

Abstract
There is a clinical need to identify new molecular targets for the treatment of osteoporosis, particularly those that simultaneously inhibit bone resorption while stimulating bone formation. We have previously shown in overexpression studies that retinoic acid receptor-related orphan receptor β (Rorβ) suppresses in vitro osteoblast differentiation. In addition, the expression of Rorβ is markedly increased in bone marrow–derived mesenchymal stromal cells with aging in both mice and humans. Here we establish a critical role for Rorβ in regulating bone metabolism using a combination of in vitro and in vivo studies. We used Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 gene editing to demonstrate that loss of Rorβ in osteoblasts enhances Wnt signaling, specifically through increased recruitment of β-catenin to T-cell factor/lymphoid enhancer factor (Tcf/Lef) DNA binding sites in the promoters of the Wnt target genes Tcf7 and Opg. This resulted in increased osteogenic gene expression and suppressed osteoclast formation through increased osteoprotegerin (OPG) secretion in Rorβ-deficient cells. Consistent with our in vitro data, genetic deletion of Rorβ in both female and male mice resulted in preserved bone mass and microarchitecture with advancing age due to increased bone formation with a concomitant decrease in resorption. The improved skeletal phenotype in the Rorβ−/− mice was also associated with increased bone protein levels of TCF7 and OPG. These data demonstrate that loss of Rorβ has beneficial skeletal effects by increasing bone formation and decreasing bone resorption, at least in part through β-catenin–dependent activation of the Wnt pathway. Thus, inhibition of Rorβ represents a novel approach to potentially prevent or reverse osteoporosis.
Keywords: AGING, OSTEOPOROSIS, OSTEOBLASTS, OSTEOCLASTS, TRANSCRIPTION FACTORS
Introduction
Osteoporosis is an enormous, and growing, public health problem.(1) Characterized by low bone mass and defects in bone microarchitecture, osteoporosis leads to increased susceptibility to fracture.(2) Proper maintenance of skeletal health throughout life relies on the coordinate and balanced actions of bone-resorbing osteoclasts and bone-forming osteoblasts, which results in net bone balance. However, with aging these actions become unbalanced (or “uncoupled”), whereby osteoclastic bone resorption exceeds osteoblastic bone formation, leading to negative bone balance and eventual bone loss.(2) Thus, achieving “positive” bone balance or net bone gain is the primary goal in treating osteoporosis.
The clinically available therapies for osteoporosis can be classified as either antiresorptive (bisphosphonates, denosu- mab, estrogen, and raloxifene; for a review, see Khosla and Hofbauer(3)), which target the bone resorbing osteoclasts, or anabolic, which increase bone formation. Currently, the only US Food and Drug Administration (FDA)-approved anabolic therapies are teriparatide (PTH 1-34) and the PTHrP analog, abaloparatide.(4) However, treatment with these agents is limited to a lifetime maximum duration of 24 months due to the increased risk of osteosarcoma,(5,6) and consequently following cessation of treatment clinicians must resort to antiresorptive drugs in treating these patients. In addition, each of these drugs either decreases bone resorption or increases bone formation; no currently available drug does both simultaneously. Thus, a more complete understanding of the fundamental molecular mechanisms and cellular pathways that regulate bone resorption and formation is needed to develop novel therapies to prevent or even reverse osteoporosis.
We have recently published evidence indicating that the retinoic acid receptor-related orphan receptor beta (Rorβ) is a novel negative regulator of osteoblastic differentiation and that Rorβ expression is highly elevated in bone marrow–derived osteoprogenitor cells isolated from old, osteoporotic mice, suggesting a potential role for Rorβ in mediating age-related bone loss.(7) Overexpression studies in the preosteoblastic mouse cell line, MC3T3-E1, demonstrated significant regulation of known osteogenic pathways,(8) supporting a key role of Rorβ for regulating osteogenesis. However, examination of the effects of Rorβ deletion in bone, either in vitro or in vivo, has not been described.
To address this issue, we generated a Rorβ -deficient MC3T3- E1 preosteoblastic cell line using the Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)/Cas9 gene editing system.(9) These cells exhibit a markedly increased osteogenic gene signature during osteoblastic differentiation with specific upregulation of osteoprotegerin (Opg, Tnfsfr11b), which suppresses osteoclast formation, as well as upregulation of Tcf7, which is part of the bone anabolic Wnt pathway. Mechanistically, we found that increased Wnt signaling in these Rorβ-deficient cells was due to enhanced β-catenin recruitment to the promoters of both Tcf7 and Opg genes, leading to increased recruitment of RNA polymerase and ultimately increased gene expression. Examination of the in vivo skeletal phenotype of the Rorβ-deficient mouse model (Rorβ−/−) revealed significantly better bone mass and microarchitecture at both the femur and spine with markedly decreased osteoclast numbers and activity as well as increased bone formation rates. Collectively, our data demonstrate that loss of Rorβ both in vitro and in vivo has beneficial effects on the skeleton by increasing bone formation and decreasing bone resorption. Our findings suggest that inhibition of Rorβ may represent a novel approach to prevent or treat osteoporosis.
Materials and Methods
Mice
Mice harboring a Rorβ 1 knockout allele (Rorβ−/−) and wild-type (WT) littermate controls (all in C57/B6 background) were housed in ventilated cages within an accredited facility under a 12-hour light/dark cycle and constant temperature (23°C), and had access to water and food ad libitum. The allele inactivated is the Rorβ 1 isoform encoded by the Rorβ genomic locus.(10) There were no adverse events reported in any experimental mouse group. All mice studies were conducted in accordance to NIH guidelines and as approved by the Institutional Animal Care and Use Committee at the Mayo Clinic (#A9715-15).
Cell culture
MC3T3-E1 mouse preosteoblasts were maintained in alpha-minimal essential growth medium (α-MEM) supplemented with 1 × antibiotic/antimycotic (ThermoFisher Scientific, Waltham, MA, USA), 1 × Glutamax, and 10% (vol/vol) fetal bovine serum (FBS; GE Healthcare Life Sciences HyClone Laboratories, Logan, UT, USA). For the osteoblast differentiation assays, growth media was supplemented with 50 mg/L ascorbic acid and 10mM β-glycerophosphate (Sigma-Aldrich, St. Louis, MO, USA). For experiments in which MC3T3-E1 cells were treated with dihydrotestosterone (DHT) or 17-β-estradiol (Sigma-Aldrich), the FBS in the cell culture medium was replaced with triple charcoal-stripped FBS (GE Healthcare Life Sciences HyClone Laboratories).
CRISPR/Cas9 deletion of the mouse Rorβ gene
Deletion of Rorβ in MC3T3-E1 cells was accomplished in vitro using the CRISPR/Cas9 system. For a complete description, see the Supporting Materials and Methods.
Transient transfection
The experimental conditions and assay details are described in full in the Supporting Material and Methods.
Lineage negative isolation
The hematopoietic cell lineage negative (lin–) population was isolated using magnetic-activated cell sorting (MACS) was performed using the mouse Lineage Cell Depletion Kit (Miltenyi Biotec, San Diego, CA, USA) as described.(11)
Western blotting
The specific conditions and procedure for the Western blot were performed as described.(12) Briefly, cytoplasmic and nuclear protein extracts were prepared(13) and equal amounts of protein were subjected to Western blot analysis. The blots were probed with activated (eg, nonphosphorylated) β-catenin (1:1000), total β-catenin (1:1000), lamin A/C (loading control for the nuclear extract; 1:1000), α-tubulin (loading control for the cytoplasmic extract; 1:100,000), and α-actin (loading control for total protein extracts). All primary antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA) and all species-specific horseradish peroxidase (HRP)-conjugated secondary antibodies (used at 1:5000) were purchased from Sigma-Aldrich. Bands were visualized and quantified using densitometry, following enhanced chemiluminescence, on the LI-COR Odyssey Fc Imaging System (LI-COR Biotechnology, Lincoln, NE, USA).
Isolation of osteocyte-enriched cells
Detailed methods and validation of our osteocyte-enriched cell isolation protocol are presented elsewhere.(14) Briefly, mouse vertebrae were stripped of muscle/connective tissues and minced into small pieces, which subsequently underwent two sequential 30-min collagenase digests (Endotoxin-free Liberase; Roche Diagnostics GmbH, Mannheim, Germany). As shown previously(14) the remaining cell fraction represents a highly enriched population of osteocytes used for quantitative polymerase chain reaction (QPCR) analyses.
