

Abstract
α1-antitrypsin (AAT) is a circulating serine protease inhibitor secreted from the liver and important in preventing proteolytic neutrophil elastase associated tissue damage, primarily in lungs. In humans, AAT is encoded by the SERPINA1 (hSERPINA1) gene in which a point mutation (commonly referred to as PiZ) causes aggregation of the miss-folded protein in hepatocytes resulting in subsequent liver damage. In an attempt to rescue the pathologic liver phenotype of a mouse model of human AAT deficiency (AATD), we used adenovirus to deliver Cas9 and a guide-RNA (gRNA) molecule targeting hSERPINA1. Our single dose therapeutic gene editing approach completely reverted the phenotype associated with the PiZ mutation, including circulating transaminase and human AAT (hAAT) protein levels, liver fibrosis and protein aggregation. Furthermore, liver histology was significantly improved regarding inflammation and overall morphology in hSERPINA1 gene edited PiZ mice. Genomic analysis confirmed significant disruption to the hSERPINA1 transgene resulting in a reduction of hAAT protein levels and quantitative mRNA analysis showed a reduction in fibrosis and hepatocyte proliferation as a result of editing. Our findings indicate that therapeutic gene editing in hepatocytes is possible in an AATD mouse model.
Keywords: α1-antitrypsin deficiency, CRISPR/Cas9 gene editing, Liver fibrosis, Protein aggregation
Highlights
-
•
α1-antitrypsin (AAT) is a circulating protein secreted from the liver and important in preventing tissue damage in lungs.
-
•
We used CRISPR/Cas9 to disrupt the gene of a mutant version of the protein to reverse liver pathology in a mouse model of human AAT deficiency (AATD)
-
•
Our gene editing approach reverted the AATD pathology and genomic analysis confirmed significant disruption to the gene.
In an attempt to treat a pathologic liver disease called α1-antitrypsin deficiency (AATD) in a mouse model of the human disease, we used CRISPR/Cas9 technology to remove a mutant form of hSERPINA1, which causes AATD in the mouse. Our gene editing approach reverted the disease associated with the mutated gene and we saw a reversal in liver fibrosis and mutant protein aggregation. Our findings in a mouse model indicate that therapeutic gene removal, by editing out a mutated form of the gene, in liver cells is possible.
1. Introduction
α1-antitrypsin (AAT) is an abundant circulating serine protease inhibitor, primarily secreted by hepatocytes, which protects against protease related lung connective tissue damage (Janciauskiene et al., 2011). In humans, homozygous inheritance of the Z-type mutant allele of AAT (PiZ) can result in AAT deficiency (AATD), which is a rare genetic condition resulting from mutations in the human SERPINA1 (hSERPINA1) gene. The PiZ mutation in the hSERPINA1 gene results in a single amino acid substitution (Glu342Lys), which causes protein miss-folding and aggregated protein retention in hepatocytes (Lomas et al., 1992; Ekeowaa et al., 2010). Around 10–20% of PiZZ homozygous AATD patients have an increased risk for clinical liver disease, including cirrhosis and hepatocellular carcinoma, due to mutant AAT protein aggregation and accumulation (Teckman and Lindblad, 2006). In addition to liver disease, AAT release from hepatocytes is impaired, resulting in lower circulating levels of AAT and consequently impaired lung function. Targeting a defective gene in order to cure inherited human disease is an attractive therapeutic option and the stable disruption of SERPINA1 variants in vivo, at the genomic or mRNA level, is a promising strategy to reduce levels of miss-folded AAT protein and ameliorate pathological findings in the liver.
A PiZ transgenic mouse model expresses the human Z-AAT protein at high levels and displays pathology associated with AATD in humans, including AAT protein aggregation and ER retention, liver fibrosis and hepatocellular carcinoma (Carlson et al., 1989; Qu et al., 1996; Teckman and Perlmutter, 1996; Wu et al., 1994; Marcus et al., 2010). The PiZ mouse has been used extensively in pre-clinical AATD research including hepatic repopulation (Ding et al., 2011) and in short interfering RNA (siRNA) and antisense oligonucleotide (ASO) approaches, which resulted in reduced Z protein production and improved liver health (Cruz et al., 2007 and Guo et al., 2015).
In this study, we explored whether targeting the hSERPINA1 gene, rather than its mRNA, can be used as an alternative therapeutic approach requiring only a single treatment. The CRISPR/Cas9 technology has been successfully used in mouse models for correcting the dystrophin gene mutation (Long et al., 2014; Zhang et al., 2017), correction of a Fah mutation in hepatocytes (Yin et al., 2014) and for excision of the HIV-1 provirus (Yin et al., 2017). CRISPR/Cas9 introduces a DNA double strand break (DSB) at predefined loci determined by a highly specific guide RNA molecule (gRNA), and is a technology which has the potential to give rise to new classes of treatments for a wide range of diseases. We designed and used a gRNA specific for hSERPINA1 expressed in the liver of PiZ mice with the aim of disrupting the gene and reversing the disease phenotype associated with the expression of the human PiZ allele. Our therapeutic gene editing approach led to a reversal of the PiZ phenotype, which included reduced circulating transaminases and Z-AAT levels, liver fibrosis and protein aggregation. Furthermore, liver histology was significantly improved in terms of inflammation and overall morphology in gene edited PiZ mice. Genomic and molecular evaluation of gene edited PiZ mice revealed major disruption of hSERPINA1 transgene sequences and reduced expression of liver fibrosis-related and proliferation-related markers.
