

Abstract
Despite recent progress in novel and targeted therapies, multiple myeloma (MM) remains a therapeutically challenging incurable disease. The regulation of important cellular processes and its link to cancer presented Src as an attractive target for MM. We suggest a novel strategy to improve the treatment of MM and overcome the drug resistance for the current therapeutic agents by specific inhibition of Src in MM cells by Tris (Dibenzylideneacetone) dipalladium (Tris DBA). Tris DBA reduces proliferation, induces G1 arrest and apoptosis in MM cells. Tris DBA showed additive effect with proteasome inhibitors reducing proliferation, cell cycle signaling, and increasing apoptosis more than each drug alone. Tris DBA overcame hypoxia-induced effects such as enhanced chemotaxis or drug resistance to proteasome inhibitors by inhibition of HIF1α expression. Moreover, we found that Tris DBA is an effective anti-myeloma agent alone or in combination with other targeted drugs and that it reverses hypoxia-induced drug resistance in myeloma.
Keywords: Tris DBA, Src, multiple myeloma, drug resistance, hypoxia
Introduction
Multiple myeloma (MM) is a plasma cell myeloma where abnormal plasma cells accumulate in the bone marrow. MM is considered to be incurable based on the high percent of patients that relapse or do not respond to treatments [1,2]. Emerging new therapies have improved survival in relapsed or refractory MM patients [3–6]. Although these novels therapies have improved outcomes for MM patients, the main goal in the MM field is to overcome the resistance to therapy.
Src is a non-receptor protein tyrosine kinase which regulates multiple fundamental cellular processes including cell growth, migration, survival and differentiation [7]. Src is expressed in many cells and tissues, and both elevated protein levels and kinase activity of Src have been detected in many cancers including breast, colon, pancreatic, lung and skin [8–13]. Activated Src in cancer lead to studies with Src as a target for anti-cancer drugs, and numerous SRC inhibitors have become available to test the importance of Src in tumor initiation and progression [14,15]. In addition, an activating mutation (Src 531) in human colon cancers with high Src activity suggests the oncogenic potential of Src [16]. Src family kinases, including Src and Lyn, have been extensively linked to aberrant signaling and progression of multiple myeloma [15,17]. Lyn was demonstrated to be involved in IL-6 mediated cell proliferation in multiple myeloma cells, and incubation with a Src family kinases specific tyrosine kinase inhibitor reduced IL-6-dependent proliferation of CD45 myeloma cells [18,19].
In MM, it has been described that in cell lines and MM patient-derived tumors, c-Src is constitutively activated [20]. This constitutive activation of c-Src promotes cell survival, proliferation, and chemoresistance, and then inhibition of Src activation enhances chemosensitization [20,21]. Thus, Src activation plays an important role in cell adhesion-mediated drug resistance and other drug resistance mechanisms in MM cells [22–24]. Src inhibitors are potentially useful as anti-drug resistance agents for the treatment of cancer.
Src requires myristoylation for its activity, conducted by the enzyme N-myristoyltransferase-1 (NMT-1). Tris (Dibenzylideneacetone) dipalladium (Tris DBA), a small-molecule palladium complex, was shown to reduce Src/NMT-1 complex in melanoma cells, as well as inhibit downstream signaling including mitogen-activated protein kinase (MAPK kinase) and phosphoinositol-3-kinase (PI3K) [25]. In this study, we characterized the role Tris DBA has on survival, cell cycle, apoptosis and migration of MM cells. We further examined how Src inhibition can influence sensitivity to proteasome inhibitors (PIs), as well as its ability to overcome hypoxia-induced drug resistance effects.
Materials and methods
Reagents
Tris (Dibenzylideneacetone) dipalladium (Tris DBA) was prepared by Dr Arbiser as previously described [25]. Propidium iodide (PI), RNAase and MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) solutions were from Sigma-Aldrich (St Louis, MO), total protein concentration was assessed by Quick Start™ Bradford dye reagent (BioRad, Hercules, CA), mAbs for Western blotting from Cell Signaling Technologies (Danvers, MA), and Annexin/PI assay from BD Biosciences (San Jose, CA). Red blood cell (RBC) lysis buffer 10 × was obtained from BioLegend (San Diego, CA). Drugs including bortezomib and carfilzomib were purchased from Selleck Chemicals (Houston, TX).
Cells
The MM cell lines (MM.1S, MM.1R, H929, RPMI-8826, and OPM2) were a kind gift from Dr Irene Ghobrial, Dana-Farber Cancer Institute, Harvard Medical School, Boston, MA. All cells were cultured at 37 °C, 5% CO2; MM cells in RPMI 1640 media (Corning CellGro, Mediatech, Manassas, VA) supplemented with 10% fetal bovine serum (FBS, Gibco, Life technologies, Grand Island, NY), 2 mmol/L of L-glutamine, 100 U/mL Penicillin and 100 μg/mL Streptomycin (CellGro, Mediatech, Manassas, VA). Cells were incubated at 37 °C under normoxic conditions (21% O2) or hypoxic conditions (1% O2) in the hypoxic chamber from Coy (Grass Lake, MI).
Peripheral blood mononuclear cells (PBMCs) were isolated from pheresis leukopaks from the Siteman Cancer Center, Washington University in Saint Louis. 1 × RBC Lysis Buffer was added to whole blood, gently vortex and incubated at room temperature, protected from light, for 10–15 min. Cell were washed and cultured in RPMI completed media. Informed consent was obtained from all patients with an approval from the Washington University Medical School IRB committee and in accord with the Declaration of Helsinki.
