

Abstract
Purpose
Epigenetic modifications, such as histone acetylation/deacetylation and DNA methylation, play a crucial role in the pathogenesis of inflammatory disorders and fibrotic diseases. The aim of this study was to study the differential gene expression of histone deacetylases (HDACs) in fibroblasts isolated from plaque tissue of Peyronie's disease (PD) or normal tunica albuginea (TA) and to examine the anti-fibrotic effect of small interfering RNA (siRNA)-mediated silencing of HDAC7 in fibroblasts derived from human PD plaque.
Materials and Methods
For differential gene expression study, we performed reverse-transcriptase polymerase chain reaction for HDAC isoforms (1–11) in fibroblasts isolated from PD plaque or normal TA. Fibroblasts isolated from PD plaque were pretreated with HDAC7 siRNA (100 pmol) and then stimulated with transforming growth factor-β1 (TGF-β1, 10 ng/mL). Protein was extracted from treated fibroblasts for Western blotting. We also performed immunocytochemistry to detect the expression of extracellular matrix proteins and to examine the effect of HDAC2 siRNA on the TGF-β1-induced nuclear translocation of Smad2/3 and myofibroblastic differentiation.
Results
The mRNA expression of HDAC2, 3, 4, 5, 7, 8, 10, and 11 was higher in fibroblasts isolated from PD plaque than in fibroblasts isolated from normal TA tissue. Knockdown of HDAC7 in PD fibroblasts inhibited TGF-β1-induced nuclear shuttle of Smad2 and Smad3, transdifferentiation of fibroblasts into myofibroblasts, and abrogated TGF-β1-induced production of extracellular matrix protein.
Conclusions
These findings suggest that specific inhibition of HDAC7 with RNA interference may represent a promising epigenetic therapy for PD.
Keywords: Extracellular matrix, Fibrosis, Histone deacetylases, Penile induration, Transforming growth factors
INTRODUCTION
Peyronie's disease (PD) is a localized fibrotic process of the tunica albuginea (TA). The fibrotic plaque impedes the expansion of the TA during erection, which results in a variety of penile deformities and often pain during erection [1,2]. Although the etiology of PD is not fully delineated, an inflammatory process and subsequent aberrant wound healing following repeated trauma to the penis during intercourse are known to be involved in fibrotic processes [3,4]. Despite promising results with intralesional injection of collagenase clostridium histolyticum [5], surgical intervention is still the only curative treatment modality that corrects penile deformities [6,7]. Therefore, the identification of novel therapeutic target involved in complex fibrogenic process of PD is required.
Epigenetic modifications, such as histone acetylation/deacetylation and DNA methylation, comprise heritable alterations in the DNA itself without changes in the nucleotide sequence. Epigenetic modifications have been shown to play a crucial role in the pathogenesis of inflammatory disorders and fibrotic diseases [8,9]. Pharmacologic inhibition of histone deacetylase (HDAC) is known to decrease fibrotic responses in a variety of conditions [9,10]. Previous study demonstrated in skin fibroblasts from patients with systemic sclerosis that silencing HDAC7, a class II HDAC, specifically reduced excessive production of extracellular matrix, which was as effective as trichostatin A (TSA), a HDAC inhibitor. However, TSA also up-regulates the expression of profibrotic factors, such as connective tissue growth factor and intracellular adhesion molecule-1, whereas silencing of HDAC7 did not influence on the expression of these profibrotic molecules [11]. Therefore, inhibition of specific HDAC isoforms by use of RNA interference technology may be more advantageous than the use of nonspecific HDAC inhibitors.
In the present study, we examined the differential expression of HDAC isoforms in fibroblasts isolated from human PD plaque or normal TA. Next, we determined the effectiveness of the knockdown of HDAC7 on the transforming growth factor-β1 (TGF-β1)-induced profibrotic responses in primary fibroblasts derived from human PD plaque.
MATERIALS AND METHODS
1. Primary fibroblast culture
We obtained plaque tissues from two patients with PD (age, 48 and 52 years, respectively) or normal TA tissues from two control patients: 1 undergoing penoplasty for congenital curvature (age, 21 years) and 1 undergoing primary repair of TA as the result of penile fracture (age, 51 years). The tissue samples were used for primary fibroblast culture as previously described [12,13]. Briefly, either plaque tissue or normal TA tissue was transferred into sterile vials containing Hank's balanced salt solution (GIBCO, Carlsbad, CA, USA) and was washed three times in phosphate-buffered saline (PBS). Biopsy tissue was minced into 1-mm2 segments and incubated in a shaker in 12.5 mL Dulbecco's modified Eagle Medium (DMEM) supplemented with 0.06% collagenase A (Sigma-Aldrich, St. Louis, MO, USA) for 1 hour. The cells and tissue fragments were collected by centrifugation (400 ×g, 5 minutes), washed in fresh culture medium, and placed in 100-mm cell culture dishes (Falcon-Becton Dickinson Labware, Franklin Lakes, NJ, USA) under standard cell culture conditions with DMEM supplemented with 10% fetal calf serum, penicillin (100 U/mL), and streptomycin (100 µg/mL). The dishes were incubated in a humidified 37℃ incubator with 5% CO2. The cells were then characterized as previously described [12,13]. Passages five to eight were used for experimentation.