2. Methods
2.1. Animal Care and Experimentation
All animal experiments were approved by the Gothenburg Ethics Committee for Experimental Animals. PiZ mice were generated as described previously (Carlson et al., 1989) and were kindly provided by Jeff Teckman of St. Louis University, USA. Experimental animals were generated by breeding heterozygous PiZ mice to C57Bl/6 N mice (Charles River). Control animals used throughout the experiments were wild type littermates of the PiZ mice. Mice were housed in a temperature controlled room (21 °C) with a 12:12 h light-dark cycle (dawn: 5.30 am, lights on: 6.00 am, dusk: 5.30 pm, lights off: 6 pm) and controlled humidity (45–55%). They had access to a normal chow diet (R36, Lactamin AB, Stockholm, Sweden) and water ad libitum and were checked daily.
Baseline plasma and tissue sampling was performed at 8 weeks of age. At 9 weeks of age female PiZ mice and female wild type littermates were injected intravenously with an amount of replication-deficient Type-5 adenovirus (deleted in the E1 and E3 regions) corresponding to 1.4 × 109 PFU of either the control Ad-CMV-eGFP-CBh-FLAG-spCas9 virus (n = 4,catalog number 1904, Vector Biolabs, Malvern, PA, USA) or the targeting Ad-U6-SERPINA1-gRNA-CBh-FLAG-spCas9 virus (n = 4, custom order, construct name: “Ad-SerpinA-gRNA-CBh-spCas9”, Vector Biolabs, Malvern, PA, USA; gRNA sequence TGCTGACCATCGACAAGAA). Blood plasma was analyzed one week prior to viral dosing and at 4, 6 and 9 weeks post-dosing through a panel of plasma chemistry parameters (human AAT, ALT and AST). At termination, mice were euthanized under isoflurane anesthesia, blood samples collected by intra-cardiac puncture in EDTA coated tubes, and livers removed, weighed and used for subsequent gene expression, protein expression and histological analyses. All tissues for histological analysis were preserved in fixatives and sectioned at 5 μm prior to staining.
2.2. hSERPINA1 Guide RNA Selection
Guide RNA sequences were designed using online software tools. Surveyor nuclease assay was used to detect indels introduced by four separate hSERPINA1 specific gRNAs. Surveyor nuclease assay using the Transgenomic SURVEYOR mutation detection kit for standard gel electrophoresis was used according to the manufacturer's instructions (Integrated DNA Technologies, Stokie, IL, USA). The human retinal pigment epithelial cell line ARPE-19, maintained in DMEM/F12 supplemented with 10% FBS, was used to transfect a plasmid construct expressing Cas9 and hSERPINA1 specific gRNAs.
2.3. Droplet Digital PCR
Genomic DNA was isolated from mouse liver obtained from 4 hSERPINA1 gRNA treated mice and 4 control treated mice by incubating approximately 10 mg liver overnight in a shaking incubator at 56 °C in lysis buffer containing 50 mM Tris at pH 8 (15504-020, Invitrogen), 25 mM EDTA at pH 8 (AM9260G, ThermoFisher), 100 mM NaCl (S7653, Sigma-Aldrich), 1% SDS (862010, Sigma-Aldrich) and 0.05 mg/ml proteinase K (P6556, Sigma-Aldrich). The DNA was extracted using isopropanol, the pellet was washed with 70% ethanol and dried before being re-suspended in ultrapure RNase and DNase free water (10977-035, Invitrogen). The DNA concentrations were measured and solutions of 3 ng/μl prepared for all samples. A FAM-labeled ddPCR-assay for the hSERPINA1 gene was designed using the BioRAD website tool. The exact sequence of the primers and probe is proprietary (custom assay number 10042958, BioRAD). A Mastermix was prepared using a final concentration of 1× ddPCR Supermix for Probes, no dUPT (186-3024, BioRad), 1× FAM-labeled human SERPINA1-assay (custom assay 10042958, BioRAD), 1× AP3B1-HEX labeled mouse reference assay (dHsaCP1000001, BioRAD) and 1/40 HaeIII (15205016, Invitrogen). 20 μl Mastermix per well to be analyzed was prepared in ultrapure RNase and DNase free water (10977–035, Invitrogen). After adding 5 μl DNA at 3 ng/μl to the 20 μl Mastermix in the semi-skirted 96-well plate (30129504, Eppendorf) followed by careful mixing, the plate was sealed (18114040, BioRAD) using a PX1™ PCR Plate Sealer (Bio-Rad, cat no. 181-4000). An automated Droplet Generator (BioRAD) was used to generate droplets in a new semi-skirted 96-well PCR plate (30129504, Eppendorf). The PCR plate was sealed (18114040, BioRAD) using a PX1™ PCR Plate Sealer (Bio-Rad, cat no. 181-4000). After sealing, the PCR plate was placed in a C1000 Touch™ Thermal Cycler (Bio-Rad, cat no. 185-1197) for PCR amplification, as detailed in Supplementary data Table 2. The droplet reading was performed with the QX 100 Droplet reader (Bio-Rad, cat no. 186-3001) using ddPCR™ Droplet Reader Oil (Bio-Rad, cat no. 186-3004). To explore the performance of the assay, an annealing (and extension) temperature gradient from 55 to 65 °C was tested to identify the temperature giving the largest separation between the four droplet populations (SERPINA1-positive, AP3B1-positive, positive for both targets, and empty droplets). Annealing temperatures of 55.0–59.0 °C worked equally well, and 57.0 °C was used in the sharp run.
