

ABSTRACT
Clostridium difficile multilocus sequence type 37 (ST37), which mainly corresponds to ribotype 017, has been a dominant genotype circulating in China. In this study, we report the use of matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) to analyze and characterize 204 C. difficile clinical isolates, including 49 ST37 and 155 non-ST37 isolates collected in China and other countries. The distributions of two major protein peaks (m/z 3,242 and 3,286) were significantly different between ST37 and non-ST37 prototype strains and clinical isolates. This difference was reproducible when analysis was performed on different colonies in different runs. This finding was repeated and confirmed by both bioMérieux Vitek MS and Bruker Microflex LT systems on isolates recovered from a variety of geographic regions worldwide. The combination of the two peaks was present in 47 of 49 ST37 isolates, resulting in a sensitivity of 95.9%. In contrast, the peak combination was absent in 153 of 155 non-ST37 isolates, resulting in a specificity of 98.7%. Our results suggest that MALDI-TOF MS is a rapid and reliable tool to identify C. difficile genotype ST37. Work is in progress to characterize the two molecules having peaks at m/z 3,242 and 3,286, which appear to be specific to C. difficile genotype ST37.
KEYWORDS: Clostridium difficile; genotype ST37; MALDI-TOF MS; m/z 3,242; m/z 3,286
INTRODUCTION
Clostridium difficile is a Gram-positive, anaerobic, spore-forming bacillus that is a major pathogenic bacterium in health care-associated diarrhea in developed countries (1, 2). Clinical symptoms associated with C. difficile infection (CDI) range from mild diarrhea to life-threatening pseudomembranous colitis (PMC) (3). Two toxins, toxin A (TcdA) and toxin B (TcdB), are primarily responsible for the pathogenicity of C. difficile (4, 5). In recent years, a toxin A-negative, toxin B-positive (A−B+) C. difficile variant, mainly belonging to sequence type 37 (ST37) by multilocus sequence type (MLST) analysis and ribotype 017 by PCR-ribotyping, has been widely distributed in China and other countries (6, 7). Previous work (8–10) has demonstrated that ST37 C. difficile is the dominant strain in adult patients with CDI, albeit with limited prevalence data for colonization in healthy individuals. ST37 isolates have been associated with increased resistance to multiple antibiotics, including fluoroquinolones, macrolides, and lincomycin (11, 12). Severe clinical symptoms, as well as outbreaks in hospitals, have been reported for these A−B+ C. difficile isolates, suggesting that this strain is a major public health problem (13–15). Thus, identification for ST37 strains of C. difficile by a rapid and accurate method is of great clinical significance for the prevention and control of CDI.
Various molecular typing methods, including PCR-ribotyping, multilocus sequence type (MLST) analysis, and pulsed-field gel electrophoresis (PFGE) typing, have been widely used for genotyping and characterization of C. difficile isolates. Unfortunately, these methods are time-consuming, relatively costly, and complex, making them difficult for routine clinical use (16). Matrix-assisted laser desorption ionization–time of flight mass spectrometry (MALDI-TOF MS) has been widely applied in clinical microbiology laboratories for identification of pathogenic bacteria (17–19). Rapid identification of microbes at species level is achieved by comparing protein fingerprint spectra, which are comprised of molecules ranging from m/z 2,000 to 20,000, with the established strain reference databases (20, 21). MALDI-TOF MS technology thus provides a rapid, accurate tool for identification of microbial pathogens (22, 23). In addition, differences between protein spectra determined by MALDI-TOF MS have been evaluated as a method for bacterial typing by identifying type-specific protein peaks in mass spectra that can be used as biomarkers to identify particular genotypes or subtypes (24, 25). This technology has also been reported to be useful for the identification of unique C. difficile genotypes, such as ribotype 001, 027, and 078/126 (26). The aim of this study was to identify and characterize potential MALDI-TOF MS biomarkers for rapid identification and characterization of C. difficile ST37 strains.
(This study was presented in part at the 10th World Congress of the World Society for Pediatric Infectious Diseases, Shenzhen, China, 2 to 5 December 2017.)