2. Transfection of small interfering RNA into cells
The fibroblasts were serum-starved for 24 hours and transfected with 100 pmol small interfering RNA (siRNA) oligonucleotides targeted specifically to HDAC7 (Santa Cruz Biotechnology, Santa Cruz, CA, USA) by using Lipofectamine 2000 (GIBCO). In parallel, 100 pmol scramble siRNA was used as a control. After transfection, cells were plated and cultured for 48 hours in DMEM. The fibroblasts were then treated with 10 ng/mL TGF-β1 (R&D Systems Inc., Minneapolis, MN, USA) for 24 hours to detect the protein expression of plasminogen activator inhibitor-1 (PAI-1), fibronectin, collagen subtypes, smooth muscle α-actin, and HDAC7.
3. Reverse-transcriptase polymerase chain reaction
Total RNA was extracted from cultured cells with Trizol (Invitrogen, Carlsbad, CA, USA) according to the manufacturer's protocols. RNA was reverse-transcribed by use of the Reverse Transcription System (Promega, Madison, WI, USA) according to the manufacturer's instructions. Reverse-transcriptase polymerase chain reaction (RT-PCR) fragments were amplified by using the AccuPower PCR premix (Bioneer, Alameda, CA, USA). The PCR reaction was performed with denaturation at 94℃ for 1 minute, annealing at 60℃ for 30 seconds, and extension at 72℃ for 30 seconds (25 cycles). For the analysis of PCR products, 10 µL of each PCR reaction was electrophoresed on a 1% agarose gel and DNA bands were visualized with a ultraviolet illuminator (Image Station IS4000R system; Kodak, Rochester, NY, USA). Glyceraldehyde 3-phosphate dehydrogenase was used as an internal control. The primer sequences are listed in Table 1.
Table 1. Primer sequences for RT-PCR.
| Primer | Sequence of PCR primers | Size (bp) | GenBank accession No. |
|---|---|---|---|
| Human HDAC1 | F: GTA CCA CAG CGA TGA CTA CAT | 420 | NT_032977 |
| R: CTG GGA AGT ACT CTC CAT ACT | |||
| Human HDAC2 | F: CAT CCC ATG AAG CCT CAT AGA ATC | 565 | NT_025741 |
| R: GCA CCA ATA TCC CTC AAG TCT CC | |||
| Human HDAC3 | F: CGC CGG CAC CAT GGC CAA GA | 361 | NT_025741 |
| R: GCT GGG TTG CTC CTT GCA GA | |||
| Human HDAC4 | F: TGT ACG ACG CCA AAG ATG AC | 50 | NT_022173 |
| R: CGG TTC AGA AGC TGT TTT CC | |||
| Human HDAC5 | F: AGT GAC ACC GTG TGG AAT GA | 244 | NT_010783 |
| R: AGT CCA CGA TGA GGA CCT TG | |||
| Human HDAC6 | F: TCA GGT CTA CTG TGG TCG TT | 151 | NT_079573 |
| R: TCT TCA CAT CTA GGA GAG CC | |||
| Human HDAC7 | F: ATG GGG GAT CCT GAG TAC CT | 51 | NT_029419 |
| R: GAT GGG CAT CAC GAC TAT CC | |||
| Human HDAC8 | F: AGA TGA AGC ATC TGG TTT TT | 558 | NT_011669 |
| R: TGG GAT CTC AGA GGA TAG TG | |||
| Human HDAC9 | F: CTG GAG CCC ATC TCA CCT T | 179 | NT_007819 |
| R: CCG TGT CAA GTT CTC ATG CT | |||
| Human HDAC10 | F: GCC GGA TAT CAC ATT GGT TC | 51 | NT_011526 |
| R: GAC GCT TCC TGT TGG ATG A | |||
| Human HDAC11 | F: GGT CAG GAA GGG GTA CAG GT | 159 | NT_022517 |
| R: TTG CAC TGA ACA GGC AAG AC | |||
| GAPDH | F: CCA CTG GCG TCT TCA CCA C | 503 | NT_006713 |
| R: CCT GCT TCA CCA CCT TCT TG |
RT-PCR: reverse-transcriptase polymerase chain reaction, HDAC: histone deacetylase, GAPDH: glyceraldehyde 3-phosphate dehydrogenase, F: forward, R: reverse.