Data acquisition and analysis was performed using the software QuantaSoft (Bio-Rad) and the “CNV2” program (two copies of the reference gene AP3B1 exist in the mouse genome). The fluorescence amplitude threshold was set manually as the midpoint between the average fluorescence amplitude of the four droplet clusters (SERPINA1-positive, AP3B1-positive, positive for both targets, and empty droplets). The same threshold was applied to all the wells of the ddPCR plate.
2.4. Quantitative Real-time PCR
RNA was extracted from 50 mg mouse liver using Qiagen RNeasy mini columns (74,104, Qiagen) in a Qiacube instrument (Qiagen) after first lysing the tissue in the presence of RLT-buffer (Qiagen) containing 20 mM dithiothreitol (43816, Sigma-Aldrich) and stainless steel beads (69989, Qiagen) in a Tissue lyser II instrument (5 min shaking at 25 Hz/s), following the instructions from the supplier. A DNase treatment step was included. Total RNA (2.5 μg) was transcribed using the High Capacity cDNA Reverse Transcription kit (4368813, Life Technologies). The cDNA was diluted 10-fold, and 12 ng was used in a total qPCR reaction volume of 10 μl. For gene expression assays used, see Supplementary table 1. The qPCR-reactions were set up in triplicate for each sample and target in 384-well format using TaqMan® Gene Expression Master Mix containing the FAM-dye reporter (4369016, ThermoFisher) and PPIA as the internal control. The Quantstudio 7 Flex instrument (Life technologies) was used to run the qPCR selecting the procedure for standard reagents. After retrieving the Ct-values, relative expression levels were determined using the 2-ΔΔCt-method (Livak et al., 2001).
2.5. Targeted Deep Sequencing
Off-target sites (up to 4 mismatches) for the hSERPINA1 gRNA in mouse genome were identified. Primers for target site and 16 potential off-target sites were designed using Primer-Blast (Ye et al., 2012) and ordered with NGS linkers. PCR products for on-target and selected off-target sites were generated using Phusion Flash High-Fidelity PCR Master Mix (ThermoFisher) from 30 ng of genomic DNA extracted from liver samples. Products were generated from 4 liver samples for each of two experimental conditions: (1) control dosed PiZ mice, (2) PiZ mice treated with hSERPINA1 gRNA. Amplified PCR products were subjected to paired-end sequencing using NextSeq500. PCR products were amplified using Illumina Nextera XT Index kit v2 adapters. Libraries were quantified with Qubit HS (ThermoFisher) and Fragment Analyzer (Advanced Analytical Technologies). Indexed libraries were pooled and sequenced on an Illumina NextSeq500 mid-output run using paired-end chemistry with 150 bp read length.
2.6. NGS Data Analysis
Sequencing reads were automatically de-multiplexed using NextSeq500 Instrument (Illumina) and paired FASTQ files were analyzed using CRISPResso (Pinello et al., 2016). Briefly, reads with a minimum average quality score of 33 were aligned to the reference sequence. A window of 50 base pair centered on the predicted cleavage site was specified for the quantification of indels. Single nucleotide variations were eliminated from analysis. The frequency of deletions and insertions among NGS reads were automatically calculated using CRISPResso. The frequency of mutated alleles was calculated based on the CRISPResso's detected alleles, “Alleles_frequency_table.txt”, in NGS data as follows. First, all of the detected alleles were imported into the Microsoft Excel program. Next, the number of reads for alleles with identical 20 nucleotides around the predicted cut site was consolidated. Alleles with a frequency of <0.01% were excluded from the analysis. Finally, the relative frequency of the 10 most frequent alleles was calculated.
2.7. Transaminase and hAAT Plasma Measurements
Alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels in mouse plasma were measured using an enzymatic ALT/AST CP assay (Horiba ABX Diagnostics, France) according to the manufacturer's instructions. Measurements were carried out in duplicates and were correlated to a 2-fold diluted standard curve generated from an ABX Pentra MultiCal solution (Horiba ABX Diagnostics, France). Human Alpha-1-Antitrypsin was measured using ELISA assay (Bethyl Laboratories, Inc. Montgomery, Texas, USA) according to the manufacturer's instructions.
2.8. Histological Staining and IHC Analysis
Mouse livers were collected, fixed in 10% buffered formalin, and paraffin embedded (FFPE). Sections were stained with hematoxylin and eosin (HE), PAS-Diastase to visualize AAT aggregates and Masson Tri-Chrome (MTC) to visualize collagen fibers using standard protocols. Paraffin embedded sections were stained with an antibody against hAAT (IR505, ready-to-use, anti-hAAT, Dako, Carpinteria, CA) on a Discovery XT Multi-Stainer (Ventana Medical Systems, Tucson, Arizona). All histological analyses were performed by a board-certified pathologist blinded to the origin of the tissue.
2.9. Quantitative Analysis
Automated IHC image analysis for hAAT positive areas in liver parenchyma were quantitated on scanned slides, using HALO software (Histologix, Nottingham, U.K.).
2.10. Statistical Analysis
All values are given as group means ± SEM. Statistical analysis was done by Student's t-test for pair-wise comparison between mice. p < 0.05 between the groups was considered to be statistically significant differences.
