

Abstract
Therapeutic proteins and peptides have revolutionized treatment for a number of diseases, and the expected increase in macromolecule-based therapies brings a new set of challenges for the pharmaceutics field. Due to their poor stability, large molecular weight, and poor transport properties, therapeutic proteins and peptides are predominantly limited to parenteral administration. The short serum half-lives typically require frequent injections to maintain an effective dose, and patient compliance is a growing issue as therapeutic protein treatments become more widely available. A number of studies have underscored the relationship of subcutaneous injections with patient non-adherence, estimating that over half of insulin-dependent adults intentionally skip injections. The development of oral formulations has the potential to address some issues associated with non-adherence including the interference with daily activities, embarrassment, and injection pain. Oral delivery can also help to eliminate the adverse effects and scar tissue buildup associated with repeated injections. However, there are several major challenges associated with oral delivery of proteins and peptides, such as the instability in the gastrointestinal (GI) tract, low permeability, and a narrow absorption window in the intestine. This review provides a detailed overview of the oral delivery route and associated challenges. Recent advances in formulation and drug delivery technologies to enhance bioavailability are discussed, including the co-administration of compounds to alter conditions in the GI tract, the modification of the macromolecule physicochemical properties, and the use of improved targeted and controlled release carriers.
KEY WORDS: Oral delivery, Hydrogels, Protein delivery, Peptide delivery, Carbohydrates, Mucoadhesion
Graphical abstract
Therapeutic proteins and peptides have revolutionized treatment for a number of diseases, and the expected increase in macromolecule-based therapies brings a new set of challenges for the pharmaceutics field. In order to fully realize the ‘holy grail’ and enable oral delivery, there are several major challenges associated that must be overcome. These include therapeutic instability in the gastrointestinal (GI) tract, low permeability, and a narrow absorption window in the intestine. This review provides a detailed discussion of the oral delivery route, associated advantages and challenges, and recent advances in delivery technology.
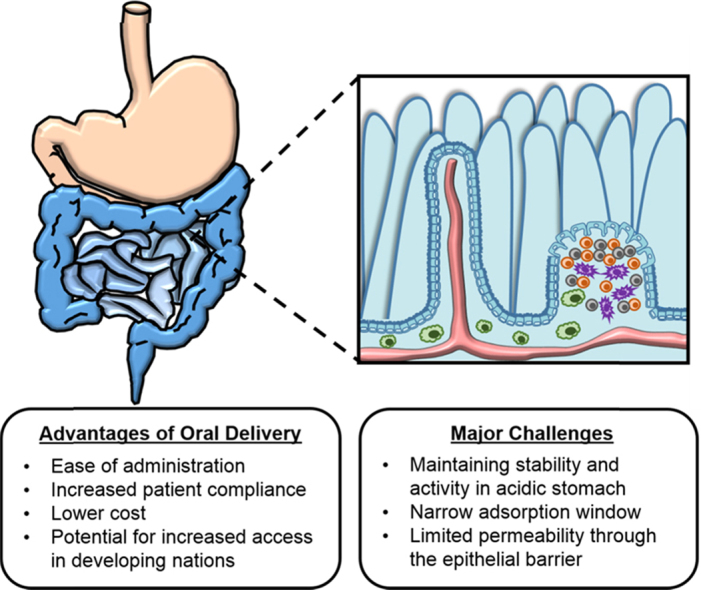
1. Introduction
Since the entrance of insulin as the first FDA-approved commercially available recombinant protein drug in 1982, protein and peptide drugs have become one of the fastest growing classes of new therapeutics. Because of the size and their stability, protein and peptide drugs are typically administered via injection. Most have short serum half-lives and, thus, need to be administered frequently or in high doses to be effective.
Alternative methods of administration which do not require injection or intravenous access are highly desirable. The oral route is the most desirable administration method for drugs as it is easy for patients and does not require injection. However, there are several significant challenges to the successful development of oral protein drug formulations: the instability of protein drugs in the gastrointestinal (GI) tract, the low permeability of protein drugs, and a narrow absorption window in the intestine. The activity of the therapeutic must be retained through the GI tract, and the active drug must reach the bloodstream at levels high enough to provide therapeutic efficacy. An oral delivery system must protect the drug from acid and enzymes in the stomach, but release the protein in the neutral environment of the small or large intestine.
This review is divided into six major sections to review the motivation, approach, and materials studied in literature to enable the oral delivery of therapeutic proteins and peptides. Detailed overviews of the oral delivery route, mechanisms, associated advantages, and major challenges are provided. Recent advances in formulation and drug delivery technologies to enhance bioavailability are discussed, including the co-administration of compounds to alter conditions in the GI tract, the modification of the macromolecule physicochemical properties, and the use of improved targeted and controlled release carriers.
2. Protein and peptide therapeutics
Protein and peptide drugs have become the fastest growing class of new pharmaceutics since the FDA approval of recombinant insulin in 1982. The field of protein and peptide therapeutics has experienced tremendous growth (Fig. 1) in part due to recombinant biotechnology but also the inherent advantages over small drugs. Proteins carry out complex functions, interact with biomolecules specifically with reduced risk of side effects, and have low immunogenicity1.
Figure 1.

Rise in FDA-approved BLAs for therapeutic biologics over the past decade. The past decade has seen a significant rise in the number of FDA-approved BLAs, and therapeutic biologics are becoming a larger percent of the total FDA-approvals (NME and BLA). The data were obtained directly from www.fda.gov (last accessed January 3, 2018).
The first accounts of protein drugs in the treatment of diseases were in the form of tissue extracts. For example, the first protein-based vaccine was developed in 1796 for small pox by Jenner using extracts from cowpox blisters of milkmaids. Over a century later in 1922, the first protein drug, insulin, was discovered by Banting et al.2 in treatment of diabetes in humans with pancreatic extracts from dog. A method for extraction of pure insulin from bovine pancreas extracts was later developed. The safety, quantity, and activity of these insulin extracts were determined by quantifying its effects on rabbit blood glucose levels. Manufacturing of insulin by synthetic chemical means was made possible by the work of Fred Sanger on sequencing insulin during the mid-1940s and -1950s.
It was not until the 1970s that the foundation for modern protein therapeutic production and engineering was established. In 1977, somatostatin was the first protein to be cloned into Escherichia coli by insertion of the somatostatin gene into the pBR322 plasmid3. A year later, the first recombinant protein, insulin, was reported by Genentech. Goeddel et al.4 synthesized insulin from two separately cloned polypeptide chains demonstrating for the first time synthetic recombinant technology to produce a therapeutic protein. Over the next decade, protein therapeutics including human growth hormone and interferon-α were reported. Meanwhile, several other major breakthroughs notably the discovery of polymerase chain reactions (PCR) for DNA amplification and the development of chemical DNA sequencing methods transformed biotechnology. These technologies along with the 1982 FDA approval of the recombinant form of insulin set the stage for protein drug development.
Recombinant DNA technology has had a significant impact on the discovery of new drugs but has also contributed to the safety and efficacy of protein drugs. For example, recombinant production of protein drugs reduced supply and immunological issues associated with protein drugs, which were previously harvested, then purified from blood or tissues. Moreover, protein engineering has led to improved drug half-lives and activity over native forms. The impact of biotechnology is already apparent in the more than 130 protein drugs approved by the Food and Drug Administration (FDA)1. Protein-based pharmaceutics are expected to continue to expand. It is estimated that by 2015 the protein drug market will exceed $150 billion US dollars (Global Industry Analysts, Inc.).
3. Advantages and challenges to oral delivery
While protein drugs have revolutionized treatment for a number of diseases, the expected increase in protein-based pharmaceutics brings a new set of challenges for the pharmaceutics field. Due to their instability, size, and poor transport, therapeutic proteins are predominantly administered by either intravenous or subcutaneous injection. Patient compliance, the extent to which a patient adheres to the treatment, is already a huge issue in diabetes treatment with insulin and is likely to become a growing issue as protein drug treatments become more widely used. A number of studies have underscored the relationship of traditional subcutaneous injections with patient non-adherence, and it is estimated that over half of insulin-dependent adults intentionally skip injections5., 6..
The development of oral formulations has the potential to address some of the issues associated with non-adherence (including the interference with daily activities, embarrassment, and injection pain) as well as the adverse effects of subcutaneous administration. Further, the successful development of oral protein delivery systems will significantly impact health care costs and the quality of patients׳ lives as small improvements in the ease of subcutaneous administration have translated to measurable improvements in patient adherence and decreased health care costs in the case of insulin pen injectors7.
Orally administered drugs follow the same route through the digestive system as food. The digestive system breaks down food, absorbs nutrients, and excretes waste. For oral drug delivery systems, the physiology of the digestive system can hinder performance but can potentially be exploited to improve drug delivery. Enzymes present throughout the GI tract can act on drugs causing a loss of bioactivity, the ability of the drug to carry out its function. On the other hand, the large surface area and high vascularization of the small intestine is advantageous in drug absorption, but the mucosa of the small intestine can serve as a barrier to drug absorption. In order for an oral delivery system to be effective, the drug of interest must retain its activity and a high bioavailability--be absorbed in quantities large enough to elicit a pharmacological effect.
Protein drugs are particularly challenging to successfully deliver orally due to their instability in acidic environments, their susceptibility to proteolysis by enzymes in the GI tract, and their large size. For example, the structure of protein drugs and the amino acid sequence of the protein drug define its bioactivity. The stability of proteins in GI tract is particularly problematic. Protein degradation can be due to either chemical or physical instability. Physical changes can result in denaturation, disruption of the secondary or tertiary structure of the protein in an irreversible or reversible process. Denaturation can occur as a result of aggregation, precipitation, adsorption, or unfolding. Chemical degradation can occur from deamination, oxidation, racemization, proteolysis, and disulfide interchange8.
The large molecular weight of protein drugs limits absorption. Protein drugs must cross the intestinal lining by penetrating an unstirred water layer and then passing across the cellular lining of the intestine. The mucus and glycocalyx of the intestine can further cause degradation of protein drugs9. Diffusion of large molecules is limited across the cell membrane. Additionally, large protein drugs cannot easily pass between cells due to tight junctions between adjacent cells. These junctions are estimated to have pore sizes as small as 10 Å. As a result, protein drug absorption is much more challenging than small molecule delivery.
3.1. The oral route
Both the anatomy and physiology of the digestive system reflect its range of functions through contrasting, tailored environments as shown in Fig. 2. The digestive system is composed of the mouth, the esophagus, the stomach, the small intestine, the large intestine, the liver, and the pancreas. This organ system functions in concert to digest food, absorb nutrients, and prevent pathogens from entering the bloodstream.
Figure 2.

Schematic of the oral delivery route and intestinal epithelium. Achieving oral delivery is ideal, but it is fraught with challenges at both the organism and tissue levels. (A) The gastrointestinal tract contains many digestive enzymes and a natural pH gradient, with a very acidic, harsh environment in the stomach and a more neutral environment in the small intestine (site of adsorption). (B) Within the small intestine, each villus has its own arteriole and venule pair as well as a lacteal. There is a thick mucosal barrier, and the cell lining is composed mostly of epithelial cells.
In the stomach, enzymes, such as pepsin and gelatinase, break down proteins and other compounds in food. Enzymatic digestion is facilitated by the acidic environment of the stomach which has a pH of approximately 2.1 in the fasted state. The time scale for gastric emptying can be as long as 2.5 h but is dependent on a number of signals.
Next, food passes to the small intestine. The small intestine is approximately 2.5 cm wide and 6.5 m long extending from the pylorus to the ileocecal valve where it connects to the large intestine. Overall, the small intestine has a relatively neutral pH between 7 and 8. The overall transit time for chyme in the small intestine varies depending on a number of factors but is on average 3–4 h.
As food passes from the stomach to small intestine through the pylorus, it enters the duodenum and is mixed with bile and pancreatic juices. The duodenum is the first 20—30 cm segment of the small intestine. The majority of the duodenum forms a loop around the pancreas. One of the functions of the duodenum is to raise the pH of chyme after it leaves the stomach. Duodenal luminal contents have a pH between 6 and 6.5. Generally, the transit time through the duodenum is rapid and can occur as fast as 1 min10.
Chyme travels from the duodenum past the duodenojejunal flexure to the jejunum, the middle section of the small intestine, which is approximately 2.5 m in length and is about 4 cm wide. The structure of the jejunum is composed of mucosal folds, valvulae conniventes. These folds increase the surface area of the jejunum to improve nutrient absorption and may also aide in the mixing of chyme. The jejunum has fewer enzymes in the luminal content than what is present in the duodenum. However, there are enzymes associated with the brush border of epithelial cells that compose the majority of the cellular lining. The jejunum is more highly vascularized than the ileum and is more absorptive than the ileum. As chyme passes through the jejunum, the pH rises to between 6.0 and 7.0. The transit time through the jejunum is considerably longer the duodenal transit time and is on average 3—4 h. The ileum is about 3.6 m in length and is narrower (3.75 cm) than the jejunum. The ileum has less pronounced valvulae conniventes and is less vascularized. Chyme then passes through the ileocecal valve to the large intestine. The pH of the digestive tract continues to rise between 7.0 and 7.5 in the large intestine, which regulates ion and fluid absorption.
