

ABSTRACT
Salmonella enterica serovar Typhimurium (Salmonella Typhimurium) and its monophasic variant (Salmonella 4,[5],12:i:−) are the major causes of gastroenteritis in both humans and animals. Pulsed-field gel electrophoresis and multilocus variable-number tandem-repeat analysis have been used widely as subtyping methods for these pathogens in molecular epidemiological analyses, but the results do not precisely reflect phylogenetic information. In this study, we performed a phylogenetic analysis of these serovars using whole-genome sequencing data and identified nine distinct genotypic clades. Then, we established an allele-specific PCR-based genotyping method detecting a clade-specific single nucleotide polymorphism to rapidly identify the clade of each isolate. Among a total of 815 isolates obtained from cattle in Japan between 1977 and 2017, clades 1, 7, and 9 contained 77% of isolates. Obvious replacement of the dominant clone was observed five times in this period, and clade 9, which mostly contains Salmonella 4,[5],12:i:−, is currently dominant. Among 140 isolates obtained from swine in Japan between 1976 and 2017, clades 3 and 9 contained 64% of isolates. Clade 9 is the latest clone as is the case in cattle isolates. Clade 9 is similar to an epidemic clone from Europe, which is characterized by sequence type 34 (ST34), chromosomal Salmonella genomic island 3, and a composite transposon containing antimicrobial resistance genes. The increased prevalence of clade 9 among food animals in Japan might be a part of the pandemic of the European Salmonella 4,[5],12:i:− clone.
KEYWORDS: Japan, Salmonella enterica, Typhimurium, food animal, monophasic variant, pandemic, phylogeny
INTRODUCTION
Salmonella enterica subsp. enterica is a major cause of gastroenteritis in both humans and animals (1, 2). Serotyping is the first step in the epidemiological investigation of salmonellosis. Each serovar is defined by the combination of the cell surface lipopolysaccharide moieties on the cell surface (O antigen) and one or two different flagellar proteins (H antigens). Many serovars can express two different flagellin proteins, although a single cell can express only one at a time (3). According to the White-Kaufmann-Le Minor scheme and supplements, more than 2,600 serovars have been reported to date (4, 5). A smaller number of these serovars are significantly associated with human and animal diseases (6).
S. enterica serovar Typhimurium has long been one of the most prevalent serovars for both human and animal infections worldwide (6, 7). Human infection by S. enterica serovar 4,[5],12:i:− (Salmonella 4,[5],12:i:−), which is believed to be a monophasic variant of Salmonella Typhimurium, has increased considerably since the mid-1990s. This serovar is currently one of the most common serovars in many countries (8–12) and is responsible for a number of outbreaks, including outbreaks in Luxembourg (13), Germany (14), France (15), and the United States (16). Many of these outbreaks were linked to the consumption of contaminated pork and other sources, such as cattle and poultry (17). Isolation of Salmonella 4,[5],12:i:− from humans and animals has also become common in Japan in recent years (10, 18, 19). However, the genetic relatedness between isolates in Japan and Western countries is not clear.
Bovine salmonellosis is an important cause of mortality and morbidity in cattle and poses a threat to both beef and daily operations (20). In Japan, most bovine salmonellosis was attributed to beef calf diarrhea in the past but was replaced by salmonellosis in adult milking cows, with severe symptoms, in the mid-1990s (21). Salmonella Typhimurium and Salmonella enterica serovar Dublin were the most prevalent serovars isolated from cattle. Most salmonellosis in adult milking cows is caused by Salmonella Typhimurium.
To investigate whether epidemic clones of Salmonella Typhimurium changed in the 1990s, we analyzed 545 Salmonella Typhimurium isolates collected between 1977 and 2009 from cattle in Japan using macrorestriction analysis of XbaI-digested genomic DNA by pulsed-field gel electrophoresis (PFGE) (22). Nine major clusters were identified from 116 PFGE patterns. Cluster I comprised 248 isolates, most of which are definitive phage type 104 (DT104), an important cause of human salmonellosis in western countries in the 1990s (23). The PFGE cluster I was dominant between 1993 and 2003, but its occurrence has since declined. Beginning in 2002, an increase was observed in the number of cluster VII isolates, consisting of 21 PFGE patterns comprising 165 isolates. Thus, a replacement of the dominant clone was observed twice among Salmonella Typhimurium isolated from cattle in Japan in this period. No information is available for the epidemic clones of Salmonella Typhimurium after 2009.
Recently, bovine salmonellosis caused by Salmonella 4,[5],12:i:− has been increasing in Japan (10, 18, 19). Molecular analysis of these isolates revealed that three different mutation patterns in the chromosomal region that defines the phase variation of flagellar antigens were the genetic basis for the monophasic phenotypes of Salmonella 4,[5],12:i:− (10). The phenotype analysis suggested that these isolates consisted of multiple distinct clones (10). However, the genetic background of these isolates remains to be elucidated.
In the present study, we performed whole-genome sequencing analysis of Salmonella Typhimurium and Salmonella 4,[5],12:i:− isolates obtained from various sources in Japan and Italy and established a novel PCR-based genotyping method (i) to elucidate the genetic relatedness between isolates in Japan and Western countries and (ii) to identify temporal changes in the genotype distribution of Salmonella Typhimurium and Salmonella 4,[5],12:i:− obtained from cattle or swine in Japan in the last 4 decades.
MATERIALS AND METHODS
Bacterial isolation, identification, and typing.
A total of 1,027 isolates of Salmonella Typhimurium and Salmonella 4,[5],12:i:− obtained in Japan (1,003 isolates) or Italy (24 isolates) were used in this study (see Table S1 in the supplemental material). The Japanese isolates were obtained from cattle, swine, birds, meats, environmental water, and humans between 1972 and 2017. These isolates were isolated by the staff of the local animal hygiene service centers or local institute of public health for diagnostic or monitoring purposes. Patient information was anonymized and deidentified prior to analysis. The isolates were epidemiologically unrelated and were isolated from different samples collected from 30 of the 47 prefectures in Japan. Of the 1,003 Japanese isolates, 576 isolates obtained between 1977 and 2009 were also used in previous studies (10, 19, 22). The Italian isolates were obtained from cattle, swine, birds, meats, and environmental water between 2009 and 2012. These isolates were isolated by the OIE and National Reference Laboratory for Salmonella (IZSVe, Legnaro, Padua, Italy). While isolates from cattle, swine, and humans were obtained from diagnostic samples, isolates from birds, meats, and environmental water were obtained from monitoring samples. Salmonella spp. were identified based on colony morphology on selective media and biochemical properties, as previously described (24). Serovar identification was performed by microtiter and slide agglutination methods according to the White-Kaufmann-Le Minor scheme (4).