Cell viability analysis
MM cell lines (MM.1S, MM.1R, H929, RPMI8826, and OPM2) and PBMCs were cultured with Tris DBA (0–10 μM) for 24 h. Cell viability was assessed using MTT solution followed by absorbance reading at 570 nm using a spectrophotometer. Briefly, the MTT solution was added to the cells 24 h after starting the treatment, and 2–4 h later the stop solution was added. In addition, cell proliferation assay of Tris DBA (0, 2 μM) with or without combination of bortezomib (0, 5 nM) or carfilzomib (0, 2.5 nM) for 24 h was analyzed on the proliferation of MM.1S cells in normoxic or hypoxic conditions (6 h in normoxia plus 18 h in hypoxia).
Cell cycle analysis
MM cell lines (MM.1S, MM.1R, H929) (1×106 cell/mL) were cultured with Tris DBA (0, 2 μM) for 24 h. Cells were washed, fixed with 70% ethanol, and washed again with PBS. RNA was degraded by incubation in RNAase for 30 min at 37°C, and the DNA was stained with PI solution for 10 min, then cells were analyzed by flow cytometry.
Apoptosis assay
MM cell lines (MM.1S, MM.1R, H929) (1×106 cell/mL) were cultured with Tris DBA (0, 2 μM) for 24 h. Cells were washed and resuspended in 1× Annexin binding buffer, incubated with Annexin for 15 min followed by staining with PI for extra 15 min, 1× binding buffer was added and the cells were analyzed with flow cytometry.
Western blotting
To test the effect of Tris DBA on proliferation, cell cycle, and apoptosis signaling, MM.1S cells were treated with Tris DBA (0, 2 μM) for 24 h. Cells were washed and lysed with 1× PMSF for 15 min. Protein concentration in the cell lysates was normalized and 50 μg of protein was loaded per lane. Electrophoresis was performed using NuPAGE 4–12% Bis-Tris gels (Novex, Life Technologies, Grand Island, NY, USA) and transferred to a nitrocellulose membrane using iBlot (Invitrogen, Life Technologies). Membranes were blocked with 5% non-fat milk in Tris-Buffered Saline/Tween20 (TBST) buffer and incubated with primary antibodies overnight at 4°C for proliferation signaling with S6R, ERK1/2, pS6R and pERK1/2; for cell cycle with Rb, pRb, p-Cyclin E, and Cyclin E; and for apoptosis with cleaved Caspase-3 and cleaved PARP. α-Tubulin was used as a loading control. The membranes were washed with TBST for 30 min, incubated for 1 h at room temperature with HRP-conjugated secondary antibody, washed, and developed using Novex ECL Chemiluminescent Substrate Reagent Kit (Invitrogen). Moreover, effect on MM.1S signaling of Tris DBA (0, 2 μM) with or without combination of bortezomib (0, 5 nM) or carfilzomib (0, 2.5 nM) for 24 h was evaluated. We performed quantitative analysis of the protein expression levels using Image Lab 4.1 Software expressed by ratio of protein/α-Tub*100 (for combination treatment) and normalized to no treatment (for proliferation, cell cycle and apoptosis). Quantitation of phospho-protein expression change was expressed as the ratios of phospho- to total protein expression levels.
Antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). S6 Ribosomal Protein (5G10) (rabbit mAb #2217), Phospho-S6 Ribosomal Protein (Ser235/236) (D57.2.2E) XP® (rabbit mAb #4858), p44/42 MAPK (Erk1/2) (137F5) (rabbit mAb #4695), Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E) XP® (rabbit mAb #4370), Rb (D20) (rabbit mAb #9313), pRb (Ser807/811) (rabbit mAb #9308), Cyclin E1 (HE12) (antibody #4129), Phospho-Cyclin E1 (Thr62) (antibody #4136), cleaved-Caspase-3 (Asp175) (5A1E) (rabbit mAb #9664), cleaved-PARP (Asp214) (D64E10) (rabbit mAb #5625), and α-Tubulin (11H10) (rabbit mAb #2125) were used at a dilution of 1:1000.
Migration assay
MM.1S cells were treated with Tris DBA (0, 2 μM) for 24 h in normoxic or hypoxic conditions (6 h in normoxia plus 18 h in hypoxia). The number of viable cells for the migration assay was adjusted to 1×106 cells/ml for all conditions. Then, cells were plated in the upper chamber of a Transwell migration plate (Costar, Corning) and were allowed to transmigrate into the lower chamber containing conditioned media of MM-derived stromal cells according to manufacturer’s instructions. After 4 h of incubation in normoxia or hypoxia, the cells which migrated to the lower chamber were counted using flow cytometry.
Expression of HIF1a
The expression of HIF1α in MM was previously reported to be affected by hypoxic conditions; therefore, we tested the effect of tris DBA on the expression of HIF1α in MM cells. MM.1S cells were treated with Tris DBA (0, 2 μM) for 24 h in normoxic or hypoxic conditions (6 h in normoxia plus 18 h in hypoxia). Then, MM cells were retrieved from 2D cultures by pipetting, washed, fixed and permeabilized (BD Cytofix/Cytoperm), incubated with AlexaFluor488-anti-HIF1α (clone H1α67; Novus Biologicals) (excitation, 488 nm; emission, 530/30 nm) on ice for 1 h, and analyzed by flow cytometry. MFI of AlexaFluor488-HIF1α was detected and normalized to normoxia untreated.
Statistical analysis
Experiments were performed in triplicates. Results are shown as mean ± standard deviation and analyzed using Student’s t-test for statistical significance, and were considered significantly different for p value less than 0.05. Combination index was calculated for the combination therapies to determine additive effect or synergy.