The microanatomy of the small intestine allows for the high absorption and transport of nutrients providing about 90% of the body's nutrients. The small intestine wall has a surface area of approximately 200 m2 (Wilson TH 1962, Intestinal Absorption Saunders, Philadelphia, USA) 600 times greater than a hollow cylinder of the same dimensions because of intestinal villi, which are finger-like projections of tissue covering the luminal surface of the small intestine. Intestinal villi are highly vascularized as shown in Fig. 2; each villus has its own capillary system feeding into an arteriole and venule pair and its own lacteal feeding into the lymphatic system. The surface area of the small intestine and its close coupling with vasculature give the small intestine its absorptive properties. Because the small intestine is the site where most absorption takes place, it is the ideal release site for orally delivered proteins.
The absorptive properties of the small intestine are counterbalanced with physiological barriers for prevention of pathogen entry. The interface of the intestinal wall and the lumen is composed of the mucosa, glycocalyx, and the brush border of the intestinal epithelial cells which is the predominant cell type in the small intestine. Columnar epithelial cells are polarized cells that have a brush border (microvilli) on their apical surface.
The main component of the mucosa is water (up to 95% by weight), mucin (less than 5% by weight), and inorganic salts (approximately 1% by weight). Mucin itself is an O-linked glycoprotein. Branched regions of mucin are covered in highly branched oligosaccharide chains (2 to 19 residues). About 25% of the amino acids in these regions are linked to an oligosaccharide chain11. Most of these oligosaccharide chains terminate with fucose, sialic acid, and sulfate esters of galactose and N-acetylglucosamine12.
Below the mucosa is the glycocalyx, a filamentous glycoprotein network that is anchored to the microvilli. The thickness of the glycocalyx ranges from 400 nm up to 500 nm13., 14.. The glycocalyx is anchored to the brush border of microvilli, the projections of the apical side of intestinal enterocytes. The brush border has mostly digestive and absorptive functions. Enzymes responsible for peptide and saccharide digestion and membrane transporters that facilitate absorption of small molecules such as glucose are closely associated with microvilli.
In addition to columnar epithelial cells, mucus-secreting goblet cells, M cells, and enteroendocrine cells are found in the cell lining of the small intestine. M cells are part of Peyer's patches which are found throughout the cell lining of the lower small intestine. These structures appear as thickened portions of the cell lining and contain lymphocytes, macrophages, and dendritic cells. Peyer's patches have distinctly different properties than surrounding tissues and have been used as specific targets for vaccine delivery applications.
3.2. Absorption mechanisms
As shown in Fig. 3, molecules that diffuse across the mucosa and the glycocalyx must cross the intestinal epithelium before entering the bloodstream. In order for absorption to take place, nutrients and even pathogens must penetrate the mucosa, which can be compared to an unstirred water layer. After passing through the mucus layer, a drug must cross through the glycocalyx, and reach the epithelial layer. Transport across the epithelium can occur by either a paracellular route, between cells, or a transcellular route, across cells.
Figure 3.

Pathways for therapeutic drug absorption via the oral route. (A) Protein therapeutic entering the bloodstream via the transcellular pathway (passing through epithelial cells). (B) By altering or disrupting the tight junctions, proteins are able to transport to the bloodstream via the paracellular pathway (in between adjacent cells). (C) Proteins may also enter the bloodstream via transcytosis or cell receptor mediated endocytosis. There is significant literature currently studying this pathway of entry. (D) By entering the M cells of the Peyer's patches, protein may be absorbed into the lymphatic system. Here, antigens can interact with antigen presenting cells (e.g., macrophages and dendritic cells), which are critical for elicitation of protective immunity.
In paracellular transport, the molecule may simply diffuse between the tight junctions of adjacent cells. Typically, paracellular transport is controlled by the permeability of the tight junctions between neighboring epithelial cells. It has been shown that the permeability of the tight junctions is dependent on divalent cations such as Ca2+ and Mg2+. As the concentration of divalent cations decreases, the permeability of the tight junctions increases.
In contrast, transcellular transport allows transport through the cell. In order for molecules to transport across the intestinal epithelium by transcellular mechanisms, the molecule must cross the cell membrane. Permeation across the epithelium of the intestinal lining is bi-directional allowing for absorption and exorption. However, flow in the intestine favors absorption rather than exorption8. The intestinal wall can be compared to a semi-permeable porous lipid membrane. Water and polar small molecules can transport easily whereas hydrophilic molecules cannot pass as easily.
There are several mechanisms by which molecules can transport across the intestinal epithelial lining. For molecules that can easily pass through lipid membrane, diffusion is the primary mechanism of diffusion, which can be described by Fick's law. However, protein drugs are large, often hydrophilic macromolecules that cannot easily diffuse across a cell membrane.
Several forms of carrier-mediated transport can occur to facilitate the transport of more polar, hydrophilic molecules. Facilitated diffusion, sometimes referred to as facilitated transport or facilitated passive transport, involves an integral membrane protein which allows the passage of molecules across the cell membrane without an energy expenditure at the cellular level. Counter transport is similar to facilitated diffusion in that a carrier protein is involved. However, in counter transport, the transport of one molecule from the luminal side of the membrane is coupled with facilitated transport of a second molecule in the opposite direction. Active transport allows facilitated passage of molecules across the cell membrane but requires energy. Generally, active transport moves a molecule against a gradient.
Pinocytosis is another mechanism by which molecules may cross the apical side of intestinal epithelium. Pinocytosis allows transport of molecules by formation of an invagination in cell membrane. The invagination engulfs nearby molecules and then breaks off the cell membrane forming a vesicle. The vesicle enters the cytoplasm and can empty its contents or can fuse with the opposite side of the cell membrane and empty its contents. Although this is a promising mechanism for transport of large molecules, the regulatory mechanisms of pinocytotic transport are not well understood.
4. Approaches to oral protein and peptide delivery
An oral formulation for a protein and peptide drug must preserve the drug's structure, protect the drug from proteolysis, and allow for the drug to be absorbed into the bloodstream. A variety of approaches to oral protein delivery have been proposed to meet these needs. Of these, there are several predominant strategies for oral protein delivery: co-administering additional compounds for altering the physiology of the GI tract, modifying the drug, and delivering the drug using a carrier. The key advantages and major challenges of each approach are summarized in Table 1.
Table 1.
Summary of common approaches to enable oral protein and peptide delivery.
| Approaches to oral protein and peptide delivery | Common example | Advantage | Major challenge |
|---|---|---|---|
| Permeation enhancers | Surfactants, fatty acids, medium chain glycerides, steroidal detergents, acylcarnitines and alkanoylcholines, N-acetylated-a-amino acids and N-acetylated non-a-amino acids, chitosans |
|
|
| Protease inhibitors | Serpin, aprotinin and soybean trypsin inhibitors, camostat mesilate, chromostatin, ovomucoids, polymer inhibitor conjugates (such as carboxymethyl cellulose-elastinal) |
|
|
| Conjugation of protein and peptide drugs | PEG, transferrin, vitamin B-12, FcRn receptor molecules |
|
|
| Enteric coatings | Eudragit® systems, hypromellose phthalate |
|
|
| Degradable polymer matrices | Poly(lactide-co-glycolide), poly(epsilon-caprolactone) |
|
|
| Mucoadhesive carriers | PEG-grafted polymers, thiomers, chitosan, lectin, sodium alginate, pectin, cellulose derivatives |
|
|
| Complexation hydrogel carriers | Poly(methacrylic acid-g-ethylene glycol), poly(methacrylic acid-co-N-vinyl pyrrolidone), poly((methacrylic acid-co-N-vinyl pyrrolidone)-g-ethylene glycol), poly(itaconic acid-co-N-vinyl pyrrolidone) |
|
|
The large size of protein drugs on the order of 10 to 100 kDa limits their ability to diffuse across GI tract tissues, as shown in Fig. 4. The permeability of the intestinal epithelium has been shown to be lowest in the colon and highest in the upper small intestine. Even in the upper small intestine, where the intestinal epithelium is most permeable and over 90% of nutrients are absorbed, protein drug absorption is still low in comparison to smaller molecular weight drug counterparts.
Figure 4.

Permeability of protein and peptide therapeutics through the intestinal epithelium. (A) Schematic diagram of therapeutic drug diffusion across the intestinal epithelial barrier, described by the equation J = P(CD–CR) (Reprinted from Ref. 15 with permission). The therapeutic drug diffusion (J) is amount per unit time and surface area as a function of the permeability coefficient (P) and the therapeutic concentration on both sides of the transport barrier (CD and CR). (B, C) Permeability of various molecular weight species through a Caco-2 cell model gastrointestinal epithelium (Reprinted from Ref. 16 with permission).
4.1. Permeation enhancers
The co-administration of compounds which alter conditions in the GI tract has been investigated for improvement of protein bioavailability. Permeation enhancers typically enhance intestinal permeability by disrupting the epithelium's tight junctions. A variety of permeation enhancers have been studied including surfactants, fatty acids, medium chain glycerides (usually monoglycerides and diglycerides of caprylic and capric acid), steroidal detergents, acylcarnitines and alkanoylcholines, N-acetylated-α-amino acids and N-acetylated non-α-amino acids, and chitosans and other mucoadhesive polymers17.
Permeation enhancers have been studied as excipients in oral protein formulations in a number of studies to allow passage of these large drugs across the epithelium. For example, oral delivery of the nonapeptide leuprolide was shown to increase 4-fold in bioavailability with the addition of sodium salicylate as a permeation enhancer18. The effect of tetradecylmaltoside (TDM) on the intestinal absorption of low molecular weight heparins increased bioavailability19. The presence of trimethyl chitosan (TMC) in gas empowered drug delivery systems was shown to improve insulin absorption in rabbits20.
Fetih et al.21 compared the effects of several permeation enhancers on carboxyfluorescein oral delivery at different sites of the intestine. Their studies showed the greatest effect of permeation enhancers on the colonic delivery. In addition, they found that N-dodecyl-β-d-maltopyranoside had the greatest effect on absorption in the colon.
While permeation enhancers have been shown to improve oral protein bioavailability22, they have also been shown to damage the intestinal epithelium. This damage has been suggested to be reversible but only after the removal of the permeation enhancer22., 23.. Yang et al.19 showed that the effect of the permeation enhancer TDM on transepithelial electrical resistance of monolayers of C2BBe1 (a Caco-2 clone) was dependent on the concentration of the permeation enhancer but full recovery took nearly 30 h after removal. The disruption of tight junctions and damage to the epithelium associated with permeation enhancers may also diminish the immunoprotective function of the intestinal epithelium in preventing pathogen entry.
4.2. Protease inhibitors
The use of protease inhibitors to reduce degradation of protein drugs in the GI tract has also been investigated. Because protease inhibitors can reduce enzymatic degradation of protein drugs, protease inhibitors have been used to improve bioavailability and by maintaining the bioactivity of more of the drug. If more bioactive protein drug is available for absorption, a higher bioavailability can be achieved.
A number of studies have shown that protease inhibitors affect bioavailability and protein degradation. For example, Aoki et al.9 demonstrated that protease inhibitors effectively reduced the activity of a variety of enzymes linked to protein drug degradation. Other studies have confirmed the effects of protease inhibitors on bioavailability in vivo. Morishita et al.24 studied the effects of several protease inhibitors on insulin delivery in the small intestine. They observed an improved hypoglycemic effect over insulin controls indicating a greater the absorption of active insulin in the presence of protease inhibitor. In contrast, Yamamoto et al.25 only observed a noticeable effect of protease inhibitor on insulin delivery in the large intestine and not the small intestine. They cited variability in enzyme presence and activity between the small and large intestine for the discrepancy between their findings and literature. They also hypothesized that a key difference in experimental methods between the studies in which the lumen was washed I one study and not washed in the other as a contributing factor in the two studies.
The use of protease inhibitors to improve oral protein formulations has several main drawbacks. First, the variability in the performance based on enzyme quantity and activity raises serious concerns over predictable dosing and patient-to-patient variability in absorption. The long-term effects of continued exposure to protease inhibitors, which may include changes to food digestion, have not been fully investigated.
4.3. Conjugation of protein and peptide drugs
Carriers such as poly(ethylene glycol) (PEG) or a transporter molecule, have been used to impart enhanced properties to the protein drug. The conjugation of PEG to a protein drug through the process of PEGylation can lead to improved resistance to degradation as well as an extended half-life in the bloodstream. PEG is relatively resistant to protein binding and hence may impart enzyme-resistant properties to a protein drug through conjugation. In general, it is thought that the hydrophilicity of PEG causes water molecules to be closely associated with the polymer forming a hydration layer. This hydration layer acts as a barrier and interferes with protein adsorption. For example, PEG-insulin conjugates have been developed for oral delivery applications to improve enzyme resistance of insulin. Tuesca et al.26 demonstrated in vivo bioactivity of PEG-insulin conjugates and a prolonged circulation time in comparison to insulin after intravenous administration.