A total of 119 isolates were selected for whole-genome sequencing (Table S2): 36 Salmonella Typhimurium isolates were obtained from cattle in Japan between 1985 and 2007 and showed different PFGE patterns (19, 22); 16 Salmonella Typhimurium isolates were obtained from cattle, swine, and birds in Japan between 1992 and 2014; 30 Salmonella 4,[5],12:i:− isolates were obtained from cattle, swine, birds, meats, environmental water, and humans in Japan between 2000 and 2009 and showed various characteristics (10); 13 Salmonella 4,[5],12:i:− isolates were obtained from cattle, swine, and birds in Japan between 2002 and 2014; and all remaining 24 isolates were isolated in Italy.
Phage typing.
Phage typing was performed using Salmonella Typhimurium typing phages according to the methods and schemes previously described by Anderson et al. (25). Phage typing was performed at the National Institute of Infectious Diseases according to the method prescribed by the Health Protection Agency, London, United Kingdom (25).
Antimicrobial susceptibility testing.
The Kirby–Bauer disc diffusion test was performed using Mueller-Hinton agar plates (Becton, Dickenson and Company, Franklin Lakes, NJ) according to Clinical and Laboratory Standards Institute standards (26, 27) using the following 17 single or types of antimicrobials: ampicillin (10 μg), streptomycin (10 μg), sulfonamides (250 μg), tetracycline (30 μg), chloramphenicol (30 μg), sulfamethoxazole-trimethoprim (23.75 and 1.25 μg, respectively), kanamycin (30 μg), gentamicin (10 μg), nalidixic acid (30 μg), ciprofloxacin (5 μg), cefazolin (30 μg), cefotaxime (30 μg), cefepime (30 μg), cefoxitin (30 μg), fosfomycin (50 μg), imipenem (10 μg), and meropenem (10 μg).
Whole-genome sequencing and genome assembly.
Chromosomal and plasmid DNAs of isolates obtained in Japan were purified from S1 nuclease-digested genomic DNA, which was separated by PFGE, as previously described (28). Genomic DNA of isolates obtained in Italy was extracted using a commercial column-based protocol (QIAamp DNA minikit; Qiagen, Hilden, Germany) by following the manufacturer's instructions. The sequencing library was prepared using a Nextera XT DNA sample preparation kit (Illumina, Inc., San Diego, CA). Sequencing was performed using an Illumina NextSeq 500 sequencer (Illumina) according to the manufacturer's instructions. The de novo assembly of all short reads was performed using A5 MiSeq program software version 20140604 (29).
In silico MLST.
Multilocus sequence typing (MLST) was performed using the nucleotide sequences of 7 housekeeping genes, aroC, dnaN, hemD, hisD, purE, sucA, and thrA, on draft genome contigs, according to the protocols available in the MLST database (http://mlst.warwick.ac.uk/mlst/dbs/Senterica).
SNP detection and phylogenetic analysis.
For sequence read alignments, paired-end Illumina sequence data were mapped onto the reference genome Salmonella Typhimurium strain LT2 (RefSeq accession number NC_003197.1) using the bwa-mem program (30). Single nucleotide polymorphisms (SNPs) were extracted using VarScan v2.3.4 (31). Repeat and prophage regions of LT2 complete genome sequences were analyzed using the nucmer (MUMmer 3.0) (32) and PHAST (33) programs, respectively, followed by exclusion for SNPs in these regions. RAxML (34) was used to construct a maximum likelihood phylogenetic tree inferred from the concatenated alignment of detected SNPs on core genome regions. The population structure was analyzed with hierBAPS software (35) using a Bayesian clustering method. Accession numbers of 14 reference strains of Salmonella Typhimurium included in the phylogenetic analysis are listed in Table S3.
Detection of horizontally acquired genetic structures.
Predicted prophage sequences were detected using the PHAST web server (a phage search tool [http://phast.wishartlab.com/]) (33). All regions detected as “intact,” “incomplete,” or “questionable” by PHAST were identified as prophage regions. Antimicrobial resistance genes were searched using the BLSTN program and the ResFinder database (https://cge.cbs.dtu.dk/services/ResFinder/) (36) with the following thresholds: minimum length coverage of ≥90% and nucleotide sequence identity of ≥96%. A homology search was performed using the NUCmer alignment program from MUMmer in GENETYX version 13 (GENETYX Co., Ltd., Tokyo, Japan) to detect the following genetic structures: Salmonella genomic island 1 (SGI1) (37), SGI3 (38), a chromosomal composite transposon reported by García et al. (39), and virulence resistance plasmid pYT1 (40). The following regions were used as query sequences: SGI1 (GenBank accession number AF261825), nucleotides (nt) 146 to 1145 and 41578 to 42577; SGI3 (GenBank accession number LN999997), nt 684938 to 685937 and 764699 to 765698; chromosomal composite transposon (GenBank accession number KR856283), nt 18591 to 25462; and pYT1 (GenBank accession number AB576781), nt 28584 to 34804. The detection thresholds were as follows: minimum length coverage of ≥80% and nucleotide sequence identity of ≥80%.
Establishment of an SNP genotyping system and analysis of wild types.
To rapidly determine to which clade a tested isolate belonged, we established an SNP genotyping system consisting of a combination of nine allele-specific PCRs (AS-PCRs) to detect clade-specific SNPs. Primers were designed as previously described (41). Briefly, in each forward primer, a complementary nucleotide for the SNP was located in the second position from the 3′ end, and a deliberate mismatch was inserted at the third or fourth position from the 3′ end. Each reverse primer was designed to be complementary to the reference strain. Designed primers are listed in Table S2 and were purchased from Hokkaido System Science Co., Ltd. (Hokkaido, Japan). A single colony (2 mm in diameter) was suspended in 50 μl of 25 mM NaOH. After being boiled for 5 min, the suspension was neutralized by adding 4 μl of 1 M Tris-HCl (pH 8.0) and centrifuged at 20,000 × g for 5 min, and 1 μl of the supernatant was used as template DNA. PCR was performed in 20-μl reaction mixtures containing template DNA, 0.3 μM each primer, 0.2 mM deoxynucleotide triphosphates (dNTPs), PCR buffer, and 0.01 U KOD -Plus- DNA polymerase (Toyobo Co., Ltd., Osaka, Japan) using 30 amplification cycles of 94°C for 15 s, 65°C for 30 s, and 68°C for 30 s. The PCR products were separated on a 2% agarose gel and visualized by staining with ethidium bromide.
Terminology.
In this report, “dominant” means that a specific clone or genotype accounted for more than half of the isolates obtained in a specific year or a period. “Clonal replacement” means a replacement of a dominant clone or genotype among isolates obtained at a specific time point.
