cells with mutant NaV1.7, large TTX‐resistant NaV currents were observed in both Ins1 and HEK cells (Fig. 2 C and D). Similar data were obtained when cells were transfected with TTX‐resistant NaV1.3 and NaV1.6 or wild‐type NaV1.5 – a naturally TTX‐resistant channel (data not shown). It was ascertained in HEK cells that making the NaV1.3, NaV1.6 and NaV1.7 channels TTX resistant did not affect voltage dependence of inactivation (not shown). From here on the channels made resistant to TTX will be referred to simply as NaV1.7, NaV1.3 and NaV1.6.
Figure 2. Endogenous NaV and mutant TTX‐resistant NaV1.7 currents in Ins1 and HEK cells.
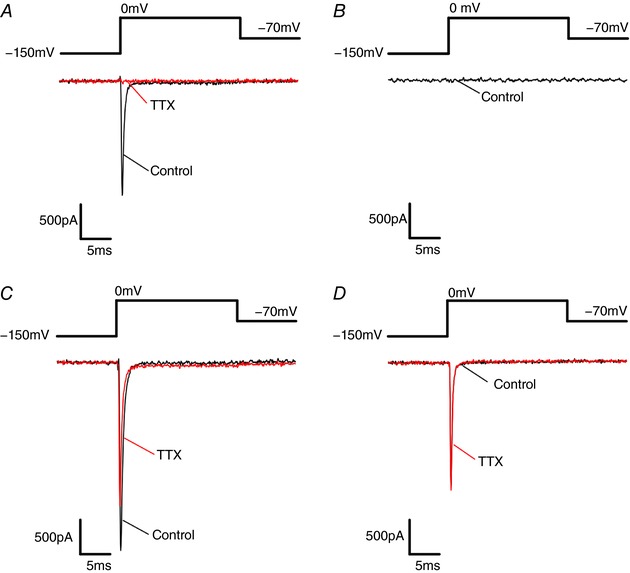
A, endogenous NaV current from Ins1 cells in response to a step depolarization from −150 to 0 mV in the absence (black; control) and presence of TTX (red; TTX treated). B, no endogenous NaV currents were evoked in HEK cells by a step depolarization from −150 to 0 mV. C, NaV currents evoked in response to a step depolarization from −150 to 0 mV in Ins1 cells co‐transfected with mutant TTX‐resistant NaV1.7 and β1‐ and β2‐subunits in the absence (black; control) and presence of TTX (red; TTX treated). The reduction of the peak current induced by TTX reflects block of endogenous NaV current. D, same as in C but expressed in HEK cells. [Color figure can be viewed at http://wileyonlinelibrary.com]
Inactivation of NaV1.3 and NaV1.7 expressed in Ins1 cells
We expressed NaV1.3 or NaV1.7 in Ins1 cells and determined their voltage dependence of activation and inactivation, which were described by fitting single Boltzmann functions to the data points (Table 1). The two types of NaV channel αsubunit exhibited rather different inactivation behaviours, and V h averaged −76 ± 2 (n = 34) and −92 ± 2 mV (n = 47) for NaV1.3 and NaV1.7, respectively (Fig. 3 A). However, it is noticeable that the inactivation of NaV1.7 in Ins1 cells shows some slight deviation from a single Boltzmann function (arrow). Although the inactivation of NaV1.3 appears to be monophasic, inactivation in individual cells was clearly best described using a double Boltzmann fit to the data. This aspect will be addressed further below (Fig. 11).
Table 1.
V h and k values of Nav channel inactivation in Ins1, HEK, CHO and αTC1‐6 cells
| NaV1.7 | NaV1.3 | NaV1.6 | NaV1.5 | |||||
|---|---|---|---|---|---|---|---|---|
| Cell type | V h (mV) | k | V h (mV) | k | V h (mV) | k | V h (mV) | k |
| Ins1 | −92 ± 2 (47) | 10 ± 0.4 (47) | −76 ± 2 (34) | 12 ± 1 (34) | −73 ± 2 (35) | 10 ± 0.3 (35) | −99 ± 2 (9) | 8 ± 0.3 (9) |
| HEK | −66 ± 2 (9) | 8 ± 0.5 (9) | −48 ± 1 (7) | 10 ± 2 (7) | −53 ± 1 (10) | 7 ± 0.4 (10) | −77 ± 1 (10) | 6 ± 0.2 (10) |
| CHO | −61 ± 3 (10) | 9 ± 1 (10) | −50 ± 2 (13) | 7 ± 0.4 (13) | n/a | n/a | n/a | n/a |
| αTC1‐6 | −68 ± 3 (10) | 7 ± 1 (10) | −59 ± 4 (9) | 6 ± 0.5 (9) | n/a | n/a | n/a | n/a |
The V h and k values of NaV α‐subunits co‐expressed with β1‐ and β2‐subunits. Data were fitted to a single Boltzmann function. Values represent means ± SEM of indicated number of cells (n).
Figure 3. Voltage dependence of inactivation and activation of NaV1.7 and NaV1.3 in Ins1 cells.
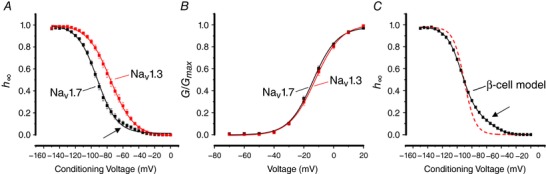
A, voltage dependence of inactivation (h ∞) of NaV1.7 (black; n = 47) and NaV1.3 (red; n = 34) when the α‐subunit is coexpressed with β1‐ and β2‐subunits in Ins1 cells. The curves represent a single Boltzmann fit to the data. Note slight deviation from inactivation at membrane potentials between −70 and −40 mV in Ins1 cells expressing NaV1.7 (arrow). B, voltage dependence of NaV1.7 (black; n = 39) and NaV1.3 (red; n = 31) current activation (G/G max) in Ins1 cells. The curves represent a single Boltzmann fit to the data. C, the black curve represents a model of β‐cell NaV current inactivation behaviour composed of 15% NaV1.3 and 85% NaV1.7, fitted to a double Boltzmann function. Superimposed is a single Boltzmann fit to the model (dashed red). The β‐cell model exhibits a clear shoulder at conditioning membrane potentials of −70 to −40 mV (arrow). See also Table 1. [Color figure can be viewed at http://wileyonlinelibrary.com]
Figure 11. Biphasic inactivation of NaV1.3, 1.6 and 1.7 currents in Ins1 cells.
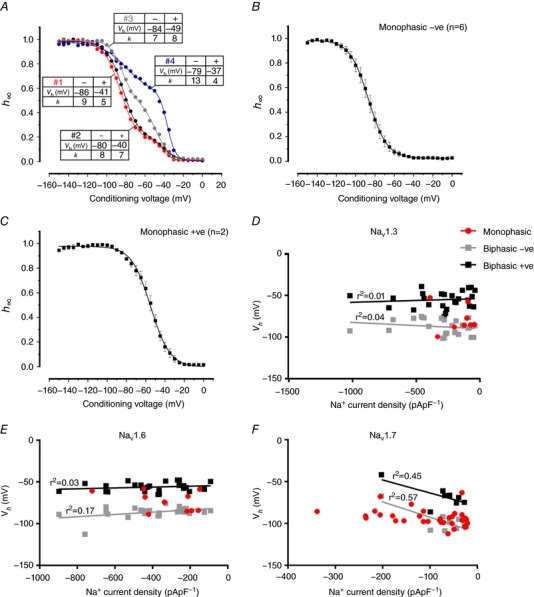
A, examples of biphasic inactivation of NaV1.3 currents when the αsubunit is coexpressed with β1 and β2 subunits in four different Ins1 cells (nos 1–4). The tables next to the curves show V h and k values for the components inactivating at negative (−) and more positive (+) membrane potentials. The curves represent a double Boltzmann fit to the data. B, average voltage dependence of inactivation of NaV1.3 currents in Ins1 that were best described with a single Boltzmann fit to the data and had a V h <−70 mV (n = 6). C, average voltage dependence of inactivation of NaV1.3 current in Ins1 cells that were best described with a single Boltzmann fit to the data that had a V h >−70 mV (n = 2). D, relationship between monophasic, biphasic negative (−ve) and biphasic positive (+ve) V h values and the peak Na+ current density of NaV1.3 in Ins1 cells. The lines represent linear regression fits to the data. The r 2 values are given next to the respective fit. E and F, same as in D but for NaV1.6 (E) and NaV1.7 (F). [Color figure can be viewed at http://wileyonlinelibrary.com]
By contrast, activation of the two α‐subunits was essentially superimposable and the half‐maximal activation is −12 ± 1 (n = 39) and −13 ± 1 mV (n = 31) for NaV1.3 and NaV1.7, respectively (Fig. 3 B).
We have previously proposed that biphasic inactivation of the Na+ current in primary β‐cells reflects the expression of NaV1.3 and NaV1.7 α‐subunits and that the two subunits underlie the positive and negative inactivation components, respectively. We modelled Na+ current inactivation behaviour in a cell containing 15% NaV1.3 and 85% NaV1.7 (the observed relative contribution of the positive and negative inactivation components in primary β‐cells (Zhang et al. 2014). As shown in Fig. 3 C, the resulting inactivation curve thus obtained exhibits a clear shoulder at conditioning membrane potentials of −70 to −40 mV (arrow). We conclude that expression of different α‐subunits may contribute to the biphasic inactivation observed in primary β‐cells. However, as will be explained below, this is not the only contributing factor.
Comparison of NaV1.3, NaV1.5, NaV1.6 and NaV1.7 inactivation in Ins1 and HEK cells
The inactivation of NaV1.7 in Ins1 cells (V h = −93 mV) is more negative than what has been reported previously in HEK cells and in neurones (Herzog et al. 2003; Eberhardt et al. 2014). We next systematically compared inactivation of NaV1.3, NaV1.5, NaV1.6 and NaV1.7 co‐expressed with β1 and β2 in Ins1 and HEK cells (Fig. 4 A–D). We found that for all αsubunits, inactivation (expressed as V h) occurred at membrane potentials 20–30 mV more negative in Ins1 cells than in HEK cells or CHO cells and that inactivation of NaV1.7 occurred at 10–20 mV more negative membrane potentials than NaV1.3 and NaV1.6 regardless of the cell type (Table 1).