4. Western blot
Equal amounts of protein from whole-cell extracts (50 µg/lane) were electrophoresed on 12% sodium dodecylsulfate-polyacrylamide gels, transferred to nitrocellulose membranes, and probed with antibody against HDAC7 (1:100; Santa Cruz Biotechnology), PAI-1 (1:300; Abcam, Cambridge, UK), fibronectin (1:300; Abcam), collagen I (1:300; Abcam), collagen IV (1:300; Abcam), smooth muscle α-actin (1:300; Sigma-Aldrich), or β-actin (1:6,000; Abcam).
5. Fluorescent immunocytochemistry
The fibroblasts were cultured on sterile cover glasses (Marienfeld Laboratory, Lauda-Königshofen, Germany) and grown until nearly confluent. The cells were washed three times with PBS and then fixed in 4% paraformaldehyde for 10 minutes at 4℃ and in 100% methanol for 10 minutes at 4℃. Individual chambers were incubated with antibody to PAI-1 (1:300; Abcam), fibronectin (1:300; Abcam), collagen I (1:300; Abcam), collagen IV (1:300; Abcam), smooth muscle α-actin (1:300; Sigma-Aldrich), F-actin (1:300; Sigma-Aldrich), or Smad2/3 (1:200; Cell Signaling, Beverly, MA, USA) overnight at 4℃ in a moist chamber. After several washes with PBS, the chambers were incubated with fluorescein isothiocyanate-conjugated (1:300; Zymed Laboratories, South San Francisco, CA, USA) or tetramethyl rhodamine isothiocyanate-conjugated (1:300; Jackson ImmunoResearch Laboratories Inc., West Grove, PA, USA) secondary antibodies for 3 hours at room temperature. Mounting medium containing 4,6-diamidino-2-phenylindole (Vector Laboratories Inc., Burlingame, CA, USA) was applied to the chamber and nuclei were labeled. Signals were visualized, and digital images were obtained with a confocal microscope (FV1000; Olympus, Tokyo, Japan) under identical exposure settings.
6. Statistical analysis
Results are expressed as the mean±standard errors. We used the Kruskal-Wallis tests for group comparison. We performed statistical analysis with SigmaStat 3.5 software (Systat Software Inc., Richmond, CA, USA). The p-values less than 5% were considered significant.
7. Ethics statement
All tissue donors provided informed consent, and the procedures were approved by the Internal Review Board of Inha University School of Medicine.
RESULTS
1. Differential gene expression of histone deacetylases in fibroblasts isolated from human Peyronie's disease plaque or from normal tunica albuginea
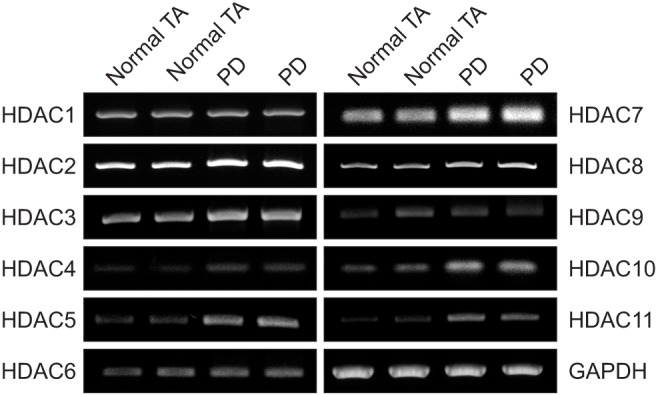
To examine the changes in gene expression of the HDAC isoforms, we performed RT-PCR. The mRNA expression of HDAC2, 3, 4, 5, 7, 8, 10, and 11 was higher in fibroblasts isolated from PD plaque than in fibroblasts isolated from normal TA. No detectable differences were noted in the gene expression of HDAC1, 6, and 9 (Fig. 1).
Fig. 1. Differential gene expression of histone deacetylases (HDACs) in fibroblasts isolated from human Peyronie's disease (PD) plaque. A representative gel picture shows the gene expression of HDACs in fibroblasts isolated from PD plaque (n=2) or from normal tunica albuginea (TA) tissue from control patients (n=2). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used as an internal control for reverse-transcriptase polymerase chain reaction. Results were similar from three independent experiments.
2. Histone deacetylase 7 knockdown inhibits extracellular matrix production induced by transforming growth factor-β1 in fibroblasts derived from human Peyronie's disease plaque
To determine the anti-fibrotic role of HDAC7, the siRNA approach was used. PD fibroblasts were transfected with siRNA specifically targeting HDAC7. Western blot analysis revealed that the treatment of PD fibroblasts with TGF-β1 induced HDAC7 expression and these expression was profoundly inhibited after treatment with HDAC7 siRNA (Fig. 2A, 2B).