Results
Tris DBA reduces myeloma cell proliferation
The effect of Tris DBA on the proliferation of MM cell lines was tested, and found that it inhibited proliferation of all MM cell lines (MM.1S, MM.1R, H929, RPMI-8826, and OPM2) tested in a similar manner (Fig. 1Ai) at an IC50 around 1.4–3 μM (Fig. 1Aii). Furthermore, we verified that Tris DBA did not affect the proliferation of PBMCs from three healthy donors at the same range dose (Fig. 1B). We corroborated that Tris DBA decreased the expression of proteins involved in survival and proliferation, including decreased pS6R and downstream signaling pERK1/2 in MM.1S cells, while total proteins were intact (Fig. 1C).
Figure 1.
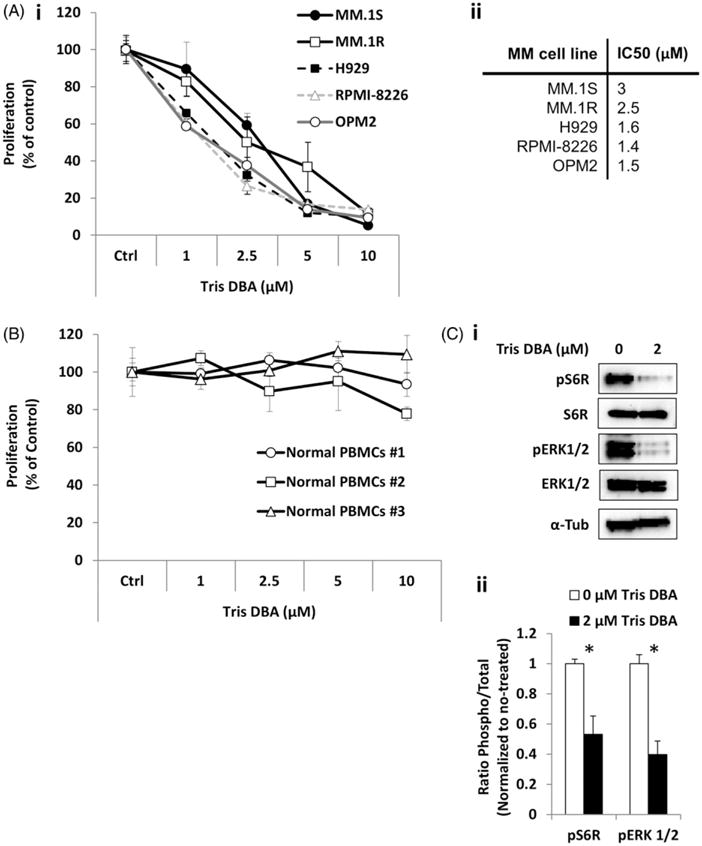
Effect of Tris DBA on MM proliferation. (A) The effect of Tris DBA (0–10 μM) on proliferation of MM.1S, MM.1R, H929, RPMI8826, and OPM2 cells for 24 h analyzed by MTT (i), and IC50 for each cell line is presented in the Table (ii). (B) The effect Tris DBA (0–10 μM) for 24 h on proliferation of normal PBMCs. (C) The effect of treatment with Tris DBA (0, 2 μM) for 24 h on proliferation signaling, S6R, pS6R, ERK1/2, and pERK1/2 in MM.1S cells by Western blotting (i), and quantification of phosphoprotein expression as ratio to total and normalized to no treatment by densitometry (ii). *p<0.05.
Tris DBA causes cell cycle arrest
We examined the role of Src inhibition in cell cycle of MM cell lines (MM.1S, H929, and MM.1R); we found that Tris DBA induced accumulation of apoptotic cells in subG1 as shown in representative flow cytometry graphs (Fig. 2A). The three cell lines showed increased sub G1 phase after 24 h of treatment (Fig. 2B). These results were confirmed by immunoblotting which showed that Tris DBA reduced the expression of phospho-proteins associated with the G1/S transition (pRb and pCyclin E), while total proteins were intact (Fig. 2C).
Figure 2.
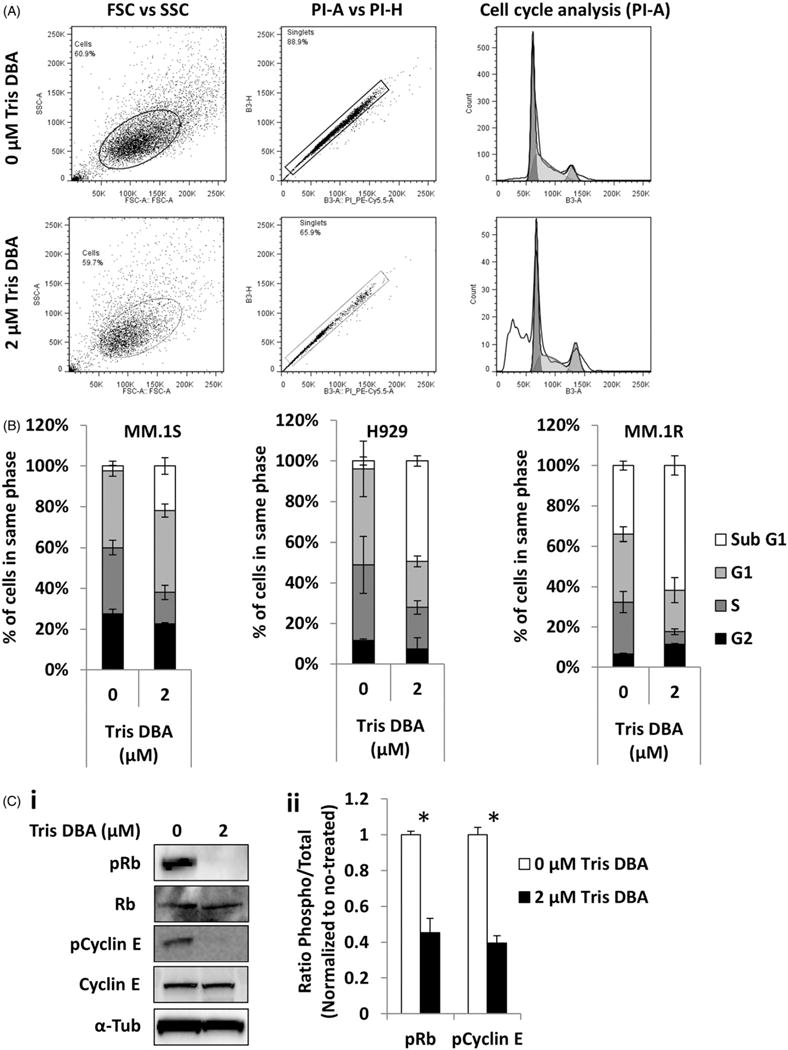
Effect of Tris DBA on cell cycle regulation. (A) Cell cycle analysis of Tris DBA (0, 2 μM) for 24 h flow cytometry graphs showing the differences. (B) MM.1S, H929, and MM.1R cells representing percent of population in each cell phase. (C) The effect of treatment for 24 h with Tris DBA (0, 2 μM) on cell cycle signaling Rb, pRb, pCyclin E, and Cyclin E in MM.1S cells by Western blotting (i), and quantification of phosphoprotein expression as ratio to total and normalized to no treatment by densitometry (ii). *p<0.05.