QPCR analysis
QPCR analysis was conducted on an ABI Prism 7900HT Real-Time System (Applied Biosystems, Carlsbad, CA, USA) using SYBR Green (QIAGEN, Valencia, CA, USA), as described.(15) The statistical method for data normalization using multiple endogenous reference genes (in this study Actb, Hprt, and Tuba1a were used) and threshold calculations are as described.(15) Primer sequences for the genes analyzed in this study were designed using the Primer Express program (Applied Biosystems) (Supporting Fig. 9).
Chromatin immunoprecipitation assay
The details of the chromatin immunoprecipitation assay (ChIP) assays, including cell lines, assay conditions, and antibodies used, are detailed in the Supporting Materials and Methods.
Skeletal phenotyping and bone histomorphometry
Complete methods for the skeletal phenotyping using peripheral quantitative computed tomography (pQCT), high-resolution micro–computed tomography (μCT) scanning, ovariectomy (OVX)/orchidectomy (ORX), and bone histomorphometry can be found in the Supporting Materials and Methods.
Osteoclast differentiation assays
The procedures are as described(16) and are described in detail in the Supporting Materials and Methods.
Immunohistochemistry
Following fixation, tibias were decalcified for 2 weeks in 12.5% (vol/vol) EDTA and paraffin embedded. Antigen retrieval was performed with Proteinase K, 20 min at 37°C. Sections were stained overnight with antibodies against mouse TCF7 (1:100, Cat#2203; Cell Signaling Technology) and OPG (15 μg/mL, AF459; R&D Systems, Minneapolis, MN, USA). TCF7 staining was performed with VECTASTAIN Elite ABC HRP Kit, Peroxidase, Rabbit IgG (Vector Laboratories, Burlingame, CA, USA). OPG staining was performed with the Anti-Goat HRP–diaminobenzidine (DAB) Cell & Tissue Staining Kit (R&D Systems). Sections were counterstained with methyl green and mounted with Eukitt Mounting Medium (Sigma). Osteocytes were scored as positive or negative using the OsteoMeasure histomorphometry system (OsteoMetrics, Decatur, GA, USA), and data are expressed as percent of total osteocytes (n = 4 per group).
Enzyme-linked immunosorbent assay and bone turnover markers
Conditioned media (CM; diluted 1:5) from MC3T3-Cont and MC3T3-ΔRorβ was assayed for OPG protein using the Mouse Osteoprotegerin/TNFRSF11B Quantikine Enzyme-Linked Immunosorbent Assay (ELISA) Kit (R&D Systems). The bone formation serum marker amino-terminal propeptide of type I collagen (P1NP) was measured using the Rat/Mouse P1NP enzyme immunoassay (EIA); whereas the bone resorption serum marker cross-linked C-telopeptide of type I collagen (CTx) was measured by the RatLaps Rat/Mouse CTx EIA kit. Both kits were purchased from Immuno Diagnostic Systems (IDS, Scottsdale, AZ, USA). Serum sclerostin levels were measured by the Mouse Sclerostin ELISA (Alpco, Salem, NH, USA) and serum DKK1 levels were measured by the DKK1 Mouse ELISA Kit (ThermoFisher Scientific). Interassay coefficients of variation (CVs) for all kits were <10%.
RNA sequencing, Ingenuity Pathway Analysis, and molecular signatures database analyses
Total RNA was isolated from the day 2 differentiated MC3T3- Cont and MC3T3-ΔRorβ cell lines (n = 3) using the RNeasy Micro Kit (QIAGEN), which included a DNase step to remove contaminating DNA (RNase-free DNase Set; QIAGEN), on a QIAcube instrument (QIAGEN). Whole-transcriptome RNA sequencing (RNAseq) was performed as described(17) using the Medical Genome Facility Genotyping Core (GTC) at the Mayo Clinic. Data were analyzed through the use of QIAGEN’s Ingenuity Pathway Analysis (IPA; QIAGEN, Valencia, CA, USA; www.qiagen.com/ingenuity). The RNA sequencing data can be downloaded at Gene Expression Omnibus (GEO Accession Number GSE107849; https://www.ncbi.nlm.nih.gov/geo/).
Statistics
Values are expressed as mean ± SE unless otherwise specified. Data were checked for outliers and normality using histograms, and all variables were tested for skewness and kurtosis. Mean values were compared between two groups, as appropriate, using the Student’s two-tailed t test after determining that the data were normally distributed and exhibited equivalent variances. For experiments involving more than two groups, after determining normality and homogeneity of variances, mean values were compared using ANOVA models followed by the Fisher’s protected least-significant difference (PLSD) post hoc test. Comparisons of QPCR values between the MC3TC-Cont and MC3T3-ΔRorβ cell lines in the longitudinal osteoblast differentiation time course and longitudinal pQCT bone imaging analyses were based on a two-way repeated measures ANOVA model. Testing was performed at a significance level of p < 0.05 (two-tailed); analyses were performed using the IBM SPSS Statistics for Windows, Version 22.0 (IBM Corp., Armonk, NY, USA). Figures were created using GraphPad Prism, version 5.03 (GraphPad Software, Inc., La Jolla, CA, USA).
The RNAseq analysis was performed as described.(17) Gene expression data with a p value ≤0.05, q value ≤0.10, and median gene counts where at least one group had >10 counts, were considered significantly altered between the MC3T3-Cont and MC3T3-ΔRorβ. For canonical pathway analysis using IPA, QIAGEN requires a gene list of 100 to 2000 genes for proper pathway identification. Therefore, we needed to increase the q value to ≤1 × 1015, which resulted in 1944 differentially expressed genes to perform IPA. We used the Benjamini-Hochberg test for multiple comparisons; pathways with a p value ≤0.05 were considered statistically significant. The transcription factor binding site analysis was performed exactly as described(17) using the C3 module (motif gene sets) from the Molecular Signatures Database (MSigDB),(18) which identifies common, statistically enriched cis-regulatory DNA motifs derived from the gene list used in the IPA analysis. These motifs are catalogued and represent known or likely DNA regulatory elements contained within 2-kilobases (kb) of DNA sequence surrounding the particular gene of interest (eg, –2 kb to +2 kb relative to the transcriptional start site).(19)
Results
Deletion of Rorβ in MC3T3 cells enhances osteoblastic gene expression
We have previously shown that constitutive Rorβ expression in the MC3T3-E1 mouse preosteoblastic cell model leads to suppressed bone marker gene expression,(7) suggesting that Rorβ inhibits osteogenic differentiation. Thus, to more definitively establish this, we performed the opposite experiment whereby we deleted Rorβ in MC3T3-E1 cells using the CRISPR/Cas9 gene editing system.(9) Figure 1A illustrates the intron/exon structure of the mouse Rorβ gene, denoting the two promoters driving expression of the Rorβ1 and Rorβ2 isoforms. We have shown that only the Rorβ1 isoform is expressed in any bone cell population (cycle threshold values for Rorβ2 are undetectable); thus, when using the term Rorβ, we are referring to the Rorβ1 isoform. The ATG start codon of Rorβ1 lies immediately at the 3′ end of exon 1; therefore, we chose to target exon 2 for CRISPR/Cas9-mediated gene editing. To accomplish this, three independent guide RNA (gRNA) sequences were designed against the DNA sequence of mouse Rorβ exon 2 (Supporting Fig. 1A–B), which encodes the N-terminal region of Rorβ (amino acids 3–28). After testing the editing efficiency of each gRNA (Supporting Fig. 1C), Rorβ gRNA1 was chosen to delete Rorβ in MC3T3-E1 cells (henceforth termed MC3T3-ΔRorβ), because of its minimal predicted off-target effects. A control cell line, “MC3T3-Cont,” was also produced using a nonspecific gRNA. Cloning and sequence analysis of the MC3T3-ΔRorβ cell line revealed a 1-basepair (bp) and 4-bp CRISPR/Cas9-mediated deletion in the mouse Rorβ alleles (Fig. 1B), which leads to frameshift mutations that ultimately resulted in nonfunctional alleles. In order to evaluate the effects of Rorβ deletion on osteoblastic gene expression, the MC3T3-Cont and MC3T3-ΔRorβ cells were differentiated in osteogenic medium for 0 to 10 days and RNA samples were collected at various time points. QPCR analyses revealed significantly increased expression of bone marker genes in MC3T3-ΔRorβ cells throughout the time course, including Bglap (Fig. 1C), Alpl (Fig. 1D), Ibsp (Fig. 1E), and Sp7 (Fig. 1F); no difference was detected for the bone-specific isoform(20,21) of Runx2 (Fig. 1G).