Accession number(s).
Raw sequence reads were deposited in the DDBJ Sequence Read Archive under accession number DRA006240 (BioProject accession number PRJDB6430, BioSample accession numbers SAMD00097407 to SAMD00097525, and Experiment DRX099907 to DRX100166) (Table S4).
RESULTS
Identification of clades.
A total of 6,205 SNPs were detected from the draft genome sequences of the 119 isolates (Data Set S1). A phylogenetic tree was generated, and nine clades were identified as discriminated from each other as genotypes for epidemiological analysis based on the following observations (Fig. 1; see also Table S2).
FIG 1.

Phylogeny of S. enterica serovar Typhimurium and its monophasic variants in Japan and Italy, isolated between 1984 and 2014. A phylogenetic tree was generated using the maximum likelihood method with 6,205 concatenated SNPs in the core genome sequences of 119 wild-type and 12 reference strains; hierBAPS clustering, sequence type, serotype, country, and source are indicated at right. Detailed information for each isolate is provided in Table S2.
The hierBAPS clustering identified four clusters among the 119 isolates. Cluster 1 was divided into clades 1, 2, and 3. Isolates of clade 1 were obtained from multiple animals. All but one clade 1 isolate consisted of Salmonella Typhimurium and corresponded to phage types DT104 and U302, which were important causes of human and bovine salmonellosis worldwide in the 1990s. Typical isolates belonging to this clade showed resistance to ampicillin, chloramphenicol, streptomycin, and sulfonamide. All clade 2 isolates were Salmonella Typhimurium, and five of the six isolates were obtained from cattle. All but 1 of the clade 3 isolates consisted of Salmonella Typhimurium, and 8 of the 11 isolates were obtained from swine.
Cluster 2 isolates exclusively consisted of Salmonella 4,[5],12:i:−, belonging to sequence type 99 (ST99) and corresponding to clade 4. Most clade 4 isolates were obtained from wild birds or environmental water. Three adjacent genes, fljA, fljB, and hin, which induce phase variation of the flagellar antigen of this clade, were detectable, with common amino acid substitutions of A46T in FljA and R140L in Hin (10). All clade 4 isolates were pansusceptible.
Cluster 3 was divided into clades 5, 6, 7, and 8. All but one of the clade 5 isolates consisted of Salmonella Typhimurium obtained from cattle, swine, and birds. All clade 5 isolates were pansusceptible. Clades 6 and 7 consisted of Salmonella Typhimurium isolated from cattle. All but one of the clade 7 isolates not phage typeable and showed resistance to all or some of the following antimicrobials: ampicillin, streptomycin, sulfonamide, tetracycline, and kanamycin. Most clade 8 isolates consisted of Salmonella 4,[5],12:i:− isolates obtained from various sources. Clades 6, 7, and 8 exclusively consisted of isolates obtained from Japan, while all other clades consisted of isolates obtained from both Japan and Italy. A reference strain, DT2, located between clades 6 and 7 in the phylogenetic tree, was omitted from the clade identification because this strain was distinct from any isolate belonging to cluster 3.
Cluster 4 corresponded to clade 9. Isolates of this clade exclusively belonged to ST34 and harbored SGI3, which encodes resistance genes against copper and arsenic and is specific to the Salmonella 4,[5],12:i:− epidemic clone in Europe (38). Most clade 9 isolates consisted of Salmonella 4,[5],12:i:− and showed resistance to all or some of the following antimicrobials: ampicillin, streptomycin, sulfonamide, and tetracycline.
Distribution of prophages.
As shown in Fig. 2, 15 different prophages were detected. Gifsy-1 was distributed in most of the 119 isolates. Gifsy-2 was also detected in most of the isolates of each clade, except for clade 9. All but one clade 9 isolate was negative for Gifsy-2. The distribution of the remaining 13 prophages was relatively limited. Fels-1 was detected only in LT2 as a reference strain. The other 12 prophages were distributed in one to five clades.
FIG 2.
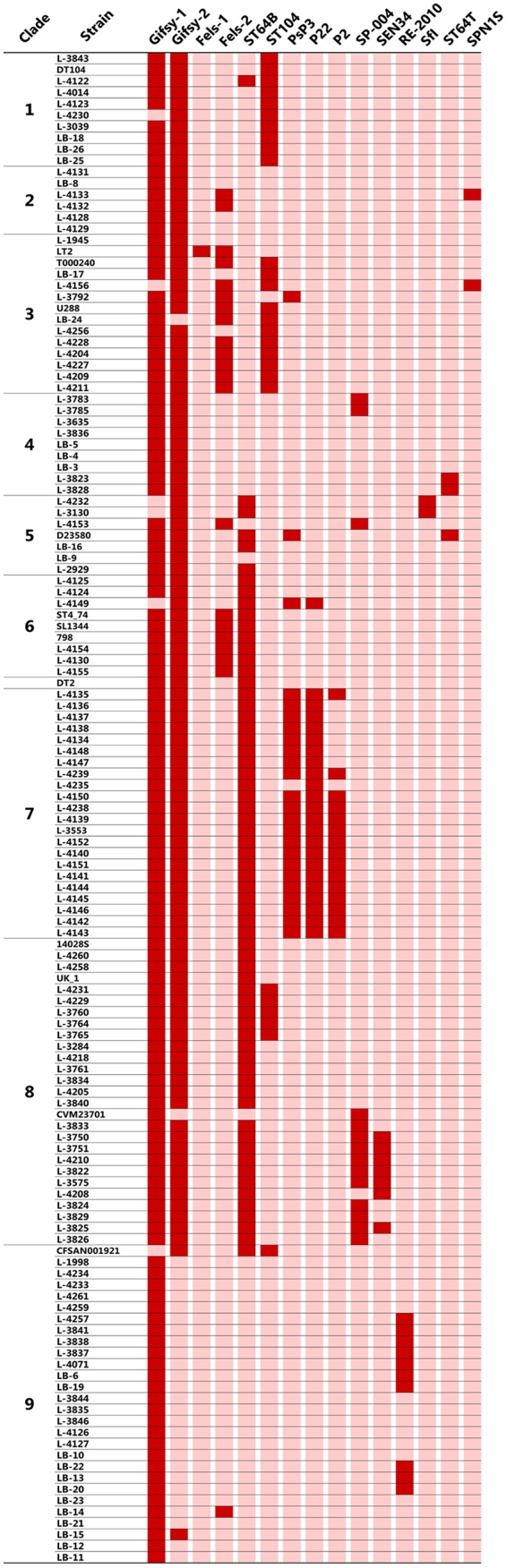
Prevalence of predicted prophage sequences among genome-sequenced isolates. Red and pink indicate presence and absence of each prophage, respectively.