The modification of protein drugs by conjugation to transporter molecules has been investigated in order to improve protein absorption. In this method, the protein drug is conjugated to molecules absorbed via receptor-mediated endocytosis. The conjugation of the transporter molecule and the protein drug allows for absorption of the protein drug by the same absorption pathway as the transporter molecule. For example, transporter molecules of transferrin and vitamin B-12 have been investigated for conjugation to protein drugs. Vitamin B-12 has been used to form conjugates with granulocyte-colony stimulating factor, erythropoietin27, leutinizing hormone28, and insulin29. It has been shown that these conjugates transport across Caco-2 cell layers using the vitamin B-12 absorption pathway27. Orally administered B-12-insulin conjugates in the diabetic rat model had a 4.7-fold greater decrease in the area under the blood glucose curve in comparison to orally administered insulin indicating that B-12 conjugation improved oral bioavailability of active insulin29.
Conjugation to transferrin has also been used to improve transport. The transferrin molecule typically transports iron across the intestinal epithelium via receptor-mediated endocytosis. Conjugates of insulin and transferrin have been shown to improve drug transport in comparison to native insulin in intestinal epithelial cell models30., 31.. Insulin-transferrin conjugation was also shown to have increased transport over insulin in a mucus-producing co-culture model of Caco-2 and HT29-MTX cells32. These studies have suggested that the ability for transferrin to be uptaken by the transferrin receptor is imparted to the conjugate by competitive inhibition experiments30., 31..
With the administration of protein-transporter conjugates, the long-term effects of chronic administration still need to be evaluated. Furthermore, the effects of administering additional transferrin and vitamin B-12 on iron absorption and vitamin B-12 absorption, respectively, have not been investigated. Transferrin and vitamin B-12 facilitated transport mechanisms could also affect cell signaling but have not yet been investigated.
4.4. Enteric coatings
Protein drugs have also been encapsulated in polymeric matrices of two kinds: enteric coatings and polymer networks. The main advantage of using a polymer coating or matrix to encapsulate a protein drug is to protect the protein drug from degradation in the stomach. Enteric coatings, such as the Eudragit® systems, are non-crosslinked polymers that dissolve to release the drug. Typically, enteric coatings dissolve at a neutral pH to allow protection of the drug in the stomach at low pH. Because dissolution of the coating occurs at specific pH values, coatings can be selected such that release occurs in one segment of the small intestine.
One of the distinct disadvantages of this strategy is that enteric coatings can only improve the amount of bioactive protein drugs available for absorption but do not facilitate the absorption process. As a result, protease inhibitors and permeation enhancers have been used in conjunction with enteric coatings to achieve improved pharmacological availabilities23., 33., 34.. A strategy combining an enteric coating and either a protease inhibitor or a permeation enhancer raises additional concerns over long-term safety.
4.5. Polymeric carriers for oral protein delivery
In order to maintain bioactivity but also improve absorption, polymeric carriers have been developed. A variety of designs have been studied including degradable matrices, thiomers, and environmentally responsive polymer networks. Typically, the protein drug is entrapped in the polymer network, and release is controlled by diffusion. Diffusion of proteins through a polymer network is strongly dependent on the mesh size (ξ) of the polymer network, the hydrodynamic radius of the protein, and the strength of polymer-drug interactions.
4.5.1. Degradable carriers
In the case of degradable polymers, drug diffusion out of the carrier is dependent on the extent of degradation. For the oral delivery of protein drugs, degradable carriers can limit release prior to degradation but allow for increased release with degradation. The ideal degradable system would remain intact in the stomach and start degrading in either the small or large intestine. Degradation products from breakdown of the carrier may interact with GI tissue, be absorbed, or be excreted. Characterization of the effect of degradation products on the GI tract is a critical challenge to oral delivery using degradable carriers.
A variety of degradable carriers have been investigated for oral protein delivery. Of the most widely studied degradable polymer carriers is poly(lactide-co-glycolide), or PLGA. For example, PEG-coated PLGA nanoparticles were shown to deliver intact tetanus toxoid orally35. Insulin delivery from PLGA particles co-loaded with insulin and magnetite nanoparticles were shown to decrease blood glucose levels even though bioavailabilities were less than 2%36. PLGA was also used to deliver an insulin-sodium oleate complex orally generating a decrease in blood glucose37. In another study, PLGA particles with and without an enteric coating of hypromellose phthalate achieved bioavailabilities of 6.27% and 3.68%38. Oral formulations composed of PLGA–COOH with olive oil filler were used to orally deliver glucagon-like peptide-1. This formulation had a positive effect after an oral glucose challenge39. These studies demonstrated that a variety of PLGA-based particles effectively deliver pharmacologically active protein drugs via the oral route.
Another degradable polymer carrier that has been studied extensively in drug delivery applications is poly(ε-caprolactone) yet there are few studies that demonstrate oral protein delivery in vivo. Poly(ε-caprolactone)/Eudragit nanoparticles were shown to decrease blood glucose levels after oral delivery of either aspart-insulin40 and insulin41. For the insulin delivery system, the bioavailability of insulin was calculated to be 13%41.
4.5.2. Mucoadhesive delivery
One strategy for improving oral delivery of challenging molecules, such as proteins, is the use of mucoadhesive drug carriers. Mucoadhesion is a form of bioadhesion that involves a mucus substrate. The use of mucoadhesive carriers to interact with mucus at the site of absorption was proposed to prolong the residence time of carriers at the site of absorption. Extension of the residence time would allow for an increase in drug bioavailabilty.
There are five main theories that describe the mechanisms of bioadhesion. Often, adhesion is described as a combination of these mechanisms. The electronic theory of adhesion involves the transfer of electrons between the polymer and the tissue. The adjacent electrons form a double layer of charge at the interface causing adhesion between the two materials based on charge attraction. Adsorption theory describes adhesion as occurring based on secondary forces such as hydrogen bonding van der Waals forces. Unlike electronic theory and adsorption theory, the theory of wettability typically describes a system in which one substrate is a liquid and the other substrate is a solid. Wettability describes adhesion based on the ability of the liquid to spread on the surface of the solid. Chain interpenetration theory describes the mechanism of adhesion between polymer gels and tissue. Polymer chains can diffuse across the interface. These interpenetrating polymer chains anchor the two substrates together causing adhesion between the polymer and biological substrate.
4.5.3. Mucoadhesive hydrogel carriers
Specifically engineered carriers with the ability to form interactions with the intestinal environment have made improvements in oral drug delivery. For example, carriers have been designed to exhibit mucoadhesive interactions with the intestinal lining. These interactions are believed to enhance the residence time of the carrier in the intestine so that there is a greater period of time for controlled release at the intestinal lining where absorption takes place. Improving the residence time of the carrier and extending the time period over which release can occur in the small intestine is desirable, because protein drug released beyond the small intestine, in the colon for example, has a lower absorption and can result in a lower overall bioavailability of the drug.
Surface decoration of the carrier has been suggested as a means to enhance cellular interactions between the carrier and the intestine by forming mucoadhesive interactions. Synthetic polymers such as PEG have been used to promote mucoadhesion by interpenetration of PEG chains into the mucosa. The ability of PEG tethers to promote adhesive-like behavior by chain interpenetration into the mucosa has been confirmed both experimentally and theoretically34., 42., 43.. Additionally, PEG chains have been used in other drug delivery applications for protecting drug delivery systems from immune recognition and shielding drugs from enzymatic degradation44., 45..
While the mechanism behind mucoadhesion of PEG tethers is due to penetration of the PEG chains into the mucosa and interactions between PEG chains and the mucosa, other systems have been studied that can form more specific interactions. For example, polymer carriers have been decorated with lectins, which are proteins that bind specific carbohydrates, in order to bind to carbohydrate residues found on mucus glycoproteins. The decoration of wheat germ agglutinin onto P(MAA-g-EG) microspheres was shown to improve the bioavailability of orally delivered insulin46. Likewise, this concept of binding components of the mucosa has been applied to forming covalent interactions. Thiol groups have been added to carriers to form thiomers that can form covalent bonds between thiol groups of the carrier and cysteine groups in mucus glycoproteins47. In vivo studies on the oral delivery of insulin showed that thiolated polymers reduced the blood glucose more than non-thiolated polymers48.
Chitosan-based drug delivery systems are some of the most studied systems for oral delivery of macromolecules and are the subject of several extensive reviews49., 50.. One of the prevailing reasons that chitosan has been the focus of so many studies is chitosan's ability to interact with intestinal mucus through charge attraction. Oral delivery of insulin using chitosan nanoparticles achieved a pharmacological activity ranging from 14% to 15.6%51. Lower pharmacological availabilities were observed for oral delivery using nanoparticles composed of a chitosan shell and alginate core. However, insulin pharmacological activity was higher when delivered using the nanoparticles (3.4% and 6.8%) in comparison to free insulin (1.6%)52. Pharmacological availabilities as high as 5.4% were obtained from insulin oral delivery using carriers of nanoparticles composed of dextran sulfate/chitosan53. Calcitonin oral delivery was also demonstrated using liposomes composed of protease inhibitor-chitosan conjugates54.
4.5.4. Complexation hydrogels
For controlled release of orally delivered protein drugs, hydrogels have become attractive candidates for carrier design. Hydrogels are crosslinked, three-dimensional networks composed of hydrophilic polymers. Hydrogels have the ability to swell and imbibe water. Hydrogels are water insoluble and typically exhibit good biocompatibility. One of the unique properties of hydrogels is that hydrogels can be tailored to swell in response to various stimuli including pH, ionic strength, electric field, and temperature. Control of swelling is synonymous with control of diffusion into and out of the hydrogel network.
Control of diffusion processes of a carrier material allows for temporal and even spatial control of the release of protein drugs. This ability to tailor hydrogels is particularly advantageous for oral protein delivery. For oral delivery of protein drugs, release of the protein drug should be minimal in the stomach where degradation is likely to occur. Drug release in the intestine at the absorption site will allow for the maximal amount of active drug to be available for absorption. As shown in Fig. 5, hydrogels with pH-dependent swelling can be used to release the protein drug based on the increase in pH between the stomach and the intestine based on complexation behavior.
Figure 5.

Complexation behavior of P(MAA-g-EG) hydrogels. In acidic conditions, P(MAA-g-EG) hydrogels are in a collapsed state with a small mesh size due to hydrogen bonding between carboxylic acid groups and etheric groups. At neutral pH, deprotonation of the carboxylic acid groups leads to electrostatic and steric repulsions which causes swelling and increases the mesh size of the polymer network.
To use this pH transition from the stomach to the small intestine as a trigger for release, anionic complexation hydrogels have been developed which rely on the complexation properties of the cross-linked polymeric carrier to control release. Anionic complexation hydrogels, such as poly(methacrylic acid-grafted-ethylene glycol) (P(MAA-g-EG)), are composed of a methacrylic acid (MAA) backbone and grafts of PEG. In acidic conditions, the carboxylic acid residues of MAA are protonated and form hydrogen bonds with the etheric groups of PEG (cite Lowman). Hydrogen bonds between adjacent polymer chains form physical crosslinks causing the polymer to be in a collapsed state. As a result, diffusion into and out of the polymer network is limited. An entrapped protein drug is shielded from degradation, and release is prevented in the stomach. The physical crosslinks formed by the hydrogen bonding breaks down in the more neutral pH values of the small intestine. With an increase in pH, the carboxylic acids become deprotonated. Deprotonation causes hydrogen bonding to break down. Meanwhile, steric repulsions between polymer chains cause the polymer network to relax, and the polymer mesh size to increase. With this process comes macroscopic swelling. The protein drug is allowed to diffuse out of the polymer network and release in the small intestine.
P(MAA-g-EG) hydrogels were developed by Peppas and co-workers and have been investigated for oral delivery. Peppas et al. have demonstrated successful loading of a number of protein drugs including insulin, calcitonin55., 56., growth hormone56, interferon-beta55, and chemotherapeutics. These hydrogels effectively protect protein drugs in acidic conditions and from proteolytic degradation57. The ability of P(MAA-g-EG) to retain protein drugs and shield them from proteolytic degradation allows for delivery of bioactive drug. Control of release based on the pH change from the stomach to the small intestine allows for site-specific delivery of the payload.
In addition to protecting the bioactivity of protein drugs, P(MAA-g-EG) carriers have been shown to improve absorption of insulin. The ability to improve absorption stems from the ability of these carriers to interact with intestinal mucosa as well as interact with the tight junctions of enterocytes. The mechanism of mucoadhesion of P(MAA-g-EG) is that of chain interpenetration where PEG diffuse into the mucosa. The ability to interact with the intestinal mucosa has been shown to be dependent on the size of the particles and the composition of the polymer. Prolonging the residence time of the carrier allows for an extended time period for the drug to release from the carrier and to be absorbed. One of the other benefits of mucoadhesive delivery systems is a reduction in the path length towards absorption.
The ability of P(MAA-g-EG) carriers to influence paracellular transport is largely based on experimental evidence from in vitro studies. Confocal studies on insulin transport across Caco-2 monolayers showed that insulin transport was predominantly paracellular58. P(MAA-g-EG) carriers can reversibly open tight junctions by chelating Ca2+. Since tight junction proteins are dependent on Ca2+, the junctions loosen with depletion of Ca2+ concentrations. Studies by Peppas and co-workers have shown the recovery of tight junction integrity using transepithelial electrical resistance (TEER)59.