Fig. 1.
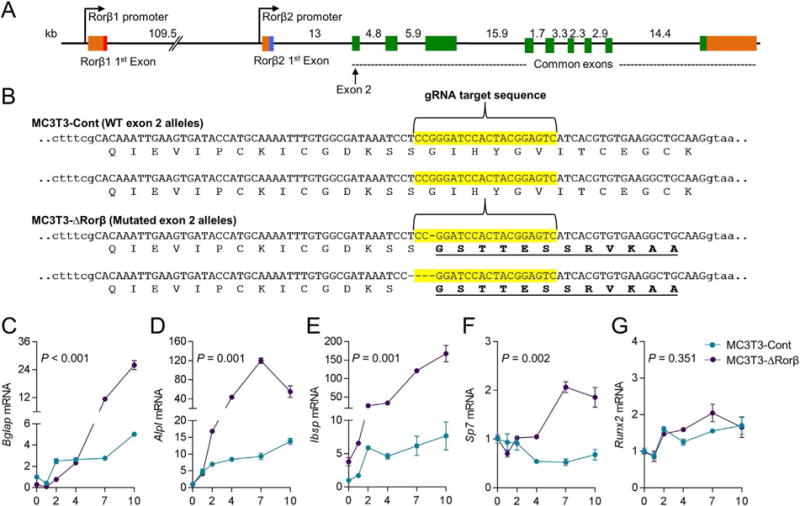
CRISPR/Cas9 deletion of Rorβ in MC3T3-E1 preosteoblastic cells. (A) The intron/exon structure of the mouse Rorβ gene is shown. (B) Exon 2 (in uppercase) of the mouse Rorβ gene was mutated using CRISPR/Cas9 gene editing methodology. The guide RNA (gRNA) sequence used to target the allele is highlighted in yellow. The translated amino acid sequence for each allele of the MC3T3-Cont and MC3T3-ΔRorβ cell lines is shown underneath the respective DNA sequence, illustrating the frameshift mutations in the latter cell line. The dashes in the DNA sequence represent single base pair deletions. (C–G) Both cell models were treated with osteogenic medium at confluence and QPCR for the denoted bone markers genes was performed on samples collected at days 0 to 10 (n = 6). Values are expressed as fold change (mean ± SE) relative to day 0. Values of p are shown for repeated-measures ANOVA.
Rorβ-deficient osteoblasts display enhanced Wnt signaling through β-catenin–dependent mechanisms
Whole RNA transcriptome sequencing (RNAseq) was performed on RNA samples isolated from MC3T3-Cont and MC3T3-ΔRorβ at day 2 of osteoblast differentiation to identify those genes and pathways that were altered early in the time course, potentially uncovering important mechanistic insights leading to the enhanced osteogenic phenotype. The RNAseq analysis and criteria used for cutoffs are detailed in Materials and Methods. Following RNAseq analysis and in order to identify canonical cellular pathways significantly altered in the MC3T3-ΔRorβ cell line, IPA was performed on a subset of these regulated genes and revealed that several pathways were significantly altered in MC3T3-ΔRorβ cells. Supporting Fig. 2 lists 10 pathways with known functions in bone metabolism or Rorβ biology.(8) Of note, a significant alteration in the Wnt/β-catenin pathway was observed and expression of several well-known regulators of this pathway were modulated in MC3T3-ΔRorβ cells, including Tcf7 (5.3-fold), Lef1 (2.7-fold), Wnt10a (1.8-fold), Wnt10b (2.3-fold), and Dkk3 (0.53-fold) (Fig. 2A, B). Examination of the cis-regulatory genomic DNA sequences of the IPA-analyzed gene set using the Molecular Signatures Database (MSigDB)(18) revealed significant enrichment in T-cell factor/lymphoid enhancer factor (Tcf/Lef) DNA binding sites (Supporting Fig. 3). These data show that loss of Rorβ in osteoblastic cells leads to alterations in the Wnt/β-catenin pathway. To better understand the cellular and molecular mechanism of increased Wnt signaling in Rorb-deficient osteoblasts, we first examined whether the subcellular localization of activated β-catenin (eg, hypophosphorylated) was altered in Rorb-deficient osteoblasts. We found no difference in subcellular localization of activated β-catenin or total cellular β-catenin, suggesting loss of Rorβ does not affect Wnt activity through altered β-catenin compartmentalization (Supporting Fig. 4A, B).
Fig. 2.
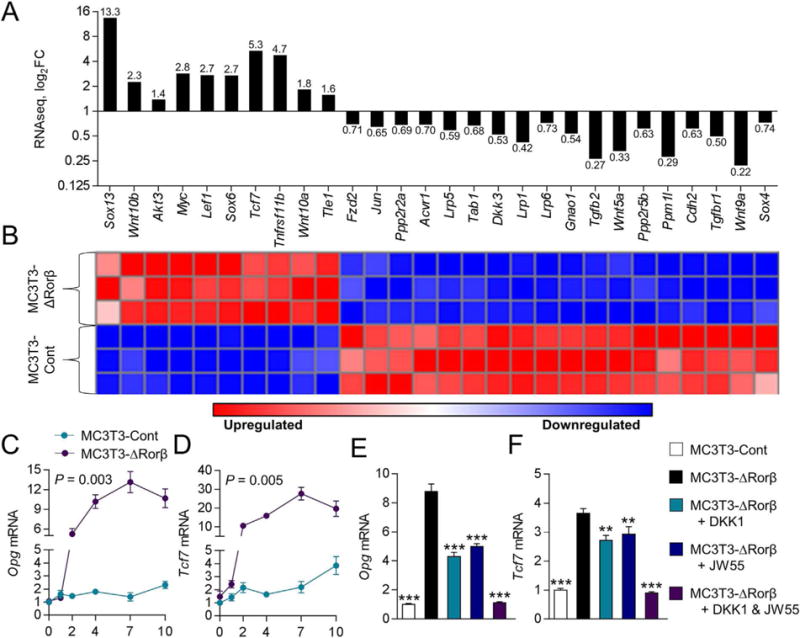
Rorβ regulates the Wnt/β-catenin pathway. (A, B) Wnt/β-catenin-regulated genes identified by RNAseq and IPA analyses are significantly regulated in MC3T3-ΔRorβ cells, and their respective fold changes (log2FC), are shown as a bar graph and heat map. (C, D) The time course from Fig. 1 was re-queried and analyzed for Opg and Tcf7 expression. Values of p are shown for repeated-measures ANOVA. (E, F) The MC3T3-Cont and MC3T3-ΔRorβ cell lines were treated with osteogenic medium at confluence and then supplemented with either Dkk1 (200 ng/mL), JW55 (10 μM), or both; cells were harvested 48 hours later and QPCR was performed for Opg and Tcf7 (n = 6). Data represent mean SE; p < 0.01; p < 0.001 (ANOVA).
To continue our analysis of altered Wnt signaling in MC3T3-ΔRorβ cells, our original osteoblast differentiation time course was requeried for regulation of two well-known Wnt target genes, Opg and Tcf7. Consistent with the RNAseq data, Opg and Tcf7 expression was significantly upregulated in Rorb-deficient cells throughout the time course (Fig. 2C, D). To further clarify the role of the Wnt pathway in Opg and Tcf7 upregulation in Rorb-deficient osteoblasts, we performed a series of inhibitor studies using either DKK1 (which binds to Lrp-containing complexes, thereby competing with extracellular Wnt ligand binding(22)), or JW55 (a small molecular inhibitor that directly binds to β-catenin and inhibits its transcriptional function(23)). Treatment of MC3T3-ΔRorβ cells with either inhibitor alone resulted in significantly reduced Opg expression, whereas treatment with both inhibitors reduced Opg expression to levels observed in the MC3T3-Cont cell line (Fig. 2E). Virtually identical changes were also observed for Tcf7 expression (Fig. 2F). These data indicate that the Wnt pathway is altered by the loss of Rorb.