Tris DBA induces apoptosis
We then tested the effect of Tris DBA on apoptosis and viability of MM cells (MM.1S, H929, and MM.1R); we found that the fraction of late apoptotic cells (Annexin+/PI+) and death cells (Annexin−/PI+) was significantly increased in the treated cells (Fig. 3Ai), which was confirmed in flow cytometry dot plots (Fig. 3Aii). We also tested the activation of apoptotic pathways, and found induction of Cleaved-Caspase-3 and Cleaved-PARP in MM.1S cells (Fig. 3B).
Figure 3.
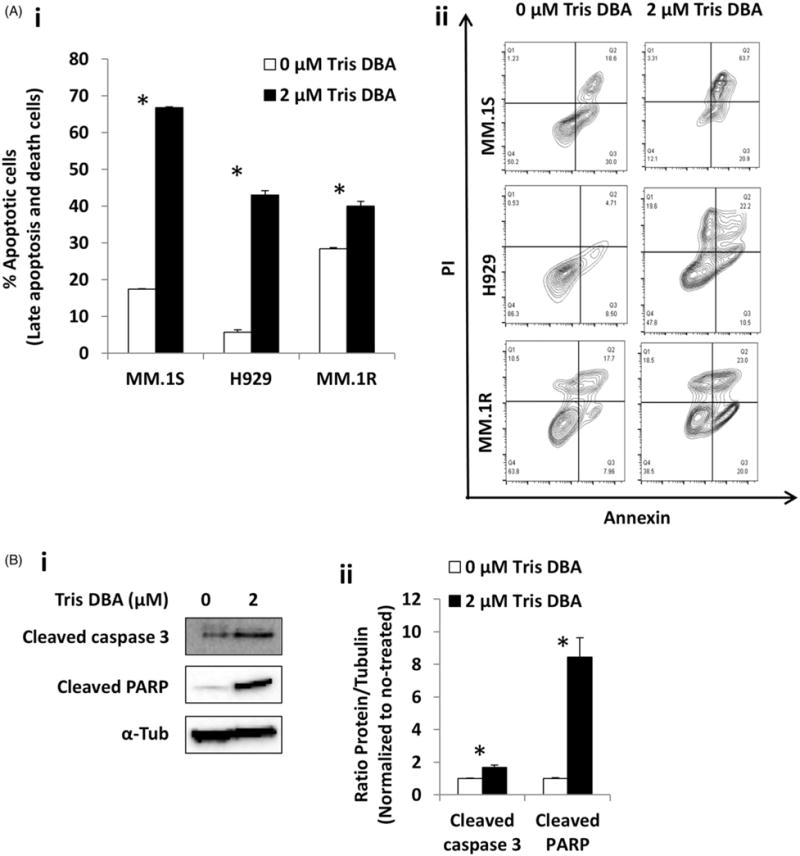
Effect of Tris DBA on apoptosis. (A) Apoptosis analysis of Tris DBA (0, 2 μM) in MM.1S, H929, and MM.1R cells for 24 h representing late apoptosis and death population (i) and flow cytometry dot plots showing the differences (ii). (B) The effect of treatment for 24 h with Tris DBA (0, 2 μM) on apoptosis signaling cleaved caspase 3 and cleaved PARP in MM.1S cells by Western blotting (i), and quantification of protein expression as ratio to tubulin and normalized to no treatment by densitometry (ii). *p<0.05.
Tris DBA shows additive effect with proteasome inhibitors and overcomes hypoxia drug effects
We further tested the effect of the combination of Tris DBA (0, 2 μM) with or without combination of bortezomib (0, 5 nM) or carfilzomib (0, 2.5 nM) for 24 h on proliferation of MM.1S cells under normoxic or hypoxic conditions. We found that the combination of Tris DBA with proteasome inhibitors (PIs) decreased the surviving fraction of MM cells more than each of the drugs alone. Under normoxic conditions, treatment with Tris DBA or bortezomib alone showed around 25–30% killing, while the combination of both drugs showed an enhanced 60% killing effect (Fig. 4Ai). Similar results in normoxia were observed for carfilzomib treatment alone or in combination (Fig. 4Aii). Additionally, compared to their normoxic control, treatments with only bortezomib (Fig. 4Ai) or carfilzomib (Fig. 4Aii) in hypoxia exhibited markedly reduced cell killing due to hypoxia-induced drug resistance of MM cells. However, the combination treatment of Tris DBA with bortezomib (Fig. 4Ai) or carfilzomib (Fig. 4Aii) were able to overcome the hypoxia-induced drug resistance to PIs.