In vivo, P(MAA-g-EG) systems have improved bioavailability over orally administered insulin. The hypoglycemic effect of orally delivered insulin from P(MAA-g-EG) was first studied by Lowman et al.60 Oral insulin bioavailability from these systems was found to be 3.40% in healthy rats and 2.44% in diabetic rats for a 25 IU/kg dose. Higher bioavailabilities of 12.8% were observed in optimized system61. The size of P(MAA-g-EG) microparticles was shown to affect drug bioavailability with smaller particles performing better61.
5. Polysaccharides in drug delivery to impart targeting characteristics
Carbohydrates are the most abundant natural biomaterials, more abundant than nucleic acids and peptides. They form polymers with a number of repeat units including monosaccharides, disaccharides, oligosaccharides, and polysaccharides. Carbohydrates decorate a variety of molecules to form glycolipids, glycosaminoglycans, glycoproteins, and proteoglycans. Additionally, they are important components of many other biomolecules including ribonucleic acid (RNA) and deoxyribonucleic acid (DNA). Because of their integration into so many biomolecules, carbohydrates play essential roles in cellular communication, inflammation, infection, development, and disease62., 63., 64., 65..
While the goals of genomics and proteomics, which look at the biological information ‘stored’ in the genetic code of organisms and the amino acid sequences of proteins, are familiar, glycomics is a much more recent topic in biotechnology. Glycomics seeks to understand glycans as bioinformatics molecules65., 66., 67.. Researchers theorize that the coding capacity of polysaccharides is far greater than that of nucleic acids and amino acids because of the numerous ways that polysaccharides can form glycosidic bonds with one another and the different isomers these molecules may assume62., 63., 65., 67.. There is a tremendous amount of information to be gained from polysaccharide sequences and their interactions with proteins and nucleic acids64. Glycomics seeks to understand the role that polysaccharides play in cellular processes for advancing biotechnology.
Many fields within biotechnology stand to gain enormous rewards from glycomics research. More information on polysaccharides' roles in cell signaling, development, inflammation, and disease states is anticipated to be generated by glycomics. The applications of this knowledge in drug delivery and tissue engineering are innumerable.
5.1. The role of carbohydrates in adhesion
Carbohydrate interactions are typically weak63., 68., 69., 70.. However, multivalent interactions where multiple saccharides are involved enhance these interactions71., 72., 73., 74.. Two phenomena are at play in multivalent carbohydrate interactions: chelate effect where multiple interactions occur after an initial interaction and ligand-induced protein clustering63., 71., 75.. These properties and effects of carbohydrate interactions with other biomolecules make polysaccharide versatile biomaterials. With recent advances in glycomics, the critical roles of carbohydrates in physiology are becoming even more apparent.
Cellular processes of adhesion and recognition are often mediated by carbohydrate-carbohydrate and carbohydrate-protein interactions76. Single carbohydrate-carbohydrate interactions are considered weak interactions. Of the few studies that have quantified these, carbohydrate-carbohydrate interactions have been shown to have dissociation constants (Kd) of 1 × 10-4 mol/L77. However, the adhesion forces of glycan-glycan interactions are considerably strong with 190—310 pN adhesion forces which are in the range of antibody-antigen interactions (244 pN)76. It is generally thought that the strength of carbohydrate interactions increases with multivalency as more carbohydrate interactions occur in close proximity78. For example, monovalent carbohydrate-protein interactions have affinities of Ka = 103 to 104 L/mol and increase 2 to 6 fold with multivalency75.
5.2. Lectins and carbohydrates
Advances in carbohydrate research have largely been driven by the use of lectins. Lectins are proteins with affinities for specific saccharide residues. Many proteins classified as lectins are found decorating cell surfaces and are involved in many cell-carbohydrate interactions. Studies have shown that selectins, a subclass of lectins, mediate lymphocyte rolling and adhesion79. In addition, lectins have been shown to play a role in cancer cell migration80., 81..
Because of their specificity for saccharide units, lectins have also been used to study the roles of carbohydrates in biology. One of the primary uses of lectins has been to understand glycosylation patterns. Labeled lectins have been used for imaging carbohydrates in cellular matrices, on the surface of cells, and intracellularly. Recent microarray technology has been used to develop lectin microarrays. These powerful new tools require only small amounts of carbohydrate ligands and can be used for quantitative, simultaneous detection of different carbohydrates. These new tools have used to study the cell surface glycome of bacteria and mammalian cells82., 83..
5.3. Carbohydrates in drug delivery
5.3.1. Carrier systems
Chitosan has been used in several successful drug delivery studies with one of the most important being gene therapy delivery systems84., 85., 86., 87.. Chitosan has the distinct advantage to complex with DNA because of its positive charge and DNA's negative charge. Roy et al.88 developed chitosan nanoparticles to deliver gene therapy-based vaccines. Chitosan particles containing a genetic vaccine (against peanut allergy) were orally administered to mice. In sensitization studies, the resulting anaphylaxis of vaccinated mice with the chitosan drug delivery system was much lower than samples which were vaccinated with only the gene (without the chitosan carrier) and controls that were not vaccinated at all88. Roy et al.88 showed promising results that chitosan drug delivery systems can successfully deliver gene-based therapies orally. Senel et al.89 examined chitosan for protein drug delivery across buccal mucosa. Their results suggested that chitosan enhanced the permeability of transforming growth factor-β89. Janes et al.90 outline several other drugs that chitosan was used as a carrier for. These included bovine serum albumin and insulin.
Alginates have been studied for drug delivery as well91. Bouhadir et al.92 oxidized sodium aliginate to form a new monomer. They were able to engineer this polysaccharide delivery system to release three different model drugs in a controlled fashion via three different mechanisms of release: diffusion, ionic dissociation, degradation of a covalent bond92. Alginate has been used to release a variety of proteins as well including but not limited to fibroblast growth factor, bovine serum albumin, nerve growth factor, and interleukin-293.
While this is just a miniscule sampling of the research on carbohydrate-based drug carriers, several reviews discuss polysaccharide drug delivery systems including amylose, dextran, and pectin in more detail94., 95..
5.3.2. Targeted delivery
Lectin—carbohydrate interactions are beginning to be exploited for targeted drug delivery. The strength of their interactions with carbohydrates increases significantly with multivalency as outlined earlier in the paper. Both lectins and their polysaccharide counterparts may be used for targeting and adhesion96. In designing these targeting modalities for drug delivery systems, carbohydrates may be used to target lectins or vice versa. The exciting research into drug polysaccharide targeting systems shows promise for transforming drug delivery.
Several lectin-mediated drug delivery systems have been developed that use lectins to target specific tissues and systems96., 97.. Umamaheshwari et al.98 demonstrated that lectin-modified nanoparticles could be used to prevent H. pylori infection of human stomach cells. Lectins have been used to target drug delivery to epithelial M-cells and Caco-2 cells46., 59., 99., 100., 101.. Wood et al.101 used wheat germ agglutinin (a lectin) to target an insulin complexation hydrogel drug delivery system to carbohydrate residues found in intestinal mucosa. Results showed enhanced permeability towards insulin transport and indicated that wheat germ agglutinin led to improved bioavailability of insulin.
Xu et al.102 also investigated lectin-mediated oral delivery of insulin. In addition to wheat germ agglutinin, Xu et al. also investigated tomato lectin and Ulex europaeus agglutinin 1 (UEA1). Liposomes were modified with lectins and loaded with insulin. In vivo drug delivery studies showed that lectins increased insulin delivery over controls. Of the lectin modifications examined by Xu et al.102, wheat germ agglutinin performed the best.
Delivery of imaging modalities to cancer via lectins has been investigated by Hama et al.103., 104.. Hama et al. showed that avidin conjugated-BODIPY could be used to bind asialo-receptors on cancer cell lines and used fluorescence emission to detect cancerous cells. Nine different cancer cell lines were observed: ovarian, colon, pancreatic, gastric, breast, and prostate. Images were observed in each culture. Therefore, Hama et al. showed that lectins may be used to target cancer cells for delivery imaging systems.
Drug delivery systems that use carbohydrates to target a drug delivery system have also been studied105., 106., 107.. Cho et al.107 have investigated galactose containing particles to target the asialoglycoprotein receptors (containing a lectin domain) on hepatic cells. Their research investigated poly(l-lactic acid) particles coated with polystyrene and galactose for targeted drug delivery of trans-retinoic acid.
Another team of researchers investigated lectin-ligand chemistry as a means to deliver anti-inflammatory drugs to the blood vessel lining by targeting P-selectin which plays a significant role in blood vessel inflammation108. P-selectin is expressed by endothelial cells and supports neutrophil rolling mediated by Sialyl-Lewisx (sLex), a sialylated and fucosylated carbohydrate. These drug delivery systems were designed to mimic this neutrophil rolling to deliver anti-inflammatory drugs. PLGA microparticles were synthesized and decorated with sLex through avidin-biotin chemistry108. Laminar flow studies showed that drug delivery system supported rolling on P-selectin coated surfaces and that it was dependent on the density of carbohydrates on the PLGA microspheres108. This study took an innovative approach to anti-inflammatory drug delivery systems and indicates that carbohydrate-decorated drug delivery systems may be used to target lectins on cell surfaces in other therapies as well. Both systems described used carbohydrates to target lectins on cell surfaces for cell-specific drug delivery.
5.3.3. Other polysaccharide-related targeting strategies
Bertozzi et al.109., 110. have proposed an innovative strategy for drug delivery using polysaccharides. They have used polysaccharides to selectively modify cell surfaces so that these modified cells may be targeted by drug delivery systems. They utilized natural oligosaccharide (metabolism and) synthesis to incorporate a ketone containing N-levulinoyl mannosamine into sialic acids on cell surfaces instead of the N-acetylmannosamine. Biotin was then bound to these ketone groups on sialic acid, and Avidin-coupled toxins were used to target and kill the cells that were biotinylated109. They suggested that their methodology for engineering the cell surface for drug targeting may be further developed to selectively target tumor cells which tend to express high levels of sialic acid.
5.4. Carbohydrate-mediated adhesion in the intestine
Studies have shown that carbohydrates are involved in adhesion processes involving both carbohydrates and proteins, and that many occur at the intestinal wall. For example, many bacteria that colonize the small intestine rely on polysaccharides on their surfaces for adhesion to the intestinal wall. Gram-negative bacteria, which are often pathogenic, adhere to the mucosa with lipolysaccharides (LPS) found on their membranes. While LPS interacts strongly with the intestinal wall, LPS have been shown to be immunogenic111., 112., 113..
O-linked polysaccharides are known to play roles in cellular adhesion especially in cancer114. Studies have suggested that O-linked polysaccharides can serve as ligands for cell binding via selectins, but to our knowledge, there are no studies linking this class of polysaccharides to adhesive interactions with the intestinal wall114.
Teichoic acids are polysaccharides found in the cell wall of Gram-positive bacteria (often non-pathogenic). Research has shown that teichoic acid is involved in the adhesion of bacteria to stomach epithelial cells. A more recent study investigating a Caco-2 model showed that lipoteichoic acids of Lactobacillus johnsii strain La1 were involved in adhesion to the intestinal epithelial cell model114. Several studies have investigated the role of teichoic acid in septic shock and immune responses highlighting that the bacterial source is a determining factor in immunogenicity115., 116., 117..
The presence of hyaluronic acid in the extracellular matrix and the expression of hyaluronic acid on a wide variety of cell types make hyaluronic acid an attractive candidate for in vivo applications. Hyaluronic acid has been shown to be a suitable tissue engineering construct for intestinal epithelial cells118. Because intestinal epithelial cells adhered to the hyaluronic acid substrate, hyaluronic acid may be a good candidate for facilitating carrier interactions with the intestinal wall. It is important to note that hyaluronic acid served as a substrate for Caco-2 cells, meaning that the basolateral side of the polarized cells adhered well to hyaluronic acid.
Drug delivery systems of pectin, which contains polygalacturonic acid, have been investigated for ocular and colonic delivery119., 120.. In addition, rheological studies have shown that pectin can interact with mucin121. These interactions depended on mucin concentration but also on the degree of esterification of pectin121.
Galectins, a type of lectin, have affinities for β-galactosides and are expressed on the apical side of the intestinal epithelium. To the best of our knowledge, the ability of β-galactosides to interact with the intestinal epithelium in a receptor-ligand fashion has not yet been investigated. However, galectin-3 is expressed on the apical side of intestinal epithelial cells and has been shown to bind glycosaminoglycans containing β-galactose122., 123., 124., 125..
Dextran and pullulan are neutral polysaccharides composed of glucose units. These polysaccharides have been used in a variety of drug delivery applications. Dextran is especially well characterized for in vivo applications and is considered non-immunogenic. These polysaccharides are not considered to be strongly mucoadhesive. However, they are typically long, mostly linear, hydrophilic chains. Their structure would allow for the tethers of dextran and pullulan to diffuse into the mucosa and promote carrier interactions with the intestinal wall via chain interpenetration.
6. Recent studies in therapeutic protein and peptide delivery
Since the development of the first oral delivery systems (capsules and nanoparticles) for oral delivery of insulin in diabetic patients, Peppas et al.59., 126., 127., 128. have further developed the oral delivery technology and applied it to a number of other applications of oral and transmucosal delivery of therapeutic agents. More recently, new platforms for the oral delivery of "high isoelectric point drugs" have been studied extensively. Such drugs include a number of well-known therapeutic agents, especially Humira for the treatment of Crohn's disease129., 130., 131..