Fig. 2. Histone deacetylase 7 (HDAC7) knockdown inhibits transforming growth factor-β1 (TGF-β1)-induced extracellular matrix protein production in fibroblasts derived from human Peyronie's disease (PD) plaque. (A) Effect of TGF-β1 on HDAC7 expression. Representative Western blot for HDAC7 protein in PD fibroblasts after specific knockdown using small interfering RNA (siRNA) or control siRNA (scramble siRNA). Data are presented as the ratio of the product of protein to that of β-actin. Fibroblasts were transfected with scramble siRNA or siRNA specific to HDAC7 by using Lipofectamine (GIBCO) reagent for 48 hours and were then treated with TGF-β1 (10 ng/mL) for 24 hours. (B) Each bar depicts the mean values (±standard error) from four experiments per group. The relative ratio measured in the no treatment group was arbitrary presented as 1. *p<0.05 compared with no treatment group, †p<0.05 compared with TGF-β1+scramble siRNA group. (C) Representative Western blot for fibronectin, plasminogen activator inhibitor-1 (PAI-1), collagen I, and collagen IV in fibroblasts. Results were similar from four independent experiments.

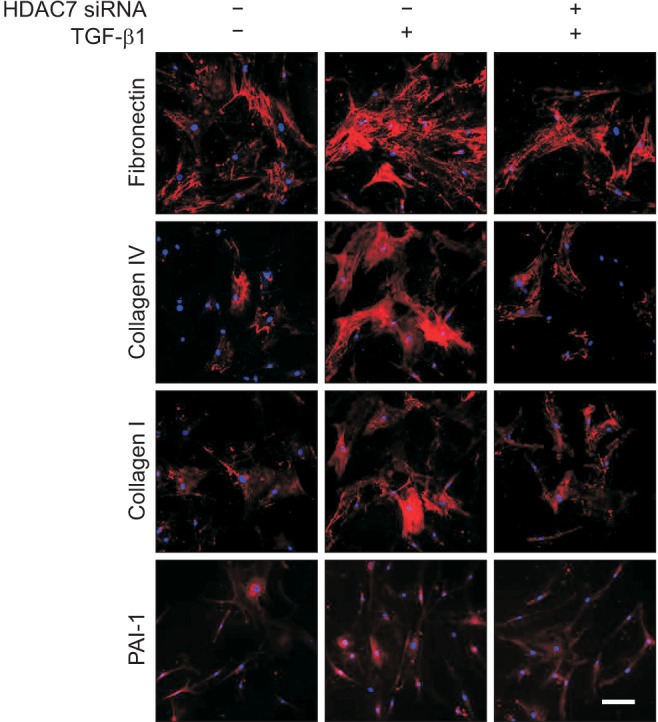
Both Western blot analysis and fluorescent immunocytochemistry showed that HDAC7 siRNA significantly inhibited TGF-β1-induced production of fibronectin, PAI-1, collagen I, and collagen IV in PD fibroblasts (Fig. 2C, 3).
Fig. 3. Fluorescent immunocytochemistry showing the inhibition of transforming growth factor-β1 (TGF-β1)-induced extracellular matrix protein expression by histone deacetylase 7 (HDAC7) small interfering RNA (siRNA) in fibroblasts derived from human Peyronie's disease plaque. The cells were washed three times with phosphate-buffered saline and then fixed in 4% paraformaldehyde for 10 minutes at 4℃ and in 100% methanol for 10 minutes at 4℃. Representative fluorescent immunocytochemistry of fibroblasts with antibody against fibronectin, plasminogen activator inhibitor-1 (PAI-1), collagen I, and collagen IV. Nuclei were labeled with the DNA dye 4,6-diamidino-2-phenylindole. Bar indicates 100 µm. Fibroblasts were transfected with scramble siRNA or siRNA specific to HDAC7 by using Lipofectamine (GIBCO) reagent for 48 hours and were then treated with TGF-β1 (10 ng/mL) for 24 hours. Results were similar from four independent experiments.