3. Results
3.1. Human SERPINA1 Transgene Analysis in the PiZ Mouse
PiZ mice were generated by random insertion of a hSERPINA1 genomic fragment carrying the PiZ mutation (Carlson et al., 1989). Because this method can result in multiple copies of the transgene being introduced within a specific loci, Targeted Locus Amplification (TLA) analysis was performed (Cergentis, Utrecht, Netherlands) to determine both the chromosomal insertion point and the approximate hSERPINA1 transgene copy number. The PiZ transgene was integrated in mouse chromosome 2 between bases 62738461–62759843, which coincides with the Kcnh7 gene. All sequences intervening the 5′ and 3′ integration sites were deleted including an exon of Kcnh7. Apart from this 21-kilobase deletion, no evidence for any additional structural variation surrounding the integration site was observed. While an exact copy number cannot be determined using TLA, an estimation can be made based on the number of integration sites, number of fusion reads, ratio of coverage on the PiZ transgene array and genome integration site, and the data suggested >10 transgene copies are present.
3.2. hSERPINA1 Specific Guide RNA Selection and Adenoviral CRISPR/Cas9 Treatment
Selection of an efficient guide RNA (gRNA) is important for successful gene editing using CRISPR/Cas9 and we designed and tested four hSERPINA1 specific guides by transfecting a human cell line (ARPE-19) to determine the efficiency of cutting for each guide using an endonuclease assay (Supplementary Fig. 1). gRNA #4 was efficient in cutting hSERPINA1 and therefore selected for further work. Adenovirus containing Cas9, FLAG and the hSERPINA1 gRNA, or Cas9 and GFP-FLAG (as control) were generated. Nine week old PiZ female mice were allocated into two cohorts and injected with either Cas9-hSERPINA1 gRNA virus or control Cas9-GFP-FLAG virus at 1.4 × 109 PFU via the tail vein. In addition, wild type littermate mice were also dosed with these virus.
3.3. Transaminase and hAAT Levels in Plasma
To assess the effectiveness of the treatment with the gRNA targeting hSERPINA1, plasma levels of aspartate transaminase (AST) and alanine transaminase (ALT) were assessed, given that elevation of these enzymes are indicative of liver damage or hepatotoxicity (Guo et al., 2015). Prior to viral dosing, plasma ALT and AST protein levels, as well as plasma human AAT (hAAT) protein levels were significantly elevated in eight week old PiZ mice compared to wild type mice (Fig. 1a). Four weeks after viral delivery, ALT and AST were significantly reduced by 74% (p < 0.001) and 60% (p < 0.001) respectively in the hSERPINA1 gRNA dosed vs. control dosed PiZ mice (Fig. 1b). In fact, the hSERPINA1 gRNA dosed mice displayed transaminase levels similar to control-dosed WT mice. In addition, circulating hAAT levels were significantly reduced by 94% (p < 0.001) in the hSERPINA1 gRNA dosed vs. control dosed PiZ mice (Fig. 1c). The hSERPINA1 gRNA dosed PiZ mice displayed a 60% (p < 0.01) plasma ALT reduction six weeks post dose and a 50% ALT reduction (p < 0.01) 9 weeks post dose compared to PiZ mice dosed with control virus (Fig. 1b). At 9 weeks post dosing the hSERPINA1 gRNA dosed mice had similar ALT levels compared to wild type mice. Plasma AST levels were not significantly different at baseline nor at 9 weeks post dose between PiZ and wild type mice. Circulating hAAT levels remained low in the hSERPINA1 gRNA virus dosed PiZ mice at the six week and nine week time-points, as expected after the genetic modification introduced by the CRISPR-therapy. In the control virus dosed PiZ mice we observed lower plasma hAAT levels at the six week time-point compared to 4 weeks post dose and a further reduction at the 9 week time-point.
Fig. 1.

Temporal changes in ALT, AST and hAAT following treatment with hSERPINA1–specific guide in PiZ mice.
Baseline levels of AST, ALT and hAAT in PiZ and wild type littermate control mice prior to treatment with a hSERPINA1 specific guideRNA (a). A time course of AST, ALT, and hAAT measurements were taken following treatment with hSERPINA1-specific gRNA, control gRNA control in PiZ and wild type mice. Plasma levels of ALT and AST (b) and hAAT (c) were determined in blood samples collected from mice treated with hSERPINA1-specific gRNA or control gRNA in PiZ and WT mice at four time-points (1 week prior to treatment and 4, 6 and 9 weeks post-treatment). Statistical analyses performed by Student's t-test *p < 0.05, **p < 0.01 and ***p < 0.001.
3.4. Liver Histology
Liver histology prior to treatment was assessed by Periodic acid–Schiff-Diastase (PAS-D) staining on formalin-fixed paraffin-embedded (FFPE) liver tissue from PiZ and wild type mice to visualize protein aggregates. PAS-D stains inclusions of alpha-1-antitrypsin with periportal red hyaline globules (representing collected alpha-1-antitrypsin not excreted from hepatocytes), characteristic for AATD. Prior to treatment we saw significant presence of protein aggregation in PAS-D stained PiZ mouse liver samples compared to WT (Fig. 2a). At termination, 9 weeks after dosing, tissues were sampled and histopathological evaluation of livers revealed significantly improved tissue morphology in the hSERPINA1 gRNA dosed PiZ vs. control dosed PiZ mice. PAS-D staining clearly demonstrated a significant reduction in protein aggregation in the hSERPINA1 gRNA dosed PiZ mice compared to control (Fig. 2b; C and D) with a dramatic decrease in size and number of hAAT aggregates in the livers of hSERPINA1 gRNA treated PiZ mice. Confirmation that protein aggregation was due to AAT was shown by hAAT immunohistochemistry (IHC) staining where a dramatic reduction in hAAT protein after hSERPINA1 gRNA treatment compared to control was seen (Fig. 2b; E and F and Fig. 2c). Furthermore, to evaluate fibrosis, the histologic liver sections were stained with Masson's Trichrome (MTC) and demonstrated significantly reduced fibrosis after hSERPINA1 gRNA treatment in PiZ mice compared to control dosed mice (Fig. 2b; G and H).