Figure 4.
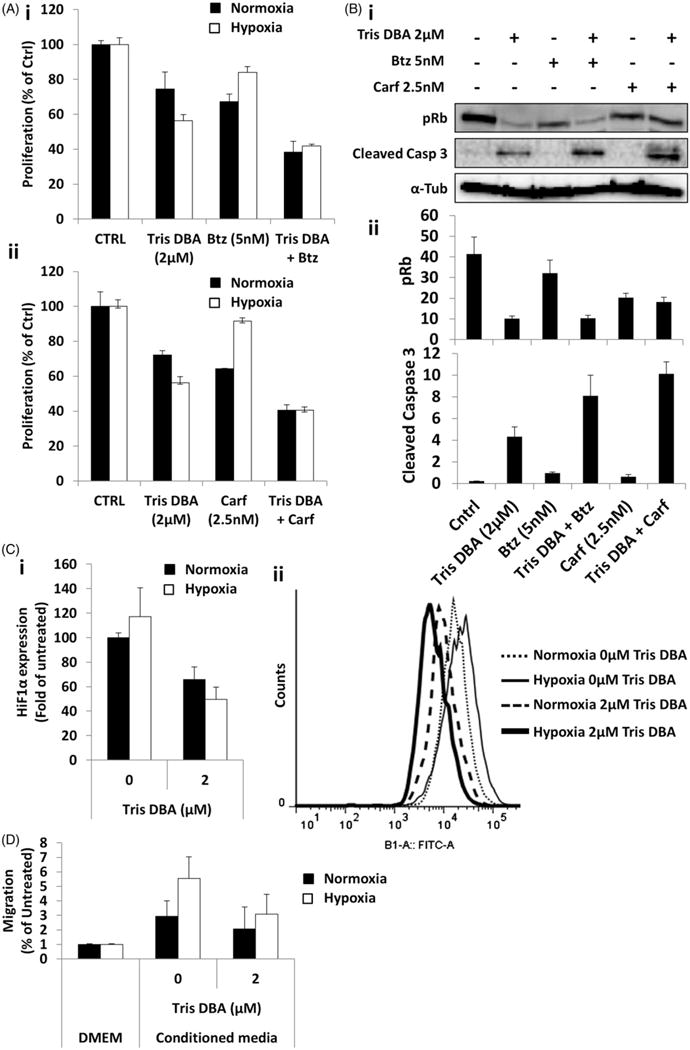
Tris DBA showed additive effect with proteasome inhibitors and overcomes hypoxia-induced effects. (A) Cell viability assay of Tris DBA (0, 2 μM) under normoxic or hypoxic conditions, with or without combination of bortezomib (0, 5 nM) (i) or carfilzomib (0, 2.5 nM) (ii) for 24 h. (B) Cell cycle signaling (pRb) and apoptosis signaling (cleaved caspase 3) in MM.1S cells by Western blotting (i), of the effect of the combination of Tris DBA (0, 2 μM), with or without Bortezomib (0, 5 nM) and Carfilzomib (0, 2.5 nM), and quantification of protein expression as ratio to tubulin by densitometry (ii). (C) HIF1 expression measured as fold of MFI of AlexaFluor488-anti-HIF1 to normoxia untreated of Tris DBA (0, 2 μM) in MM.1S cells for 24 h under normoxic or hypoxic conditions (i), and flow cytometry representative histogram of HIF1α expression (AlexaFluor488) (ii). (D) Migration assay of Tris DBA (0, 2 μM) in MM.1S cells for 24 h under normoxic or hypoxic conditions.
Moreover, we tested the effect of combination treatment on cell cycle and apoptosis signaling under normoxic conditions. We found that the combination of Tris DBA and PIs decreased the expression of pRb which is involved in cell cycle transition from G1 to S phase, and also increased caspase-3 cleavage more than either drug alone (Fig. 4Bi). Quantification of densitometry bands showed the differences between the combination treatments or each of the drugs alone (Fig. 4Bii).
We then evaluated the effect of Tris DBA on HIF1α expression under normoxic or hypoxic conditions. Tris DBA decreased the expression of HIF1α in normoxic and hypoxic conditions (Fig. 4Ci). Flow histograms showed a clear shift of HIF1α expression after treatment (Fig. 4Cii). Finally, to test the effect of Tris DBA in hypoxia-derived effects on MM cells, we examined migration and drug resistance of MM.1S cells treated under normoxic or hypoxic conditions. We found that chemotaxis of MM.1S cells towards conditioned media from MM-derived stromal cells was reduced with Tris DBA treatment (Fig. 4D). While hypoxia induced approximately a 3-fold increase in migration compared to normoxic conditions, Src inhibition from Tris DBA was able to significantly decrease the hypoxic-induced migration.