3. Histone deacetylase 7 knockdown inhibits myofibroblastic differentiation induced by transforming growth factor-β1 in fibroblasts derived from human Peyronie's disease plaque
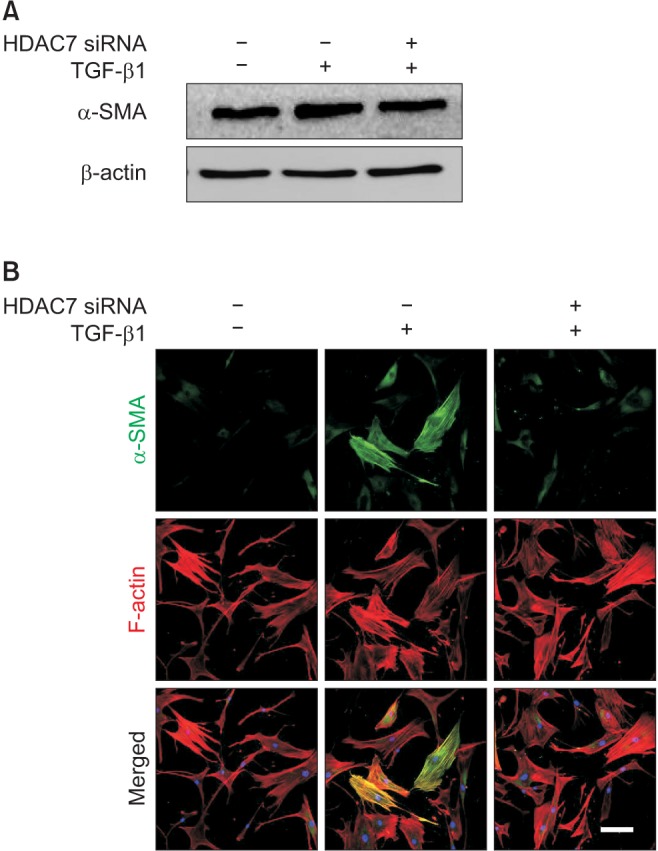
The expression of smooth muscle α-actin, a marker for myofibroblasts, at the protein level was determined with Western blot analysis. The treatment of PD fibroblasts with TGF-β1 resulted in an increase in smooth muscle α-actin expression, which was attenuated after treatment with HDAC7 siRNA (Fig. 4A). Fluorescent immunocytochemistry also revealed that HDAC7 siRNA inhibited TGF-β1-stimulated α-actin fiber formation (Fig. 4B).
Fig. 4. Histone deacetylase 7 (HDAC7) knockdown inhibits transforming rowth factor-β1 (TGF-β1)-induced myofibroblastic differentiation in fibroblasts derived from human Peyronie's disease plaque. (A) Representative Western blot for alpha-smooth muscle actin (α-SMA). Fibroblasts were transfected with scramble small interfering RNA (siRNA) or siRNA specific to HDAC7 by using Lipofectamine (GIBCO) reagent for 48 hours and were then treated with TGF-β1 (10 ng/mL) for 24 hours. (B) The fibroblasts were serum-starved for 24 hours and transfected with 100 pmol siRNA oligonucleotides targeted specifically to HDAC7 by using Lipofectamine 2000. After transfection, cells were plated and cultured for 48 hours in Dulbecco's modified Eagle's medium. The fibroblasts were then treated with 10 ng/mL TGF-β1 for 24 hours. The cells were washed three times with phosphate-buffered saline and then fixed in 4% paraformaldehyde for 10 minutes at 4℃ and in 100% methanol for 10 minutes at 4℃. Representative fluorescent immunocytochemistry of fibroblasts with antibody against α-SMA (a myofibroblast marker) and F-actin (a cytoskeleton marker). Nuclei were labeled with the DNA dye 4,6-diamidino-2-phenylindole. Bar indicates 100 µm. Results were similar from four independent experiments.
4. Histone deacetylase 7 knockdown inhibits nuclear translocation of Smad2/3 induced by transforming growth factor-β1 in fibroblasts derived from human Peyronie's disease plaque
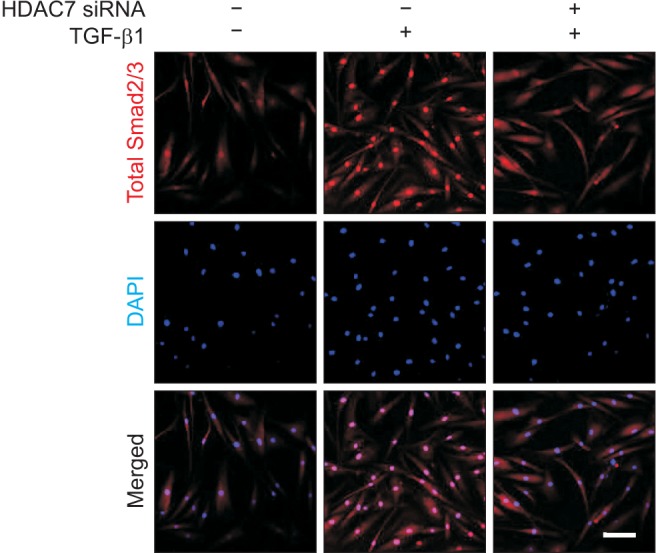
TGF-β1 has been shown to induce the translocation of Smad2/3 proteins from the cytoplasm to the nucleus [14]. In order to evaluate whether HDAC7 inhibition affects on TGF-β1-induced nuclear shuttling of Smad2/3, we performed immunofluorescent staining of fibroblasts with antibody against total Smad2/3. The treatment of PD fibroblasts with TGF-β1 profoundly induced nuclear translocation of Smad2/3. HDAC7 siRNA reduced TGF-β1-induced nuclear accumulation of Smad proteins (Fig. 5).