Figure 4. Voltage dependence of inactivation of NaV1.7, NaV1.3, NaV1.6 and NaV1.5 currents in Ins1 cells compared to HEK cells.
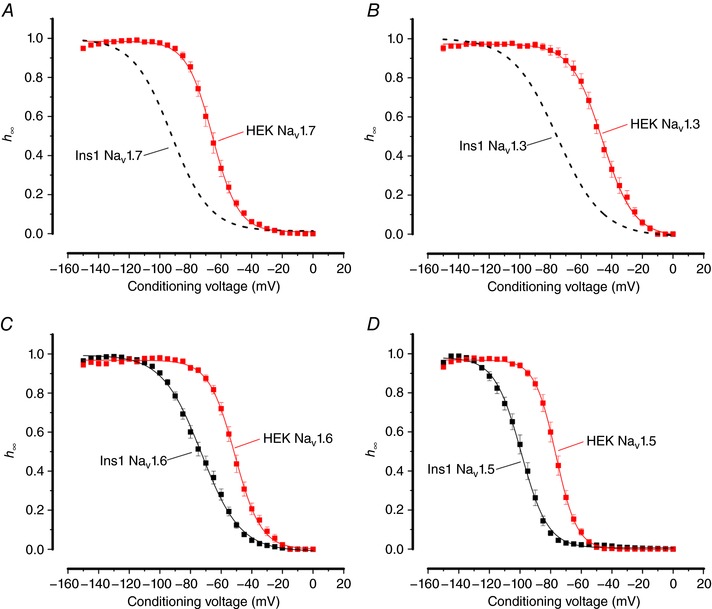
A, voltage dependence of NaV1.7 current inactivation (h ∞) when the α‐subunit is co‐expressed with β1‐ and β2‐subunits in Ins1 (same data as in Fig. 3 A: dashed curve) and HEK cells (red; n = 9). The curve represents a single Boltzmann fit to the data. B, as in A but for NaV1.3 (dashed curve same data as in Fig. 3 A; red, n = 7). C, as in A but for NaV1.6 (black, n = 35; red, n = 10). D, as in A but for NaV1.5 (black, n = 9; red, n = 10). See also Table 1. [Color figure can be viewed at http://wileyonlinelibrary.com]
Inactivation properties of both NaV1.3 and NaV1.7 expressed in CHO cells were similar to that observed in HEK cells (Fig. 5 A and B and Table 1). In the glucagonsecreting cell line αTC1‐6, inactivation of both NaV1.3 and NaV1.7 was more similar to that found in HEK and CHO cells than in the insulin‐secreting cells (Fig. 5 A and B and Table 1).
Figure 5. Voltage dependence of inactivation of NaV1.7 and NaV1.3 currents in αTC1‐6 and CHO cells.
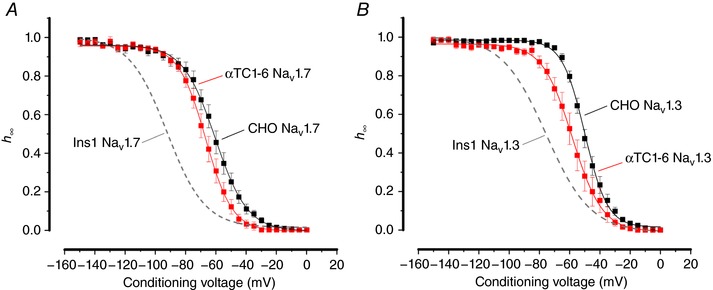
A, voltage dependence of inactivation of NaV1.7 currents when the α‐subunit is coexpressed with β1‐ and β2‐subunits in αTC1‐6 (red; n = 10) and CHO (black; n = 10) cells. The curve represents a single Boltzmann fit to the data and the grey dashed line represents the inactivation of NaV1.7 expressed in Ins1 cells (shown in Fig. 3). B, same as in A, but experiments were conducted with NaV1.3 in αTC16 (red; n = 9) and CHO cells (black; n = 13). See also Table 1. [Color figure can be viewed at http://wileyonlinelibrary.com]
Impact of β‐subunits on NaV inactivation
The NaV channel β‐subunits modulate inactivation of NaV α‐subunits (Cummins et al. 2001; Calhoun & Isom, 2014). Moreover, as the β‐subunits are prone to cell‐specific post‐translational modulation, their effect on NaV inactivation may be cell specific, i.e. the same β‐subunit can have different effects on NaV inactivation in different cell types (Isom et al. 1992, Moran et al. 2000; Meadows & Isom, 2005). To test whether modulation of β‐subunits is the mechanism by which NaV currents in Ins1 cells inactivate at more hyperpolarized potentials compared to other cells types, siRNA knockdown was used to ablate β‐subunit expression in Ins1 cells. The only β‐subunit expressed endogenously in Ins1 cells is the β3subunit (Fig. 6 A). At the mRNA level, 71% knockdown of Scn3b was achieved with no compensatory increase in other β‐subunits (i.e. Scn1b, 2b or 4b). Down‐regulation of Scn3b only marginally affected the inactivation of the endogenous Na+ current in Ins1 cells (Fig. 6 B, Table 2). The inactivation of the endogenous NaV current was clearly biphasic but this was not affected by down‐regulating the β3subunit.
Figure 6. Effect of β‐subunits on NaV current inactivation in Ins1 and HEK cells.
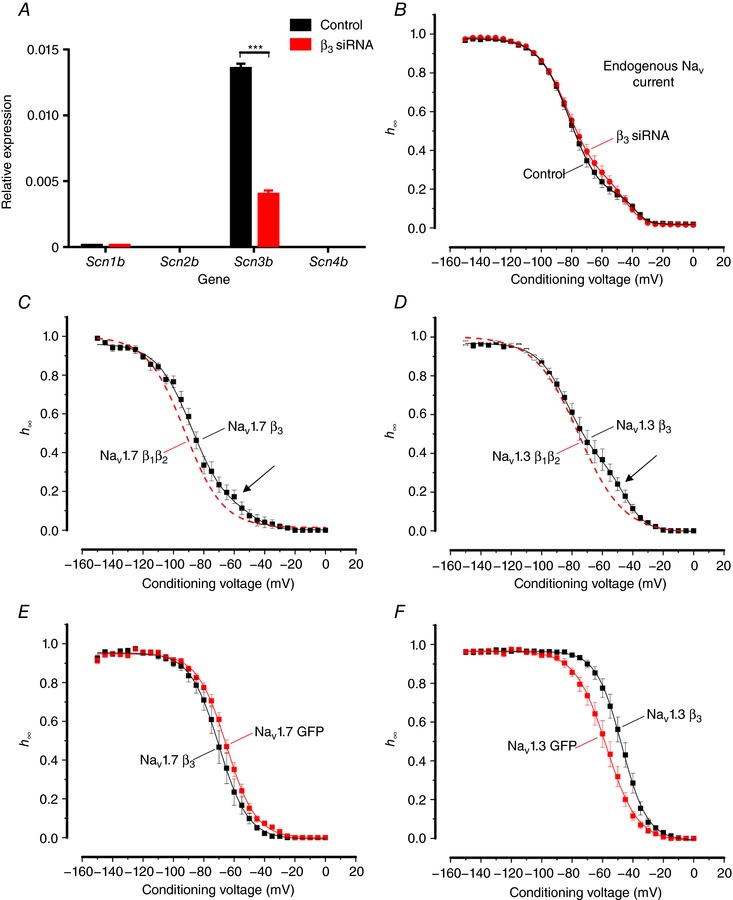
A, relative mRNA expression of Scn1b, Scn2b, Scn3b and Scn4b in control Ins1 cells transfected with a scramble siRNA or β3 siRNA (n = 3 preparations). B, voltage dependence of inactivation (h ∞) of endogenous NaV currents from control (black; n = 22, from 3 preparations) and β3 siRNA treated Ins1 cells (red; n = 26 from 3 preparations). The curves represent a double Boltzmann fit to the data. C, voltage dependence of NaV1.7 current inactivation when the α‐subunit is co‐expressed with β3‐subunit (black; n = 9) in Ins1 cells. Dashed red curve represents voltage dependence of NaV1.7 current inactivation when the α‐subunit is co‐expressed with β1‐ and β2‐subunits in Ins1 cells (data shown in Fig. 3). The curves represent a double Boltzmann fit to the data. Arrow indicates a component of inactivation occurring at positive membrane potentials. D, as in C but for NaV1.3 (black; n = 11). E, voltage dependence of NaV1.7 current inactivation when the αsubunit is coexpressed with β3‐subunit (black; n = 9) or GFP alone (red; n = 12) in HEK cells. The curves represent a single Boltzmann fit to the data. F, as in E, but for NaV1.3 (black, n = 12; red; n = 10). V h and k value for the respective conditions are given in Table 2. [Color figure can be viewed at http://wileyonlinelibrary.com]
Table 2.
Effect of βsubunits on NaV current inactivation in Ins1 and HEK cells
| Monophasic | Biphasic | ||||||
|---|---|---|---|---|---|---|---|
| Cell type | NaV current | V h (mV) | k | V h1 (mV) | k 1 | V h2 (mV) | k 2 |
| Ins1 | Endogenous control | −81 ± 2 (11) | 11 ± 1 (11) | −81 ± 1 (11) | 9 ± 1 (11) | −43 ± 3 (11) | 6 ± 2 (11) |
| Ins1 | Endogenous β3 siRNA | −82 ± 3 (9) | 10 ± 1 (9) | −83 ± 2 (17) | 10 ± 1 (17) | −47 ± 2 (17) | 6 ± 1 (17) |
| Ins1 | NaV1.7 + β3 | −89 ± 2 (8) | 11 ± 1 (8) | −86 (1) | 6 ± 1 (1) | −68 (1) | 14 (1) |
| Ins1 | NaV1.3 + β3 | −72 ± 10 (3) | 9 ± 2 (3) | −84 ± 2 (8) | 11 ± 1 (8) | −46 ± 2 (8) | 5 ± 1 (8) |
| HEK | NaV1.7 + β3 | −70 ± 3 (9) | 8 ± 1 (9) | ||||
| HEK | NaV1.7 + GFP | −66 ± 2 (12) | 9 ± 1 (12) | ||||
| HEK | NaV1.3 + β3 | −48 ± 2 (12) | 7 ± 1 (12) | ||||
| HEK | NaV1.3 + GFP | −59 ± 3 (10) | 8 ± 1 (10) | ||||
Values represent means ± SEM of indicated number of cells (n). See Fig. 6. V h represents membrane potential at which inactivation is half‐maximal in cells with monophasic inactivation and k represents the slope factor. In cells with biphasic inactivation, V h1 and V h2 represent the membrane potential at which inactivation is half‐maximal for the current components inactivating at negative and positive membrane potentials, respectively. k 1 and k 2 represent the respective slope factors for the currents inactivating with V h1 and V h2.