To further understand the mechanism of β-catenin in regulation of the Opg and Tcf7 genes, we next performed ChIP assays on the MC3T3-Cont and MC3T3-ΔRorβ cells. In both unstimulated as well as Wnt3a-treated cells, we found that β-catenin and RNA polymerase (RNAP) recruitment was increased at Tcf/Lef DNA binding sites located within the promoters of the Opg and Tcf7 genes(24,25) in the Rorb-deficient cells (Fig. 3 A–D). Conversely, using ChIP assays of transiently transfected, myc-tagged β-catenin and 6x-His-tagged-Rorβ constructs, we found that Rorβ expression inhibits the recruitment of β-catenin to these DNA same binding sites (Supporting Fig. 5A,B). Because these data demonstrate that under normal physiological conditions Rorβ inhibits Wnt transcriptional activity though β-catenin antagonism, we further verified this by showing that Rorβ inhibits the transcriptional activity of a constitutively active form of β-catenin (ca-βcat) on the TOP-FLASH Wnt-reporter construct (Fig. 3E).(12,26) Collectively, these data show that Rorβ inhibits Wnt activity through the inhibition of β-catenin recruitment to Tcf/Lef binding sites in the promoters of Wnt- responsive genes in osteoblasts.
Fig. 3.
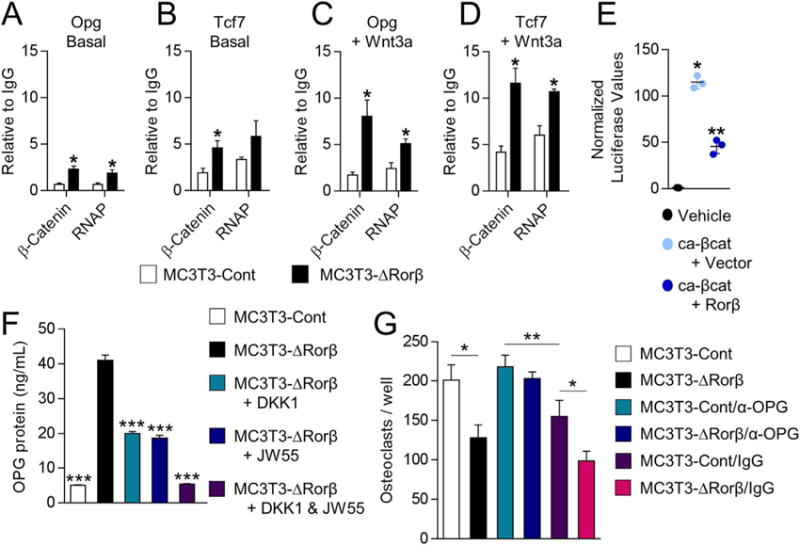
Rorb-deletion enhances β-catenin recruitment to the promoters of Wnt-responsive genes. (A–D) ChIP assays were performed on the MC3T3-Cont and MC3T3-ΔRorβ cell lines following 6-hour treatment with 10 ng/mL Wnt3a, or untreated (Basal). IPs were performed for β-catenin and RNAP and QPCR specific for the Tcf/Lef DNA binding site in the promoters of the Opg and Tcf7 gene. The data are presented relative to an IgG negative control IP. Data represent mean SE; p < 0.05. (E) ca-βcat and TOP-FLASH were co-transfected –/+ Rorβ and luciferase activity was measured 48 hours later (n = 6). Data represent mean SE; p < 0.001 (ANOVA), p < 0.01 (ANOVA compared to vector + ca-βcat). (F) CM from the treated cells in Fig. 2E, F was collected and OPG protein levels were determined by ELISA (n = 5). (G) Bone marrow–derived osteoclast precursors were incubated with osteoclastogenic-supportive medium supplemented with 1% of the same CM described in F (n = 6 to 7). In some wells, both cell models were cultured in the presence of either a mouse neutralizing antibody for osteoprotegerin (α-OPG; 1 mg/mL) or a control goat IgG. On day 4 of treatment, cells were fixed, stained for TRAP activity, and counted. Values represent mean ±SE; p < 0.05; p < 0.01 (ANOVA). ChIP = chromatin immunoprecipitation; RNAP = RNA polymerase II; CM = conditioned medium; ca-βcat = constitutively active β-catenin; TRAP = tartrate-resistant acid phosphatase.
To assess the implications of increased OPG on osteoclastogenesis, we first measured secreted OPG protein levels using ELISA on the CM isolated from the previously treated MC3T3-Cont and MC3T3-ΔRorβ cells (see Fig. 2E, F). Secreted OPG protein followed a pattern nearly identical to the Opg mRNA (Fig. 3F). Because increased OPG inhibits the development of mature osteoclasts, we hypothesized that CM from these cells would inhibit osteoclast differentiation in vitro. Bone marrow–derived osteoclast precursors were incubated with CM derived from MC3T3-Control or MC3T3-ΔRorβ cells; CM from MC3T3-ΔRorβ cells significantly reduced mature osteoclast formation as compared to control CM (Fig. 3G, black bar). Furthermore, incubation of MC3T3-ΔRorβ –derived CM with an OPG neutralizing antibody reversed this effect to levels observed using MC3T3-Cont–derived CM (Fig. 3G, dark blue bar). These findings show that Rorb-deficient cells are capable of suppressing osteoclastogenesis through increased OPG production and secretion.
Rorβ expression increases in mouse bone in vivo throughout the lifespan
We previously found(8) that the expression of Rorβ was increased in bone biopsies obtained from elderly postmenopausal women as compared to young premenopausal women. To corroborate these findings in mice, Rorβ expression was measured in bone marrow-derived lin– cells (an osteoprogenitor-enriched population(27)) where it was significantly increased (fivefold) in 12-month-old versus 6-month-old WT mice (Supporting Fig. 6A). Rorβ expression was also significantly increased (fourfold) in osteocyte-enriched bone samples(14) from 12-month-old versus 6-month-old mice (Supporting Fig. 6B).
Skeletal phenotype of Rorβ-knockout (Rorβ −/−) mice
Based on these findings, it was hypothesized that Rorβ is a negative regulator of osteogenesis and therefore is causally implicated in age-related bone loss. To test this hypothesis, the skeletal phenotype of a Rorβ mouse knockout (Rorb−/−) model that lacked the major Rorb1 isoform encoded by the Rorβ gene(10) (Fig. 4A) was examined throughout the first year of life (Fig. 4B), because previous work has established that normal chronologically aged mice experience substantial trabecular bone loss prior to this period.(28,29) In vivo pQCT scanning at the tibial metaphysis revealed that trabecular volumetric bone mineral density (Tb.vBMD) was not substantially different at 1 and 3 months of age, but was significantly higher in female Rorβ−/− mice at 6, 9, and 12 months as compared to age- and sex-matched WT littermate controls (Fig. 4C). High-resolution μCT scanning of the lumbar spine (Fig. 4D, E) and femoral metaphysis (Fig. 4K–L) at one year of age revealed markedly higher bone mass in female Rorβ−/− mice as compared to age- and sex-matched WT littermate controls. Further μCT analysis at the lumbar spine revealed significantly (all p < 0.05) better trabecular bone micro- architectural parameters in the female Rorβ−/− mice as compared to WT controls (ie, increased bone volume fraction [BV/TV; Fig. 4F], increased trabecular number [Tb.N; Fig. 4G], no change in trabecular thickness [Tb.Th; Fig. 4H], decreased trabecular separation [Tb.Sp; Fig. 4I], and a lower [“better/stronger”] structural model index [SMI; Fig. 4J]). Consistent with this, virtually identical findings were observed at the femoral metaphysis of female mice (Fig. 4M–Q). μCT analyses at the lumbar spine (Fig. 5A–G) and femoral metaphysis (Fig. 5H–N) in males revealed a virtually identical phenotype to that observed in the females, whereby at 1 year of age male Rorβ−/− mice had substantially improved bone mass and microarchitecture as compared to age/sex-matched WT littermate controls. These data show that deletion of Rorβ in vivo prevents the natural loss of bone mass and micro-architecture in both sexes with advancing age.
Fig. 4.