Distribution of antimicrobial resistance genes.
As shown in Fig. 3, multiple resistance genes were detected from all isolates of clades 1 and 7. Most clade 9 isolates also harbored multiple resistance genes. All but one clade 1 isolate harbored the SGI1-containing resistance genes aadA2, blaPSE-1, floR, and tet(G) in the chromosome. The resistance genes of clade 7 isolates, including aadA1, aph(3′)-Ia, blaTEM-1B, sul1, and tet(A), were located mainly in a pYT1-like virulence resistance plasmid (40), but some isolates harbored the same resistance genes in the chromosome. Most clade 9 isolates harbored a composite transposon (39) containing all or some of the following resistance genes in the chromosome: blaTEM-1B, strA, strB, sul2, and tet(B). Some clade 3 isolates harbored several resistance genes in the chromosome.
FIG 3.

Distribution of antimicrobial resistance genes in chromosomes and plasmids. Red and pink indicate the presence and absence of each gene, respectively. Gray indicates a lack of applicability due to lack of plasmid genome data.
Validation of the established SNP genotyping system.
Nine AS-PCRs were established to detect a distinctive SNP in each clade (Table S5; Fig. S1). Isolates positive for clade 1-specific SNP were designated SNP genotype 1. Other SNP genotypes were determined in the same manner. We performed nine AS-PCRs to determine the SNP genotype of each isolate. The 119 genome-sequenced isolates of Salmonella Typhimurium and Salmonella 4,[5],12:i:− were subjected to this system to verify the accuracy. Only one of the clade-specific SNPs was positive for all isolates, and no inconsistency was detected between the results of SNP genotyping and the clade classification.
Comparison of the results of SNP genotyping and other typing methods.
In the previous study, we subtyped 545 Salmonella Typhimurium isolates obtained from cattle in Japan between 1977 and 2009 using PFGE and multilocus variable-number tandem-repeat analysis (MLVA). Thirty-six of the 545 isolates were included in the 119 genome-sequenced isolates. Nine PFGE clusters were observed among the 545 isolates, while five MLVA clusters were identified among 495 of the 545 isolates (19, 22). We subjected the 545 isolates to SNP genotyping. As shown in Table 1, all but one isolate were sorted into one of the eight SNP genotypes. SNP genotype 4 was not detected among the isolates. No amplification was observed in the nine AS-PCRs for the remaining isolate. All isolates belonging to the three SNP genotypes grouped into a single PFGE cluster. All SNP genotype 1 isolates grouped into PFGE cluster I and vice versa. All SNP genotype 7 isolates grouped into PFGE cluster VII, but 3 of 165 PFGE cluster VII isolates grouped into SNP genotype 5 or 6. All SNP genotype 9 grouped into PFGE cluster III and vice versa, although only two isolates were obtained in this period. Isolates belonging to SNP genotypes 2, 3, 5, 6, and 8 grouped into two to four PFGE clusters. All isolates belonging to SNP genotypes 1, 2, 6, 7, and 8 grouped into MLVA clusters A, B, D, C, and E, respectively, and vice versa.
TABLE 1.
Relationships of typing results by SNP genotyping, PFGE, and MLVA for isolates of S. enterica serovar Typhimurium and its monophasic variant
| Clustering | No. of isolates with indicated SNP genotype |
Total no. of isolates | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | ||
| PFGE | ||||||||||
| I | 248 | 248 | ||||||||
| II | 5 | 3 | 8 | 17 | 3 | 36 | ||||
| III | 2 | 2 | ||||||||
| IV | 36 | 36 | ||||||||
| V | 5 | 1 | 6 | 12 | ||||||
| VI | 29 | 2 | 31 | |||||||
| VII | 2 | 1 | 162 | 165 | ||||||
| VIII | 12 | 1 | 13 | |||||||
| IX | 1 | 1 | ||||||||
| Total | 248 | 75 | 7 | 0 | 10 | 36 | 162 | 4 | 2 | 544 |
| MLVA | ||||||||||
| A | 239 | 239 | ||||||||
| B | 75 | 75 | ||||||||
| C | 162 | 162 | ||||||||
| D | 16 | 16 | ||||||||
| E | 3 | 3 | ||||||||
| Total | 239 | 75 | 0 | 0 | 0 | 16 | 162 | 3 | 0 | 495 |
Temporal change in the distribution of SNP genotypes.
To detect temporal changes in SNP genotype distribution among isolates obtained from cattle and swine in Japan, we performed SNP genotyping on an additional 399 isolates. A total of 1,027 isolates were subjected to SNP genotyping in this study. Among them, 1,006 isolates were sorted into one of the nine SNP genotypes. No amplification was observed among the remaining 21 isolates (9 isolates from cattle and 12 isolates from swine). Among the 976 isolates originating from cattle or swine in Japan between 1976 and 2017, the SNP genotypes of 955 isolates were successfully determined.
Table 2 shows the distribution of SNP genotypes among 815 isolates obtained from cattle between 1977 and 2017. SNP genotypes 1, 7, and 9 constituted 77.2% of the isolates. Before 1984, all but one isolate belonged to SNP genotype 6, although the total number of isolates was relatively limited. An increase in SNP genotype 2 was observed in 1985, and this type was dominant until 1992. A dominant clone was clearly replaced by SNP genotype 1 in 1993 and SNP genotype 7 in 2004. In 2008, approximately half of the isolates were from SNP genotype 8, but this type did not become dominant after that. An increase in SNP genotype 9 was observed in 2012, and this type is still dominant.
TABLE 2.