Fig. 5. Histone deacetylase 7 (HDAC7) knockdown suppresses transforming growth factor-β1 (TGF-β1)-induced Smad2/3 nuclear translocation in fibroblasts derived from human Peyronie's disease plaque. Representative fluorescent immunocytochemistry of primary human fibroblasts with antibody against total Smad2/3. Fibroblasts were transfected with scramble small interfering RNA (siRNA) or siRNA specific to HDAC7 by using Lipofectamine (GIBCO) reagent for 48 hours and were then treated with TGF-β1 (10 ng/mL) for 1 hour. The cells were washed three times with phosphate-buffered saline and then fixed in 4% paraformaldehyde for 10 minutes at 4℃ and in 100% methanol for 10 minutes at 4℃. Nuclei were labeled with the DNA dye 4,6-diamidino-2-phenylindole (DAPI). Bar indicates 100 µm. Results were similar from four independent experiments.
DISCUSSION
Among the 11 HDACs tested in the present study, the expression of HDAC2, 3, 4, 5, 7, 8, 10, and 11 transcripts was higher in fibroblasts isolated from PD plaque than in fibroblasts isolated from normal TA. However, this result is limited by the small sample size, and further studies in a larger study population are required for further validation. It was also reported that the expression of both class I HDACs (HDAC1, HDAC2, HDAC3, HDAC8) and class II HDACs (HDAC4, HDAC 5, HDAC 7, HDAC 9) were significantly elevated in lung tissues from patients with idiopathic pulmonary fibrosis compared with those from non-diseased controls [15]. Moreover, siRNA mediated silencing of HDAC7 corrects the ΔF508 mutation in cystic fibrosis transmembrane conductance regulator that is responsible for premature lung failure and reduced lifespan in patients with cystic fibrosis [16]. HDAC7 is also known to be involved in the hepatic fibrosis by binding promoter region of hepatocyte growth factor (HGF) and limits the antifibrotic function of HGF [17]. These findings led us to investigate whether and how HDAC7 exerts its antifibrotic effects in fibroblasts isolated from human PD plaque.
Here, it was shown that siRNA-mediated knockdown of HDAC7 successfully ameliorated the TGF-β1-induced accumulation of extracellular matrix in human PD fibroblasts by blocking nuclear translocation of Smad2 and Smad3, the crucial step for TGF-β-mediated fibrosis, and by inhibiting TGF-β1-induced myofibroblastic differentiation.
In the present study, treatment of PD fibroblasts with TGF-β1 significantly induced HDAC7 protein expression. The specific gene knockdown of HDAC7 with siRNA significantly decreased the TGF-β1-induced accumulation of extracellular matrix proteins, such as fibronectin, PAI-1, collagen I, and collagen IV. Similar to the results from ours, silencing of HDAC7 in skin fibroblasts from patients with systemic sclerosis also decreased constitutive and cytokine (TGFβ1)-induced production of type I and type III collagen on both the mRNA and protein levels [11], suggesting HDAC7 as a potential therapeutic target in a variety of fibrotic diseases.
Accumulating evidences suggest that HDACs are involved in the cytokine-induced differentiation of fibroblast into myofibroblast [13,18,19,20,21]. Treatment of rat renal interstitial fibroblasts with TSA, a nonspecific HDAC inhibitor, decreased the expression of smooth muscle α-actin and deposition of extracellular matrix [20]. We recently reported in human PD fibroblasts that inhibition of HDAC2 abrogated TGF-β1-induced transdifferentiation of fibroblasts into myofibroblasts [13]. Knockdown of HDAC4 is also known to inhibit TGF-β1-stimulated smooth muscle α-actin expression in normal human lung fibroblasts [18]. However, the role of HDAC7 in myofibroblastic differentiation is largely unknown. In the present study, treatment of PD fibroblasts with HDAC7 siRNA significantly reduced TGF-β1-induced fibroblast-to-myofibroblast transition. Because activation of fibroblasts into myofibroblasts is responsible for increased production of extracellular matrix, HDAC7 siRNA-mediated decrease in myofibroblastic differentiation is a key mechanism for halting fibrotic processes in the TA.