More specifically, Koetting et al.129., 130. have developed a new generation of oral delivery systems for delivery of calcitonin for possible treatment of osteoporosis in postmenopausal women. The authors synthesized pH-responsive hydrogels of itaconic acid (IA) copolymerized with N-vinyl pyrrolidone (NVP) or poly(ethylene glycol) methacrylate, designated P(IA-co-NVP) or P(IA-g-PEG), respectively. In pH-dependent swelling studies, the carriers displayed favorable swelling characteristics for delivery in the small intestine, achieving up to 10 times greater swelling ratios within a therapeutic time frame than previously studied P(MAA-g-EG) or P(MAA-co-NVP) hydrogels. Using in vitro loading and release studies, it was shown that the carriers were capable of delivering therapeutic levels of calcitonin, although methacrylic acid-based hydrogels exhibit greater overall release. Finally, it was demonstrated that improved delivery of calcitonin could be achieved via from simple optimization of the protein loading solution. Utilizing solutions of reduced ionic strength allows for up to 100% improvement in the fraction of loaded drug that is released and up to a 16-fold improvement in drug delivered per unit weight of polymer vehicle. Further, the authors demonstrated that up to 170% improvement can be obtained in the in vitro delivery capability of the high pI protein salmon calcitonin (sCT) resulting from use of itaconic acid as the pH-responsive moiety.
Two additional proteins have been evaluated: anti-TNF-α monoclonal antibody (MAb) and growth hormone (GH)131., 132., 133.. Besides their therapeutic relevance for the treatment of rheumatoid arthritis and growth hormone deficiency, respectively, these proteins have been selected because they exhibit a completely different window of stability, and have different size, solubility and isoelectric points than insulin. The hydrogel chemistries were rationally designed tailored to the specific protein properties. A reformulation of hydrogels was necessary to incorporate N-vinyl pyrrolidone (NVP), (P((MAA-co-NVP)-g-EG)) for the delivery of GH, and both P(MAA-g-EG) and P(MAA-co-NVP) hydrogels were synthesized using different crosslinkers were evaluated for the release and stability of the high molecular weight anti-TNF-α MAb.
Polymer disks were prepared by UV-initiated free radical solution polymerization, and crushed and sieved into microparticles of 50–150 μm in diameter. Dynamic and equilibrium pH swelling studies were completed to determine the behavior of the compositions in gastric and intestinal conditions. For the delivery of anti-TNF-α MAb, Fig. 6 illustrates the dynamic swelling behavior of both P(MAA-g-EG) and P(MAA-co-NVP) hydrogels synthesized using different crosslinkers (PEGDMA-400, TEGDMA, and PEGDMA-1000)131. All hydrogel formulations demonstrated an increase in the volume swelling ratio with an increase in pH, and the presence of EG dramatically increased the swelling potential of the system as compared to P(MAA-co-NVP) compositions. Additionally, incorporation of crosslinkers with increased length and hydrophilicity led to a significantly increased hydrogel swelling ratio in both the P(MAA-g-EG) and P(MAA-co-NVP) formulations. Further, a detailed evaluation on protein structure and in vitro bioactivity was performed in order to determine any instability caused by encapsulation and release from hydrogel microparticles. The primary, secondary, and tertiary structure of anti-TNF-α Ab was evaluated using SDS-PAGE, circular dichroism, and fluorescence spectroscopy. The biological function of the released anti- TNF-α was determined by measuring the neutralizing ability to the cytotoxic effect of TNF-α MAb in L929 cells. As shown in Fig. 7, a slight change in the tertiary structure (i.e., decrease in fluorescence intensity) of anti-TNF-α Ab was observed when released from P(MAA-co-NVP) hydrogels131.
Figure 6.

Dynamic weight swellg ratio, q, of P(MAA-g-EG) (A) and P(MAA-co-NVP) (B) disks with varying crosslinker composition (PEGDMA-400, TEGDMA, and PEGDMA-1000), in DMGA/sodium hydroxide buffers (Reprinted from Ref. 131 with permission).
Figure 7.

The secondary and tertiary structure of anti-TNF-α Ab released from P(MAA-g-EG) and P(MAA-co-NVP) microparticles (Reprinted from Ref. 131 with permission). (A) Changes in secondary structure from unencapsulated protein were monitored using circular dichroism spectroscopy. (B) Changes in tertiary structure from unencapsulated protein were monitored using fluorescence spectroscopy.
More recently, Horava et al.134., 135., 136. completed pioneering new work on the development of one of the first oral delivery systems for hemophiliac factor IX (FIX), an agent whose absence from the blood is a main cause of Hemophilia B. The pH-responsive microcarrier system is an anionic complexation hydrogel comprised of a copolymer of methacrylic acid and poly(ethylene glycol) dimethacrylate (PEGDMA). This polymer network is physically crosslinked with an ethylene glycol-containing agent. Therapeutically relevant protein, FIX, is encapsulated in the hydrogel post polymerization. Due to the pH-responsive swelling of the hydrogel, the microcarrier protects the protein from the harsh environment of the stomach and releases the protein in the small intestine, which is the desired site of absorption. The mesh size of the P(MAA-g-EG) system was tailored for the loading and release of FIX by crosslinking with poly(ethylene glycol) dimethacrylate of increasing molecular weight (MW=400, MW=600, or MW=1000). The MW 1000 PEGDMA crosslinking agent has a longer chain length, allowing for a significantly larger mesh size when compared to the MW 400 (78.9 ± 5.1 Å vs. 103.3 ± 5.2 Å). Increasing the PEGDMA length led to improved loading and protection of active FIX protein in simulated in vitro conditions. Further, the authors demonstrated enhanced in vitro intestinal absorption of FIX and showed that the presence of particles increased FIX permeability.
Peppas et al.137., 138., 139., 140., 141., 142., 143., 144., 145., 146., 147., 148. have developed a new generation of therapeutic formulations that can be used for the release of siRNA and microRNAs. Here, the authors designed a two-stage, multi-component delivery system of a hydrogel nanoparticle encapsulated within a microparticle. The particles were designed to have an inverse pH-responsive behavior to first allow the cargo to safely traverse the harsh environment of the GI tract through an anionic pH-responsive microparticle, and subsequently facilitate intracellular delivery of the siRNA payload through a cationic pH-responsive nanogel that exploits the natural pH gradient found during endosomal trafficking. Such systems show value in treating Crohn's disease, ulcerative colitis, and celiac disease. This platform technology has also applicability in designing a new generation of oral delivery vaccines149., 150.. The two-stage delivery holds can effectively address a few challenges in addition to those generally associated with oral delivery–including that protein antigens are not highly immunogenic and require both spatial and temporal presentation of the antigens to the immune system. Further, Schoener et al.151., 152., 153., 154. have studied oral delivery systems for chemotherapeutic agents for treatment of certain types of cancers.
7. Conclusions and future perspective
With the completion of the human genome project and advances in protein engineering, therapeutic proteins and peptides are poised to become more widely used for disease treatment. Protein drugs, such as insulin, hemophilia factors VIII and VIIII, calcitonin, growth hormone, and interferon-β, have traditionally been administered by injection because of their large molecular weights and the dependence of their bioactivity on structure.
While parenteral administration has been an effective route, alternative routes of administration are of interest to increase patient adherence, reduce waste, and eliminate adverse effects associated with repeated injections. As described in this review, the oral administration of protein drugs has major challenges. The activity of the protein drug must be maintained in the GI tract, and therapeutic levels of the active drug must make their way into the bloodstream leading to a therapeutic bioavailability. To meet these challenges, an oral delivery system must protect the drug in the stomach from proteolytic enzymes and acid but allow absorption of the drug in either the small or large intestine. The target site of absorption is the small intestine which is typically preferred for absorption of hydrophilic proteins due to a more permeable epithelium than the colon.
Co-administration of certain molecules in formulations, such as permeation enhancers and protease inhibitors, has improved the ability to orally deliver therapeutic proteins and peptides. However, there are still significant challenges associated with the approaches. Permeation enhancers do not protect the therapeutic from the harsh conditions of the GI tract and, thus, may result in significant therapeutic degradation before intestinal absorption. The use of protease inhibitors to alter conditions in the GI tract carries significant concerns, such as the long-term effects of continuous exposure and patient-to-patient variability in absorption.
Anionic complexation hydrogels demonstrate pH-responsive swelling behavior and have shown potential for use in such an oral protein and peptide formulation. Poly(methacrylic acid-grafted-ethylene glycol), P(MAA-g-EG), gels are particularly suited for oral protein delivery because they have the ability to swell dependent on pH allowing for drug release to be triggered by a change in pH. In more acidic environments, diffusion into and out of P(MAA-g-EG) gels is limited due to physical crosslinks composed of hydrogen bonds between methacrylic acid and ethylene glycol. In the neutral conditions of the small intestine P(MAA-g-EG) gels swell allowing for diffusion of entrapped drugs out of the gel. These properties allow for protection of the protein in the stomach but release in the small intestine.
In the case of oral delivery, the bioactivity of the therapeutic must not only be preserved in the GI tract, but sufficient absorption of the drug into the bloodstream must take place. A promising strategy to further improve bioavailability is to enhance the interactions between oral delivery carriers and the intestinal wall. A biomimetic approach based on the mediation of carbohydrates and polysaccharides in many adhesion and recognition processes has shown potential to improve the interactions. Improved interactions between the carrier and the intestinal wall will increase the residence time of the carrier allowing more time for the protein drug to release at the site of absorption. A better understanding of these mechanisms as well as other muscus-penetrating strategies will help in the design and development of the next generation of intelligent biocompatible carriers for peptide and protein delivery.
Acknowledgments
The work was supported in part by a grant from the National Institutes of Health (R01-EB-00246020) and the Cockrell Family Regents Chair. Angela M. Wagner was supported by a National Science Foundation Graduate Research Fellowship (DGE-1610403), the S.E.S.H.A. Endowed Graduate Fellowship in Engineering, and the Philanthropic Educational Organization Scholar Award.
Footnotes
Peer review under responsibility of Institute of Materia Medica, Chinese Academy of Medical Sciences and Chinese Pharmaceutical Association.