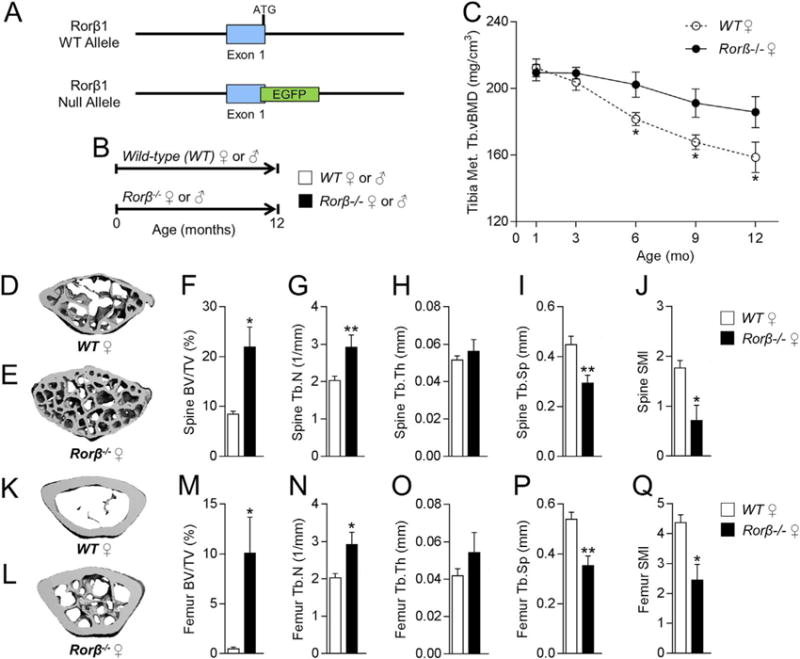
Female Rorβ−/− mice exhibit high bone mass and improved bone microarchitecture during aging. (A) Strategy for deletion of Rorβ1 in mice by insertion of EGFP at the 3′ of exon 1. Both the WT and the null allele are shown. (B) Timeline of the mouse phenotypic studies. (C) Female Rorβ−/− and WT control mice were scanned in vivo using pQCT at 1, 3, 6, 9, and 12 months of age. (D, E) Representative μCT 3D reconstructions of the lumbar spine of 12-month-old female WT and Rorβ−/− mice. (F–J) μCT-derived bone parameters measured at the lumbar spine (n = 5); (F) BV/TV (%), (G) Tb.N (1/mm), (H) Tb.Th (mm), (I) Tb.Sp (mm), and (J) SMI. (K, L) Representative μCT 3D reconstruction of 12-month-old female WT and Rorβ−/− mice at the femoral metaphysis. (M–Q) μCT-derived bone (n = 5), parameters at the femoral metaphysis. Data for all panels represent mean ±SE; p < 0.05; p < 0.01; p < 0.001 (independent samples t test). pQCT = peripheral quantitative computed tomography; BV/TV = bone volume/tissue volume; Tb. N = trabecular number; Tb.Th = trabecular thickness; Tb.Sp = trabecular separation; SMI = structure model index.
Fig. 5.
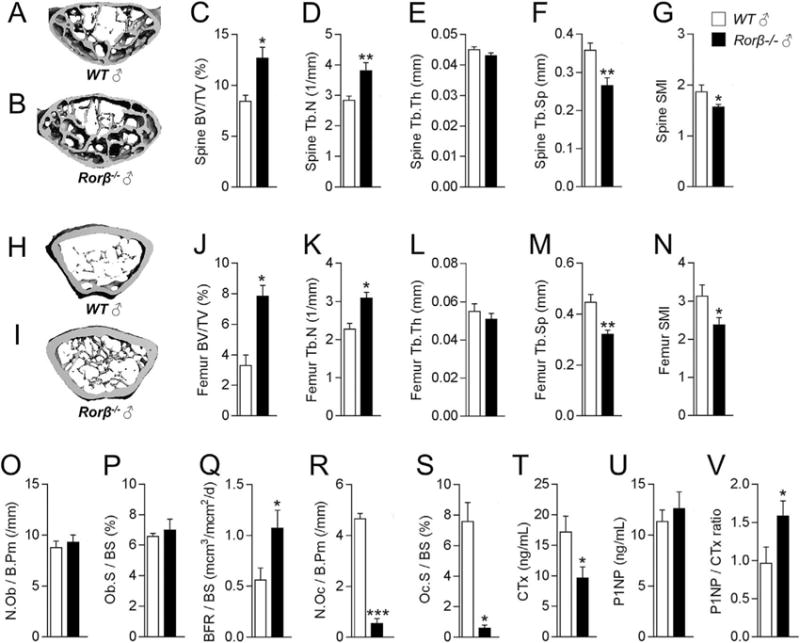
Male Rorβ−/− mice also exhibit high bone mass, and dynamic bone histology reveals bone anabolic effects in Rorβ−/− mice. (A, B) Representative μCT 3D reconstructions of the lumbar spine of 12-month-old male WT and Rorβ−/− mice. (C–G) μCT-derived bone parameters measured at the lumbar spine (n = 5); as described in Fig. 4F–J. (H, I) Representative μCT 3D reconstruction of 12-month-old male WT and Rorβ −/− mice at the femoral metaphysis. (J–N) μCT-derived bone parameters measured at the femoral metaphysis (n = 5). (O–S) Femurs from 12-month-old female WT and Rorβ−/− mice (n = 3 to 4) were processed for analyses of static and dynamic bone histomorphometric parameters: (O) N.Ob/B.Pm, (P) Ob.S/BS, (Q) BFR/BS, (R) N.Oc/B.Pm, and (S) Oc.S/BS. The serum bone turnover markers (T) CTx, (U) P1NP, and (V) P1NP/CTx ratio were measured. Data for all panels represent mean ±SE; p < 0.05; p < 0.01; p < 0.001 (independent samples t test). N.Ob/B.Pm = osteoblast number/bone perimeter; Ob.S/BS = osteoblast surface/bone surface; BFR/BS = bone formation rate/bone surface; N.Oc/B.Pm = osteoclast number/bone perimeter; Oc.S/BS = osteoclast surface/bone surface.
Bone histomorphometric analyses were next performed to examine the underlying cellular mechanisms responsible for the improved skeletal phenotype in the Rorβ−/− mice. Static osteoblast parameters, including osteoblast number per bone perimeter (N.Ob./B.Pm) and osteoblast surface per bone surface (Ob.S/BS), were not substantially different between the groups (Fig. 5O, P). However, osteoblast activity quantified by the bone formation rate per bone surface (BFR/BS) was significantly increased in the Rorβ−/− mice (Fig. 5Q). In contrast, osteoclast parameters including osteoclast number per bone perimeter (N. Oc/B.Pm; Fig. 5R) and osteoclast surface per bone surface (Oc.S/BS; Fig. 5S) were markedly decreased in the Rorβ−/−mice. Consistent with the bone histomorphometry data, the bone resorption serum marker CTx (Fig. 5T) was significantly decreased, while the bone formation serum marker P1NP (Fig. 5U) was unchanged, leading to a significant increase in the P1NP/CTx ratio (Fig. 5V). DKK1 and sclerostin (both well- established circulating Wnt inhibitors) were not substantially affected in Rorβ−/− mice (Supporting Fig. 7A, B). We also examined the potential role of sex steroid involvement in Rorβ biology. In vitro studies showed that Rorβ mRNA levels were not affected by either 17-b-estradiol or DHT treatment in MC3T3-E1 cells (Supporting Fig. 8A, B). In addition, we performed an in vivo OVX and ORX study in females and males, respectively, which showed that loss of Rorβ did not protect against sex steroid- induced bone loss (Supporting Fig. 8C, D).
TCF7 and OPG protein expression is increased in Rorβ−/− mice
TCF7 is a major transcriptional effector of the Wnt pathway that is increased in response to Wnt signaling activation.(30) Thus, to determine whether Wnt activity was increased in the Rorβ−/− mice, as observed in the MC3T3-ΔRorβ cells (Fig. 2), in vivo TCF7 protein levels were assessed in osteocytes using immunohistochemistry (IHC) in bone sections from Rorβ−/− mice and age-matched littermate controls at 1 year of age. These analyses revealed that compared to WT littermate control mice, TCF7 staining was enhanced in osteocytes of Rorβ−/− mice (Fig. 6A, B). Quantification of the percentage of TCF7-positive (+) osteocytes (Fig. 6C) revealed that this parameter was significantly increased in the Rorβ−/− mice as compared to WT littermate control mice. Similarly, the percentage of OPG+ osteocytes (Fig. 6D–F) was significantly increased in Rorβ−/− mice. These data show that Rorβ−/− mice have enhanced in vivo Wnt pathway activation and increased OPG production in bone, which is consistent with our in vitro findings in Rorb-deleted osteoblastic cells.
Fig. 6.