Discussion
Despite recent progress in novel and targeted therapies, MM remains a therapeutically challenging incurable disease. The regulation of important cellular processes and its link to cancer presented Src as an attractive target for MM. Additionally, given its role in bone resorption, Src inhibitors may hold great promise for inhibiting progression of tumors with a propensity to metastasize to the bone, such as multiple myeloma. Multiple signaling pathways are activated in MM. N- or K-ras oncogenes are mutated frequently in myeloma [26], resulting in activation of MAPK signaling [27]. The PI3K-AKT pathway mediates proliferative and anti-apoptotic signals in MM, and its activity increases with progression of the disease [3,28]. Cell signaling targeted monotherapies (PI3K/AKT/mTOR, MAPK pathways) have yielded promising results in the MM field; however, their use alone is clearly insufficient to cure myeloma. Dasatinib, a multi-kinase inhibitor of several multiple myeloma-related targets (including Src family kinases, c-Kit, PDGFR, Bcr-Abl and ephrins), has shown preclinical activity, including antitumor synergy with bortezomib [20,29]. Tris DBA has previously been reported to have potent antitumor activity against B16 murine and A375 human melanoma in vivo by inhibiting several important pathways in melanoma, including MAPK and PI3K activation. In addition, Tris DBA was well tolerated in vivo [25]. In the present study, we suggest a novel strategy for improving MM treatment by overcoming the drug resistance common to first line MM therapies. To accomplish this, the specific inhibition of Src in MM cells by the organopalladium compound Tris DBA will be evaluated to reverse hypoxia-induced effects.
The Src inhibitor Tris DBA reduced the proliferation of all MM cell lines tested with an IC50 of about 1.5–3 μM after 24 h treatment as a single agent, while none of the normal PBMC controls showed effect on their proliferation in the same dose range. These results were consistent with the decreased expression of proliferation signaling proteins from MAPK pathways (pERK), as well as PI3K (pS6R), previously shown to be affected by Tris DBA in melanoma [25]. It is important to note that Tris DBA inhibits multiple genetic subtypes of myeloma cells; the chosen cell lines represented RAS wild-type (OPM2), as well as KRAS (MM.1S, RPMI-8226) and NRAS mutated cell lines (H929) [30,31]. Taken together, these data suggest that there is no relationship between RAS status of MM cells and the sensitivity of the cell lines to Tris DBA. Src inhibition also led to the induction of a sub-G1 peak, which indicated accumulating apoptotic cells shown by DNA staining with PI. Apoptosis was then analyzed by Annexin/PI and confirmed by cleavage of caspase-3 and PARP. Our data demonstrate that inhibition of Src in MM cells affects proliferation, cell cycle and apoptosis as a single agent therapy.
Proteasome inhibitors, such as bortezomib and carfilzomib significantly improved the treatment of MM. Carfilzomib has durable anticancer activity in patients with relapsed/refractory MM, including those previously treated with bortezomib; however, bortezomib studies showed that 60% of patients will develop resistance to the treatment [4,32]. In the current study we suggested combining Src inhibition with bortezomib or carfilzomib treatments to overcome the observed resistance when using proteasome inhibitors in MM. We found that Tris DBA showed additive effect with bortezomib and carfilzomib by inhibiting proliferation of MM cells and reducing cell cycle protein signaling more than either of the drugs alone. Moreover, the Tris DBA/Bortezomib or Tris DPA/Carfilzomib combination therapies significantly increased apoptosis by caspase-3 cleavage more than treatment with either proteasome inhibitor individually.
The bone marrow of MM patients was shown to be hypoxic [33,34]. Tumor growth in MM was found to induce hypoxic conditions in the BM with a direct correlation, and in turn results in hypoxia induced dissemination and spread of MM [33]. Several studies have indicated that the hypoxic microenvironment contributes to cancer progression and hypoxia was also related to induced drug resistance [35–37]. We have recently found that hypoxia promotes stem cell-like properties and resistance to proteasome inhibitors in MM and other hematologic malignancies [38–40]. Moreover, it was recently shown that inhibition of HIF1α restores sensitivity to therapeutic agents such as bortezomib [41]. Our results confirmed that Tris DBA inhibited HIF1α expression in both normoxic and hypoxic conditions. HIF1α is an important target for hypoxia-driven drug resistance. Our studies confirmed hypoxia promoted faster chemotaxis of MM cells towards the chemo-attractants found in stromal cell conditioned media, and that Tris DBA treatment could overcome this hypoxia-induced effect. In addition, the development of hypoxia-induced drug resistance to individual bortezomib or carfilzomib treatment was overcome with combination treatment of Tris DBA under hypoxic conditions.
Tris DBA is a novel effective anti-myeloma agent with significant antitumor activity and ability to overcome hypoxia mediated drug resistance. Tris DBA is potentially useful as anti-drug resistance agent for the treatment of MM. Further preclinical evaluation of Tris DBA is warranted for future clinical application.
Conclusions
In summary, Tris DBA reduces proliferation and induces G1 arrest and apoptosis in MM cells. Tris DBA showed additive effect with proteasome inhibitors reducing proliferation and cell cycle signaling, as well as increasing apoptosis more than each drug alone. Tris DBA overcame hypoxia-induced effects such as enhanced chemotaxis or drug resistance to proteasome inhibitors by inhibition of HIF1α expression. Moreover, we found that Tris DBA is an effective anti-myeloma agent alone or in combination with other targeted drugs and that it reverses hypoxia-induced drug resistance in myeloma. These results suggest the use of Tris DBA as a new therapeutic agent in relapsed refractory myeloma.
Acknowledgments
Dr Arbiser is listed as inventor on US patent 8030299. Dr Azab receives research support from Verastem, Selexys, Karyopharm and Cell Works, and is the founder and owner of Targeted Therapeutics LLC.
Footnotes
Authors’ contributions
P.P. designed the study, performed experiments, analyzed, interpreted the data and wrote the manuscript; F.A. performed experiments and evaluated the data; B.M. and M.L. evaluated the data, and reviewed the manuscript; A.K.A and J.A designed and supervised the study, interpreted the data and edited the manuscript. All authors reviewed and approved the manuscript.