We also compared the inactivation of the expressed human NaV1.3 and NaV1.7 when co‐expressed with β3 (rather than β1 and β2, as in previous experiments) in Ins1 cells (Fig. 6 C and D). In both cases, overexpression of the β3‐subunit made biphasic inactivation more apparent (arrows).
Finally, we compared NaV1.3 and NaV1.7 inactivation when co‐expressed with or without β3 in HEK cells. Whereas the inactivation of NaV1.7 was shifted by 4 mV towards more negative voltages in the presence of β3, inactivation of NaV1.3 was shifted 11 mV towards more depolarized membrane potentials (Fig. 6 E and F). It was ascertained that HEK cells do not express any endogenous βsubunits (not shown).
Collectively, these data suggest that the widely different inactivation behaviours of NaV1.3 and NaV1.7 when expressed in Ins1 and HEK cells cannot be attributed to any of the differential modulation of βsubunits in a cell‐specific manner, expression of a different endogenous β‐subunit complement in Ins1 cells or the αsubunits forming heterodimers with different β‐subunits (in which case overexpression of β3 should have made biphasic inactivation less apparent). Rather the data suggest that the hyperpolarized inactivation observed in β‐cells is due to a direct effect on the NaV channel α‐subunit. Moreover, we conclude that the biphasic inactivation of the endogenous Na+ current in Ins1 cells need not reflect the presence of multiple α‐subunits as biphasic inactivation was also observed in the experiments involving expression of NaV1.3 and NaV1.7 alone.
Negative NaV inactivation in Ins1 cells is not due to cytosolic diffusible factor
We hypothesized that the inactivation of NaV1.3 and NaV1.7 is shifted towards more negative membrane potentials in Ins1 cells (and mouse β‐cells) compared to other cells (dorsal root ganglion neurones, HEK, CHO, αTC1‐6) because of the presence of a cytoplasmic factor in β‐cells that is not present in other cells. To explore this possibility, NaV1.3 and NaV1.7 were expressed in Ins1 cells and CHO cells and inactivation was measured in cell‐attached and subsequently in inside‐out patch configuration (to promote rapid ‘wash‐out’ of any attached modulators). These experiments required channel expression high enough to allow measurements of macroscopic (‘whole‐cell’) currents in cell‐attached patches (Fig. 7 A and B).
Figure 7 C shows inactivation curves for NaV1.3 currents recorded in cell‐attached patches and at various times after excision and formation of inside‐out patches in Ins1 cells. In the cell‐attached configuration, V h for NaV1.7 was −92 ± 3 mV (n = 23) and −74 ± 4 mV (n = 15) in Ins1 and CHO cells, respectively. The corresponding values for NaV1.3 were −69 ± 2 (n = 18) and −56 ± 5 mV (n = 6) in Ins1 and CHO cells, respectively. We acknowledge that these values are not identical to those measured in the wholecell configuration. In the cell‐attached experiments, the membrane potential of the cell was assumed to be 0 mV when the cells were immersed in the high‐[K+]o medium, but it is possible that a slight voltage difference remains. This idea is supported by the observation that following excision of the patches, there was an immediate ∼10 mV shift in inactivation in the hyperpolarizing direction for both NaV1.3 and NaV1.7 expressed in either cell type.
In the cell‐attached patches, inactivation of NaV1.3 was invariably monophasic. However, the distribution at which NaV1.3 inactivated in the cell‐attached configuration appears to have two distinct components. Figure 7 F shows the normalized cumulative distribution of V h measured in 18 membrane patches (each represented by a single point) in Ins1 cells expressing NaV1.3. The continuous curve represents a double Boltzmann fit to data points with V h values of −74 mV and −58 mV. Five of the 18 patches (28%) had V h values more positive than −60 mV. For NaV1.7 (black squares and line), the cumulative distribution for 23 patches was essentially monophasic with a V h of −93 mV with only two patches (9%) showing a V h at membrane potentials more positive than −65 mV.
If there is a diffusible modulator of NaV inactivation present in β‐cells that shifts inactivation, then patch excision would result in a positive shift of inactivation (i.e. towards that observed in CHO cells). As already remarked, patch excision resulted in an immediate ∼10 mV shift of inactivation towards more hyperpolarized membrane potentials for both NaV1.3 and NaV1.7 and for both Ins1 and CHO cells. Following patch excision there was then a time‐dependent additional and parallel negative shift of V h in both cell types until V h eventually settled at −120 mV in Ins1 cells and −100 mV in CHO cells (Fig. 7 D). Similar time‐ and cell‐dependent changes were observed for NaV1.3 channels (Fig. 7 E). If diffusible factors modulating NaV current inactivation were present in Ins1, then the curves would have been expected to converge. In addition, there was a time‐dependent decrease in current amplitude. We attribute this to rundown of channel activity, a process observed for many types of channels (Becq, 1996). Importantly, the shift in inactivation was nearly maximal 2 min after patch excision when effects on current kinetics were moderate (compare red and blue traces in Fig. 7 A and B).
Activation of the NaV1.3 and NaV1.7 channels underwent similar time‐dependent changes in the hyperpolarizing direct following patch excision (not shown).
We also tested the alternative possibility that inactivation is more negative in Ins1 cells because something present in all other cells is missing in these cells. This was tested by an approach similar to that in Fig. 7 but instead of leaving the patch in the bath medium after excision, the patch was ‘crammed’ (Kramer, 1990) into a neighbouring HEK cell (Fig. 8 A). However, as shown in Fig. 8 B and C, the time‐dependent shift of V h towards more negative membrane potentials persisted and was as pronounced after cramming the electrode into a HEK cell as when the patch was left in the bath medium; inactivation still underwent a 25 mV shift towards more negative membrane potentials 5–10 min after patch excision and cramming into the neighbouring HEK cell (Fig. 8 D).
Figure 8. NaV1.7 expressed in Ins1 cells crammed into HEK cells.
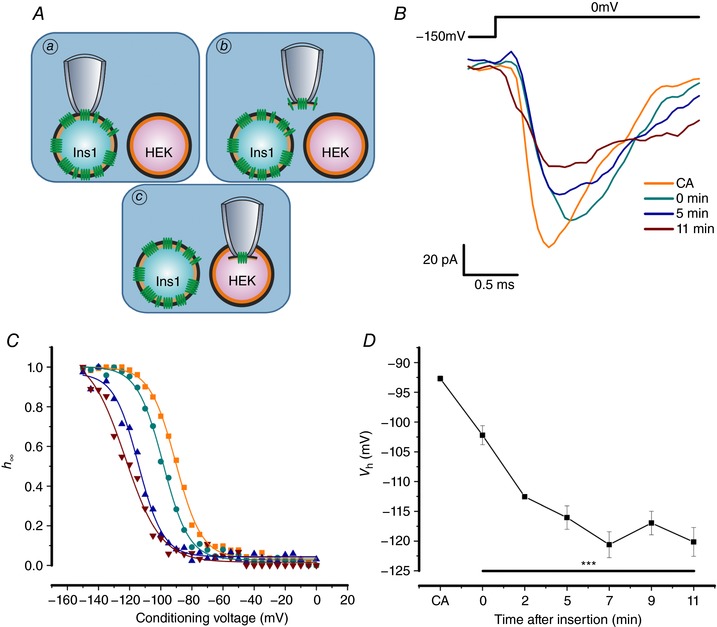
A, schematic representation of patch cramming procedure. NaV1.7 channels were expressed in Ins1 cells and a cell‐attached patch was formed (a); the patch of membrane was excised (b) and immediately crammed into HEK cells (c). B, NaV1.7 currents when the α‐subunit is co‐expressed with β1‐ and β2subunits in response to a step depolarization from −150 to 0 mV in cell‐attached (CA) configuration in Ins1 cells, immediately after excision and cramming into HEK cells (0 min) and 5 and 11 min after insertion of the membrane into HEK cells. C, voltage dependence of inactivation of NaV1.7 currents shown in B with same colour coding. D, summarized voltage dependence of inactivation of NaV1.7 currents in cell‐attached configuration and after cramming into HEK cells (n = 4). *** P < 0.001 for each time point compared to CA, using a one‐way ANOVA. [Color figure can be viewed at http://wileyonlinelibrary.com]
Cytoplasmic domains of NaV1.3 and NaV1.7 do not confer different inactivation behaviours
The data of Figs 7 and 8, suggest that the differences in inactivation of NaV channels between Ins1 and HEK and CHO cells is unlikely to be attributable to freely diffusible factors present in the cytosol of either cell type. However, we acknowledge that the change in inactivation may result from firmer interaction between the NaV αsubunits and another factor (protein) at the time they are inserted into the plasmalemma.
The schematic diagram in Fig. 9 A shows the topology of the NaV1.7 channels and highlights the areas of divergence in the amino acid sequence between NaV1.3 and NaV1.7. As indicated, the greatest differences are found in the N‐terminus, the C‐terminus and the cytoplasmic loops L1 and L2. We hypothesized that the sequence variations contribute both to differences in inactivation between (i) NaV1.3 and NaV1.7 and (ii) Ins1 and HEK cells. We acknowledge that this assumes that the differences in the intracellular/cytoplasmic milieu are transduced via the cytoplasmic domains of the channels. It follows from these premises that substituting the cytoplasmic domains of NaV1.7 for NaV1.3 will reduce the differences in V h between the two αsubunits (i.e. change V h from −92 mV to −76 mV in Ins1 cells; Table 1).