Fig. 2.
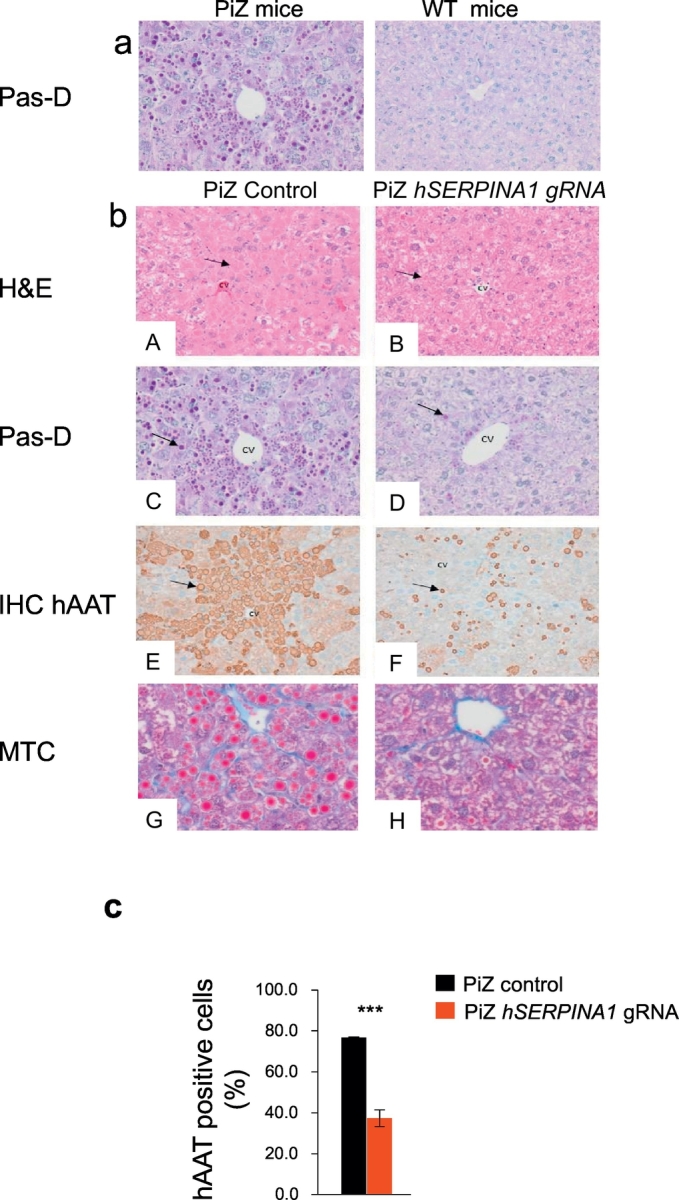
hSERPINA1-specific gRNA mediated reversal of liver globule accumulation and reduction of hAAT in livers of PiZ mice.
Baseline levels of PAS-D staining (a) in PiZ and wild type littermate control mice prior to treatment with a hSERPINA1 specific gRNA. (b) PiZ mice and wild type mice were treated once with hSERPINA1 specific gRNA virus or control virus via a tail vein injection and tissues and blood sampled after 9 weeks. A and B shows H&E and C and D PAS-D staining of liver sections from hSERPINA1 gRNA virus and control virus treated PiZ livers. Arrows in A and B indicates globules within hepatocytes, which are very distinct and present in controls but almost undetectable in hSERPINA1 gRNA treated PiZ mice. Arrows in C and D indicate globule-containing and globule-devoid areas of liver. E and F show sections analyzed by immuno-histochemistry with an anti-hAAT antibody and arrows indicate a reduction in hAAT protein aggregation in hSERPINA1 gRNA treated PiZ mice. G and H shows MTC staining of virus or control virus treated PiZ mice. CV indicates central vein. Automated quantification of IHC for hAAT is shown in (c), results are expressed as percent positive cells per scanned area. Statistical analyses performed by Student's t-test, ***p < 0.001.
3.5. Quantification of Liver Fibrosis and Cell Proliferation by qPCR
To confirm reversal of the liver fibrosis phenotype after the CRISPR/Cas9-treatment, we measured the expression of fibrosis-related markers by quantitative PCR (qPCR). The liver expression levels of Timp1, Col1A1, Col3A1 and Mmp2 were all significantly reduced (p < 0.05) by at least 2-fold (3.3, 4.8, 2.3 and 2.6 fold respectively) compared to control treated PiZ mice (Fig. 3a). These data further confirmed that treatment with hSERPINA1 gRNA prevents the development of fibrosis in the livers of PiZ mice as evidenced in our histological data. Furthermore, PiZ mice have an enhanced regeneration capacity leading to increased levels of hepatocyte proliferation, and this proliferative effect is thought to be derived from hepatocytes devoid of PAS-D positive globules (Rudnick et al., 2004). If this assumption would be true, we would expect that the reduction we observed in PAS-D positive globules following treatment with the hSERPINA1 gRNA (Fig. 2) would translate to a reduced hepatocyte proliferative potential of hSERPINA1 gRNA treated PiZ mice. Indeed, qPCR analysis of specific cell proliferation marker genes indicated a significant (p < 0.05) reduction in the liver expression levels of Pcna, Mki67, and Top2A of 1.3, 2.8 and 2.5 fold respectively, and a reduction of 3.8 fold for Ccnb1 (p = 0.07) compared to control treated PiZ mice (Fig. 3b).