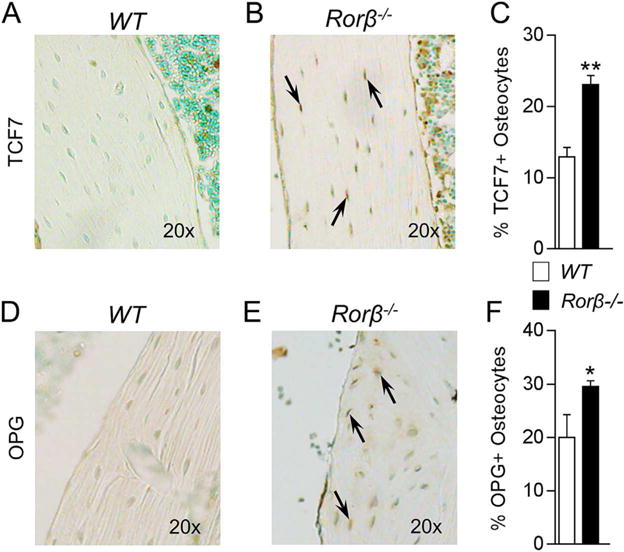
Rorβ−/− mice display increased TCF7 and OPG expression in bone. (A, B) Female WT and Rorβ−/− control mice (n = 4) were euthanized at 12 months of age and femurs were fixed, embedded, and sectioned. IHC for mouse TCF7 protein levels was performed. TCF7-positive staining (dark brown) is denoted by arrows. (C) TCF7-positive (+) cells were counted using OsteoMeasure software and plotted as a percentage of TCF7+ cells per total osteocytes. (D–F) These are the same samples and procedures as described in A–C; however, IHC was performed for OPG protein levels and plotted in the same manner. Data represent mean SE; p < 0.05; p < 0.01 (independent samples t test).
Discussion
Osteoporosis is a chronic age-associated disease that occurs when the normally coupled processes of osteoblastic bone formation and osteoclastic bone resorption becomes unbalanced, resulting in a net loss of bone over time. Antiresorptive therapies target the osteoclastic lineage and result in decreases in resorption; however, due to coupling between osteoclasts and osteoblasts,(31) these drugs also result in decreased bone formation, leading to low bone turnover. Currently available anabolic therapies for osteoporosis have limited efficacy as well as higher risk for undesirable secondary outcomes.(5,6) Therefore, there is an increasing need to identify cellular pathways that increase bone formation and perhaps simultaneously inhibit bone resorption in an effort to develop new therapies to combat age-related osteoporosis. Data from the present study suggest that Rorβ may be a novel and viable target.
The regulation of osteoblast differentiation requires the coordinate activities of numerous transcription factors. Al- though factors such as Runx2 and osterix are essential to the formation of calcified bone,(32–35) others act to fine-tune the response to specific external cues. In a previous report,(7) we discovered that the orphan nuclear receptor Rorβ, was sharply downregulated during the process of osteoblast differentiation, Rorβ overexpression inhibited mineralization, and that Rorβ was upregulated in osteoblast progenitors during aging. These observations lead us to formulate a preliminary hypothesis that Rorβ may be a repressor of bone metabolism, because its expression appeared inversely correlated with osteogenic potential. However, our previous study relied on an in vitro overexpression system, which has the limitation of potentially displaying inherent nonphysiological effects. Another limitation of previous work was the lack of in vivo data. To overcome these limitations in our current study, we used both a novel in vitro cell system (CRISPR/Cas9) to delete Rorβ in osteoblasts and an in vivo Rorβ−/− mouse model to examine the roles of Rorβ in osteogenesis and bone mass acquisition, respectively.
We first examined whether loss of Rorβ in an osteoblastic cell model results in an increased osteoblastic phenotype. To accomplish this, we used the CRISPR/Cas9 genome editing system to delete Rorβ in the MC3T3-E1 osteoblastic cell line. Our data show that Rorβ deletion increased the expression of several bone marker genes during osteoblastic differentiation, which supports our previous work showing that Rorβ is an inhibitor of osteoblastic function.(7) To better understand the underlying mechanism(s) involved in enhanced osteoblastic function in MC3T3-ΔRorβ cells, we performed whole-transcriptome RNAseq analysis, which revealed that deletion of Rorβ led to enhancement of the Wnt pathway, which is a well-established, important regulator of bone formation(22) and resorption.(36) Indeed, over the past several decades, the importance of Wnt signaling has become increasingly clear since alterations in Wnt pathway components have profound skeletal effects in humans.(37–41) Further, numerous mouse models have also demonstrated that loss of various Wnt ligands leads to bone loss.(42–44) TCF7 is a known transcriptional effector of the Wnt pathway, complexing with active β-catenin in the nucleus to drive gene expression of Wnt target genes.(45) The Tcf7 transcript was markedly upregulated during osteoblast differentiation in the MC3T3-ΔRorβ cells, and this was due to increased Wnt activity because DKK1, a known inhibitor of the Wnt pathway,(22) and the small molecule Wnt-inhibitor JW55(23) significantly attenuated Tcf7 expression. We further showed that in Rorb-deficient cells, the recruitment of β-catenin to both the Tcf7 and Opg promoters is enhanced, providing a plausible explanation for the mechanism of the increased expression of these Wnt-responsive genes observed in these cells. Although the loss of Rorβ affects β-catenin recruitment, we cannot rule out the possibility that the noncanonical Wnt pathway may also contribute to the observed phenotype. Further studies will be needed to address the potential contribution of the noncanonical Wnt pathway.
We observed a significant increase in Opg gene expression and OPG protein secretion, another important Wnt target,(46) in differentiating MC3T3-ΔRorβ cells. OPG acts as a decoy receptor, preventing receptor activator of nuclear factor kappa b-ligand (RANKL) from binding RANK on the surface of osteoclast precursors.(47) Indeed, our data demonstrate that incubation of conditioned media from MC3T3-ΔRorβ cells inhibits the formation of osteoclasts in vitro. This is consistent with the observed skeletal phenotype in the Rorβ−/− mice, in which we found markedly decreased osteoclast numbers and osteoclast surfaces. Collectively, these results demonstrate that bone resorption is reduced in these animals. In combination with the observed increase in Tcf7 mRNA expression in the MC3T3-ΔRorβ cell model and increased TCF7 protein expression in bones of Rorβ−/− mice, a significant anabolic bone response was observed. This is an important distinction from currently available antiresorptive therapies (eg, bisphosphonates, denosumab, estrogen, raloxifene) where although bone resorption is decreased, bone formation also decreases due to coupling.(31)
The physiological context for the potential role of Rorβ in age-related osteoporosis comes from our observations that Rorβ levels are highly elevated in the bone marrow–derived lin– cells and osteocyte-enriched bone samples isolated from 12-month- old WT C57BL/6 mice, as compared to 6-month-old mice. Previous work has clearly established that both female and male C57BL/6 mice lose bone, particularly from trabecular skeletal compartments, between 6 and 12 months of age.(28,29) This correlation of high Rorβ expression at a period in the murine lifespan when trabecular bone mass is declining is clearly suggestive of an important role of Rorβ in the etiology of age- related bone loss. Indeed, similar observations were seen in humans, where we found that the expression of Rorβ was increased in bone biopsies obtained from elderly postmenopausal women as compared to young premenopausal women, suggesting that our findings in mice may translate to humans.(8)
Another important finding is that Rorβ deletion prevents the normal bone loss observed in mice up to 12 months of age. High-resolution μCT analyses also clearly show an improvement in trabecular bone microarchitecture, suggesting that a proanabolic bone environment is maintained in the absence of the normally increasing Rorβ levels during aging in WT mice. We also noted a significant reduction in osteoclasts, yet no difference in osteoblasts, along with the significantly higher bone formation rates, showing that deletion of Rorβ in mice leads to a marked reduction in bone resorption but without the “coupled” reduction in bone formation. Collectively, this results in a positive “anabolic” net bone balance, leading to the improved skeletal phenotype in the Rorβ−/− mice. Thus, high levels of Rorβ in the adult skeleton appear to be detrimental to the normal function of osteoblastic cells. Indeed, our data showing that Rorβ deletion results in a considerably healthier skeleton with age establishes a potential causal role for Rorβ in bone loss with aging. A potential limitation of the current study is the use of a global Rorβ knockout model. Thus, future studies are needed to explore the effects of tissue/cell-specific Rorβ deletion on bone metabolism.