Figure 9. NaV1.7–NaV1.3 chimeras expressed in Ins1 and HEK cells.
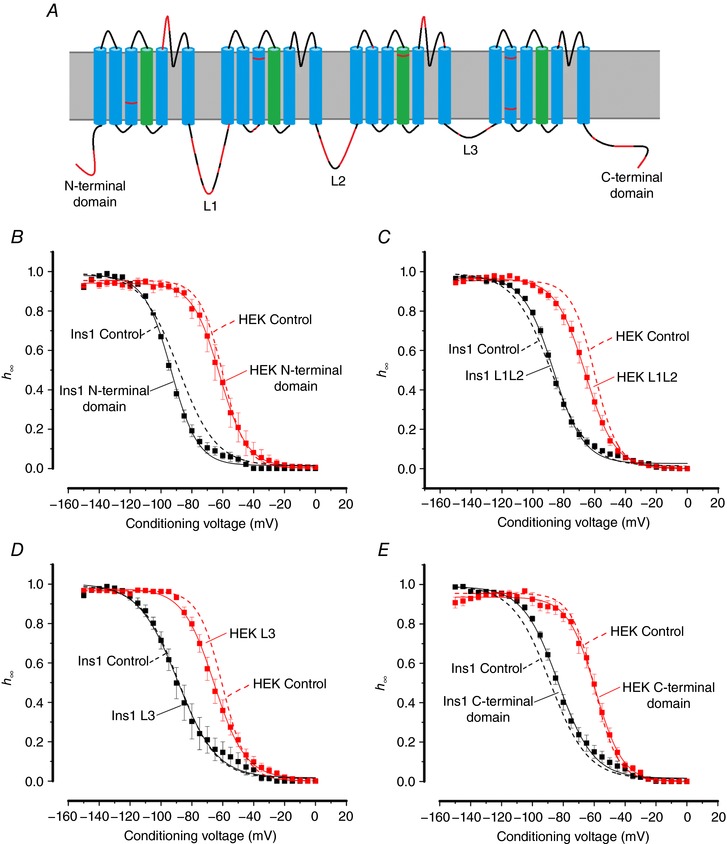
A, schematic representation of the structure of the NaV1.7 channel. Highlighted in red are the areas of divergence in the amino acid sequence to NaV1.3. Sequence alignment was performed using the Clustal Omega program (http://www.uniprot.org/align/), using the NCBI reference sequences NP_002968.1 and NP_008853.3 for human NaV1.7 and NaV1.3, respectively. B, voltage dependence of inactivation of N‐terminal chimera currents when the α‐subunit is co‐expressed with β1‐ and β2‐subunits in Ins1 (black; n = 9) and HEK cells (red; n = 6); and control TTX‐resistant NaV1.7 currents when the α‐subunit is co‐expressed with β1‐ and β2subunits in Ins1 (dashed black line; n = 37) and HEK cells (dashed red line; n = 6). C, as in B but for L1L2 chimera currents (black, n = 15; red, n = 9) with the same controls as in B. D, as in B but for L3 chimera currents (black, n = 16; red, n = 11) with the same controls as in B. E, as in B but for C‐terminal chimera currents (black, n = 7; red, n = 8) with the same controls as in B. V h and k values are given in Table 3. [Color figure can be viewed at http://wileyonlinelibrary.com]
We generated chimeric channels based on the NaV1.7 backbone by replacing the N‐terminal domain, L1 and L2, the C‐terminal domain or L3 sequences with the corresponding sequences of NaV1.3 and analysed their properties in Ins1 (black) and HEK cells (red) (Fig. 9 B–E: see also Table 3). For comparison, the inactivation curves for wild‐type NaV1.7 in Ins1 and HEK cells are also shown (dashed black and red lines). None of the substitutions significantly affected NaV1.7 inactivation. Taken together with the data of Figs 7 and 8, it seems unlikely that the negative shift is caused by an intracellular factor interacting with the cytoplasmic domains of the NaV1.7 channels.
Table 3.
NaV1.7–NaV1.3 chimeras expressed in Ins1 and HEK cells
| Monophasic | Biphasic | ||||||
|---|---|---|---|---|---|---|---|
| Cell type | NaV current of NaV1.7–1.3 chimeras | V h (mV) | k | V h1 (mV) | k 1 | V h2 (mV) | k 2 |
| Ins1 | NaV1.7 Control | −90 ± 2 (28) | 9 ± 1 (28) | −94 ± 4 (9) | 9 ± 1 (9) | −63 ± 5 (9) | 8 ± 1 (9) |
| Ins1 | Nterminal domain | −95 ± 1 (7) | 7 ± 1 (7) | −98 ± 1 (2) | 6 ± 1 (2) | −67 ± 1 (2) | 12 ± 2 (2) |
| Ins1 | L1L2 | −88 ± 1 (12) | 9 ± 1 (12) | −86 ± 1 (3) | 7 ± 1 (3) | −49 ± 5 (3) | 13 ± 7 (3) |
| Ins1 | L3 | −96 ± 3 (12) | 8 ± 1 (12) | −98 ± 3 (4) | 9 ± 2 (4) | −50 ± 5 (4) | 9 ± 2 (4) |
| Ins1 | C‐terminal domain | −86 ± 1 (5) | 9 ± 1 (5) | −84 ± 1 (2) | 10 ± 1 (2) | −50 ± 6 (2) | 10 ± 3 (2) |
| HEK | NaV1.7 Control | −60 ± 1 (6) | 8 ± 1 (6) | n/a | n/a | n/a | n/a |
| HEK | N‐terminal domain | −61 ± 4 (6) | 8 ± 1 (6) | n/a | n/a | n/a | n/a |
| HEK | L1L2 | −66 ± 2 (9) | 9 ± 1 (9) | n/a | n/a | n/a | n/a |
| HEK | L3 | −67 ± 3 (11) | 12 ± 1 (11) | n/a | n/a | n/a | n/a |
| HEK | C‐terminal domain | −60 ± 2 (8) | 9 ± 1 (8) | n/a | n/a | n/a | n/a |
V h and k value for the indicated NaV currents. Values represent means ± SEM of indicated number of cells (n). Inactivation curves are presented in Fig. 9. V h represents membrane potential at which inactivation is half‐maximal in cells with monophasic inactivation and k represents the slope factor. In cells with biphasic inactivation, V h1 and V h2 represent the membrane potential at which inactivation is half‐maximal for the current components inactivating at negative and positive membrane potentials, respectively. k 1 and k 2 represent the respective slope factors for the currents inactivating with V h1 and V h2. See Fig. 1 for generation of constructs.
Biphasic inactivation of endogenous NaV currents in Ins1 cells
We examined the expression of NaV channel αsubunits in Ins1 cells and found that these cells express high levels of Scn3a (NaV1.3) and low levels of Scn2a (NaV1.2) and Scn8a (NaV1.6) (Fig. 10 A). We measured the inactivation of the endogenous NaV currents, and the average inactivation curve (n = 24) is shown in Fig. 10 B. We found that whereas inactivation was monophasic in 50% of the cells (12 of 24 cells) with a V h of −88 ± 2 mV (Fig. 10 C), it was clearly biphasic in the remaining cells (n = 12; Fig. 10 D). In this subgroup of cells, the major component (comprising 71 ± 6%) was similar to that in cells with monophasic inactivation and had a V h of −87 ± 3 mV. In addition, there was a smaller component that accounted for 29 ± 6% of the total with a V h of −43 ± 2 mV. This characteristic inactivation appears to be unique to βcells because αTC1‐6 cells, which also express NaV1.2, NaV1.3, NaV1.6 and NaV1.7, displayed only a single component of inactivation with a V h of −55 ± 4 mV (n = 6; Fig. 10 E and F).
Figure 10. Characterization of endogenous NaV currents in Ins1 and αTC1‐6 cells.
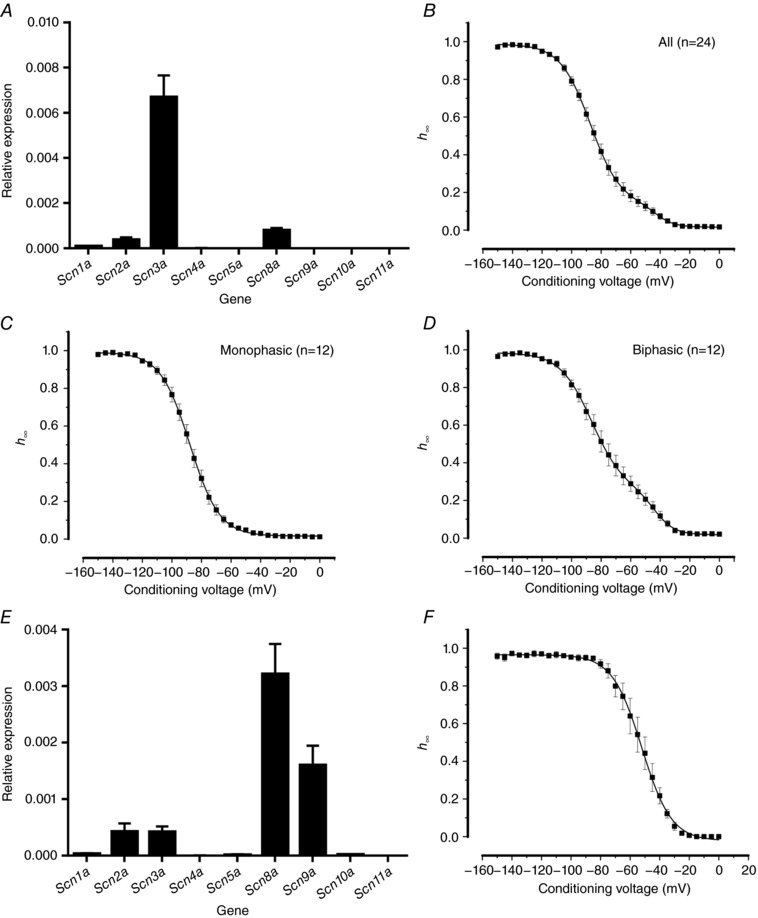
A, relative mRNA expression of NaV channel α‐subunits in Ins1 cells (n = 2 preparations). B, voltage dependence of inactivation of endogenous Ins1 NaV currents, average of all cells (n = 24). Curve represents a double Boltzmann fit to the data. C, as in B but for cells with NaV inactivation best fitted to a single Boltzmann function (i.e. had a monophasic inactivation; n = 12). D, as in C but for cells where inactivation was best fitted to a double Boltzmann function (i.e. had a biphasic inactivation; n = 12). E, relative mRNA expression of NaV channel α‐subunits in αTC1‐6 cells (n = 3 preparations). F, voltage dependence of inactivation of endogenous αTC1‐6 NaV currents (n = 6).