SNP genotypes identified from 815 isolates of S. enterica serovar Typhimurium and its monophasic variant from cattle between 1977 and 2017
| Yr of isolation | No. of isolates with indicated SNP genotype |
Total no. of isolates | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | ||
| 1977 | 1 | 1 | ||||||||
| 1980 | 2 | 2 | ||||||||
| 1981 | 1 | 1 | ||||||||
| 1982 | 4 | 4 | ||||||||
| 1983 | 1 | 1 | ||||||||
| 1984 | 1 | 3 | 4 | |||||||
| 1985 | 6 | 4 | 10 | |||||||
| 1986 | 1 | 8 | 1 | 10 | ||||||
| 1987 | 9 | 9 | ||||||||
| 1988 | 7 | 7 | ||||||||
| 1989 | 7 | 2 | 9 | |||||||
| 1990 | 1 | 4 | 5 | |||||||
| 1991 | 13 | 13 | ||||||||
| 1992 | 4 | 12 | 1 | 17 | ||||||
| 1993 | 27 | 3 | 30 | |||||||
| 1994 | 38 | 1 | 1 | 40 | ||||||
| 1995 | 22 | 2 | 24 | |||||||
| 1996 | 12 | 1 | 13 | |||||||
| 1997 | 21 | 21 | ||||||||
| 1998 | 19 | 1 | 1 | 2 | 23 | |||||
| 1999 | 28 | 1 | 1 | 30 | ||||||
| 2000 | 9 | 1 | 1 | 1 | 2 | 14 | ||||
| 2001 | 13 | 1 | 1 | 1 | 1 | 1 | 18 | |||
| 2002 | 13 | 1 | 3 | 10 | 27 | |||||
| 2003 | 18 | 3 | 3 | 12 | 37 | |||||
| 2004 | 8 | 2 | 19 | 1 | 30 | |||||
| 2005 | 3 | 2 | 2 | 1 | 1 | 46 | 2 | 57 | ||
| 2006 | 1 | 2 | 3 | 38 | 45 | |||||
| 2007 | 11 | 3 | 2 | 46 | 1 | 64 | ||||
| 2008 | 3 | 1 | 1 | 5 | 9 | 1 | 21 | |||
| 2009 | 2 | 1 | 2 | 1 | 4 | 4 | 14 | |||
| 2010 | 3 | 2 | 3 | 2 | 10 | |||||
| 2011 | 1 | 1 | 1 | 2 | 5 | |||||
| 2012 | 1 | 1 | 2 | 2 | 6 | 12 | ||||
| 2013 | 3 | 1 | 6 | 18 | 28 | |||||
| 2014 | 2 | 1 | 2 | 26 | 31 | |||||
| 2015 | 2 | 58 | 60 | |||||||
| 2016 | 3 | 7 | 5 | 54 | 69 | |||||
| 2017 | 3 | 3 | ||||||||
| Total | 261 | 90 | 12 | 1 | 11 | 39 | 192 | 33 | 176 | 815 |
Table 3 shows the distribution of SNP genotypes among 140 isolates obtained from swine between 1976 and 2017. SNP genotypes 3 and 9 constituted 63.6% of the isolates. SNP genotype 3 was detected in 1977 for the first time, but 42 of the 55 isolates were detected between 2006 and 2015. SNP genotype 9 was detected in 2002 for the first time, and 27 of the 34 isolates were detected after 2011. Sixteen isolates of SNP genotype 2 were detected before 1990. Fifteen isolates of SNP genotype 1 were detected between 1991 and 2011, while 12 isolates of SNP genotype 8 were detected between 2001 and 2013.
TABLE 3.
SNP genotypes identified from 140 isolates of S. enterica serovar Typhimurium and its monophasic variant from swine between 1976 and 2017
| Yr of isolation | No. of isolates with indicated SNP genotype |
Total no. of isolates | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | ||
| 1976 | 4 | 4 | ||||||||
| 1977 | 1 | 1 | 2 | |||||||
| 1978 | 1 | 1 | ||||||||
| 1982 | 2 | 4 | 6 | |||||||
| 1983 | 1 | 1 | ||||||||
| 1988 | 4 | 5 | 9 | |||||||
| 1989 | 4 | 4 | ||||||||
| 1991 | 1 | 1 | ||||||||
| 1992 | 3 | 3 | ||||||||
| 1993 | 1 | 1 | ||||||||
| 1994 | 3 | 3 | ||||||||
| 1995 | 3 | 3 | ||||||||
| 1999 | 0 | |||||||||
| 2000 | 2 | 2 | ||||||||
| 2001 | 1 | 1 | 1 | 3 | ||||||
| 2002 | 1 | 1 | ||||||||
| 2003 | 2 | 1 | 3 | |||||||
| 2004 | 1 | 1 | ||||||||
| 2005 | 1 | 4 | 5 | |||||||
| 2006 | 2 | 3 | 1 | 6 | ||||||
| 2007 | 1 | 3 | 1 | 5 | ||||||
| 2008 | 1 | 3 | 1 | 2 | 7 | |||||
| 2009 | 2 | 1 | 3 | 6 | ||||||
| 2010 | 4 | 4 | ||||||||
| 2011 | 1 | 7 | 2 | 10 | ||||||
| 2012 | 5 | 2 | 7 | |||||||
| 2013 | 7 | 2 | 5 | 14 | ||||||
| 2014 | 2 | 4 | 6 | |||||||
| 2015 | 6 | 4 | 10 | |||||||
| 2016 | 11 | 11 | ||||||||
| 2017 | 1 | 1 | ||||||||
| Total | 15 | 16 | 55 | 2 | 2 | 4 | 0 | 12 | 34 | 140 |
DISCUSSION
PFGE and MLVA have long been used as subtyping methods for Salmonella Typhimurium in molecular epidemiological analyses due to their high discriminatory power and reproducibility (42–44). PFGE is time- and labor-intensive, and the macrorestriction pattern is not suitable for strain comparison between laboratories. The discriminatory power of MLVA is reported to be equal or superior to that of PFGE, and data are comparable between laboratories. PFGE pattern and MLVA profile are defined by the number and location of XbaI sites in the genome and the number of tandem repeats in five specific loci in the genome, respectively. Neither method reflects genome-wide information precisely; they are both difficult to use for phylogenetic analysis of Salmonella Typhimurium. In contrast, the SNP genotyping method established in this study is based on whole-genome sequencing data and reflects the phylogeny of Salmonella Typhimurium. Moreover, SNP genotypes can be determined by a combination of nine different AS-PCRs, and thus, this method is time- and labor-saving compared to PFGE and MLVA.
The SNP genotypes of 1,006 of the 1,027 isolates used in this study were successfully determined. No amplification was observed in the SNP genotyping of the remaining 21 isolates, which were obtained from cattle or swine. The MLVA clusters of these isolates were not sorted into the previously determined clusters (19) (Fig. S2), while each MLVA cluster corresponded to a single SNP genotype (Table 1), suggesting that the genetic background of these isolates was different from the 9 SNP genotypes.
Epidemic SNP genotypes that have dominated for 1 to 11 years in the following order since 1980 are SNP genotypes 6, 2, 1, 7, 8, and 9. Although it is possible that potentially confounding variables affect this variation, SNP genotypes 1, 7, 8, and 9 occupied more than 80% of the cattle isolates, and these isolates were obtained continuously throughout a relatively limited period, suggesting the existence of some epidemiological background. Isolates of SNP genotypes 1 and 7 solely include PFGE types 1 and 7, respectively (Table 1). The corresponding clades occupied distinct positions in the phylogenetic tree (Fig. 1), suggesting that the clonal replacement of PFGE type 1 with type 7 observed in 2002 was a dissemination of an unprecedented clone among cattle populations, rather than evolution of PFGE type 1 to type 7. All SNP genotype 7 isolates consisted of Salmonella Typhimurium and were obtained from cattle only, while most SNP genotype 8 isolates consisted of Salmonella 4,[5],12:i:− obtained from various sources (Fig. 1; Table 2). SNP genotypes 7 and 8 were detected in almost the same period (Tables 2 and 3) and occupied nearby positions in the phylogenetic tree (Fig. 1). However, prophage distribution of both SNP genotypes was clearly different (Fig. 2). Thus, it is unlikely that SNP genotype 8 is a monophasic variant of SNP genotype 7.