Figure 7 summarizes our current understanding of the osteoprotective effects of the deletion of Rorb, which triggers a multifaceted response in the bone microenvironment. Deletion of Rorβ enhances bone formation through increased Tcf7 and Wnt signaling responses, in both in vitro and in vivo models. Concurrently, Rorβ deletion affects bone resorption through increased production and secretion of OPG, which reduces osteoclast numbers and osteoclast activity. Collectively, the increase in bone formation and decrease in bone resorption result in the prevention of bone loss.
Fig. 7.
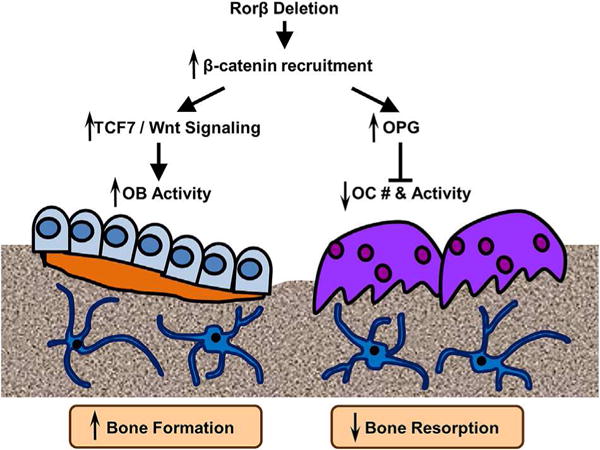
Model of Rorβ action in bone. Rorβ deletion leads to increased β-catenin recruitment to Wnt-responsive promoters, which leads to increases in Tcf7 and Wnt activities in bone and increased bone formation and osteoblastic activity (left side of figure). Concomitantly, Rorβ deletion also increases expression and secretion of OPG, leading to suppression of osteoclastogenesis (right side of figure). Collectively, these events contribute to increased bone mass.
Because Rorβ expression is highly elevated in bone marrow– derived lin– cells and osteocyte-enriched bone digests isolated from aged mice, therapies involving inhibition of either Rorβ expression or RORβ protein in individuals with osteoporosis may represent a novel avenue for treatment. This is supported by our previous work showing that Rorβ expression is also increased in bone biopsies obtained from elderly postmenopausal women as compared to young premenopausal women.(8) Furthermore, Rorβ is particularly amenable to small molecule drug targeting, because of the presence of a ligand binding domain, which is capable of binding small molecules.(48) In fact, all-trans retinoic acid (ATRA) was identified as a functional ligand for Rorβ that inhibited Rorb-induced transcriptional activation.(49) However, ATRA also binds Rorα and Rorγ, as well as all three members of the retinoic acid receptor (RAR) family, and therefore does not display selectivity.(50) Finally, the expression pattern of Rorβ throughout the lifespan is particularly unique in that during development Rorβ is highly expressed in the brain and eye(50); however, with aging Rorβ expression appears to be more restricted to bone. Given this restricted expression pattern during aging, direct inhibition of Rorβ represents an effective target to induce bone anabolism. Future work is necessary to identify novel Rorb-specific inhibitory compounds that may pave the way toward the development of more diseases.
Supplementary Material
Acknowledgments
This work was supported by NIH grants R01 AR068275 (DGM), P01 AG004875 (SK/DGM), R01 AG048792 (SK/DGM), K01 AR070241 (JNF), K01 AR070281 (MMW), R01 DE14036 (JRH); career development awards (JNF, MMW) from the Mayo Clinic Robert and Arlene Kogod Center on Aging; and by the Intramural Research Program at NIDDK at NIH (HL, DF). We thank Ms. Glenda Evans for technical assistance and data collection, as well as Mr. James Peterson and Ms. Elizabeth Atkinson for RNAseq and data analyses.
Authors’ roles: DGM, JNF, and SK conceived and directed the project. DGM, JNF, DF, HL, SK, and MMW designed the experiments and interpreted the data. JNF, MMW, KMN, BAN, JLO, BST, DGF, MR, JRH, and DGM performed the experiments. DGM, JNF, MMW, and SK wrote the manuscript. All authors reviewed the manuscript.
Footnotes
Additional Supporting Information may be found in the online version of this article.
Disclosures
All authors state that they have no conflicts of interest.
References
- 1.Wright NC, Looker AC, Saag KG, et al. The recent prevalence of osteoporosis and low bone mass in the United States based on bone mineral density at the femoral neck or lumbar spine. J Bone Miner Res. 2014;29(11):2520–6. doi: 10.1002/jbmr.2269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Khosla S. Pathogenesis of age-related bone loss in humans. J Gerontol A Biol Sci Med Sci. 2013;68(10):1226–35. doi: 10.1093/gerona/gls163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Khosla S, Hofbauer LC. Osteoporosis treatment: recent developments and ongoing challenges. Lancet Diabetes Endocrinol. 2017;5(11):898–907. doi: 10.1016/S2213-8587(17)30188-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leder BZ. Parathyroid hormone and parathyroid hormone-related protein analogs in osteoporosis therapy. Curr Osteoporos Rep. 2017;15(2):110–9. doi: 10.1007/s11914-017-0353-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harper KD, Krege JH, Marcus R, Mitlak BH. Osteosarcoma and teriparatide? J Bone Miner Res. 2007;22(2):334. doi: 10.1359/jbmr.061111. [DOI] [PubMed] [Google Scholar]
- 6.Watanabe A, Yoneyama S, Nakajima M, et al. Osteosarcoma in Sprague-Dawley rats after long-term treatment with teriparatide (human parathyroid hormone (1–34)) J Toxicol Sci. 2012;37(3):617–29. doi: 10.2131/jts.37.617. [DOI] [PubMed] [Google Scholar]
- 7.Roforth MM, Liu G, Khosla S, Monroe DG. Examination of nuclear receptor expression in osteoblasts reveals Rorbeta as an important regulator of osteogenesis. J Bone Miner Res. 2012;27(4):891–901. doi: 10.1002/jbmr.1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roforth MM, Khosla S, Monroe DG. Identification of Rorbeta targets in cultured osteoblasts and in human bone. Biochem Biophys Res Commun. 2013;440(4):768–73. doi: 10.1016/j.bbrc.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Komor AC, Badran AH, Liu DR. CRISPR-based technologies for the manipulation of eukaryotic genomes. Cell. 2017;168(1–2):20–36. doi: 10.1016/j.cell.2016.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu H, Kim SY, Fu Y, et al. An isoform of retinoid-related orphan receptor beta directs differentiation of retinal amacrine and horizontal interneurons. Nat Commun. 2013;4:1813. doi: 10.1038/ncomms2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Syed FA, Fraser DG, Monroe DG, Khosla S. Distinct effects of loss of classical estrogen receptor signaling versus complete deletion of estrogen receptor alpha on bone. Bone. 2011;49(2):208–16. doi: 10.1016/j.bone.2011.03.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Modder UI, Oursler MJ, Khosla S, Monroe DG. Wnt10b activates the Wnt, notch, and NFkappaB pathways in U2OS osteosarcoma cells. J Cell Biochem. 2011;112(5):1392–402. doi: 10.1002/jcb.23048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cicek M, Fukuyama R, Welch DR, Sizemore N, Casey G. Breast cancer metastasis suppressor 1 inhibits gene expression by targeting nuclear factor-kappaB activity. Cancer Res. 2005;65(9):3586–95. doi: 10.1158/0008-5472.CAN-04-3139. [DOI] [PubMed] [Google Scholar]
- 14.Farr JN, Fraser DG, Wang H, et al. Identification of senescent cells in the bone microenvironment. J Bone Miner Res. 2016;31(11):1920–9. doi: 10.1002/jbmr.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Modder UI, Roforth MM, Hoey K, et al. Effects of estrogen on osteoprogenitor cells and cytokines/bone regulatory factors in postmenopausal women. Bone. 2011;49:202–7. doi: 10.1016/j.bone.2011.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gingery A, Bradley E, Shaw AC, Oursler MJ. Phosphatidylinositol 3- kinase coordinately activates the MEK/ERK and AKT/NFkappaB pathways to maintain osteoclast survival. J Cell Biochem. 2003;89:165–79. doi: 10.1002/jcb.10503. [DOI] [PubMed] [Google Scholar]