Fig. 3.

hSERPINA1-specific guide treatment reduces the expression of liver fibrosis and cellular proliferation marker genes in the liver of PiZ mice.
Liver fibrosis-related (a) and proliferation–related (b) gene mRNA levels quantified by qPCR. Average gene expression levels as compared to peptidylprolyl isomerase A (Ppia) for each mice group (n = 4 for each of the two virus-treated groups, n = 3 for wild type mice). Standard errors indicate SEM. Significant reduction (p < 0.05; Student's t-test) in gene expression after treatment with hSERPINA1-targeting virus as compared to control virus was observed for all genes except Ccnb1 (p = 0.07). Statistical analyses performed by Student's t-test, *p < 0.05.
3.6. Droplet Digital PCR and Next Generation Sequencing Analysis
To determine the extent to which we disrupted the hSERPINA1 transgene array we performed a range of molecular analyses. As mentioned previously an exact copy number cannot be determined using TLA. Therefore, Droplet Digital PCR (ddPCR) was performed on liver samples from hSERPINA1 gene edited PiZ mice and on control treated PiZ mice to more accurately determine transgene copy number (Fig. 4a). In an effort to obtain an as accurate result as possible regarding the remaining hSERPINA1 copy number, the primers and probes used in the ddPCR assay were designed to bind in an area 305–182 bp upstream of the gRNA. If we would instead have placed the assay closer to the gRNA, the frequent indels arising close to the gRNA binding and Cas9 cut site could have prevented the primers or probe from binding, resulting in an underestimated remaining copy number. Based on ddPCR results, approximately 8 copies of the hSERPINA1 gene are present in untreated PiZ mice which is lower than the previous data (including our own TLA analysis) which has suggested up to 11 copies. On average, hSERPINA1 gRNA dosed mice were reduced in copy number from 7.6 to 6.0 (Fig. 4a). This may seem low considering the major changes we observed in the phenotype of the PiZ mice upon hSERPINA1 gRNA treatment. However, as long as the primers and probe used in ddPCR-assay still recognize the DNA sequence, a PCR product is expected. Thus, this result rather suggests that few large deletions or rearrangements in the transgene array had occurred. Instead, cutting by CRISPR/Cas9 in the tandem array of hSERPINA1 genes may preferentially generate smaller genetic modifications, including insertions and deletions (indels).
Fig. 4.

Genomic analysis of hSERPINA1 transgene sequences following hSERPINA1-specific guide treatment.
(a) Reduction in hSERPINA1 copy number observed with Droplet Digital PCR after using hSERPINA1-targeting virus as compared to control virus in PiZ-expressing mice. (b) Deep targeted sequencing analysis for on-target and off-target sites in PiZ mice treated with hSERPINA1 gRNA. Sequence of off-target sites and number of mismatches in each off-target site is shown. Primers for OT2 amplicon amplify six target sites simultaneously. PAM is shown in blue. Mismatches are indicated as lowercase red letters. (c) Targeting efficiency for on-target and 11 off-targets in liver samples are shown. Error bars indicate SEM (n = 4). (d) Most frequent alleles following Cas9 cleavage at hSERPINA1 target site in one of the liver samples are shown. On the wild type sequence, the Cas9 target site is shown in red and PAM is shown in green and underlined. ∆: deletion. +: insertion.
In order to assess whether smaller genetic changes occurred, a Next Generation Sequencing (NGS) approach was applied to look for on-target changes to hSERPINA1 to determine how many copies had been affected. It was challenging to determine the exact effect on hSERPINA1 disruption at the genetic level given that we analyzed a piece of liver and within that biopsy there will be wide variations in the genomic re-arrangement that have occurred at the single cell level, particularly given that we have multiple copies of the hSERPINA1 gene to begin with. NGS showed that 30% of the reads were modified, suggesting 30% targeting efficiency in terms of introducing indels (Fig. 4c). However, given that we have multiple copies of hSERPINA1 in tandem, those few copies which based on ddPCR analysis were perfectly deleted (via non-homologous end joining (NHEJ) between adjacent or distal hSERPINA1 genes) were not detected in our NGS analysis. Therefore, the actual targeting efficiency was likely somewhat higher than 30%.
To assess potential off-target DNA editing we amplified the genomic regions surrounding 11 predicted off-target sites in the mouse liver samples and analyzed the outcome by NGS. Compared to control-virus treated mice, we detected no off-target activity of the hSERPINA1 gRNA (Fig. 4c). A representative example of allelic modifications introduced by targeting the hSERPINA1 gene from one liver sample is shown in Fig. 4d.