Multiple Salmonella 4,[5],12:i:− clones have been reported previously (38, 45–47). Among them, two major clones appear to have spread worldwide. The “Spanish clone” emerged in 1997 and spread in European countries and the United States (45, 46). This clone was characterized by a deletion in the allantoin-glyoxylate operon and the fljAB operon, phage type U302, and ST19, and it harbored an IncA/C plasmid that defined resistance to ampicillin, chloramphenicol, sulfonamide, gentamicin, streptomycin, tetracycline, and trimethoprim. In our study, a Salmonella 4,[5],12:i:− isolate, L-3843, was isolated from human in 2002 and grouped into clade 1; this isolate harbored an IncA/C plasmid defining resistance to ampicillin, sulfonamide, gentamicin, streptomycin, tetracycline, and trimethoprim. Assuming that this isolate is a Spanish clone, the SNP genotyping system does not discriminate the Spanish clone from SNP genotype 1, which corresponds to phage types DT104 and U302.
The “European clone” was first reported in approximately 2005 and spread across European countries (38). This clone is characterized by ST34, and a genetic region containing the fljAB-hin operon is replaced by a composite transposon insertion in the chromosome containing antimicrobial resistance genes, consistent with the resistance profile of this clone [strA, strB, sul2, tet(B), and blaTEM-1]. Possession of Salmonella genomic island 3 (SGI3), which encodes resistance to heavy metals, including copper and zinc, is also characteristic of this clone. In our study, clade 9 isolates showed characteristics similar to those of the European clone. All clade 9 isolates grouped into ST34 and showed resistance to all or some of the following antimicrobials: ampicillin, streptomycin, sulfonamide, and tetracycline. The presence of SGI3 was confirmed in most clade 9 isolates (Table S2). The first detections of clade 9 isolates from cattle and swine were in 1998 and 2002, respectively. Since then, clade 9 isolates spread among cattle and swine populations and have become dominant. Increased detection of SNP genotype 9 in Japan might be a part of a pandemic of the European clone.
Clade 9 includes four isolates of Salmonella Typhimurium, including two Japanese isolates obtained from cattle in 1998 (L-4126 and L-4127) and two Italian isolates obtained from poultry and swine in 2011 and 2012 (LB-14 and LB-15). Antimicrobial resistance genes were located on the chromosome (Fig. 3), but the fljAB-hin regions were intact in these isolates. SGI3 was also detected from these isolates, except for isolate LB-14. The genetic relationship of these isolates to other isolates in the same clade remains to be elucidated.
Clade 4 was a remaining major clade of Salmonella 4,[5],12:i:−, and these isolates were obtained from various samples in Japan and Italy. These isolates harbored common amino acid substitutions of A46T in FljA and R140L in Hin, suggesting the high clonality of these isolates. Interestingly, four and three of the nine clade 4 isolates were obtained from birds and environmental water, respectively. Wild birds may have contributed to the worldwide dissemination of this clone.
We also determined the SNP genotypes of 140 isolates obtained from swine in Japan between 1976 and 2017. Dominant clones were not clear compared to those from cattle isolates. However, SNP genotypes 2, 1, 8, 3, and 9 were detected for more than a continuous 4-year period, in this order. In particular, SNP genotypes 3 and 9 occupied 63.6% of the swine isolates. Fifty-five isolates of SNP genotype 3 were detected in swine, while 12 isolates were detected from cattle, suggesting that this genotype is more adapted to swine.
The driving force of the clonal replacement was not clear from this study. The common characteristic of the dominant clones during and after the 1990s was a multidrug resistance defined by chromosomal islands or a plasmid. For example, most clade 1 isolates harbored the SGI1-containing resistance genes, aadA2, blaPSE-1, floR, sul1, and tet(G). Most clade 7 isolates harbored a virulence resistance plasmid encoding aadA1, aph(3′)-Ia, blaTEM-1, sul1, and tet(A) (22, 40). Most clade 9 isolates also harbored multiple antimicrobial resistance genes in the chromosomal composite transposon (39). However, antimicrobial resistance phenotypes of these SNP genotypes were similar, and it is unlikely that antimicrobial use is the sole selection pressure for these clones.
Clade 9 isolates harbored SGI3, which contains genes for resistance to copper and zinc. These heavy metals are generally added to swine feed as micronutrients and general antimicrobials. Petrovska et al. reported that the European clone harbored SGI3 and showed a higher MIC for copper sulfate in rich broth culture (38). As we confirm the existence of SGI3 in clade 9 isolates, heavy metals as feed additives might be one selection pressure for this clone.
In conclusion, multiple distinctive clades were identified among isolates of Salmonella Typhimurium and Salmonella 4,[5],12:i:−, which were obtained mainly in food animals in Japan. Among cattle isolates, the replacement of a major epidemic clone was observed five times over the last 4 decades. The duration of domination for a clade was 1 to 11 years. Clade 9 is the current predominant clone, and these isolates have characteristics similar to those of the European clone of Salmonella 4,[5],12:i:−. The increased prevalence of this clone among food animals in Japan might be a part of the pandemic of the specific clone of Salmonella 4,[5],12:i:−.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to the staff at the prefectural livestock hygiene service centers for providing Salmonella isolates.
This study was supported by a grant-in-aid for the Research Program on Emerging and Reemerging Infectious Diseases (15fk0108021h0002) from the Japan Agency for Medical Research and Development, AMED.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/JCM.01758-17.