TGF-β1 is one of the most studied cytokine associated with PD and the expression of TGF-β1 and its downstream signaling pathway, such as Smad2 and Smad3 transcriptional factors, are known to be up-regulated in human PD plaque [12,22,23]. TSA is known to inhibit nuclear translocation and DNA binding of Smad transcription factors in skin fibroblasts from patients with systemic sclerosis [10]. Similar to recent studies by us that showed a decrease in the nuclear translocation of Smad2 and Smad3 in human PD fibroblasts in vitro [13] and in PD rats in vivo [24], when HDAC2 was knockdown, treatment of PD fibroblasts with HDAC7 siRNA also prevented nuclear accumulation of Smad proteins. Therefore, suppression of the activation of Smad2 and Smad3 proteins is another mechanism by which silencing HDAC7 ameliorates fibrotic responses in PD fibroblasts. Further studies are required to determine the anti-fibrotic role of HDAC7 knockdown in PD models in vivo. It is also necessary to examine the efficacy of other HDAC isoforms in PD models.
In spite of distinctive physiologic function of each HDAC isoforms, most known HDAC inhibitors target multiple isoforms, which greatly limit their therapeutic utility. Therefore, dichotomizing individual function of HDAC isoforms by use of RNA interference technology may open new avenues for developing specific and safe treatment modality for PD.
CONCLUSIONS
The specific gene knockdown of HDAC7 in PD fibroblasts successfully attenuated TGF-β1-induced extracellular matrix production by inhibiting transdifferentiation of fibroblasts into myofibroblasts and by blocking activation of Smad2/3 pathway. Inhibition of HDAC7 with RNA interference may represent a promising epigenetic therapy for PD.
ACKNOWLEDGEMENTS
This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIP) (Jun-Kyu Suh, 2016R1A2B4013130).
Footnotes
Disclosure: The authors have no potential conflicts of interest to disclose.
Author Contribution: Research conception & design: Kang DH, Yin GN, Ryu JK, Suh JK. Performing the experiments: Choi MJ, Song KM. Data acquisition: Ghatak K, Minh NN. Data analysis and interpretation: Yin GN, Choi MJ. Statistical analysis: Kang DH, Kwon MH, Seong DH. Drafting of the manuscript: Kang DH, Yin GN. Critical revision of the manuscript: Ryu JK, Suh JK. Receiving grant: Ryu JK. Approval of final manuscript: all authors.
References
- 1.Gholami SS, Gonzalez-Cadavid NF, Lin CS, Rajfer J, Lue TF. Peyronie's disease: a review. J Urol. 2003;169:1234–1241. doi: 10.1097/01.ju.0000053800.62741.fe. [DOI] [PubMed] [Google Scholar]
- 2.Hellstrom WJ, Bivalacqua TJ. Peyronie's disease: etiology, medical, and surgical therapy. J Androl. 2000;21:347–354. [PubMed] [Google Scholar]
- 3.Devine CJ, Jr, Somers KD, Jordan SG, Schlossberg SM. Proposal: trauma as the cause of the Peyronie's lesion. J Urol. 1997;157:285–290. doi: 10.1016/s0022-5347(01)65361-8. [DOI] [PubMed] [Google Scholar]
- 4.Jarow JP, Lowe FC. Penile trauma: an etiologic factor in Peyronie's disease and erectile dysfunction. J Urol. 1997;158:1388–1390. doi: 10.1016/s0022-5347(01)64222-8. [DOI] [PubMed] [Google Scholar]
- 5.Gabrielson AT, Alzweri LM, Hellstrom WJ. Collagenase clostridium histolyticum in the treatment of Peyronie's disease: review of a minimally invasive treatment option. World J Mens Health. 2017;35:134–145. doi: 10.5534/wjmh.17033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Joice GA, Burnett AL. Nonsurgical interventions for Peyronie's disease: update as of 2016. World J Mens Health. 2016;34:65–72. doi: 10.5534/wjmh.2016.34.2.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Segal RL, Burnett AL. Surgical management for Peyronie's disease. World J Mens Health. 2013;31:1–11. doi: 10.5534/wjmh.2013.31.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McKinsey TA. Targeting inflammation in heart failure with histone deacetylase inhibitors. Mol Med. 2011;17:434–441. doi: 10.2119/molmed.2011.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pang M, Zhuang S. Histone deacetylase: a potential therapeutic target for fibrotic disorders. J Pharmacol Exp Ther. 2010;335:266–272. doi: 10.1124/jpet.110.168385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huber LC, Distler JH, Moritz F, Hemmatazad H, Hauser T, Michel BA, et al. Trichostatin A prevents the accumulation of extracellular matrix in a mouse model of bleomycin-induced skin fibrosis. Arthritis Rheum. 2007;56:2755–2764. doi: 10.1002/art.22759. [DOI] [PubMed] [Google Scholar]