4. Discussion
Our aim of this study was to assess the effectiveness of treatment with a gRNA targeting hSERPINA1 in the PiZ mouse model of AATD. Our results indicate that hSERPINA1 gRNA dosed PiZ mice, when compared to PiZ mice receiving control virus, display a reverted pathological liver phenotype due to a reduction in globule-induced liver injury and associated liver fibrosis. Targeting human SERPINA1 using CRISPR/Cas9 reduces plasma AST, ALT and plasma and liver hAAT protein levels in a mouse model of AATD liver disease. In addition, levels of aggregated hAAT protein was significantly reduced ameliorating hepatic fibrosis, indicating the potential benefit of inactivating a disease causing mutant allele in humans. The decrease in plasma hAAT levels may be due to increased AAT protein aggregation in the liver of the PiZ mice or alternatively a gradual repopulation of cells with lower levels of transgene expression. Liver histology clearly indicated that hSERPINA1 gene editing reduced liver protein aggregation and fibrosis in PiZ mice. Comparing the effects of hSERPINA1 gRNA treatment to baseline PAS-D measurements and control treated animals, it is clear the treatment prevented accumulation of globules over the course of the 9-week study. In addition, quantification of liver fibrosis and cell proliferation by qPCR confirmed that treatment with hSERPINA1 gRNA prevents globule formation and AAT aggregation in the livers of PiZ mice as evidenced in our histological data. NGS and ddPCR analysis, when taken together, suggest that we have caused a sufficiently large re-arrangement in the hSERPINA1 transgene array throughout the tissue sampled to revert the liver disease phenotype in PiZ transgenic mice. One important consideration before CRISPR/Cas9 can be used in a human therapeutic context is to ensure the absence of off-target DNA editing and in this study we detected no off-target activity associated with the treatment. However, we are aware that for human gene editing, off-target studies are only valid in the context of the human genome. In the study we do not address the pathogenesis of AATD liver disease and whether this can be reversed. This is an important aspect to consider in the context of therapeutic intervention and our future studies will focus on the effect of therapy on factors such as ER stress and AAT polymerization.
While the outcome of this study opens up the door for exploring whether directly treating AATD-patients with a guide RNA targeting hSERPINA1 can reduce mutant protein production, potential risks with such a treatment need to be carefully considered. For example, PiZZ patients have approximately 15% AAT levels in serum compared to normal patients and there are conflicting reports of whether residual Z protein may or may not protect against lung disease (Abboud et al., 2011). Currently, available treatment options for patients with AATD are limited and include protein replacement therapy using plasma-derived AAT in lung disease and, in the case of advanced cirrhosis, liver transplantation. AATD has been a target for gene therapy, but progress has been slow with recent non-viral and viral based clinical trials failing to achieve protective levels of AAT in serum (reviewed in Wozniak et al., 2015). Therefore, developing novel therapies targeting AATD is crucial and our study, where CRISPR/Cas9 editing of hSERPINA1 in PiZ mice effectively reversed the disease phenotype, supports the potential use of genome editing for the treatment of liver disease in AATD patients.
Author Contribution
Conceptualization: JW, MB, MB-Y, LM.
Methodology: JW, MB, RN, MM, ATG, FS, EE.
Validation: JW, EE, ATG, MB, MB-Y.
Investigation: JW, EE, TA, MJP, LMag, FS, MC, LA, MB, MB-Y.
Writing: Original Draft: JW, MB.
Writing: Review & Editing: JW, MB, MM, EE, ATG.
Supervision: MB, MB-Y, LMay, JW.
Funding Sources
All authors are employees of AstraZeneca.
Conflicts of Interest
We declare no conflict of interest.
Acknowledgements
We would like to thank Kerstin Magnell and Anna-Lena Loyd (Discovery Sciences, Innovative Medicines and Early Development Biotech Unit, AstraZeneca) for re-derivation of the PiZ model. Caroline Rhoden and Marie Johansson (Drug Safety and Metabolism, Innovative Medicines and Early Development Biotech Unit, AstraZeneca) for animal husbandry and technical support and Mike Firth (Bioinformaticians, Discovery Sciences, Innovative Medicines and Early Development Biotech Unit, AstraZeneca) for bioinformatics support.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ebiom.2018.02.015.
Appendix A. Supplementary Data
Supplementary material
References
- Abboud R.T., Nelson T.N., Jung B., Mattman A. Alpha1-antitrypsin deficiency: a clinical-genetic overview. Appl. Clin. Genet. 2011;2011(4):55–65. doi: 10.2147/TACG.S10604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson J.A., Rogers B.B., Sifers R.N., Finegold M.J., Clift S.M., DeMayo F.J., Bullock D.W., Woo S.L. Accumulation of PiZ α-1-antitrypsin causes liver damage in transgenic mice. J. Clin. Invest. 1989;83(4):1183–1190. doi: 10.1172/JCI113999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz P.E., Mueller C., Cossette T.L., Golant A., Qiushi Tang Q., Beattie S.G., Brantly M., Campbell-Thompson M., Blomenkamp K.S., Teckman J.H., Flotte T.R. In vivo post-transcriptional gene silencing of α-1 antitrypsin by adeno-associated virus vectors expressing siRNA. Lab. Investig. 2007;87(9):893–902. doi: 10.1038/labinvest.3700629. [DOI] [PubMed] [Google Scholar]