MATERIALS AND METHODS
C. difficile strains and clinical isolates.
C. difficile prototype strains, including ATCC BAA-1870 (ST1), 9689 (ST3), 43598 (ST37), 43255 (ST46), BAA-1812 (ST53), BAA-1382 (ST54) and BAA-1804 (ST63), were obtained from the American Type Culture Collection (Manassas, VA). A total of 204 C. difficile isolates were recovered from fecal specimens of patients with diarrhea or from healthy donors in China and other countries as previously described (8, 10, 27) (Table 1). All prototype strains and isolates were stored at −70°C in brain heart infusion broth with 10% glycerol until subsequent analyses. For subsequent experiments, all strains and clinical isolates were retrieved on Columbia blood agar (CBA; Oxoid, Cambridge, UK) plates and incubated at 37°C for 48 h under anaerobic conditions. A single bacterial colony with typical morphology was subcultured again on CBA plates and incubated under the same conditions for 48 h.
TABLE 1.
Sources and sequence types of clinical isolates recruited in this study
| Geographic origin | Sequence type | Total no. of isolates |
|---|---|---|
| Hebei, China | 37 | 31 |
| 3 | 29 | |
| 54 | 23 | |
| 2 | 14 | |
| 35 | 13 | |
| 8 | 5 | |
| 26 | 5 | |
| 48 | 5 | |
| 205 | 5 | |
| Other STs | 48 | |
| Hangzhou, China | 37 | 5 |
| Hong Kong, China | 37 | 2 |
| New York City, USA | 37 | 8 |
| 3 | 6 | |
| South Korea | 37 | 2 |
| Singapore | 37 | 1 |
| 35 | 1 | |
| Australia | 2 | 1 |
DNA extraction, toxin gene analysis, and MLST analysis.
DNA was extracted from a single isolated colony using a TIANamp bacteria DNA kit (Tiangen Biotech, Beijing, China) according to the manufacturer's instructions. DNA samples were stored at −20°C until use. Toxin genes tcdA and tcdB were tested as described by Lemee et al. (28). MLST analysis was performed as previously described (16). In brief, fragments from seven genes (adk, atpA, dxr, glyA, recA, sodA, and tpi) were amplified and sequenced at Sangon Biotech (Shanghai, China) and analyzed at the Beijing Genomics Institute (Beijing, China). The DNA sequences of the seven genes were submitted to the MLST database (https://pubmlst.org/cdifficile/) to obtain the ST genotype.
MALDI-TOF sample preparation.
An ethanol/formic acid method was used for extraction of bacterial proteins, in which one-half to one 10-μl inoculation loop of fresh bacterial culture was thoroughly suspended in 300 μl of molecular-grade water and 900 μl ethanol was added. The components were mixed well and centrifuged at 13,000 × g for 2 min. After centrifugation, the supernatant was removed, and the pellets were air dried. Then, 20 μl of formic acid (70% in water) was added to the pellets and mixed. Finally, 20 μl of acetonitrile was added and mixed thoroughly. After centrifugation at 13,000 × g for 2 min, the supernatant containing the bacterial protein extract was transferred to a clean tube.
MALDI-TOF MS data acquisition.