Potential conflict of interest: Disclosure forms provided by the authors are available with the full text of this article at http://dx.doi.org/10.3109/10428194.2015.1099645.
Other authors state no conflicts of interest.
References
- 1.Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc. 2003;78:21–33. doi: 10.4065/78.1.21. [DOI] [PubMed] [Google Scholar]
- 2.Jemal A, Murray T, Ward E, et al. Cancer statistics, 2005. CA Cancer J Clin. 2005;55:10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 3.de la Puente P, Muz B, Azab F, et al. Molecularly targeted therapies in multiple myeloma. Leuk Res Treatment. 2014;2014:976567. doi: 10.1155/2014/976567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.de la Puente P, Azab AK. Contemporary drug therapies for multiple myeloma. Drugs Today (Barc) 2013;49:563–573. doi: 10.1358/dot.2013.49.9.2020941. [DOI] [PubMed] [Google Scholar]
- 5.Kumar SK, Rajkumar SV, Dispenzieri A, et al. Improved survival in multiple myeloma and the impact of novel therapies. Blood. 2008;111:2516–2520. doi: 10.1182/blood-2007-10-116129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mitsiades CS, Hayden PJ, Anderson KC, et al. From the bench to the bedside: emerging new treatments in multiple myeloma. Best Pract Res Clin Haematol. 2007;20:797–816. doi: 10.1016/j.beha.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene. 2004;23:7906–7909. doi: 10.1038/sj.onc.1208160. [DOI] [PubMed] [Google Scholar]
- 8.Mazurenko NN, Kogan EA, Zborovskaya IB, et al. Expression of pp60c-src in human small cell and non-small cell lung carcinomas. Eur J Cancer. 1992;28:372–377. doi: 10.1016/s0959-8049(05)80056-5. [DOI] [PubMed] [Google Scholar]
- 9.Irby RB, Yeatman TJ. Role of Src expression and activation in human cancer. Oncogene. 2000;19:5636–5642. doi: 10.1038/sj.onc.1203912. [DOI] [PubMed] [Google Scholar]
- 10.Egan C, Pang A, Durda D, et al. Activation of Src in human breast tumor cell lines: elevated levels of phosphotyrosine phosphatase activity that preferentially recognizes the Src carboxy terminal negative regulatory tyrosine 530. Oncogene. 1999;18:1227–1237. doi: 10.1038/sj.onc.1202233. [DOI] [PubMed] [Google Scholar]
- 11.Cartwright CA, Meisler AI, Eckhart W. Activation of the pp60c-src protein kinase is an early event in colonic carcinogenesis. Proc Natl Acad Sci USA. 1990;87:558–562. doi: 10.1073/pnas.87.2.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lutz MP, Esser IB, Flossmann-Kast BB, et al. Overexpression and activation of the tyrosine kinase Src in human pancreatic carcinoma. Biochem Biophys Res Commun. 1998;243:503–508. doi: 10.1006/bbrc.1997.8043. [DOI] [PubMed] [Google Scholar]
- 13.Bjorge JD, O’Connor TJ, Fujita DJ. Activation of human pp60c-src. Biochem Cell Biol. 1996;74:477–484. doi: 10.1139/o96-052. [DOI] [PubMed] [Google Scholar]
- 14.Belsches-Jablonski AP, Demory ML, Parsons JT, et al. The Src pathway as a therapeutic strategy. Drug Discov Today: Therapeut Strat. 2005;2:313–321. [Google Scholar]
- 15.Summy J, Gallick G. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003;22:337–358. doi: 10.1023/a:1023772912750. [DOI] [PubMed] [Google Scholar]
- 16.Irby RB, Mao W, Coppola D, et al. Activating SRC mutation in a subset of advanced human colon cancers. Nat Genet. 1999;21:187–190. doi: 10.1038/5971. [DOI] [PubMed] [Google Scholar]
- 17.Gertz MA. New targets and treatments in multiple myeloma: Src family kinases as central regulators of disease progression. Leuk Lymphoma. 2008;49:2240–2245. doi: 10.1080/10428190802475311. [DOI] [PubMed] [Google Scholar]
- 18.Hallek M, Neumann C, Schaffer M, et al. Signal transduction of interleukin-6 involves tyrosine phosphorylation of multiple cytosolic proteins and activation of Src-family kinases Fyn, Hck, and Lyn in multiple myeloma cell lines. Exp Hematol. 1997;25:1367–1377. [PubMed] [Google Scholar]
- 19.Ishikawa H, Tsuyama N, Abroun S, et al. Requirements of src family kinase activity associated with CD45 for myeloma cell proliferation by interleukin-6. Blood. 2002;99:2172–2178. doi: 10.1182/blood.v99.6.2172. [DOI] [PubMed] [Google Scholar]
- 20.Coluccia AML, Cirulli T, Neri P, et al. Validation of PDGFRb and c-Src tyrosine kinases as tumor/vessel targets in patients with multiple myeloma: preclinical efficacy of the novel, orally available inhibitor dasatinib. Leuk Lymphoma. 2008;112:1346–1356. doi: 10.1182/blood-2007-10-116590. [DOI] [PubMed] [Google Scholar]
- 21.Sandur SK, Pandey MK, Sung B, et al. 5-Hydroxy-2-methyl-1,4-naphthoquinone, a vitamin K3 analogue, suppresses STAT3 activation pathway through induction of protein tyrosine phosphatase, SHP-1: potential role in chemosensitization. Molec Cancer Res. 2010;8:107–118. doi: 10.1158/1541-7786.MCR-09-0257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu YX, Zhang XR, Li QS, et al. Observation of the operation of epiphora caused by plica semilunaris laxation. Int J Ophthalmol. 2010;10:145–146. [Google Scholar]