- Ding J., Yannam G.R., Roy-Chowdhury N., Hidvegi T., Basma H., Rennard S.I., Wong R.J., Avsar Y., Guha C., Perlmutter D.H., Fox I.J., Roy-Chowdhury J. Spontaneous hepatic repopulation in transgenic mice expressing mutant human α-1 antitrypsin by wild-type donor hepatocytes. J. Clin. Invest. 2011;121(5):1930–1934. doi: 10.1172/JCI45260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekeowaa U.I., Freekeb J., Mirandaa E., Gooptuc B., Bushd M.F., Péreze J., Teckman J., Robinson C.V., Lomasa D.A. Defining the mechanism of polymerization in the serpinopathies. Proc. Natl. Acad. Sci. U. S. A. 2010;107(40):17146–17151. doi: 10.1073/pnas.1004785107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S., Booten S.L., Aghajan M., Hung G., Zhao C., Blomenkamp K., Gattis D., Watt A., Freier S.M., Teckman J.H., McCaleb M.L., Monia B.P. Antisense oligonucleotide treatment ameliorates alpha-1 antitrypsin–related liver disease in mice. J. Clin. Invest. 2015;124(1):251–261. doi: 10.1172/JCI67968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janciauskiene S.M., Bals R., Koczulla R., Vogelmeier C., Kohnlein T., Welte T. The discovery of alpha1-antitrypsin and its role in health and disease. Respir. Med. 2011;105(8):1129–1139. doi: 10.1016/j.rmed.2011.02.002. [DOI] [PubMed] [Google Scholar]
- Livak K.J., Thomas D., Schmittgen T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 22DDCT method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lomas D.A., Evans D.L., Finch J.T., Carrell R.W. The mechanism of Z α-1-antitrypsin accumulation in the liver. Nature. 1992;357(6379):605–607. doi: 10.1038/357605a0. [DOI] [PubMed] [Google Scholar]
- Long C., McAnally J.R., Shelton J.M., Mireault A.A., Bassel-Duby R., Olson E.N. Prevention of muscular dystrophy in mice by CRISPR/Cas9–mediated editing of germline DNA. Science. 2014;345:1184–1188. doi: 10.1126/science.1254445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus N.Y., Brunt E.M., Blomenkamp K., Ali F., Rudnick D.A., Ahmad M., Teckman J.H. Characteristics of hepatocellular carcinoma in a murine model of α-1-antitrypsin deficiency. Hepatol. Res. 2010;40(6):641–653. doi: 10.1111/j.1872-034X.2010.00663.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinello L., Canver M.C., Hoban M.D., Orkin S.H., Kohn D.B., Bauer D.E., Yuan G.C. Analyzing CRISPR genome-editing experiments with CRISPResso. Nat. Biotechnol. 2016;34:695–697. doi: 10.1038/nbt.3583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu D., Teckman J.H., Omura S., Perlmutter D.H. Degradation of a mutant secretory protein, α-1-antitrypsin Z, in the endoplasmic reticulum requires proteasome activity. J. Biol. Chem. 1996;271(37):22791–22795. doi: 10.1074/jbc.271.37.22791. [DOI] [PubMed] [Google Scholar]
- Rudnick D.A., Liao Y., An J.K., Muglia L.J., Perlmutter D.H., Teckman J.H. Analyses of hepatocellular proliferation in a mouse model of α-1-antitrypsin deficiency. Hepatology. 2004;39(4):1048–1055. doi: 10.1002/hep.20118. [DOI] [PubMed] [Google Scholar]
- Teckman J.H., Lindblad D. α-1-antitrypsin deficiency:diagnosis, pathophysiology, and management. Curr. Gastroenterol. Rep. 2006;8(1):14–20. doi: 10.1007/s11894-006-0059-8. [DOI] [PubMed] [Google Scholar]
- Teckman J.H., Perlmutter D.H. The endoplasmic reticulum degradation pathway for mutant secretory proteins α-1-antitrypsin Z and S is distinct from that for an unassembled membrane protein. J. Biol. Chem. 1996;271(22):13215–13220. doi: 10.1074/jbc.271.22.13215. [DOI] [PubMed] [Google Scholar]
- Wozniak J., Wandtke T., Kopinski P., Chorostowska-Wynimko J. Challenges and prospects for Alpha-1 antitrypsin deficiency gene therapy. Hum. Gene Ther. 2015;26(11):709–718. doi: 10.1089/hum.2015.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y., Whitman I., Molmenti E., Moore K., Hippenmeyer P., Perlmutter D.H. A lag in intracellular degradation of mutant α-1-antitrypsin correlates with the liver disease phenotype in homozygous PiZZalpha 1-antitrypsin deficiency. Proc. Natl. Acad. Sci. U. S. A. 1994;91(19):9014–9018. doi: 10.1073/pnas.91.19.9014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J., Coulouris G., Zaretskaya I., Cutcutache I., Rozen S., Madden T.L. Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012;13:134. doi: 10.1186/1471-2105-13-134. http://www.biomedcentral.com/1471-2105/13/134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin H., Xue W., Chen S., Bogorad R.L., Benedetti E., Grompe M., Koteliansky V., Sharp P.A., Jacks T., Anderson D.G. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat. Biotechnol. 2014;32(6):551–553. doi: 10.1038/nbt.2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin C., Zhang T., Qu X., Zhang Y., Putatunda R., Xiao X., Li F., Xiao W., Zhao H., Dai S., Qin X., Mo X., Young W.B., Khalili K., Hu W. In vivo excision of HIV-1 provirus by saCas9 and multiplex single-guide RNAs in animal models. Mol. Ther. 2017;25(5):1168–1186. doi: 10.1016/j.ymthe.2017.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Long C., Li H., McAnally J.R., Baskin K.K., Shelton J.M., Bassel-Duby R., Olson E.N. CRISPR-Cpf1 correction of muscular dystrophy mutations in human cardiomyocytes and mice. Sci. Adv. 2017 doi: 10.1126/sciadv.1602814. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material