- 17.Farr JN, Roforth MM, Fujita K, et al. Effects of age and estrogen on skeletal gene expression in humans as assessed by RNA sequencing. PLoS One. 2015;10(9):e0138347. doi: 10.1371/journal.pone.0138347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome- wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–50. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xie X, Lu J, Kulbokas EJ, et al. Systematic discovery of regulatory motifs in human promoters and 3′ UTRs by comparison of several mammals. Nature. 2005;434(7031):338–45. doi: 10.1038/nature03441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujiwara M, Tagashira S, Harada H, et al. Isolation and characterization of the distal promoter region of mouse Cbfa1. Biochim Biophys Acta. 1999;1446:265–72. doi: 10.1016/s0167-4781(99)00113-x. [DOI] [PubMed] [Google Scholar]
- 21.Park MH, Shin HI, Choi JY, et al. Differential expression patterns of Runx2 isoforms in cranial suture morphogenesis. J Bone Miner Res. 2001;16(5):885–92. doi: 10.1359/jbmr.2001.16.5.885. [DOI] [PubMed] [Google Scholar]
- 22.Monroe DG, McGee-Lawrence ME, Oursler MJ, Westendorf JJ. Update on Wnt signaling in bone cell biology and bone disease. Gene. 2012;492(1):1–18. doi: 10.1016/j.gene.2011.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Waaler J, Machon O, Tumova L, et al. A novel tankyrase inhibitor decreases canonical Wnt signaling in colon carcinoma cells and reduces tumor growth in conditional APC mutant mice. Cancer Res. 2012;72(11):2822–32. doi: 10.1158/0008-5472.CAN-11-3336. [DOI] [PubMed] [Google Scholar]
- 24.Sato MM, Nakashima A, Nashimoto M, Yawaka Y, Tamura M. Bone morphogenetic protein-2 enhances Wnt/beta-catenin signaling- induced osteoprotegerin expression. Genes Cells. 2009;14(2):141–53. doi: 10.1111/j.1365-2443.2008.01258.x. [DOI] [PubMed] [Google Scholar]
- 25.Wu JQ, Seay M, Schulz VP, et al. Tcf7 is an important regulator of the switch of self-renewal and differentiation in a multipotential hematopoietic cell line. PLoS Genet. 2012;8(3):e1002565. doi: 10.1371/journal.pgen.1002565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fahnert B, Veijola J, Roel G, et al. Murine Wnt-1 with an internal c-myc tag recombinantly produced in Escherichia coli can induce intracellular signaling of the canonical Wnt pathway in eukaryotic cells. J Biol Chem. 2004;279(46):47520–7. doi: 10.1074/jbc.M403207200. [DOI] [PubMed] [Google Scholar]
- 27.Itoh S, Aubin JE. A novel purification method for multipotential skeletal stem cells. J Cell Biochem. 2009;108:368–77. doi: 10.1002/jcb.22262. [DOI] [PubMed] [Google Scholar]
- 28.Glatt V, Canalis E, Stadmeyer L, Bouxsein ML. Age-related changes in trabecular architecture differ in female and male C57BL/6J mice. J Bone Miner Res. 2007;22:1197–207. doi: 10.1359/jbmr.070507. [DOI] [PubMed] [Google Scholar]
- 29.Hamrick MW, Ding KH, Pennington C, et al. Age-related loss of muscle mass and bone strength in mice is associated with a decline in physical activity and serum leptin. Bone. 2006;39:845–53. doi: 10.1016/j.bone.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 30.Waterman ML. Lymphoid enhancer factor/T cell factor expression in colorectal cancer. Cancer Metastasis Rev. 2004;23(1–2):41–52. doi: 10.1023/a:1025858928620. [DOI] [PubMed] [Google Scholar]
- 31.Khosla S. Odanacatib: location and timing are everything. J Bone Miner Res. 2012;27(3):506–8. doi: 10.1002/jbmr.1541. [DOI] [PubMed] [Google Scholar]
- 32.Ducy P, Zhang R, Geoffroy V, Ridall AL, Karsenty G. Osf2/Cbfa1: a transcriptional activator of osteoblast differentiation. Cell. 1997;89(5):747–54. doi: 10.1016/s0092-8674(00)80257-3. [DOI] [PubMed] [Google Scholar]
- 33.Otto F, Thornell AP, Crompton T, et al. Cbfa1, a candidate gene for cleidocranial dysplasia syndrome, is essential for osteoblast differentiation and bone development. Cell. 1997;89(5):765–71. doi: 10.1016/s0092-8674(00)80259-7. [DOI] [PubMed] [Google Scholar]
- 34.Komori T, Yagi H, Nomura S, et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89(5):755–64. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 35.Nakashima K, Zhou X, Kunkel G, et al. The novel zinc finger- containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108(1):17–29. doi: 10.1016/s0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- 36.Weivoda MM, Ruan M, Hachfeld CM, et al. Wnt signaling inhibits osteoclast differentiation by activating canonical and noncanonical cAMP/PKA pathways. J Bone Miner Res. 2016;31(1):65–75. doi: 10.1002/jbmr.2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kiel DP, Demissie S, Dupuis J, et al. Genome-wide association with bone mass and geometry in the Framingham Heart Study. BMC Med Genet. 2007;8(Suppl 1):S14. doi: 10.1186/1471-2350-8-S1-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Riancho JA, Olmos JM, Pineda B, et al. Wnt receptors, bone mass, and fractures: gene-wide association analysis of LRP5 and LRP6 polymorphisms with replication. Eur J Endocrinol. 2011;164(1):123–31. doi: 10.1530/EJE-10-0582. [DOI] [PubMed] [Google Scholar]
- 39.Rivadeneira F, Styrkarsdottir U, Estrada K, et al. Twenty bone-mineral-density loci identified by large-scale meta-analysis of genome-wide association studies. Nat Genet. 2009;41(11):1199–206. doi: 10.1038/ng.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sims AM, Shephard N, Carter K, et al. Genetic analyses in a sample of individuals with high or low BMD shows association with multiple Wnt pathway genes. J Bone Miner Res. 2008;23(4):499–506. doi: 10.1359/jbmr.071113. [DOI] [PubMed] [Google Scholar]
- 41.van Meurs JB, Trikalinos TA, Ralston SH, et al. Large-scale analysis of association between LRP5 and LRP6 variants and osteoporosis. JAMA. 2008;299(11):1277–90. doi: 10.1001/jama.299.11.1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bennett CN, Longo KA, Wright WS, et al. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc Natl Acad Sci U S A. 2005;102:3324–9. doi: 10.1073/pnas.0408742102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bennett CN, Ouyang H, Ma YL, et al. Wnt10b increases postnatal bone formation by enhancing osteoblast differentiation. J Bone Miner Res. 2007;22(12):1924–32. doi: 10.1359/jbmr.070810. [DOI] [PubMed] [Google Scholar]
- 44.Day TF, Guo X, Garrett-Beal L, Yang Y. Wnt/beta-catenin signaling in mesenchymal progenitors controls osteoblast and chondrocyte differentiation during vertebrate skeletogenesis. Dev Cell. 2005;8:739–50. doi: 10.1016/j.devcel.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 45.Cadigan KM, Waterman ML. TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb Perspect Biol. 2012;4(11) doi: 10.1101/cshperspect.a007906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.De Toni EN, Thieme SE, Herbst A, et al. OPG is regulated by beta- catenin and mediates resistance to TRAIL-induced apoptosis in colon cancer. Clin Cancer Res. 2008;14(15):4713–8. doi: 10.1158/1078-0432.CCR-07-5019. [DOI] [PubMed] [Google Scholar]
- 47.Khosla S. Minireview: the OPG/RANKL/RANK system. Endocrinology. 2001;142:5050–5. doi: 10.1210/endo.142.12.8536. [DOI] [PubMed] [Google Scholar]
- 48.Stehlin C, Wurtz JM, Steinmetz A, et al. X-ray structure of the orphan nuclear receptor RORbeta ligand-binding domain in the active conformation. EMBO J. 2001;20(21):5822–31. doi: 10.1093/emboj/20.21.5822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Stehlin-Gaon C, Willmann D, Zeyer D, et al. All-trans retinoic acid is a ligand for the orphan nuclear receptor ROR beta. Nat Struct Biol. 2003;10(10):820–5. doi: 10.1038/nsb979. [DOI] [PubMed] [Google Scholar]
- 50.Jetten AM. Retinoid-related orphan receptors (RORs): critical roles in development, immunity, circadian rhythm, and cellular metabolism. Nucl Recept Signal. 2009;7:e003. doi: 10.1621/nrs.07003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.