References
- 1.Leader B., Baca Q.J., Golan D.E. Protein therapeutics: a summary and pharmacological classification. Nat Rev Drug Discov. 2008;7:21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 2.Banting F.G., Best C.H., Collip J.B., Campbell W.R., Fletcher A.A. Pancreatic extracts in the treatment of diabetes mellitus. Can Med Assoc J. 1922;12:141–146. [PMC free article] [PubMed] [Google Scholar]
- 3.Itakura K., Hirose T., Crea R., Riggs A.D., Heyneker H.L., Bolivar F. Expression in Escherichia coli of a chemically synthesized gene for the hormone somatostatin. Science. 1977;198:1056–1063. doi: 10.1126/science.412251. [DOI] [PubMed] [Google Scholar]
- 4.Goeddel D.V., Kleid D.G., Bolivar F., Heyneker H.L., Yansura D.G., Crea R. Expression in Escherichia coli of chemically synthesized genes for human insulin. Proc Natl Acad Sci U S A. 1979;76:106–110. doi: 10.1073/pnas.76.1.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peyrot M., Rubin R.R., Kruger D.F., Travis L.B. Correlates of insulin injection omission. Diabetes Care. 2010;33:240–245. doi: 10.2337/dc09-1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fu A.Z., Qiu Y., Radican L. Impact of fear of insulin or fear of injection on treatment outcomes of patients with diabetes. Curr Med Res Opin. 2009;25:1413–1420. doi: 10.1185/03007990902905724. [DOI] [PubMed] [Google Scholar]
- 7.Lee W.C., Balu S., Cobden D., Joshi A.V., Pashos C.L. Medication adherence and the associated health-economic impact among patients with type 2 diabetes mellitus converting to insulin pen therapy: an analysis of third-party managed care claims data. Clin Ther. 2006;28:1712–1725. doi: 10.1016/j.clinthera.2006.10.004. [DOI] [PubMed] [Google Scholar]
- 8.Csáky T.Z. Intestinal permeation and permeability: an overview. In: Czáky T.Z., editor. Pharmacology of intestinal permeation I. Springer-Verlag; Berlin, Heidelberg: 1984. pp. 51–59. [Google Scholar]
- 9.Aoki Y., Morishita M., Takayama K. Role of the mucous/glycocalyx layers in insulin permeation across the rat ileal membrane. Int J Pharm. 2005;297:98–109. doi: 10.1016/j.ijpharm.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 10.Hwang S.J., Park H., Park K. Gastric retentive drug-delivery systems. Crit Rev Ther Drug Carr Syst. 1998;15:243–284. [PubMed] [Google Scholar]
- 11.Shogren R., Gerken T.A., Jentoft N. Role of glycosylation on the conformation and chain dimensions of O-linked glycoproteins: light-scattering studies of ovine submaxillary mucin. Biochemistry. 1989;28:5525–5536. doi: 10.1021/bi00439a029. [DOI] [PubMed] [Google Scholar]
- 12.MacAdam A. The effect of gastro-intestinal mucus on drug absorption. Adv Drug Deliv Rev. 1993;11:201–220. [Google Scholar]
- 13.Maury J., Bernadac A., Rigal A., Maroux S. Expression and glycosylation of the Filamentous Brush Border Glycocalyx (FBBG) during rabbit enterocyte differentiation along the crypt-villus axis. J Cell Sci. 1995;108:2705–2713. doi: 10.1242/jcs.108.7.2705. [DOI] [PubMed] [Google Scholar]
- 14.Frey A., Giannasca K.T., Weltzin R., Giannasca P.J., Reggio H., Lencer W.I. Role of the glycocalyx in regulating access of microparticles to apical plasma membranes of intestinal epithelial cells: implications for microbial attachment and oral vaccine targeting. J Exp Med. 1996;184:1045–1059. doi: 10.1084/jem.184.3.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Goldberg M., Gomez-Orellana I. Challenges for the oral delivery of macromolecules. Nat Rev Drug Discov. 2003;2:289–295. doi: 10.1038/nrd1067. [DOI] [PubMed] [Google Scholar]
- 16.Uskoković V., Lee K., Lee P.P., Fischer K.E., Desai T.A. Shape efect in the design of nanowire-coated microparticles as transepithelial drug delivery devices. ACS Nano. 2012;6:7832–7841. doi: 10.1021/nn3019865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aungst B.J. Intestinal permeation enhancers. J Pharm Sci. 2000;89:429–442. doi: 10.1002/(SICI)1520-6017(200004)89:4<429::AID-JPS1>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 18.Zheng Y., Qiu Y., Lu M.Y., Hoffman D., Reiland T.L. Permeability and absorption of leuprolide from various intestinal regions in rabbits and rats. Int J Pharm. 1999;185:83–92. doi: 10.1016/s0378-5173(99)00146-5. [DOI] [PubMed] [Google Scholar]
- 19.Yang T., Arnold J.J., Ahsan F. Tetradecylmaltoside (TDM) enhances in vitro and in vivo intestinal absorption of enoxaparin, a low molecular weight heparin. J Drug Target. 2005;13:29–38. doi: 10.1080/10611860400020191. [DOI] [PubMed] [Google Scholar]
- 20.Sadeghi A.M., Avadi M.R., Ejtemaimehr S., Abashzadeh S., Partoazar A., Dorkoosh F. Development of a gas empowered drug delivery system for peptide delivery in the small intestine. J Control Release. 2009;134:11–17. doi: 10.1016/j.jconrel.2008.10.012. [DOI] [PubMed] [Google Scholar]
- 21.Fetih G., Lindberg S., Itoh K., Okada N., Fujita T., Habib F. Improvement of absorption enhancing effects of n-dodecyl-β-d-maltopyranoside by its colon-specific delivery using chitosan capsules. Int J Pharm. 2005;293:127–135. doi: 10.1016/j.ijpharm.2004.12.017. [DOI] [PubMed] [Google Scholar]
- 22.Swenson E.S., Milisen W.B., Curatolo W. Intestinal permeability enhancement: efficacy, acute local toxicity, and reversibility. Pharm Res. 1994;11:1132–1142. doi: 10.1023/a:1018984731584. [DOI] [PubMed] [Google Scholar]
- 23.Hastewell J., Lynch S., Fox R., Williamson I., Skelton-Stroud P., Mackay M. Enhancement of human calcitonin absorption across the rat colon in vivo. Int J Pharm. 1994;101:115–120. [Google Scholar]
- 24.Morishita M., Morishita I., Takayama K., Machida Y., Nagai T. Novel oral microspheres of insulin with protease inhibitor protecting from enzymatic degradation. Int J Pharm. 1992;78:1–7. [Google Scholar]
- 25.Yamamoto A., Taniguchi T., Rikyuu K., Tsuji T., Fujita T., Murakami M. Effects of various protease inhibitors on the intestinal absorption and degradation of insulin in rats. Pharm Res. 1994;11:1496–1500. doi: 10.1023/a:1018968611962. [DOI] [PubMed] [Google Scholar]
- 26.Tuesca A.D., Reiff C., Joseph J.I., Lowman A.M. Synthesis, characterization and in vivo efficacy of PEGylated insulin for oral delivery with complexation hydrogels. Pharm Res. 2009;26:727–739. doi: 10.1007/s11095-008-9816-8. [DOI] [PubMed] [Google Scholar]
- 27.Russell-Jones G.J., Westwood S.W., Habberfield A.D. Vitamin B12 mediated oral delivery systems for granulocyte-colony stimulating factor and erythropoietin. Bioconjugate Chem. 1995;6:459–465. doi: 10.1021/bc00034a016. [DOI] [PubMed] [Google Scholar]
- 28.Russell-Jones G.J., Westwood S.W., Farnworth P.G., Findlay J.K., Burger H.G. Synthesis of LHRH antagonists suitable for oral administration via the vitamin B12 uptake system. Bioconjugate Chem. 1995;6:34–42. doi: 10.1021/bc00031a600. [DOI] [PubMed] [Google Scholar]
- 29.Petrus A.K., Vortherms A.R., Fairchild T.J., Doyle R.P. Vitamin B12 as a carrier for the oral delivery of insulin. ChemMedChem. 2007;2:1717–1721. doi: 10.1002/cmdc.200700239. [DOI] [PubMed] [Google Scholar]
- 30.Shah D., Shen W.C. Transcellular delivery of an insulin-transferrin conjugate in enterocyte-like Caco-2 cells. J Pharm Sci. 1996;85:1306–1311. doi: 10.1021/js9601400. [DOI] [PubMed] [Google Scholar]
- 31.Kavimandan N.J., Losi E., Peppas N.A. Novel delivery system based on complexation hydrogels as delivery vehicles for insulin-transferrin conjugates. Biomaterials. 2006;27:3846–3854. doi: 10.1016/j.biomaterials.2006.02.026. [DOI] [PubMed] [Google Scholar]
- 32.Shofner J.P., Phillips M.A., Peppas N.A. Cellular evaluation of synthesized insulin-transferrin bioconjugates for oral insulin delivery using intelligent complexation hydrogels. Macromol Biosci. 2010;10:299–306. doi: 10.1002/mabi.200900223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Morishita I., Morishita M., Takayama K., Machida Y., Nagai T. Enteral insulin delivery by microspheres in 3 different formulations using eudragit L100 and S100. Int J Pharm. 1993;91:29–37. [Google Scholar]
- 34.Sahlin J.J., Peppas N.A. Enhanced hydrogel adhesion by polymer interdiffusion: use of linear poly(ethylene glycol) as an adhesion promoter. J Biomater Sci Polym Ed. 1997;8:421–436. doi: 10.1163/156856297x00362. [DOI] [PubMed] [Google Scholar]
- 35.Tobío M., Sánchez A., Vila A., Soriano I., Evora C., Vila-Jato J.L. The role of PEG on the stability in digestive fluids and in vivo fate of PEG-PLA nanoparticles following oral administration. Colloids Surf B Biointerfaces. 2000;18 doi: 10.1016/s0927-7765(99)00157-5. :315--23. [DOI] [PubMed] [Google Scholar]
- 36.Cheng J., Teply B.A., Jeong S.Y., Yim C.H., Ho D., Sherifi I. Magnetically responsive polymeric microparticles for oral delivery of protein drugs. Pharm Res. 2006;23:557–564. doi: 10.1007/s11095-005-9444-5. [DOI] [PubMed] [Google Scholar]
- 37.Sun S., Liang N., Piao H., Yamamoto H., Kawashima Y., Cui F. Insulin-S.O (sodium oleate) complex-loaded PLGA nanoparticles: formulation, characterization and in vivo evaluation. J Microencapsul. 2010;27:471–478. doi: 10.3109/02652040903515490. [DOI] [PubMed] [Google Scholar]
- 38.Cui F.D., Tao A.J., Cun D.M., Zhang L.Q., Shi K. Preparation of insulin loaded PLGA-Hp55 nanoparticles for oral delivery. J Pharm Sci. 2007;96:421–427. doi: 10.1002/jps.20750. [DOI] [PubMed] [Google Scholar]
- 39.Joseph J.W., Kalitsky J., St-Pierre S., Brubaker P.L. Oral delivery of glucagon-like peptide-1 in a modified polymer preparation normalizes basal glycaemia in diabetic db/db mice. Diabetologia. 2000;43:1319–1328. doi: 10.1007/s001250051529. [DOI] [PubMed] [Google Scholar]
- 40.Damgé C., Socha M., Ubrich N., Maincent P. Poly(ε-caprolactone)/eudragit nanoparticles for oral delivery of aspart-insulin in the treatment of diabete. J Pharm Sci. 2010;99:879–889. doi: 10.1002/jps.21874. [DOI] [PubMed] [Google Scholar]
- 41.Damgé C., Maincent P., Ubrich N. Oral delivery of insulin associated to polymeric nanoparticles in diabetic rats. J Control Release. 2007;117:163–170. doi: 10.1016/j.jconrel.2006.10.023. [DOI] [PubMed] [Google Scholar]
- 42.Huang Y., Szleifer I., Peppas N.A. A molecular theory of polymer gels. Macromolecules. 2002;35:1373–1380. [Google Scholar]
- 43.Huang Y., Leobandung W., Foss A., Peppas N.A. Molecular aspects of muco- and bioadhesion: tethered structures and site-specific surfaces. J Control Release. 2000;65:63–71. doi: 10.1016/s0168-3659(99)00233-3. [DOI] [PubMed] [Google Scholar]
- 44.Molineux G. Pegylation: engineering improved pharmaceuticals for enhanced therapy. Cancer Treat Rev. 2002;28:13–16. doi: 10.1016/s0305-7372(02)80004-4. [DOI] [PubMed] [Google Scholar]
- 45.Owens D.E., III, Peppas N.A. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307:93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 46.Wood K.M., Stone G.M., Peppas N.A. Wheat germ agglutinin functionalized complexation hydrogels for oral insulin delivery. Biomacromolecules. 2008;9:1293–1298. doi: 10.1021/bm701274p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Leitner V.M., Marschütz M.K., Bernkop-Schnürch A. Mucoadhesive and cohesive properties of poly(acrylic acid)-cysteine conjugates with regard to their molecular mass. Eur J Pharm Sci. 2003;18:89–96. doi: 10.1016/s0928-0987(02)00245-2. [DOI] [PubMed] [Google Scholar]
- 48.Deutel B., Greindl M., Thaurer M., Bernkop-Schnürch A. Novel insulin thiomer nanoparticles: in vivo evaluation of an oral drug delivery system. Biomacromolecules. 2008;9:278–285. doi: 10.1021/bm700916h. [DOI] [PubMed] [Google Scholar]
- 49.George M., Abraham T.E. Polyionic hydrocolloids for the intestinal delivery of protein drugs: alginate and chitosan–a review. J Control Release. 2006;114:1–14. doi: 10.1016/j.jconrel.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 50.Bowman K., Leong K.W. Chitosan nanoparticles for oral drug and gene delivery. Int J Nanomed. 2006;1:117–128. doi: 10.2147/nano.2006.1.2.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pan Y., Li Y.J., Zhao H.Y., Zheng J.M., Xu H., Wei G. Bioadhesive polysaccharide in protein delivery system: chitosan nanoparticles improve the intestinal absorption of insulin in vivo. Int J Pharm. 2002;249:139–147. doi: 10.1016/s0378-5173(02)00486-6. [DOI] [PubMed] [Google Scholar]