The spectra were collected using two mass spectrometry systems, the bioMérieux Vitek MS and the Bruker Microflex LT. For analysis with the bioMérieux Vitek MS, 1 μl bacterial protein extract was transferred to a Vitek MS disposable slide (bioMérieux SA, Marcy-l'Étoile, France) and allowed to dry before overlaying with 1 μl of a saturated solution of α-cyano-4-hydroxycinnamic acid (α-CHCA) in 28% acetonitrile (CHCA matrix; bioMérieux SA) and dried in ambient air. Four spots were prepared for each sample. The Vitek MS MALDI-TOF MS was equipped with an N2 laser (λ = 337 nm). Spectra were collected in linear SARAMIS RUO mode, in the range of m/z 2,000 to m/z 20,000. The software used for data acquisition was LaunchPad (version 2.8; bioMérieux). Spectra were acquired in automatic mode by accumulating 100 profiles. Spectral data were automatically processed and exported as peak lists for analysis in SARAMIS. Escherichia coli strain ATCC 8739 was used for mass calibration instrument parameter optimization. For analysis with the Bruker Microflex LT, 1 μl bacterial protein extract was transferred to an MSP 96 ground steel sample target (Bruker Daltonik, Bremen, Germany) and allowed to dry at room temperature. Subsequently, the sample was overlaid with 1 μl of matrix (a saturated solution of α-cyano-4-hydroxycinnamic acid in 50% acetonitrile and 2.5% trifluoroacetic acid) and dried in ambient air. Twelve spots were prepared for each sample. The Microflex LT MALDI-TOF MS system was equipped with a 337-nm N2 laser. The molecular ions were measured in the linear positive-ion mode. The software used for the data acquisition was flexControl (version 3.0; Bruker Daltonik) software. The parameters used were as follows: mass range, 2,000 to 20,000 Da; ion source 1, 20 kV; ion source 2, 18.5 kV; lens, 8.45 kV; pulsed ion extraction, 320 ns; laser frequency, 20.0 Hz. A Bruker bacterial test standard (BTS255343; Bruker Daltonik) was used for mass calibration instrument parameter optimization. A peak was considered to be present when it was detected by the automated spectrum processing performed by the respective software programs.
Data analysis.
Data were analyzed using Statistical Package for Social Sciences (SPSS; Chicago, IL) version 21.0. As all peak values were nonnormally distributed, the Mann-Whitney U test was used to analyze correlations between peak values and STs. P values were calculated to assess the differences between spectra of ST37 and non-ST37 isolates, and a P value of ≤0.05 was considered statistically significant.
RESULTS
A total of 211 C. difficile isolates/strains, including seven ATCC reference strains and 204 clinical isolates, were included in the study. Among the 204 clinical isolates, 178 (87.3%), 5 (2.5%), 2 (1.0%), 2 (1.0%), 2 (1.0%), 1 (0.5%), and 14 (6.9%) were collected from Hebei, Hangzhou, Hong Kong, Singapore, South Korea, Australia, and New York City, respectively (Table 1).
We first compared C. difficile whole-cell mass spectra for ST37 and non-ST37 genotypes, using ATCC reference strains and several additional clinical isolates (Fig. 1). The mass spectra were acquired in the mass range of m/z 2,000 to 20,000. Approximately 80 peaks were detected per spectrum, with the highest peaks concentrated in the range of m/z 3,000 to 10,000. Based on the data analysis, two protein peaks (m/z 3,242 and 3,286) were identified as correlated with genotype ST37. Peak m/z 3,286 was present in the spectra of ST37 isolates and absent in those of non-ST37 isolates; peak m/z 3,242 of ST37 isolate spectra had significantly higher intensity values compared with those of non-ST37 isolates (Fig. 2B). This observation was repeated and confirmed using both bioMérieux Vitek MS and Bruker Microflex LT systems.
FIG 1.