Although we could not rule out that the expression of α‐subunits other than NaV1.3 explains the biphasic inactivation behaviour in Ins1 cells, we think that this is unlikely given the very low expression of Scn2a and Scn8a and we instead hypothesize that NaV1.3 channels expressed in the same cell may undergo inactivation with different voltage dependences.
Figure 11 A shows inactivation in four different Ins1 cells in which inactivation of expressed NaV1.3 channels was clearly biphasic with values of V h ranging between −86 and −37 mV. Such cells accounted for 70% of all cells tested. In these 26 cells (of a total of 34 cells), the negative and positive components of inactivation contributed 57 ± 5% and 43 ± 5% of the total current, respectively. The corresponding values of V h were −87 ± 2 and −55 ± 2 mV (Table 4). In addition, there were cells in which inactivation was monophasic (n = 8; Table 4): in most of these cells, inactivation occurred at negative voltages (V h = −90 mV) (Fig. 11 B) but in two cells inactivation was instead at positive voltages (V h = −50 mV) (Fig. 11 C). Similarly, in cells expressing NaV1.6, 25 cells out of a total of 35 cells exhibited biphasic inactivation (Table 4). For NaV1.7, biphasic inactivation was observed in 9 out of 47 cells (P < 0.001 by χ2 vs. both NaV1.3 and NaV1.6). None of the αsubunits showed biphasic inactivation when expressed in HEK, CHO or αTC16 cells.
Table 4.
Monophasic and biphasic inactivation of NaV1.7, 1.3 and 1.6 in Ins1 cells
| Monophasic | Biphasic | |||||
|---|---|---|---|---|---|---|
| NaV current | V h (mV) | k | V h1 (mV) | k 1 | V h2 (mV) | k 2 |
| NaV1.3 | −79 ± 6 (8) | 9 ± 1 (8) | −87 ± 2 (26) | 9 ± 1 (26) | −55 ± 2 (26) | 7 ± 1 (26) |
| NaV1.6 | −73 ± 4 (10) | 8 ± 1 (10) | −86 ± 1 (25) | 8 ± 1 (25) | −56 ± 1 (25) | 6 ± 1 (25) |
| NaV1.7 | −94 ± 2 (38) | 9 ± 1 (38) | −97 ± 4 (9) | 8 ± 1 (9) | −67 ± 4 (9) | 10 ± 2 (9) |
Same data as in Table 1, except cells are divided between those that exhibited NaV current inactivation that were best described using a single Boltzmann function (monophasic) and those that were best described using a double Boltzmann function (biphasic) (Akaike information criterion test). Values represent means ± SEM of indicated number of cells (n). V h represents membrane potential at which inactivation is half‐maximal in cells with monophasic inactivation and k represents the slope factor. In cells with biphasic inactivation, V h1 and V h2 represent the membrane potential at which inactivation is half‐maximal for the current components inactivating at negative and positive membrane potentials, respectively. k 1 and k 2 represent the respective slope factors for the currents inactivating with V h1 and V h2.
It is worth noticing that the fraction of cells showing biphasic Na+ current inactivation was not increased by substitution of cytoplasmic NaV1.7 for NaV1.3 domains (Table 3).
Figure 11 D shows the relationship between Na+ current density and V h of the negative and positive components fitted to a linear regression with r 2 values of 0.04 and 0.01, respectively. It is clear that there is no impact of current density on V h. However, there are clearly two distinct groups that inactivate at distinct membrane potentials, separated by approximately 30 mV. Interestingly, cells that displayed monophasic inactivation fall into either of these distinct groups. Similar data were obtained for NaV1.6 and NaV1.7 channels expressed in Ins1 cells (Fig. 11 E and F).
Modulation of inactivation
Protein kinase C (PKC) phosphorylation of NaV1.7 exhibits a depolarizing effect on the voltage dependence of inactivation in some (Tan et al. 2014) but not all cell types (Vijayaragavan et al. 2004). Moreover, NaV1.3 and NaV1.6 contain a putative PKC phosphorylation site in the same L3 region. We therefore tested whether there was acute activation or inhibition by PKC of NaV1.7 inactivation by application of PMA (10 nm) or the inhibitor BIM23056 (100 nm). However, neither compound affected NaV1.7 inactivation (Fig. 12 A and Table 5). Down‐regulation of PKC (by long‐term exposure to PMA) was likewise without effect on inactivation of endogenous NaV currents in Ins1 cells (Fig. 12 B) and it did not affect the biphasic pattern of inactivation. Consistent with earlier reports in the rat insulinoma RINm5F (Rorsman et al. 1986), chronic exposure to PMA reduced the amplitude of the NaV current (not shown).
Figure 12. Modulation of NaV inactivation in Ins1 cells.
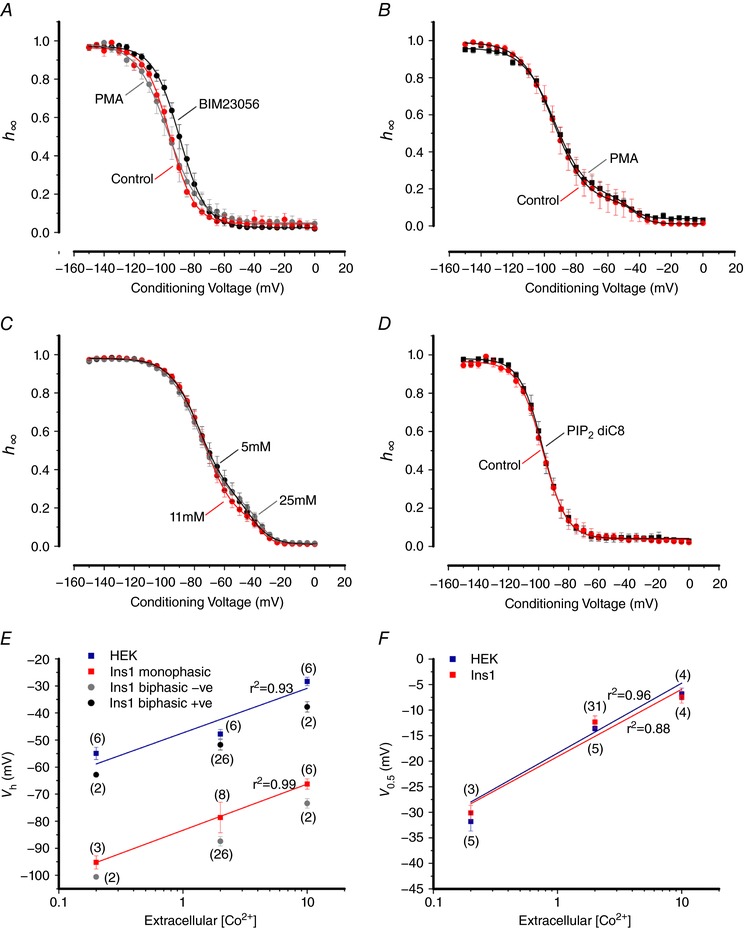
A, voltage dependence of inactivation of NaV1.7 α‐subunit co‐expressed with β1‐ and β2‐subunits in control (red; n = 5), 100 nm BIM23056 treated (black; n = 5) and 10 nm PMA treated (grey; n = 4) Ins1 cells. The curves represent a single Boltzmann fit to the data. B, voltage dependence of inactivation of endogenous NaV currents in control Ins1 cells (red; n = 4) and Ins1 cells chronically (48 h) treated with 10 nm PMA (black; n = 11). The curves represent a double Boltzmann fit to the data. C, voltage dependence of inactivation of endogenous NaV currents in Ins1 cells in response to a 48 h chronic incubation in 5 mm (black; n = 22), 11 mm (red; n = 22) and 25 mm (grey; n = 18) glucose. D, same as in A, but for control (red; n = 5) and 50 μm PIP2 diC8 treated (black; n = 5) Ins1 cells. E, summarized voltage dependence of inactivation of NaV1.3 currents in Ins1 (red, grey and black) and HEK cells (blue) of indicated number of cells (n) at increasing concentrations of extracellular Co2+ (0.2, 2 and 10 mm). The lines represent linear regression fits to the data. The r 2 values are given next to the respective fit. F, same as in E but for the voltage dependence of activation of NaV1.3 currents in Ins1 (red) and HEK cells (blue). [Color figure can be viewed at http://wileyonlinelibrary.com]
Table 5.