- 52.Sarmento B., Ribeiro A., Veiga F., Sampaio P., Neufeld R., Ferreira D. Alginate/chitosan nanoparticles are effective for oral insulin delivery. Pharm Res. 2007;24:2198–2206. doi: 10.1007/s11095-007-9367-4. [DOI] [PubMed] [Google Scholar]
- 53.Sarmento B., Ribeiro A., Veiga F., Ferreira D., Neufeld R. Oral bioavailability of insulin contained in polysaccharide nanoparticles. Biomacromolecules. 2007;8:3054–3060. doi: 10.1021/bm0703923. [DOI] [PubMed] [Google Scholar]
- 54.Werle M., Takeuchi H. Chitosan-aprotinin coated liposomes for oral peptide delivery: development, characterisation and in vivo evaluation. Int J Pharm. 2009;370:26–32. doi: 10.1016/j.ijpharm.2008.11.013. [DOI] [PubMed] [Google Scholar]
- 55.Kamei N., Morishita M., Chiba H., Kavimandan N.J., Peppas N.A., Takayama K. Complexation hydrogels for intestinal delivery of interferon β and calcitonin. J Control Release. 2009;134:98–102. doi: 10.1016/j.jconrel.2008.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Carr D.A., Gómez-Burgaz M., Boudes M.C., Peppas N.A. Complexation hydrogels for the oral delivery of growth hormone and salmon calcitonin. Ind Eng Chem Res. 2010;49:11991–11995. doi: 10.1021/ie1008025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yamagata T., Morishita M., Kavimandan N.J., Nakamura K., Fukuoka Y., Takayama K. Characterization of insulin protection properties of complexation hydrogels in gastric and intestinal enzyme fluids. J Control Release. 2006;112:343–349. doi: 10.1016/j.jconrel.2006.03.005. [DOI] [PubMed] [Google Scholar]
- 58.Kavimandan N.J., Peppas N.A. Confocal microscopic analysis of transport mechanisms of insulin across the cell monolayer. Int J Pharm. 2008;354:143–148. doi: 10.1016/j.ijpharm.2007.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wood K.M., Stone G.M., Peppas N.A. The effect of complexation hydrogels on insulin transport in intestinal epithelial cell models. Acta Biomater. 2010;6:48–56. doi: 10.1016/j.actbio.2009.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lowman A.M., Morishita M., Kajita M., Nagai T., Peppas N.A. Oral delivery of insulin using pH-responsive complexation gels. J Pharm Sci. 1999;88:933–937. doi: 10.1021/js980337n. [DOI] [PubMed] [Google Scholar]
- 61.Morishita M., Goto T., Peppas N.A., Joseph J.I., Torjman M.C., Munsick C. Mucosal insulin delivery systems based on complexation polymer hydrogels: effect of particle size on insulin enteral absorption. J Control Release. 2004;97:115–124. doi: 10.1016/j.jconrel.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 62.Rudiger H., Siebert H.C., Solis D., Jimenez-Barbero J.I., Romero A., Von Der Lieth C.W. Medicinal chemistry based on the sugar code: fundamentals of lectinology and experimental strategies with lectins as targets. Curr Med Chem. 2000;7:389–416. doi: 10.2174/0929867003375164. [DOI] [PubMed] [Google Scholar]
- 63.Bertozzi C.R., Kiessling L.L. Chemical glycobiology. Science. 2001;291:2357–2364. doi: 10.1126/science.1059820. [DOI] [PubMed] [Google Scholar]
- 64.Ratner D.M., Adams E.W., Disney M.D., Seeberger P.H. Tools for glycomics: mapping interactions of carbohydrates in biological systems. ChemBioChem. 2004;5:1375–1383. doi: 10.1002/cbic.200400106. [DOI] [PubMed] [Google Scholar]
- 65.Hirabayashi J., Arata Y., Kasai K.I. Glycome project: concept, strategy and preliminary application to Caenorhabditis elegans. Proteomics. 2001;1:295–303. doi: 10.1002/1615-9861(200102)1:2<295::AID-PROT295>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 66.Freeze H.H. Genetic defects in the human glycome. Nat Rev Genet. 2006;7:537–551. doi: 10.1038/nrg1894. [DOI] [PubMed] [Google Scholar]
- 67.Gabius H.J., Siebert H.C., André S., Jiménez-Barbero J., Rüdiger H. Chemical biology of the sugar code. ChemBioChem. 2004;5:741–764. doi: 10.1002/cbic.200300753. [DOI] [PubMed] [Google Scholar]
- 68.Spillmann D., Burger M.M. Carbohydrate-carbohydrate interactions in adhesion. J Cell Biochem. 1996;61:562–568. doi: 10.1002/(sici)1097-4644(19960616)61:4<562::aid-jcb9>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 69.David A., Kopečková P., Minko T., Rubinstein A., Kopeček J. Design of a multivalent galactoside ligand for selective targeting of HPMA copolymer-doxorubicin conjugates to human colon cancer cells. Eur J Cancer. 2004;40:148–157. doi: 10.1016/j.ejca.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 70.Roy R. Syntheses and some applications of chemically defined multivalent glycoconjugates. Curr Opin Struct Biol. 1996;6:692–702. doi: 10.1016/s0959-440x(96)80037-6. [DOI] [PubMed] [Google Scholar]
- 71.Mammen M., Choi S.K., Whitesides G.M. Polyvalent interactions in biological systems: implications for design and use of multivalent ligands and inhibitors. Angew Chem Int Ed. 1998;37:2754–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 72.Yarema K.J., Bertozzi C.R. Chemical approaches to glycobiology and emerging carbohydrate-based therapeutic agents. Curr Opin Chem Biol. 1998;2:49–61. doi: 10.1016/s1367-5931(98)80035-5. [DOI] [PubMed] [Google Scholar]
- 73.Mann D.A., Kanai M., Maly D.J., Kiessling L.L. Probing low affinity and multivalent interactions with surface plasmon resonance: ligands for concanavalin A. J Am Chem Soc. 1998;120:10575–10582. [Google Scholar]
- 74.Sigal G.B., Mammen M., Dahmann G., Whitesides G.M. Polyacrylamides bearing pendant α-sialoside groups strongly inhibit agglutination of erythrocytes by influenza virus: the strong inhibition reflects enhanced binding through cooperative polyvalent interactions. J Am Chem Soc. 1996;118:3789–3800. [Google Scholar]
- 75.Burke S.D., Zhao Q., Schuster M.C., Kiessling L.L. Synergistic formation of soluble lectin clusters by a templated multivalent saccharide ligand. J Am Chem Soc. 2000;122:4518–4519. [Google Scholar]
- 76.Bucior I., Scheuring S., Engel A., Burger M.M. Carbohydrate-carbohydrate interaction provides adhesion force and specificity for cellular recognition. J Cell Biol. 2004;165:529–537. doi: 10.1083/jcb.200309005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Patel T.R., Harding S.E., Ebringerova A., Deszczynski M., Hromadkova Z., Togola A. Weak self-association in a carbohydrate system. Biophys J. 2007;93:741–749. doi: 10.1529/biophysj.106.100891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bucior I., Burger M.M. Carbohydrate-carbohydrate interaction as a major force initiating cell--cell recognition. Glycoconj J. 2004;21:111–123. doi: 10.1023/B:GLYC.0000044843.72595.7d. [DOI] [PubMed] [Google Scholar]
- 79.Lawrence M.B., Springer T.A. Neutrophils roll on E-Selectin. J Immunol. 1993;151:6338–6346. [PubMed] [Google Scholar]
- 80.Camby I., Le Mercier M., Mathieu V., Ingrassia L., Lefranc F., Kiss R. Galectin-1 as potential therapeutic target for cancer progression. Drug Future. 2008;33:1057–1069. [Google Scholar]
- 81.Thijssen V.L., Postel R., Brandwijk R.J., Dings R.P., Nesmelova I., Satijn S. Galectin-1 is essential in tumor angiogenesis and is a target for antiangiogenesis therapy. Proc Natl Acad Sci U S A. 2006;103:15975–15980. doi: 10.1073/pnas.0603883103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hsu K.L., Pilobello K.T., Mahal L.K. Analyzing the dynamic bacterial glycome with a lectin microarray approach. Nat Chem Biol. 2006;2:153–157. doi: 10.1038/nchembio767. [DOI] [PubMed] [Google Scholar]
- 83.Pilobello K.T., Slawek D.E., Mahal L.K. A ratiometric lectin microarray approach to analysis of the dynamic mammalian glycome. Proc Natl Acad Sci U S A. 2007;104:11534–11539. doi: 10.1073/pnas.0704954104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dang J.M., Leong K.W. Natural polymers for gene delivery and tissue engineering. Adv Drug Deliv Rev. 2006;58:487–499. doi: 10.1016/j.addr.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 85.Green D.W., Mann S., Oreffo R.O. Mineralized polysaccharide capsules as biomimetic microenvironments for cell, gene and growth factor delivery in tissue engineering. Soft Matter. 2006;2:732–737. doi: 10.1039/b604786f. [DOI] [PubMed] [Google Scholar]
- 86.Prabaharan M., Mano J.F. Chitosan-based particles as controlled drug delivery systems. Drug Deliv. 2005;12:41–57. doi: 10.1080/10717540590889781. [DOI] [PubMed] [Google Scholar]
- 87.Lim S.T., Martin G.P., Berry D.J., Brown M.B. Preparation and evaluation of the in vitro drug release properties and mucoadhesion of novel microspheres of hyaluronic acid and chitosan. J Control Release. 2000;66:281–292. doi: 10.1016/s0168-3659(99)00285-0. [DOI] [PubMed] [Google Scholar]
- 88.Roy K., Mao H.Q., Huang S.K., Leong K.W. Oral gene delivery with chitosan-DNA nanoparticles generates immunologic protection in a murine model of peanut allergy. Nat Med. 1999;5:387–391. doi: 10.1038/7385. [DOI] [PubMed] [Google Scholar]
- 89.S,enel S., Kremer M.J., Kas, S., Wertz P.W., Hıncal A.A., Squier C.A. Enhancing effect of chitosan on peptide drug delivery across buccal mucosa. Biomaterials. 2000;21:2067–2071. doi: 10.1016/s0142-9612(00)00134-4. [DOI] [PubMed] [Google Scholar]
- 90.Janes K.A., Calvo P., Alonso M.J. Polysaccharide colloidal particles as delivery systems for macromolecules. Adv Drug Deliv Rev. 2001;47:83–97. doi: 10.1016/s0169-409x(00)00123-x. [DOI] [PubMed] [Google Scholar]
- 91.Augst A.D., Kong H.J., Mooney D.J. Alginate hydrogels as biomaterials. Macromol Biosci. 2006;6:623–633. doi: 10.1002/mabi.200600069. [DOI] [PubMed] [Google Scholar]
- 92.Bouhadir K.H., Alsberg E., Mooney D.J. Hydrogels for combination delivery of antineoplastic agents. Biomaterials. 2001;22:2625–2633. doi: 10.1016/s0142-9612(01)00003-5. [DOI] [PubMed] [Google Scholar]
- 93.Gombotz W.R., Wee S.F. Protein release from alginate matrices. Adv Drug Deliv Rev. 1998;31:267–285. doi: 10.1016/s0169-409x(97)00124-5. [DOI] [PubMed] [Google Scholar]
- 94.Sinha V.R., Kumria R. Polysaccharides in colon-specific drug delivery. Int J Pharm. 2001;224:19–38. doi: 10.1016/s0378-5173(01)00720-7. [DOI] [PubMed] [Google Scholar]
- 95.Vandamme T.F., Lenourry A., Charrueau C., Chaumeil J.C. The use of polysaccharides to target drugs to the colon. Carbohydr Polym. 2002;48:219–231. [Google Scholar]
- 96.Gabor F., Wirth M. Lectin-mediated drug delivery: fundamentals and perspectives. STP Pharma Sci. 2003;13:3–16. [Google Scholar]
- 97.Gabor F., Stangl M., Wirth M. Lectin-mediated bioadhesion: binding characteristics of plant lectins on the enterocyte-like cell lines Caco-2, HT-29 and HCT-8. J Control Release. 1998;55:131–142. doi: 10.1016/s0168-3659(98)00043-1. [DOI] [PubMed] [Google Scholar]
- 98.Umamaheshwari R.B., Jain N.K. Receptor mediated targeting of lectin conjugated gliadin nanoparticles in the treatment of Helicobacter pylori. J Drug Target. 2003;11:415–424. doi: 10.1080/10611860310001647771. [DOI] [PubMed] [Google Scholar]
- 99.Jepson M.A., Clark M.A., Hirst B.H. M cell targeting by lectins: a strategy for mucosal vaccination and drug delivery. Adv Drug Deliv Rev. 2004;56:511–525. doi: 10.1016/j.addr.2003.10.018. [DOI] [PubMed] [Google Scholar]
- 100.Clark M.A., Hirst B.H., Jepson M.A. Lectin-mediated mucosal delivery of drugs and microparticles. Adv Drug Deliv Rev. 2000;43:207–223. doi: 10.1016/s0169-409x(00)00070-3. [DOI] [PubMed] [Google Scholar]
- 101.Wood K.M. The University of Texas at Austin; Austin: 2006. Molecular design of advanced oral protein delivery systems using complexation hydrogels [dissertation] [Google Scholar]
- 102.Zhang N., Ping Q.N., Huang G.H., Xu W.F. Investigation of lectin-modified insulin liposomes as carriers for oral administration. Int J Pharm. 2005;294:247–259. doi: 10.1016/j.ijpharm.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 103.Hama Y., Urano Y., Koyama Y., Kamiya M., Bernardo M., Paik R.S. In vivo spectral fluorescence imaging of submillimeter peritoneal cancer implants using a lectin-targeted optical agent. Neoplasia. 2006;8:607–612. doi: 10.1593/neo.06268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hama Y., Urano Y., Koyama Y., Choyke P.L., Kobayashi H. Targeted optical imaging of cancer cells using lectin-binding BODIPY conjugated avidin. Biochem Biophys Res Commun. 2006;348:807–813. doi: 10.1016/j.bbrc.2006.07.169. [DOI] [PubMed] [Google Scholar]