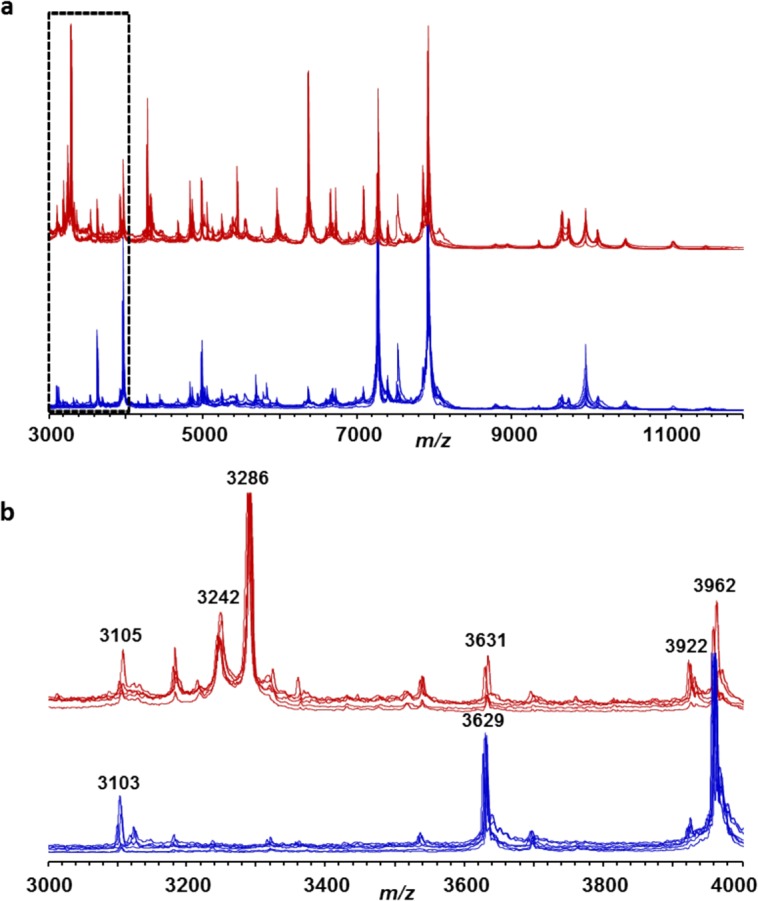
Exemplary whole-cell mass spectra of C. difficile strains and isolates of ST37 (ATCC 43598, CD013, CD015, CD017, CD026, and CD126; red lines) and non-ST37 (ATCC BAA-1382, ATCC BAA-1804, ATCC BAA-1870, ATCC BAA-1812, CD105 (ST3), and CD151 (ST2); blue lines). The lower panel (b) shows an enlarged section of the full upper spectra (a).
FIG 2.

Cluster analysis of spectra of 49 ST37 isolates of different geographic origins. The dendrogram was computed in SARAMIS with a single-linkage agglomerative algorithm-based relative spectrum similarity (percentage of matching peaks within a tolerance of 0.08%). The dotted purple line indicates 65% similarity, which is generally the threshold for intraspecies similarity. Yellow, Hebei; green, Hangzhou; blue, New York; black, ATCC 43598.
We then assessed the reproducibility of our observations for an additional three ST37 and three non-ST37 isolates. Values of m/z 3,370 (ST37-nonspecific) and 3,242 and 3,286 (ST37-specific) were included for intra- and interexperimental variability analysis. Each of the six isolates was run either four or nine times in three experiments run on different days by three experimental operators. High intraexperimental coefficient of variation (CV) values were revealed for both ST37 (117 to 209%) and non-ST37 isolates (Table 2), while relatively low interexperimental CV values were noticed for both ST37 (51% for m/z 3,242, 44% for m/z 3,286, and 64% for m/z 3,370) and non-ST37 isolates (58% for m/z 3,242, 173% for m/z 3,286, and 48% for m/z 3,370). As presented in Table 2, in all three experiments, all three ST37 isolates possessed significantly higher intensity values at m/z 3,242 (P < 0.001) and at m/z 3,286 (P < 0.001) than non-ST37 isolates. In contrast, there was no statistical difference between ST37 and non-ST37 isolates in m/z 3,370 peak values (P > 0.05).
TABLE 2.