- 11.Hemmatazad H, Rodrigues HM, Maurer B, Brentano F, Pileckyte M, Distler JH, et al. Histone deacetylase 7, a potential target for the antifibrotic treatment of systemic sclerosis. Arthritis Rheum. 2009;60:1519–1529. doi: 10.1002/art.24494. [DOI] [PubMed] [Google Scholar]
- 12.Piao S, Choi MJ, Tumurbaatar M, Kim WJ, Jin HR, Shin SH, et al. Transforming growth factor (TGF)-β type I receptor kinase (ALK5) inhibitor alleviates profibrotic TGF-β1 responses in fibroblasts derived from Peyronie's plaque. J Sex Med. 2010;7:3385–3395. doi: 10.1111/j.1743-6109.2010.01753.x. [DOI] [PubMed] [Google Scholar]
- 13.Ryu JK, Kim WJ, Choi MJ, Park JM, Song KM, Kwon MH, et al. Inhibition of histone deacetylase 2 mitigates profibrotic TGF-β1 responses in fibroblasts derived from Peyronie's plaque. Asian J Androl. 2013;15:640–645. doi: 10.1038/aja.2013.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoodless PA, Haerry T, Abdollah S, Stapleton M, O'Connor MB, Attisano L, et al. MADR1, a MAD-related protein that functions in BMP2 signaling pathways. Cell. 1996;85:489–500. doi: 10.1016/s0092-8674(00)81250-7. [DOI] [PubMed] [Google Scholar]
- 15.Korfei M, Skwarna S, Henneke I, MacKenzie B, Klymenko O, Saito S, et al. Aberrant expression and activity of histone deacetylases in sporadic idiopathic pulmonary fibrosis. Thorax. 2015;70:1022–1032. doi: 10.1136/thoraxjnl-2014-206411. [DOI] [PubMed] [Google Scholar]
- 16.Hutt DM, Herman D, Rodrigues AP, Noel S, Pilewski JM, Matteson J, et al. Reduced histone deacetylase 7 activity restores function to misfolded CFTR in cystic fibrosis. Nat Chem Biol. 2010;6:25–33. doi: 10.1038/nchembio.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pannem RR, Dorn C, Hellerbrand C, Massoumi R. Cylindromatosis gene CYLD regulates hepatocyte growth factor expression in hepatic stellate cells through interaction with histone deacetylase 7. Hepatology. 2014;60:1066–1081. doi: 10.1002/hep.27209. [DOI] [PubMed] [Google Scholar]
- 18.Glenisson W, Castronovo V, Waltregny D. Histone deacetylase 4 is required for TGFbeta1-induced myofibroblastic differentiation. Biochim Biophys Acta. 2007;1773:1572–1582. doi: 10.1016/j.bbamcr.2007.05.016. [DOI] [PubMed] [Google Scholar]
- 19.Guo W, Shan B, Klingsberg RC, Qin X, Lasky JA. Abrogation of TGF-beta1-induced fibroblast-myofibroblast differentiation by histone deacetylase inhibition. Am J Physiol Lung Cell Mol Physiol. 2009;297:L864–L870. doi: 10.1152/ajplung.00128.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noh H, Oh EY, Seo JY, Yu MR, Kim YO, Ha H, et al. Histone deacetylase-2 is a key regulator of diabetes- and transforming growth factor-beta1-induced renal injury. Am J Physiol Renal Physiol. 2009;297:F729–F739. doi: 10.1152/ajprenal.00086.2009. [DOI] [PubMed] [Google Scholar]
- 21.Pang M, Kothapally J, Mao H, Tolbert E, Ponnusamy M, Chin YE, et al. Inhibition of histone deacetylase activity attenuates renal fibroblast activation and interstitial fibrosis in obstructive nephropathy. Am J Physiol Renal Physiol. 2009;297:F996–F1005. doi: 10.1152/ajprenal.00282.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.El-Sakka AI, Hassoba HM, Pillarisetty RJ, Dahiya R, Lue TF. Peyronie's disease is associated with an increase in transforming growth factor-beta protein expression. J Urol. 1997;158:1391–1394. [PubMed] [Google Scholar]
- 23.Haag SM, Hauck EW, Szardening-Kirchner C, Diemer T, Cha ES, Weidner W, et al. Alterations in the transforming growth factor (TGF)-beta pathway as a potential factor in the pathogenesis of Peyronie's disease. Eur Urol. 2007;51:255–261. doi: 10.1016/j.eururo.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 24.Kwon KD, Choi MJ, Park JM, Song KM, Kwon MH, Batbold D, et al. Silencing histone deacetylase 2 using small hairpin RNA induces regression of fibrotic plaque in a rat model of Peyronie's disease. BJU Int. 2014;114:926–936. doi: 10.1111/bju.12812. [DOI] [PubMed] [Google Scholar]