REFERENCES
- 1.Hohmann EL. 2001. Nontyphoidal salmonellosis. Clin Infect Dis 32:263–269. doi: 10.1086/318457. [DOI] [PubMed] [Google Scholar]
- 2.Majowicz SE, Musto J, Scallan E, Angulo FJ, Kirk M, O'Brien SJ, Jones TF, Fazil A, Hoekstra RM. 2010. The global burden of nontyphoidal Salmonella gastroenteritis. Clin Infect Dis 50:882–889. doi: 10.1086/650733. [DOI] [PubMed] [Google Scholar]
- 3.Iino T. 1969. Genetics and chemistry of bacterial flagella. Bacteriol Rev 33:454–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grimont P, Weil F. 2007. Antigenic formulae of the Salmonella serovars, 9th ed World Health Organization Centre for Reference and Research on Salmonella. Pasteur Institute, Paris, France. [Google Scholar]
- 5.Issenhuth-Jeanjean S, Roggentin P, Mikoleit M, Guibourdenche M, de Pinna E, Nair S, Fields PI, Weill FX. 2014. Supplement 2008-2010 (no. 48) to the White-Kauffmann-Le Minor scheme. Res Microbiol 165:526–530. doi: 10.1016/j.resmic.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 6.Foley SL, Lynne AM. 2008. Food animal-associated Salmonella challenges: pathogenicity and antimicrobial resistance. J Anim Sci 86:E173–E187. doi: 10.2527/jas.2007-0447. [DOI] [PubMed] [Google Scholar]
- 7.Herikstad H, Motarjemi Y, Tauxe RV. 2002. Salmonella surveillance: a global survey of public health serotyping. Epidemiol Infect 129:1–8. doi: 10.1017/S0950268802006842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dionisi AM, Graziani C, Lucarelli C, Filetici E, Villa L, Owczarek S, Caprioli A, Luzzi I. 2009. Molecular characterization of multidrug-resistant strains of Salmonella enterica serotype Typhimurium and monophasic variant (S. 4,[5],12:i:–) isolated from human infections in Italy. Foodborne Pathog Dis 6:711–717. doi: 10.1089/fpd.2008.0240. [DOI] [PubMed] [Google Scholar]
- 9.Gallati C, Stephan R, Hachler H, Malorny B, Schroeter A, Nuesch-Inderbinen M. 2013. Characterization of Salmonella enterica subsp. enterica serovar 4,[5],12:i:– clones isolated from human and other sources in Switzerland between 2007 and 2011. Foodborne Pathog Dis 10:549–554. doi: 10.1089/fpd.2012.1407. [DOI] [PubMed] [Google Scholar]
- 10.Ido N, Lee K, Iwabuchi K, Izumiya H, Uchida I, Kusumoto M, Iwata T, Ohnishi M, Akiba M. 2014. Characteristics of Salmonella enterica serovar 4,[5],12:i:– as a monophasic variant of serovar Typhimurium. PLoS One 9:e104380. doi: 10.1371/journal.pone.0104380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Majtan V, Majtanova L, Majtan J. 2011. Phenotypic and molecular characterization of human Salmonella enterica serovar 4,[5],12:i:– isolates in Slovakia. Curr Microbiol 63:491–495. doi: 10.1007/s00284-011-0010-6. [DOI] [PubMed] [Google Scholar]
- 12.Switt AI, Soyer Y, Warnick LD, Wiedmann M. 2009. Emergence, distribution, and molecular and phenotypic characteristics of Salmonella enterica serotype 4,5,12:i. Foodborne Pathog Dis 6:407–415. doi: 10.1089/fpd.2008.0213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mossong J, Marques P, Ragimbeau C, Huberty-Krau P, Losch S, Meyer G, Moris G, Strottner C, Rabsch W, Schneider F. 2007. Outbreaks of monophasic Salmonella enterica serovar 4,[5],12:i:– in Luxembourg, 2006. Euro Surveill 12:E11–E12. doi: 10.2807/esm.12.06.00719-en. [DOI] [PubMed] [Google Scholar]
- 14.Trüpschuch S, Laverde Gomez JA, Ediberidze I, Flieger A, Rabsch W. 2010. Characterisation of multidrug-resistant Salmonella Typhimurium 4,[5],12:i:– DT193 strains carrying a novel genomic island adjacent to the thrW tRNA locus. Int J Med Microbiol 300:279–288. doi: 10.1016/j.ijmm.2010.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Bone A, Noel H, Le Hello S, Pihier N, Danan C, Raguenaud ME, Salah S, Bellali H, Vaillant V, Weill FX, Jourdan-da Silva N. 2010. Nationwide outbreak of Salmonella enterica serotype 4,12:i:- infections in France, linked to dried pork sausage, March–May 2010. Euro Surveill 15(24):19592. [PubMed] [Google Scholar]
- 16.CDC. 2007. Investigation of outbreak of human infections caused by Salmonella I 4,[5],12:i:–. CDC, Department of Health and Human Services, Atlanta, GA. [Google Scholar]
- 17.Bonardi S. 2017. Salmonella in the pork production chain and its impact on human health in the European Union. Epidemiol Infect 145:1513–1526. doi: 10.1017/S095026881700036X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ido N, Kudo T, Sasaki K, Motokawa M, Iwabuchi K, Matsudate H, Seimiya YM, Akiba M. 2011. Molecular and phenotypic characteristics of Salmonella enterica serovar 4,5,12:i:- isolated from cattle and humans in Iwate Prefecture, Japan. J Vet Med Sci 73:241–244. doi: 10.1292/jvms.10-0140. [DOI] [PubMed] [Google Scholar]
- 19.Kurosawa A, Imamura T, Tanaka K, Tamamura Y, Uchida I, Kobayashi A, Hata E, Kanno T, Akiba M, Yukawa S, Tamura Y. 2012. Molecular typing of Salmonella enterica serotype Typhimurium and serotype 4,5,12:i:- isolates from cattle by multiple-locus variable-number tandem-repeats analysis. Vet Microbiol 160:264–268. doi: 10.1016/j.vetmic.2012.05.023. [DOI] [PubMed] [Google Scholar]
- 20.Wray C, Davies RH. 2000. Salmonella infections in cattle, p 169–190. In Wray C, Wray A (ed), Salmonella in domestic animals. CABI Publishing, Wallingford, United Kingdom. [Google Scholar]
- 21.Tamada Y, Nakaoka Y, Nishimori K, Doi A, Kumaki T, Uemura N, Tanaka K, Makino SI, Sameshima T, Akiba M, Nakazawa M, Uchida I. 2001. Molecular typing and epidemiological study of Salmonella enterica serotype Typhimurium isolates from cattle by fluorescent amplified-fragment length polymorphism fingerprinting and pulsed-field gel electrophoresis. J Clin Microbiol 39:1057–1066. doi: 10.1128/JCM.39.3.1057-1066.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tamamura Y, Uchida I, Tanaka K, Okazaki H, Tezuka S, Hanyu H, Kataoka N, Makino S, Kishima M, Kubota T, Kanno T, Hatama S, Ishihara R, Hata E, Yamada H, Nakaoka Y, Akiba M. 2011. Molecular epidemiology of Salmonella enterica serovar Typhimurium isolates from cattle in hokkaido, Japan: evidence of clonal replacement and characterization of the disseminated clone. Appl Environ Microbiol 77:1739–1750. doi: 10.1128/AEM.01910-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helms M, Ethelberg S, Molbak K. 2005. International Salmonella Typhimurium DT104 infections, 1992–2001. Emerg Infect Dis 11:859–867. doi: 10.3201/eid1106.041017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Edwards PR, Ewing WH. 1986. Edwards and Ewing's identification of Enterobacteriaceae, 4th ed Elsevier Science Publishing Co, Inc, New York, NY. [Google Scholar]