Experimental variability of peak height values in spectra of ST37 and non-ST37 isolatesa
| Expt | m/z | Mean peak ht value (integrated in thousands) ± SD for following isolate of ST37 genotype: |
Mean ± SD | CV (%) | Mean peak ht value (integrated in thousands) ± SD for following isolate of non-ST37 genotype: |
Mean ± SD | CV (%) | ST37/non-ST37 ratio | Mann-Whitney U | P | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CD001 | CD038 | CD090 | CD003 | CD045 | CD086 | |||||||||
| 1 | 3,242 | 1,219 ± 1,035 | 5,881 ± 5,483 | 1,516 ± 1,722 | 2,871 ± 3,899 | 136 | 25 ± 49 | 15 ± 26 | 8 ± 14 | 16 ± 32 | 200 | 179 | 51 | <0.001 |
| 3,286 | 6,325 ± 6,151 | 19,169 ± 16,845 | 5,854 ± 6,131 | 10,449 ± 12,249 | 117 | 452 ± 861 | 0 | 0 | 151 ± 525 | 348 | 69 | 17 | <0.001 | |
| 3,370 | 14 ± 36 | 142 ± 203 | 280 ± 293 | 145 ± 227 | 157 | 5 ± 9 | 70 ± 99 | 41 ± 64 | 31 ± 56 | 181 | 5 | 296 | 0.193 | |
| 2 | 3,242 | 941 ± 1,177 | 1,370 ± 3,142 | 832 ± 800 | 1,048 ± 1,928 | 184 | 11 ± 10 | 5 ± 7 | 7 ± 14 | 8 ± 11 | 138 | 131 | 16 | <0.001 |
| 3,286 | 4,076 ± 5,084 | 5,082 ± 10,544 | 3,548 ± 3,215 | 4,236 ± 6,765 | 160 | 0 | 0 | 0 | 0 | NA | NA | <0.001 | <0.001 | |
| 3,370 | 54 ± 86 | 10 ± 19 | 20 ± 31 | 31 ± 56 | 181 | 9 ± 12 | 4 ± 7 | 29 ± 28 | 13 ± 20 | 154 | 2 | 287 | 0.305 | |
| 3 | 3,242 | 659 ± 693 | 9,448 ± 1,076 | 204 ± 144 | 3,437 ± 7,173 | 209 | 39 ± 50 | 26 ± 20 | 18 ± 28 | 28 ± 33 | 118 | 123 | 1 | <0.001 |
| 3,286 | 1,266 ± 2,111 | 17,937 ± 15,370 | 459 ± 288 | 6,554 ± 11,682 | 178 | 0 | 0 | 0 | 0 | NA | NA | <0.001 | <0.001 | |
| 3,370 | 197 ± 105 | 63 ± 127 | 17 ± 23 | 93 ± 118 | 127 | 24 ± 29 | 6 ± 7 | 17 ± 25 | 16 ± 22 | 138 | 6 | 50 | 0.182 | |
For experimental conditions and the no. of replicates, see the text. NA, not applicable; SD, standard deviation; CV, coefficient of variation.
A total of 204 C. difficile clinical isolates collected from different geographic regions in China and other countries was analyzed for tcdA and tcdB patterns (Table 3). In addition to ST37, these isolates included 35 ST3, 24 ST54, 15 ST2, 14 ST35, 5 ST8, 5 ST26, 5 ST48, and 5 ST205 genotypes, as well as 34 other rare ST genotypes. For the 49 ST37 isolates, the sole tcdA or tcdB pattern was A−B+. However, the 155 non-ST37 isolates were comprised of 124 (80.0%) A+B+, 1 (0.6%) A−B+, and 30 (19.4%) A−B−. Notably, the only A−B+ variant found to be a non-ST37 isolate was an ST81 genotype, which was recovered in Hebei, China (Table 3).
TABLE 3.
Molecular characteristics of ST37 and non-ST37 C. difficile isolates
| Genotype | Geographic origin | No. of isolates tested | MSa peak (no. present [%]) at m/z: |
tcdA or tcdB phenotypeb |
||||
|---|---|---|---|---|---|---|---|---|
| 3,242 | 3,286 | 3,370 | A+B+ | A−B+ | A−B− | |||
| ST37 | Hebei, China | 31 | 31 (100) | 30 (96.8) | 30 (96.8) | 0 (0) | 31 (100) | 0 (0) |
| Hangzhou, China | 5 | 5 (100) | 4 (80.0) | 5 (100) | 0 (0) | 5 (100) | 0 (0) | |
| Asia Pacific region | 5 | 5 (100) | 5 (100) | 5 (100) | 0 (0) | 5 (100) | 0 (0) | |
| New York, USA | 8 | 8 (100) | 8 (100) | 1 (12.5) | ND | ND | ND | |
| All ST37 origins | 49 | 49 (100) | 47 (95.9) | 41 (83.7) | ||||
| Non-ST37 | Hebei, China | 147 | 62 (42.2) | 1 (0.7) | 135 (91.8) | 116 (78.9) | 1 (0.7) | 30 (20.4) |
| Asia Pacific region | 2 | 0 (0) | 0 (0) | 2 (100) | 2 (100) | 0 (0) | 0 (0) | |
| New York, USA | 6 | 2 (33.3) | 1 (16.7) | 0 (0) | ND | ND | ND | |
| All non-ST37 origins | 155 | 64 (41.3) | 2 (1.3) | 137 (88.4) | ||||
MS, mass spectrometry.