- 105.Eniola A.O., Hammer D.A. Characterization of biodegradable drug delivery vehicles with the adhesive properties of leukocytes II: effect of degradation on targeting activity. Biomaterials. 2005;26:661–670. doi: 10.1016/j.biomaterials.2004.03.003. [DOI] [PubMed] [Google Scholar]
- 106.Park I.K., Kim T.H., Park Y.H., Shin B.A., Choi E.S., Chowdhury E.H. Galactosylated chitosan-graft-poly(ethylene glycol) as hepatocyte-targeting DNA carrier. J Control Release. 2001;76:349–362. doi: 10.1016/s0168-3659(01)00448-5. [DOI] [PubMed] [Google Scholar]
- 107.Nishikawa M., Kamijo A., Fujita T., Takakura Y., Sezaki H., Hashida M. Synthesis and pharmacokinetics of a new liver-specific carrier, glycosylated carboxymethyl-dextran, and its application to drug targeting. Pharm Res. 1993;10:1253–1261. doi: 10.1023/a:1018949109004. [DOI] [PubMed] [Google Scholar]
- 108.Eniola A.O., Hammer D.A. Artificial polymeric cells for targeted drug delivery. J Control Release. 2003;87:15–22. doi: 10.1016/s0168-3659(02)00346-2. [DOI] [PubMed] [Google Scholar]
- 109.Mahal L.K., Yarema K.J., Bertozzi C.R. Engineering chemical reactivity on cell surfaces through oligosaccharide biosynthesis. Science. 1997;276:1125–1128. doi: 10.1126/science.276.5315.1125. [DOI] [PubMed] [Google Scholar]
- 110.Luchansky S.J., Goon S., Bertozzi C.R. Expanding the diversity of unnatural cell-surface sialic acids. ChemBioChem. 2004;5:371–374. doi: 10.1002/cbic.200300789. [DOI] [PubMed] [Google Scholar]
- 111.Raetz C.R., Whitfield C. Lipopolysaccharide endotoxins. Annu Rev Biochem. 2002;71:635–700. doi: 10.1146/annurev.biochem.71.110601.135414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ulevitch R.J., Tobias P.S. Receptor-dependent mechanisms of cell stimulation by bacterial endotoxin. Annu Rev Immunol. 1995;13:437–457. doi: 10.1146/annurev.iy.13.040195.002253. [DOI] [PubMed] [Google Scholar]
- 113.Wells C.L., Jechorek R.P., Olmsted S.B., Erlandsen S.L. Effect of LPS on epithelial integrity and bacterial uptake in the polarized human enterocyte-like cell-line Caco-2. Circ Shock. 1993;40:276–288. [PubMed] [Google Scholar]
- 114.Fukuda M. Roles of mucin-type O-glycans in cell adhesion. Biochim Biophys Acta. 2002;1573:394–405. doi: 10.1016/s0304-4165(02)00409-9. [DOI] [PubMed] [Google Scholar]
- 115.Granato D., Perotti F., Masserey I., Rouvet M., Golliard M., Servin A. Cell surface-associated lipoteichoic acid acts as an adhesion factor for attachment of Lactobacillus johnsonii La1 to human enterocyte-like Caco-2 cells. Appl Environ Microbiol. 1999;65:1071–1077. doi: 10.1128/aem.65.3.1071-1077.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Finney S.J., Anning P.B., Cao T.V., Perretti M., Evans T.W., Burke-Gaffney A. Butanol-extracted lipoteichoic acid induces in vivo leukocyte adhesion. Biochem Biophys Res Commun. 2007;364:831–837. doi: 10.1016/j.bbrc.2007.10.088. [DOI] [PubMed] [Google Scholar]
- 117.Schär-Zammaretti P., Ubbink J. The cell wall of lactic acid bacteria: surface constituents and macromolecular conformations. Biophys J. 2003;85:4076–4092. doi: 10.1016/S0006-3495(03)74820-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Esposito A., Mezzogiorno A., Sannino A., De Rosa A., Menditti D., Esposito V. Hyaluronic acid based materials for intestine tissue engineering: a morphological and biochemical study of cell-material interaction. J Mater Sci Mater Med. 2006;17:1365–1372. doi: 10.1007/s10856-006-0612-x. [DOI] [PubMed] [Google Scholar]
- 119.Bigucci F., Luppi B., Monaco L., Cerchiara T., Zecchi V. Pectin-based microspheres for colon-specific delivery of vancomycin. J Pharm Pharmacol. 2009;61:41–46. doi: 10.1211/jpp/61.01.0006. [DOI] [PubMed] [Google Scholar]
- 120.Sriamornsak P., Wattanakorn N. Rheological synergy in aqueous mixtures of pectin and mucin. Carbohydr Polym. 2008;74:474–481. [Google Scholar]
- 121.Ludwig A. The use of mucoadhesive polymers in ocular drug delivery. Adv Drug Deliv Rev. 2005;57:1595–1639. doi: 10.1016/j.addr.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 122.Gunning A.P., Bongaerts R.J., Morris V.J. Recognition of galactan components of pectin by galectin-3. FASEB J. 2009;23:415–424. doi: 10.1096/fj.08-106617. [DOI] [PubMed] [Google Scholar]
- 123.Iwaki J., Minamisawa T., Tateno H., Kominami J., Suzuki K., Nishi N. Desulfated galactosaminoglycans are potential ligands for galectins: evidence from frontal affinity chromatography. Biochem Biophys Res Commun. 2008;373:206–212. doi: 10.1016/j.bbrc.2008.05.190. [DOI] [PubMed] [Google Scholar]
- 124.Delacour D., Cramm-Behrens C.I., Drobecq H., Le Bivic A., Naim H.Y., Jacob R. Requirement for galectin-3 in apical protein sorting. Curr Biol. 2006;16:408–414. doi: 10.1016/j.cub.2005.12.046. [DOI] [PubMed] [Google Scholar]
- 125.Delacour D., Greb C., Koch A., Salomonsson E., Leffler H., Le Bivic A. Apical sorting by galectin-3-dependent glycoprotein clustering. Traffic. 2007;8:379–388. doi: 10.1111/j.1600-0854.2007.00539.x. [DOI] [PubMed] [Google Scholar]
- 126.Carr D.A., Peppas N.A. Assessment of poly(methacrylic acid-co-N-vinyl pyrrolidone) as a carrier for the oral delivery of therapeutic proteins using Caco-2 and HT29-MTX cell lines. J Biomed Mater Res Part A. 2010;92A:504–512. doi: 10.1002/jbm.a.32395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Liechty W.B., Kryscio D.R., Slaughter B.V., Peppas N.A. Polymers for drug delivery systems. Annu Rev Chem Biomol Eng. 2010;1:149–173. doi: 10.1146/annurev-chembioeng-073009-100847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Morishita M., Peppas N.A. Advances in oral drug delivery: improved bioavailability of poorly absorbed drugs by tissue and cellular optimization. Adv Drug Deliv Rev. 2012;64:479. doi: 10.1016/j.addr.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 129.Koetting M.C., Guido J.F., Gupta M., Zhang A., Peppas N.A. pH-Responsive and enzymatically-responsive hydrogel microparticles for the oral delivery of therapeutic proteins: effects of protein size, crosslinking density, and hydrogel degradation on protein delivery. J Control Release. 2016;221:18–25. doi: 10.1016/j.jconrel.2015.11.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Koetting M.C., Peppas N.A. pH-Responsive poly(itaconic acid-co-N-vinylpyrrolidone) hydrogels with reduced ionic strength loading solutions offer improved oral delivery potential for high isoelectric point-exhibiting therapeutic proteins. Int J Pharm. 2014;471:83–91. doi: 10.1016/j.ijpharm.2014.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Carrillo-Conde B.R., Brewer E., Lowman A., Peppas N.A. Complexation hydrogels as oral delivery vehicles of therapeutic antibodies: an in vitro and ex vivo evaluation of antibody stability and bioactivity. Ind Eng Chem Res. 2015;54:10197–10205. doi: 10.1021/acs.iecr.5b01193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.O'Connor C., Steichen S., Peppas N.A. Development and characterization of stimuli responsive hydrogel microcarriers for oral protein delivery. J Biomed Mater Res Part A. 2017;105:1243–1251. doi: 10.1002/jbm.a.36030. [DOI] [PubMed] [Google Scholar]
- 133.Steichen S., O'Connor C., Peppas N.A. Development of a P((MAA-co-NVP)-g-EG) hydrogel platform for oral protein delivery: effects of hydrogel composition on environmental response and protein partitioning. Macromol Biosci. 2016;17:1600266. doi: 10.1002/mabi.201600266. [DOI] [PubMed] [Google Scholar]
- 134.Horava S.D., Moy K.J., Peppas N.A. Biodegradable hydrophilic carriers for the oral delivery of hematological factor IX for hemophilia B treatment. Int J Pharm. 2016;514:220–228. doi: 10.1016/j.ijpharm.2016.05.056. [DOI] [PubMed] [Google Scholar]
- 135.Horava S.D., Peppas N.A. Design of pH-responsive biomaterials to enable the oral route of hematological factor IX. Ann Biomed Eng. 2016;44:1970–1982. doi: 10.1007/s10439-016-1566-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Horava S.D., Peppas N.A. Recent advances in hemophilia B therapy. Drug Deliv Transl Res. 2017;7:359–371. doi: 10.1007/s13346-017-0365-8. [DOI] [PubMed] [Google Scholar]
- 137.Creixell M., Peppas N.A. Co-delivery of siRNA and therapeutic agents using nanocarriers to overcome cancer resistance. Nano Today. 2012;7:367–379. doi: 10.1016/j.nantod.2012.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Fisher O.Z., Khademhosseini A., Peppas N.A. Drug delivery: nanoscale devices. In: Buschow K.H., Cahn R.W., Flemings M.C., Ilschner B., Kramer E.J., Mahajan S., editors. Encyclopedia of materials: science and technology. Elsevier; Oxford: 2010. pp. 1–9. [Google Scholar]
- 139.Forbes D.C., Creixell M., Frizzell H., Peppas N.A. Polycationic nanoparticles synthesized using ARGET ATRP for drug delivery. Eur J Pharm Biopharm. 2013;84:472–478. doi: 10.1016/j.ejpb.2013.01.007. [DOI] [PubMed] [Google Scholar]
- 140.Forbes D.C., Peppas N.A. Oral delivery of small RNA and DNA. J Control Release. 2012;162:438–445. doi: 10.1016/j.jconrel.2012.06.037. [DOI] [PubMed] [Google Scholar]
- 141.Forbes D.C., Peppas N.A. Differences in molecular structure in cross-linked polycationic nanoparticles synthesized using ARGET ATRP or UV-initiated polymerization. Polymer. 2013;54:4486–4492. [Google Scholar]
- 142.Forbes D.C., Peppas N.A. Polycationic nanoparticles for siRNA delivery: comparing ARGET ATRP and UV-initiated formulations. ACS Nano. 2014;8:2908–2917. doi: 10.1021/nn500101c. [DOI] [PubMed] [Google Scholar]
- 143.Knipe J.M., Chen F., Peppas N.A. Enzymatic biodegradation of hydrogels for protein delivery targeted to the small intestine. Biomacromolecules. 2015;16:962–972. doi: 10.1021/bm501871a. [DOI] [PubMed] [Google Scholar]
- 144.Knipe J.M., Peppas N.A. Multi-responsive hydrogels for drug delivery and tissue engineering applications. Regen Biomater. 2014;1:57–65. doi: 10.1093/rb/rbu006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Knipe J.M., Strong L.E., Peppas N.A. Enzyme- and pH-responsive microencapsulated nanogels for oral delivery of siRNA to induce TNF-α knockdown in the intestine. Biomacromolecules. 2016;17:788–797. doi: 10.1021/acs.biomac.5b01518. [DOI] [PubMed] [Google Scholar]
- 146.Liechty W.B., Caldorera-Moore M., Phillips M.A., Schoener C., Peppas N.A. Advanced molecular design of biopolymers for transmucosal and intracellular delivery of chemotherapeutic agents and biological therapeutics. J Control Release. 2011;155:119–127. doi: 10.1016/j.jconrel.2011.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Liechty W.B., Peppas N.A. Expert opinion: responsive polymer nanoparticles in cancer therapy. Eur J Pharm Biopharm. 2012;80:241–246. doi: 10.1016/j.ejpb.2011.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Phillips M.A., Gran M.L., Peppas N.A. Targeted nanodelivery of drugs and diagnostics. Nano Today. 2010;5:143–159. doi: 10.1016/j.nantod.2010.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sharpe L.A., Daily A.M., Horava S.D., Peppas N.A. Therapeutic applications of hydrogels in oral drug delivery. Expert Opin Drug Deliv. 2014;11:901–915. doi: 10.1517/17425247.2014.902047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Vela Ramirez J.E., Sharpe L.A., Peppas N.A. Current state and challenges in developing oral vaccines. Adv Drug Deliv Rev. 2017;114:116–131. doi: 10.1016/j.addr.2017.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Puranik A.S., Pao L.P., White V.M., Peppas N.A. Synthesis and characterization of pH-responsive nanoscale hydrogels for oral delivery of hydrophobic therapeutics. Eur J Pharm Biopharm. 2016;108:196–213. doi: 10.1016/j.ejpb.2016.09.007. [DOI] [PubMed] [Google Scholar]
- 152.Schoener C.A., Hutson H.N., Peppas N.A. pH-Responsive hydrogels with dispersed hydrophobic nanoparticles for the delivery of hydrophobic therapeutic agents. Polym Int. 2012;61:874–879. doi: 10.1002/pi.4219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Schoener C.A., Hutson H.N., Peppas N.A. pH-Responsive hydrogels with dispersed hydrophobic nanoparticles for the oral delivery of chemotherapeutics. J Biomed Mater Res Part A. 2012;101A:2229–2236. doi: 10.1002/jbm.a.34532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Schoener C.A., Hutson H.N., Peppas N.A. Amphiphilic interpenetrating polymer networks for the oral delivery of chemotherapeutics. AIChE J. 2013;59:1472–1478. [Google Scholar]