- 23.Nowak D, Boehrer S, Hochmuth S, et al. Src kinase inhibitors induce apoptosis and mediate cell cycle arrest in lymphoma cells. Anti-Cancer Drugs. 2007;18:981–995. doi: 10.1097/CAD.0b013e3281721ff6. [DOI] [PubMed] [Google Scholar]
- 24.Tsubaki M, Komai M, Itoh T, et al. By inhibiting Src, verapamil and dasatinib overcome multidrug resistance via increased expression of Bim and decreased expressions of MDR1 and survivin in human multidrug-resistant myeloma cells. Leuk Res. 2014;38:121–130. doi: 10.1016/j.leukres.2013.10.017. [DOI] [PubMed] [Google Scholar]
- 25.Bhandarkar SS, Bromberg J, Carrillo C, et al. Tris (dibenzylideneacetone) dipalladium, a N-myristoyltransferase-1 inhibitor, is effective against melanoma growth in vitro and in vivo. Clin Cancer Res. 2008;14:5743–5748. doi: 10.1158/1078-0432.CCR-08-0405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Neri A, Murphy JP, Cro L, et al. Ras oncogene mutation in multiple myeloma. J Exp Med. 1989;170:1715–1725. doi: 10.1084/jem.170.5.1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Roberts PJ, Der CJ. Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene. 2007;26:3291–3310. doi: 10.1038/sj.onc.1210422. [DOI] [PubMed] [Google Scholar]
- 28.Hsu J, Shi Y, Krajewski S, et al. The AKT kinase is activated in multiple myeloma tumor cells. Blood. 2001;98:2853–2855. doi: 10.1182/blood.v98.9.2853. [DOI] [PubMed] [Google Scholar]
- 29.Luo FR, Barrett YC, Yang Z, et al. Identification and validation of phospho-SRC, a novel and potential pharmacodynamic biomarker for dasatinib (SPRYCEL), a multi-targeted kinase inhibitor. Cancer Chemother Pharmacol. 2008;62:1065–1074. doi: 10.1007/s00280-008-0699-5. [DOI] [PubMed] [Google Scholar]
- 30.Chesi M, Brents LA, Ely SA, et al. Activated fibroblast growth factor receptor 3 is an oncogene that contributes to tumor progression in multiple myeloma. Leuk Lymphoma. 2001;97:729–736. doi: 10.1182/blood.v97.3.729. [DOI] [PubMed] [Google Scholar]
- 31.Steinbrunn T, Stuhmer T, Gattenlohner S, et al. Mutated RAS and constitutively activated Akt delineate distinct oncogenic pathways, which independently contribute to multiple myeloma cell survival. Blood. 2011;117:1998–2004. doi: 10.1182/blood-2010-05-284422. [DOI] [PubMed] [Google Scholar]
- 32.San Miguel JF, Schlag R, Khuageva NK, et al. Bortezomib plus melphalan and prednisone for initial treatment of multiple myeloma. N Engl J Med. 2008;359:906–917. doi: 10.1056/NEJMoa0801479. [DOI] [PubMed] [Google Scholar]
- 33.Azab AK, Hu J, Quang P, et al. Hypoxia promotes dissemination of multiple myeloma through acquisition of epithelial to mesenchymal transition-like features. Blood. 2012;119:5782–5794. doi: 10.1182/blood-2011-09-380410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Asosingh K, De Raeve H, de Ridder M, et al. Role of the hypoxic bone marrow microenvironment in 5T2MM murine myeloma tumor progression. Haematologica. 2005;90:810–817. [PubMed] [Google Scholar]
- 35.Unruh A, Ressel A, Mohamed HG, et al. The hypoxia-inducible factor-1 alpha is a negative factor for tumor therapy. Oncogene. 2003;22:3213–3220. doi: 10.1038/sj.onc.1206385. [DOI] [PubMed] [Google Scholar]
- 36.Rohwer N, Cramer T. Hypoxia-mediated drug resistance: novel insights on the functional interaction of HIFs and cell death pathways. Drug Resist Updat. 2011;14:191–201. doi: 10.1016/j.drup.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 37.Brown JM, Siim BG. Hypoxia-specific cytotoxins in cancer therapy. Semin Radiat Oncol. 1996;6:22–36. doi: 10.1053/SRAO0060022. [DOI] [PubMed] [Google Scholar]
- 38.Muz B, de la Puente P, Azab F, et al. Hypoxia promotes dissemination and colonization in new bone marrow niches in Waldenström macroglobulinemia. Mol Cancer Res. 2015;13:263–272. doi: 10.1158/1541-7786.MCR-14-0150. [DOI] [PubMed] [Google Scholar]
- 39.Muz B, de la Puente P, Azab F, et al. Hypoxia promotes stem cell-like phenotype in multiple myeloma cells. Blood Cancer J. 2014;4:e262. doi: 10.1038/bcj.2014.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Muz B, de la Puente P, Azab F, et al. The role of hypoxia and exploitation of the hypoxic environment in hematologic malignancies. Mol Cancer Res. 2014;12:1347–1354. doi: 10.1158/1541-7786.MCR-14-0028. [DOI] [PubMed] [Google Scholar]
- 41.Maiso P, Huynh D, Moschetta M, et al. Metabolic signature identifies novel targets for drug resistance in multiple myeloma. Cancer Res. 2015;75:2071–2082. doi: 10.1158/0008-5472.CAN-14-3400. [DOI] [PMC free article] [PubMed] [Google Scholar]