- 25.Anderson ES, Ward LR, Saxe MJ, de Sa JD. 1977. Bacteriophage-typing designations of Salmonella typhimurium. J Hyg (Lond) 78:297–300. doi: 10.1017/S0022172400056187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Clinical and Laboratory Standards Institute. 2012. Performance standards for antimicrobial disk susceptibility tests; approved standard, 11th ed, document M02-A11. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 27.Clinical and Laboratory Standards Institute. 2014. Performance standards for antimicrobial susceptibility testing; 24th informational supplement, document M100-S24. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 28.Shahada F, Sekizuka T, Kuroda M, Kusumoto M, Ohishi D, Matsumoto A, Okazaki H, Tanaka K, Uchida I, Izumiya H, Watanabe H, Tamamura Y, Iwata T, Akiba M. 2011. Characterization of Salmonella enterica serovar Typhimurium isolates harboring a chromosomally encoded CMY-2 beta-lactamase gene located on a multidrug resistance genomic island. Antimicrob Agents Chemother 55:4114–4121. doi: 10.1128/AAC.00560-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Coil D, Jospin G, Darling AE. 2015. A5-miseq: an updated pipeline to assemble microbial genomes from Illumina MiSeq data. Bioinformatics 31:587–589. doi: 10.1093/bioinformatics/btu661. [DOI] [PubMed] [Google Scholar]
- 30.Li H, Durbin R. 2010. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koboldt DC, Chen K, Wylie T, Larson DE, McLellan MD, Mardis ER, Weinstock GM, Wilson RK, Ding L. 2009. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics 25:2283–2285. doi: 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, Salzberg SL. 2004. Versatile and open software for comparing large genomes. Genome Biol 5:R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. 2011. PHAST: a fast phage search tool. Nucleic Acids Res 39:W347–W352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cheng L, Connor TR, Siren J, Aanensen DM, Corander J. 2013. Hierarchical and spatially explicit clustering of DNA sequences with BAPS software. Mol Biol Evol 30:1224–1228. doi: 10.1093/molbev/mst028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, Aarestrup FM, Larsen MV. 2012. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother 67:2640–2644. doi: 10.1093/jac/dks261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Boyd D, Peters GA, Cloeckaert A, Boumedine KS, Chaslus-Dancla E, Imberechts H, Mulvey MR. 2001. Complete nucleotide sequence of a 43-kilobase genomic island associated with the multidrug resistance region of Salmonella enterica serovar Typhimurium DT104 and its identification in phage type DT120 and serovar Agona. J Bacteriol 183:5725–5732. doi: 10.1128/JB.183.19.5725-5732.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petrovska L, Mather AE, AbuOun M, Branchu P, Harris SR, Connor T, Hopkins KL, Underwood A, Lettini AA, Page A, Bagnall M, Wain J, Parkhill J, Dougan G, Davies R, Kingsley RA. 2016. Microevolution of monophasic Salmonella Typhimurium during epidemic, United Kingdom, 2005–2010. Emerg Infect Dis 22:617–624. doi: 10.3201/eid2204.150531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.García P, Malorny B, Rodicio MR, Stephan R, Hachler H, Guerra B, Lucarelli C. 2016. Horizontal acquisition of a multidrug-resistance module (R-type ASSuT) is responsible for the monophasic phenotype in a widespread clone of Salmonella serovar 4,[5],12:i. Front Microbiol 7:680. doi: 10.3389/fmicb.2016.00680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tamamura Y, Tanaka K, Akiba M, Kanno T, Hatama S, Ishihara R, Uchida I. 2013. Complete nucleotide sequences of virulence-resistance plasmids carried by emerging multidrug-resistant Salmonella enterica serovar Typhimurium isolated from cattle in Hokkaido, Japan. PLoS One 8:e77644. doi: 10.1371/journal.pone.0077644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cha RS, Zarbl H, Keohavong P, Thilly WG. 1992. Mismatch amplification mutation assay (MAMA): application to the c-H-ras gene. PCR Methods Appl 2:14–20. [DOI] [PubMed] [Google Scholar]
- 42.Lindstedt BA, Vardund T, Aas L, Kapperud G. 2004. Multiple-locus variable-number tandem-repeats analysis of Salmonella enterica subsp. enterica serovar Typhimurium using PCR multiplexing and multicolor capillary electrophoresis. J Microbiol Methods 59:163–172. doi: 10.1016/j.mimet.2004.06.014. [DOI] [PubMed] [Google Scholar]
- 43.Lindstedt BA, Heir E, Gjernes E, Kapperud G. 2003. DNA fingerprinting of Salmonella enterica subsp. enterica serovar Typhimurium with emphasis on phage type DT104 based on variable number of tandem repeat loci. J Clin Microbiol 41:1469–1479. doi: 10.1128/JCM.41.4.1469-1479.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wattiau P, Boland C, Bertrand S. 2011. Methodologies for Salmonella enterica subsp. enterica subtyping: gold standards and alternatives. Appl Environ Microbiol 77:7877–7885. doi: 10.1128/AEM.05527-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Soyer Y, Moreno Switt A, Davis MA, Maurer J, McDonough PL, Schoonmaker-Bopp DJ, Dumas NB, Root T, Warnick LD, Grohn YT, Wiedmann M. 2009. Salmonella enterica serotype 4,5,12:i:−, an emerging Salmonella serotype that represents multiple distinct clones. J Clin Microbiol 47:3546–3556. doi: 10.1128/JCM.00546-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Laorden L, Herrera-Leon S, Martinez I, Sanchez A, Kromidas L, Bikandi J, Rementeria A, Echeita A, Garaizar J. 2010. Genetic evolution of the Spanish multidrug-resistant Salmonella enterica 4,5,12:i:- monophasic variant. J Clin Microbiol 48:4563–4566. doi: 10.1128/JCM.00337-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mourão J, Machado J, Novais C, Antunes P, Peixe L. 2014. Characterization of the emerging clinically-relevant multidrug-resistant Salmonella enterica serotype 4,[5],12:i:– (monophasic variant of S. Typhimurium) clones. Eur J Clin Microbiol Infect Dis 33:2249–2245. doi: 10.1007/s10096-014-2180-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.