Modulation of NaV inactivation in Ins1 cells
| Monophasic | Biphasic | ||||||
|---|---|---|---|---|---|---|---|
| NaV current | Treatment | V h (mV) | k | V h1 (mV) | k 1 | V h2 (mV) | k 2 |
| NaV1.7 | Control | −96 ± 1 (5) | 8 ± 1 (5) | n/a | n/a | n/a | n/a |
| NaV1.7 | 100 nm BIM23056 | −90 ± 2 (5) | 8 ± 1 (5) | n/a | n/a | n/a | n/a |
| NaV1.7 | 10 nm PMA | −96 ± 4 (4) | 10 ± 1 (4) | n/a | n/a | n/a | n/a |
| Ins1 endogenous | Control | −94 ± 6 (2) | 10 ± 1 (2) | −94 ± 1 (2) | 10 ± 1 (2) | −45 ± 5 (2) | 4 ± 1 (2) |
| Ins1 endogenous | 10 nm PMA (chronic) | −91 ± 2 (6) | 11 ± 1 (6) | −92 ± 2 (5) | 10 ± 1 (5) | −46 ± 2 (5) | 5 ± 1 (5) |
| Ins1 endogenous | 5 mm glucose | −70 ± 3 (16) | 9 ± 1 (16) | −80 ± 4 (6) | 9 ± 1 (6) | −41 ± 3 (6) | 5 ± 1 (6) |
| Ins1 endogenous | 11 mm glucose | −74 ± 2 (13) | 9 ± 1 (13) | −73 ± 6 (9) | 9 ± 1 (9) | −43 ± 4 (9) | 5 ± 1 (9) |
| Ins1 endogenous | 25 mm glucose | −72 ± 4 (7) | 9 ± 1 (7) | −78 ± 2 (11) | 11 ± 1 (11) | −39 ± 2 (11) | 5 ± 2 (11) |
| NaV1.7 | Control | −99 ± 1 (5) | 7 ± 1 (5) | n/a | n/a | n/a | n/a |
| NaV1.7 | 50 μm PIP2 diC8 | −97 ± 2 (5) | 7 ± 1 (5) | n/a | n/a | n/a | n/a |
| NaV1.7 | Control | −98 ± 3 (7) | 11 ± 2 (7) | n/a | n/a | n/a | n/a |
| NaV1.7 | 50 μm neomycin | −98 ± 1 (7) | 8 ± 1 (7) | n/a | n/a | n/a | n/a |
V h and k value for the indicated NaV currents and treatment conditions. Values represent means ± SEM of indicated number of cells (n). Inactivation curves are presented in Fig. 12. V h represents membrane potential at which inactivation is half‐maximal in cells with monophasic inactivation and k represents the slope factor. In cells with biphasic inactivation, V h1 and V h2 represent the membrane potential at which inactivation is half‐maximal for the current components inactivating at negative and positive membrane potentials, respectively. k 1 and k 2 represent the respective slope factors for the currents inactivating with V h1 and V h2.
NaV current inactivation has been reported to be regulated by the intracellular ATP concentration (Zou et al. 2013) suggestive of metabolic regulation. We therefore tested the effect of metabolic regulation by culturing cells at 5, 11 or 25 mm glucose for 48 h. No effects on the inactivation of endogenous NaV currents was observed (Fig. 12 C). We also tested the acute effects of elevating glucose from 1 to 20 mm on NaV1.3 and NaV1.7 currents, but no effects on inactivation were observed over 10 min in perforated patch recordings (not shown).
Insulin‐secreting cells will be exposed to high concentrations of insulin (a biologically very active molecule). We tested the potential long‐term role of insulin signalling by treating Ins1 cells expressing NaV1.7 with KATP channel activator diazoxide (100 μm for 48 h) or a receptor antagonist S961 (1 μm for 24 h) (Schaffer et al. 2008) to inhibit its release or its action, respectively. Again, no impact on NaV1.7 inactivation was observed.
It has been proposed that β‐cells contain higher levels of PIP2 than other cells and that this, via modulation of the ATP sensitivity, explains how KATP channels remain active in intact β‐cells (Baukrowitz et al. 1998; Shyng & Nichols, 1998). We therefore considered the possibility that differences in PIP2 content could underlie the differences in NaV1.7 inactivation in Ins1 and HEK cells but inclusion of PIP2 diC8 (50 μm) into the pipette solution was without effect on NaV1.7 inactivation (Fig. 12 D). Likewise, neomycin (50 μm), which would shield the negative charges of PIP2 (MacGregor et al. 2002; Bista et al. 2015), was without effect on NaV1.7 expressed in Ins1 cells (not shown). Thus, differences in PIP2 concentrations in Ins1 and HEK cells is unlikely to cause of the different NaV current inactivation properties.
Next we tested whether there was a difference in surface charge between Ins1 and HEK cells that might influence NaV channel inactivation. One major source of negative charge is due to glycosylation of extracellular NaV channel domains that consist of the carbohydrate derivative Nacetylneuraminic acid or sialic acid (Ednie & Bennett, 2012). To explore differences in surface charge we generated inactivation curves for NaV1.3 in the presence of 0.2, 2 and 10 mm extracellular Co2+ in Ins1 and HEK cells (Bennett et al. 1997; Bennett, 2002). Figure 12 E shows the relationship between the extracellular Co2+ concentration and V h in Ins1 and HEK cells. However, increasing the Co2+ concentration produces the same ∼25 mV shift in Ins1 and HEK cells but the difference between the two cells persists (Fig. 12 E). It is also evident that both the positive and negative components of NaV1.3 inactivation in Ins1 cells are shifted by ∼25 mV and that V h values for the positive components superimpose on those obtained in HEK cells for all Co2+ concentrations. Collectively, these data suggest that it is unlikely that differences in extracellular surface charges account for the negative NaV current inactivation in Ins1 cells. We also analysed the impact of increasing Co2+ on the voltage dependence of activation (V 0.5) of NaV1.3 currents. Notably, there is no difference in the V 0.5 values at any of the Co2+ concentrations and there was a ∼20 mV shift between 0.2 and 10 mm Co2+.
Biphasic inactivation of NaV1.3 in primary β‐cells
Finally we expressed NaV1.3 in primary mouse β‐cells. Although the transfection rate was low, large TTX‐resistant NaV1.3 currents were observed in three β‐cells (Fig. 13 A). The average current amplitude was −1 ± 0.1 nA. These currents underwent voltage‐dependent inactivation with a V h of −88 mV. However, in two of the three cells, there was biphasic inactivation with a small (43% and 22%) component that inactivated at −95 and −103 mV and a larger (57% and 78%) component that inactivated at −73 mV in both cells.
We have previously reported that ablation of Scn3a leads to the loss of NaV current components that inactivate at positive membrane potentials (Zhang et al. 2014). We have now reanalysed these data and expressed current amplitudes in absolute (in pA) rather than relative (normalized to maximum current) terms. Figure 13 B shows NaV currents recorded in control (Scn3a+/−) and knockout Scn3a−/− β‐cells. In agreement with the previous conclusion, ablation of Scn3a was associated with the loss of the NaV current inactivating at positive membrane potentials (Fig. 13 B, shaded area). However, there was also a large reduction (∼200 pA) of NaV current at negative membrane potentials. Figure 13 C shows the net NaV1.3 current isolated by subtracting the currents in Scn3a −/− from those measured in Scn3a +/− mice. The net current shows biphasic inactivation and can be described as the sum of two Boltzmann functions with values of V h of −86 and −57 mV that comprised 76% and 24%, respectively. These values are close to those observed for the isolated NaV1.3 current and similar to the two components of inactivation observed in Ins1 cells expressing NaV1.3 (Table 4).
Discussion
We have compared the inactivation of NaV channels in insulin‐secreting Ins1 and three other non‐β‐cell types (HEK, CHO and αTC1‐6 cells). To this end, we generated mutant TTX‐resistant NaV channels, which allowed us to isolate the expressed channels by blocking the endogenous NaV channels with TTX.
NaV channels inactivate at negative membrane potentials in Ins1 cells
We showed that NaV channels inactivate at ∼30 mV more negative membrane potentials when expressed in Ins1 cells compared to what is seen in HEK cells and dorsal root ganglion neurones (Herzog et al. 2003; Eberhardt et al. 2014). We also confirmed that inactivation of NaV1.7 occurs at more negative membrane potentials than NaV1.3, NaV1.5 and NaV1.6 when these channels are expressed in Ins1 cells. Thus, although the NaV α‐subunit expressed makes a significant difference to voltage dependence of inactivation, it appears that there is something special about Ins1 cells (echoing what is observed in primary β‐cells) that shifts inactivation to functionally irrelevant membrane potentials.
Individual NaV subtypes exhibit two distinct inactivation behaviours
In >75% of the experiments with NaV1.3, two distinct components of inactivation (each accounting for – on average – ∼50% of the total current) separated by ∼30 mV were observed. Biphasic inactivation was also observed for NaV1.7 (in 20% of experiments) and NaV1.6 (60%). Importantly, biphasic inactivation was not observed when these channels were instead expressed in HEK, CHO or αTC16 cells. It should also be noted that the voltage dependence for the component inactivating at more depolarized membrane potentials is very similar to that found in HEK or CHO cells (in which biphasic inactivation was never observed). In cells that showed monophasic inactivation, it proceeded at either positive or negative voltages but not in between. Likewise, when the measurements were done in cell‐attached patches, inactivation again tended to proceed at either negative or positive voltages but was never biphasic.
It appears that the data obtained in Ins1 cells can be extended to primary β‐cells and the net current that can be isolated by subtracting the currents recorded from the NaV1.3‐deficient cells from control cells likewise exhibited a biphasic voltage dependence of inactivation. Thus, it appears that although different αsubunits contribute to the biphasic inactivation, there is also an additional cellspecific modulation whereby a single αsubunit may exhibit two distinct voltage dependences of inactivation.
Negative inactivation does not result from a diffusible factor
We explored the possibility that inactivation at negative membrane potentials reflects the interaction between the NaV αsubunits and an intracellular diffusible factor. This was done by recording macroscopic NaV currents in membrane patches before and after patch excision, the rationale being that wash‐out of any such factor would be more efficient in the inside‐out configuration than in the whole‐cell configuration. If such an intracellular factor shifting the inactivation of NaV currents towards a more negative membrane potential exists in β‐cells, then we would expect patch excision to be associated with a change in inactivation towards more positive voltages. However, such a jump was not observed. By contrast, in both Ins1 and CHO cells for NaV1.3 and NaV1.7 channels alike, patch excision resulted in a time‐dependent shift of inactivation towards more negative membrane potentials. Why this negative shift occurs is not immediately clear but has been observed by others and might reflect the time‐dependent loss of charged molecules that affect the transmembrane voltage sensed by the inactivation particle(s) (Cachelin et al. 1983; Jo & Bean, 2014). However, in the context of the current study, the key observation here is the absence of even a transient shift towards more depolarized membrane potentials. Thus, we argue that there is no diffusible modulator of channel modulation. The fact that substituting the cytoplasmic domains of NaV1.7 for NaV1.3 is also without effect on the voltage dependence of inactivation also suggests that the chief determinant of NaV current inactivation lies within the transmembrane domains.
We can discount the possibility that βsubunits account for the negative inactivation of NaV in insulin‐secreting cells as (1) Ins1 cells and primary mouse βcells express different β‐subunits (β1 and β3, respectively) and yet show the same negative inactivation in both cell types; (2) expressing NaV with β3 instead of β1 and β2 has only a small (∼5 mV) effects on channel inactivation; and (3) knockdown of β‐subunits in Ins1 cells yielded the same effect on inactivation as control, suggesting that the β‐subunit is not modulated in a cell‐specific manner to modulate channel gating.
Biphasic inactivation in Ins1 cells: possible interpretation of the data
An explanation of these data would have to account both for the fact that inactivation of all studied NaV subtypes is shifted by 20–30 mV in Ins1 cells relative to that seen in other cell types (HEK, CHO, etc.) and the finding that biphasic Na+ current inactivation is observed also when a single NaV subtype is expressed.
We considered the possibility that inactivation of the NaV currents is more negative in Ins1 cells than in HEK cells as a result of surface charge effects due to differential sialylation of the channels and/or membrane. We tested this using increasing concentrations of extracellular Co2+. Hypothetically, NaV channels expressed in Ins1 cells might carry more negative charges than channels expressed in HEK cells (for example due to increased sialylation of the extracellular domains). Accordingly, the voltage difference sensed by the inactivation gate would be reduced and this might explain the shift in inactivation towards more negative voltages. If this were the case, then increasing the divalent cationic strength would be expected to produce a greater shift in Ins1 cells (because there are more negative charges to shield). However, we think that this possibility can be discarded because varying the extracellular cationic strength had the same effects in both cell types and both negative and positive components of inactivation in Ins1 cells. Moreover, the voltage dependence of activation was superimposable in Ins1 and HEK cells and was invariably monophasic in both cell types. For both inactivation and activation, there was the same 20–25 mV shift in gating for a 50‐fold increase in extracellular Co2+. Altogether, these observations make it less likely that the widely different voltage dependences of inactivation in Ins1 and HEK cells can be attributed to variable surface charge effects.
Rather, we favour the idea that the differences between Ins1 and other cell types is likely to reside within the plasma membrane itself and does not involve cytoplasmic factors. The plasma membrane lipidome is extremely complex and contains >2000 lipid subtypes (Galbiati et al. 2001; Simons & Ehehalt, 2002). The heterogeneous distribution of lipids within the plasma membrane is not uniform but shows considerable regional differences. Indeed, the plasma membrane has been described as a ‘patchwork of different lipid environments’ (Edidin, 1993). Perhaps the best known example of such lipid aggregates is the lipid rafts that are enriched in cholesterol. There are many examples of lipid microdomains and cholesterol affecting ion channel function (Dart, 2010). For example, in β‐cells, cholesterol depletion causes a hyperpolarizing shift in the inactivation curve of KV2.1 channels (Xia et al. 2004). We therefore propose that the lipidome of the β‐cell plasma membrane is different from that of HEK and most other cells and that the β‐cell membrane contains specialized lipid domains that shift inactivation into very negative membrane potentials. Na+ channels outside these domains behave exactly as in other cell types. Indeed, in cells showing biphasic NaV current inactivation, the positive component shows essentially the same voltage dependence as in non‐β‐cells (see schematic representation in Fig. 14).
Figure 14. Biphasic inactivation in Ins1 cells.
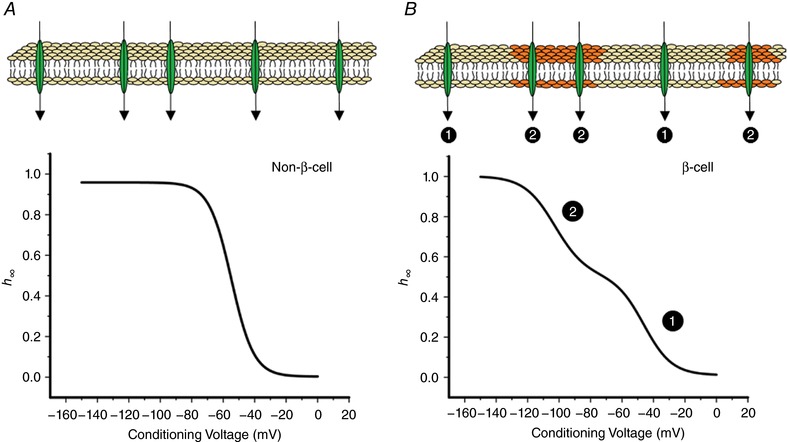
Schematic representation of NaV channel inactivation in non‐β‐cells (A) and β‐cells (B). In nonβcells, the plasma membrane is uniform in terms of impact on NaV current inactivation and the relationship between membrane potential and the fraction of activatable NaV channels (h ∞). However, β‐cells contain specialized domains that differ from the rest of the plasma membrane. NaV channels within these domains undergo inactivation at more negative membrane potentials whereas NaV channels outside these regions inactivate with the same voltage dependence as the corresponding NaV channels when expressed in non‐β‐cells. Thus, the Ins1 cell will contain two electrophysiologically distinct populations of NaV channels (❶ and ❷), accounting for the biphasic NaV current inactivation. [Color figure can be viewed at http://wileyonlinelibrary.com]
The true nature of these specialized domains remains to be established but that such domains exist is suggested by the cell‐attached measurements in which a small part of the cell (1 μm2) is isolated and where channel inactivation was invariably monophasic but with V h values varying between −80 and −55 mV. We speculate that this is because all channels within an individual patch reside in a uniform membrane environment, which may differ between patches. It is interesting that the ‘inactivation curve’ for NaV1.3 channels reconstructed from the cumulative distribution of V h in cell‐attached patches was biphasic (with V h values of −75 and −55 mV) and resembled that which can be recorded from individual cells expressing this αsubunit. For NaV1.7, the cumulative distribution was monophasic with a V h of ∼−90 mV. Notably, 10% of the patches had V h values ∼30 mV more positive. This is in agreement with the whole‐cell data indicating biphasic inactivation in <20% of the cells expressing NaV1.7. The idea that negative and positive inactivation results from insertion of the α‐subunits into membrane domains of different composition might seem in conflict with the observation that the fraction of NaV channels inactivating at negative and positive membrane potentials varied for NaV1.3, NaV1.6 and NaV1.7. However, it is possible that the intracellular trafficking and the association of NaV channels with distinct membrane domains is α‐subunit‐dependent.
The idea that NaV channel inactivation is influenced by the lipid composition of the plasma membrane also raises the interesting possibility that differences in diet may explain why the same NaV αsubunit inactivates at widely different voltages in rodent, dog, pig and human β‐cells (Rorsman & Ashcroft, 2018).
Harnessing the properties of NaV channels in β‐cells for therapeutic use?
It is clear that inactivation of NaV channels in rodent β‐cells is special in that it proceeds at membrane potentials 20–30 mV more negative than in other cell types. In the case of NaV1.7 channels, this effect may be particularly dramatic in as far as all channels will have undergone inactivation at the physiological membrane potentials (−70 mV and above). If the negative shift of inactivation in β‐cells could be harnessed (i.e. by topical/local application of agents modifying the membrane), it may provide a novel means of regulating Na+ channel activity. NaV1.7 channels play an important role in pain perception (Cox et al. 2006; Dib‐Hajj et al. 2013). If the inactivation of the NaV1.7 channels in nociceptive neurons became more like in β‐cells, then the channels would inactivate and thus be unavailable for action potential propagation, which may represent a novel means of pain relief, especially in disease states associated with NaV1.7 hyperexcitability (Dib‐Hajj et al. 2008).
Additional information
Competing interests
The authors declare that they have no conflict of interest.
Author contributions
M.G. performed all electrophysiological experiments and data analysis, except those with the Scn3a KO mice, which were performed by Q.Z. M.V.C. performed all molecular biology manipulations with the channels. M.G., M.V.C. and P.R. designed the experiments and interpreted the data. M.G., M.V.C. and P.R. wrote the paper. All authors have read and approved the final version of this manuscript and agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
Financial support was provided by a Wellcome Senior Investigator Award to P.R. M.G. was supported by an RDM Scholars DPhil studentship. Q.Z. is a Diabetes UK RD Lawrence Fellow.
Acknowledgements
We thank Dr F. Reimann (University of Cambridge, UK) for generously providing some of the Na+ channel cDNAs, Dr L. Briant (University of Oxford, UK) for help with statistical analysis and Professor Frances M. Ashcroft and Dr Gregor Sachse for valuable discussions. We also acknowledge that some of the data of Fig. 13 are based on reanalysis of data originally published in Zhang et al. (J Physiol. 592, 4677–96, 2014) and we are grateful to our co‐authors (Dr M. A. Nassar and Professor J. N. Wood) for the provision of the mouse model.
Biography
Mahdieh Godazgar is a final year DPhil student in the laboratory of Professor Patrik Rorsman at The University of Oxford. She obtained her BSc in Physiology from King's College London. She hopes to continue research as a postdoctoral fellow after she completes her DPhil.

Edited by: Ian Forsythe & Fiona Gribble
This is an Editor's Choice article from the 1 May 2018 issue.
Contributor Information
Margarita V. Chibalina, Email: margarita.chibalina@ocdem.ox.ac.uk
Patrik Rorsman, Email: patrik.rorsman@drl.ox.ac.uk.
References
- Adriaenssens AE, Svendsen B, Lam BYH, Yeo GSH, Holst JJ, Reimann F & Gribble FM (2016). Transcriptomic profiling of pancreatic alpha, beta and delta cell populations identifies delta cells as a principal target for ghrelin in mouse islets. Diabetologia 59, 2156–2165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baukrowitz T, Schulte U, Oliver D, Herlitze S, Krauter T, Tucker SJ, Ruppersberg JP & Fakler B (1998). PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science 282, 1141–1144. [DOI] [PubMed] [Google Scholar]
- Becq F (1996). Ionic channel rundown in excised membrane patches. Biochim Biophys Acta 1286, 53–63. [DOI] [PubMed] [Google Scholar]
- Benner C, Van der Meulen T, Caceres E, Tigyi K, Donaldson CJ & Huising MO (2014). The transcriptional landscape of mouse beta cells compared to human beta cells reveals notable species differences in long non‐coding RNA and protein‐coding gene expression. BMC Genomics 15, 620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett E, Urcan MS, Tinkle SS, Koszowski AG & Levinson SR (1997). Contribution of sialic acid to the voltage dependence of sodium channel gating: a possible electrostatic mechanism. J Gen Physiol 109, 327–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett ES (2002). Isoform‐specific effects of sialic acid on voltage‐dependent Na+ channel gating: functional sialic acids are localized to the S5–S6 loop of domain I. J Physiol 538, 675–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bista P, Pawlowski M, Cerina M, Ehling P, Leist M, Meuth P, Aissaoui A, Borsotto M, Heurteaux C, Decher N, Pape HC, Oliver D, Meuth SG & Budde T (2015). Differential phospholipase C‐dependent modulation of TASK and TREK two‐pore domain K+ channels in rat thalamocortical relay neurons. J Physiol 593, 127–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun M, Ramracheya R, Bengtsson M, Zhang Q, Karanauskaite J, Partridge C, Johnson PR & Rorsman P (2008). Voltage‐gated ion channels in human pancreatic beta‐cells: electrophysiological characterization and role in insulin secretion. Diabetes 57, 1618–1628. [DOI] [PubMed] [Google Scholar]
- Cachelin AB, De Peyer JE, Kokubun S & Reuter H (1983). Sodium channels in cultured cardiac cells. J Physiol 340, 389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun JD & Isom LL (2014). The role of non‐pore‐forming β subunits in physiology and pathophysiology of voltage‐gated sodium channels. Handb Exp Pharmacol 221, 51–89. [DOI] [PubMed] [Google Scholar]
- Catterall WA, Wisedchaisri G & Zheng N (2017). The chemical basis for electrical signaling. Nat Chem Biol 13, 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox JJ, Reimann F, Nicholas AK, Thornton G, Roberts E, Springell K, Karbani G, Jafri H, Mannan J, Raashid Y, Al‐Gazali L, Hamamy H, Valente EM, Gorman S, Williams R, Mchale DP, Wood JN, Gribble FM & Woods CG (2006). An SCN9A channelopathy causes congenital inability to experience pain. Nature 444, 894–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Aglieco F, Renganathan M, Herzog RI, Dib‐Hajj SD & Waxman SG (2001). Nav1.3 sodium channels: rapid repriming and slow closed‐state inactivation display quantitative differences after expression in a mammalian cell line and in spinal sensory neurons. J Neurosci 21, 5952–5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dart C (2010). Lipid microdomains and the regulation of ion channel function. J Physiol 588, 3169–3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Marinis YZ, Salehi A, Ward CE, Zhang Q, Abdulkader F, Bengtsson M, Braha O, Braun M, Ramracheya R, Amisten S, Habib AM, Moritoh Y, Zhang E, Reimann F, Rosengren A, Shibasaki T, Gribble F, Renstrom E, Seino S, Eliasson L & Rorsman P (2010). GLP‐1 inhibits and adrenaline stimulates glucagon release by differential modulation of N‐ and L‐type Ca2+ channel‐dependent exocytosis. Cell Metab 11, 543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dib‐Hajj SD, Yang Y, Black JA & Waxman SG (2013). The NaV1.7 sodium channel: from molecule to man. Nat Rev Neurosci 14, 49–62. [DOI] [PubMed] [Google Scholar]
- Dib‐Hajj SD, Yang Y & Waxman SG (2008). Genetics and molecular pathophysiology of Nav1.7‐related pain syndromes. Adv Genet 63, 85–110. [DOI] [PubMed] [Google Scholar]
- Digruccio MR, Mawla AM, Donaldson CJ, Noguchi GM, Vaughan J, Cowing‐Zitron C, Van der Meulen T & Huising MO (2016). Comprehensive alpha, beta and delta cell transcriptomes reveal that ghrelin selectively activates delta cells and promotes somatostatin release from pancreatic islets. Mol Metab 5, 449–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt M, Nakajima J, Klinger AB, Neacsu C, Huhne K, O'reilly AO, Kist AM, Lampe AK, Fischer K, Gibson J, Nau C, Winterpacht A & Lampert A (2014). Inherited pain: sodium channel Nav1.7 A1632T mutation causes erythromelalgia due to a shift of fast inactivation. J Biol Chem 289, 1971–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edidin M (1993). Patches and fences: probing for plasma membrane domains. J Cell Sci Suppl 17, 165–169. [DOI] [PubMed] [Google Scholar]
- Ednie AR & Bennett ES (2012). Modulation of voltage‐gated ion channels by sialylation. Compr Physiol 2, 1269–1301. [DOI] [PubMed] [Google Scholar]
- Fernandez JM, Fox AP & Krasne S (1984). Membrane patches and whole‐cell membranes: a comparison of electrical properties in rat clonal pituitary (GH3) cells. J Physiol 356, 565–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galbiati F, Razani B & Lisanti MP (2001). Emerging themes in lipid rafts and caveolae. Cell 106, 403–411. [DOI] [PubMed] [Google Scholar]
- Gonoi T & Hille B (1987). Gating of Na channels. Inactivation modifiers discriminate among models. J Gen Physiol 89, 253–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopel S, Kanno T, Barg S, Galvanovskis J & Rorsman P (1999). Voltage‐gated and resting membrane currents recorded from B‐cells in intact mouse pancreatic islets. J Physiol 521, 717–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog RI, Cummins TR, Ghassemi F, Dib‐Hajj SD & Waxman SG (2003). Distinct repriming and closed‐state inactivation kinetics of Nav1.6 and Nav1.7 sodium channels in mouse spinal sensory neurons. J Physiol 551, 741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B (2001). Ion Channels of Excitable Membranes. Sinauer, Sunderland, MA, USA. [Google Scholar]
- Hiriart M & Matteson DR (1988). Na channels and two types of Ca channels in rat pancreatic B cells identified with the reverse hemolytic plaque assay. J Gen Physiol 91, 617–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isom LL, De Jongh KS, Patton DE, Reber BF, Offord J, Charbonneau H, Walsh K, Goldin AL & Catterall WA (1992). Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science 256, 839–842. [DOI] [PubMed] [Google Scholar]
- Jo S & Bean BP (2014). Sidedness of carbamazepine accessibility to voltage‐gated sodium channels. Mol Pharmacol 85, 381–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer RH 1990. Patch cramming: Monitoring intracellular messengers in intact cells with membrane patches containing detector inn channels. Neuron 4, 335–341. [DOI] [PubMed] [Google Scholar]
- Kruger LC & Isom LL (2016). Voltage‐gated Na+ channels: Not just for conduction. Cold Spring Harb Perspect Biol 8, a029264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou XL, Yu X, Chen XK, Duan KL, He LM, Qu AL, Xu T & Zhou Z (2003). Na+ channel inactivation: a comparative study between pancreatic islet β‐cells and adrenal chromaffin cells in rat. J Physiol 548, 191–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macgregor GG, Dong K, Vanoye CG, Tang L, Giebisch G & Hebert SC (2002). Nucleotides and phospholipids compete for binding to the C terminus of KATP channels. Proc Natl Acad Sci USA 99, 2726–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meadows LS & Isom LL (2005). Sodium channels as macromolecular complexes: implications for inherited arrhythmia syndromes. Cardiovasc Res 67, 448–458. [DOI] [PubMed] [Google Scholar]
- Moran O, Nizzari M & Conti F (2000). Endogenous expression of the β1A sodium channel subunit in HEK‐293 cells. FEBS Lett 473, 132–134. [DOI] [PubMed] [Google Scholar]
- Plant TD (1988). Na+ currents in cultured mouse pancreatic B‐cells. Pflugers Arch 411, 429–435. [DOI] [PubMed] [Google Scholar]
- Rorsman P, Arkhammar P & Berggren PO (1986). Voltage‐activated Na+ currents and their suppression by phorbol ester in clonal insulin‐producing RINm5F cells. Am J Physiol 251, C912–C919. [DOI] [PubMed] [Google Scholar]
- Rorsman P & Ashcroft FM (2018). Pancreatic β‐cell electrical activity and insulin secretion: of mice and men. Physiol Rev 98, 117–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffer L, Brand CL, Hansen BF, Ribel U, Shaw AC, Slaaby R & Sturis J (2008). A novel high‐affinity peptide antagonist to the insulin receptor. Biochem Biophys Res Commun 376, 380–383. [DOI] [PubMed] [Google Scholar]
- Shyng SL & Nichols CG (1998). Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science 282, 1138–1141. [DOI] [PubMed] [Google Scholar]
- Simons K & Ehehalt R (2002). Cholesterol, lipid rafts, and disease. J Clin Invest 110, 597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan ZY, Priest BT, Krajewski JL, Knopp KL, Nisenbaum ES & Cummins TR (2014). Protein kinase C enhances human sodium channel hNav1.7 resurgent currents via a serine residue in the domain III–IV linker. FEBS Lett 588, 3964–3969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignali S, Leiss V, Karl R, Hofmann F & Welling A (2006). Characterization of voltage‐dependent sodium and calcium channels in mouse pancreatic A‐ and B‐cells. J Physiol 572, 691–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayaragavan K, Boutjdir M & Chahine M (2004). Modulation of Nav1.7 and Nav1.8 peripheral nerve sodium channels by protein kinase A and protein kinase C. J Neurophysiol 91, 1556–1569. [DOI] [PubMed] [Google Scholar]
- Xia F, Gao X, Kwan E, Lam PP, Chan L, Sy K, Sheu L, Wheeler MB, Gaisano HY & Tsushima RG (2004). Disruption of pancreatic β‐cell lipid rafts modifies Kv2.1 channel gating and insulin exocytosis. J Biol Chem 279, 24685–24691. [DOI] [PubMed] [Google Scholar]
- Zhang Q, Chibalina MV, Bengtsson M, Groschner LN, Ramracheya R, Rorsman NJ, Leiss V, Nassar MA, Welling A, Gribble FM, Reimann F, Hofmann F, Wood JN, Ashcroft FM & Rorsman P (2014). Na+ current properties in islet α‐ and β‐cells reflect cell‐specific Scn3a and Scn9a expression. J Physiol 592, 4677–4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou N, Wu X, Jin YY, He MZ, Wang XX, Su LD, Rupnik M, Wu ZY, Liang L & Shen Y (2013). ATP regulates sodium channel kinetics in pancreatic islet beta cells. J Membr Biol 246, 101–107. [DOI] [PubMed] [Google Scholar]