ND, not done.
The presence and absence of the m/z 3,242 and 3,286 peaks were then analyzed in the 204 clinical isolates using the C. difficile-universal m/z 3,370 peak as the control (Table 3). The m/z 3,370 peak was detected in majority of isolates tested. Among the 49 ST37 isolates, all but two isolates possessed both m/z 3,242 and 3,286 peaks. In contrast, among the 155 non-ST37 isolates, the combination of peaks m/z 3,242 and 3,286 was only detected in two isolates, namely, one ST81 isolate from Hebei and one ST3 isolate from New York. A cluster analysis based on full spectra of ST37 isolates revealed no significant difference in spectra of strains from different geographic origins (Fig. 2). The differing distributions of m/z 3,242 and 3,286 peaks between ST37 and non-ST37 isolates was consistent irrespective of the geographic origin of the strains. Accordingly, the combination of two peaks (m/z 3,242 and 3,286) had a sensitivity of 95.9% and a specificity of 98.7% for identification and characterization of C. difficile genotype ST37 by MALDI-TOF MS.
DISCUSSION
In this study, we describe a new strategy, based on MALDI-TOF MS, for the rapid identification of C. difficile genotype ST37. A set of particular peaks (m/z 3,242 and 3,286) appeared to be somewhat specific to ST37 C. difficile. Using these two peaks, the ST37 genotype can be quickly identified and differentiated from non-ST37 genotypes. In comparison to MLST technology, the method for identification of ST37 C. difficile based on MALDI-TOF MS is rapid, accurate, and cost-effective.
The peak combination of m/z 3,242 and 3,286 as a biomarker for ST37 strains has high specificity and sensitivity. Two widely used MALDI-TOF MS systems in clinical microbiology laboratories were included in this study; both produced satisfactory results for identification of the C. difficile ST37 genotype. Among the 204 clinical isolates studied, the two-peak combination was detected in one ST3 isolate, as well as in one ST81 isolate. It is worth noting that both ST81 and ST37 belong to clade 4, so the two genotypes may not be distinguishable because of their highly similar protein spectra. According to MLST analysis, there is only one allele variation between ST37 and ST81. Moreover, our previous data showed that the ribotype of the ST81 isolate was 017. Among the 31 ST37 isolates tested by PCR-ribotyping, all but one (CD044) were ribotype 017 (data not shown). It was reported in a general teaching hospital in Shanghai that, like ST37, the ST81 genotype had a much higher resistance rate to clindamycin and moxifloxacin (29). According to a recent report from China (30), non-ST37 isolates belonging to clade 4 are also seen in a certain proportion in the population of persons in China with C. difficile infection. Larger studies in China are being planned in order to include more non-ST37 isolates belonging to clade 4.
MALDI-TOF MS-based typing techniques have been used for the typing of many bacterial species, such as Escherichia coli (25, 31, 32), Staphylococcus aureus (33, 34), Streptococcus agalactiae (35), Streptococcus pneumoniae (36), Salmonella enterica (37), and Acinetobacter baumannii (38). Reil et al. successfully identified the C. difficile strains RT001, 027, and 078/126 using an extended MALDI-TOF MS system in 2011, while the RT017 strain, due to the low number of isolates, could not be identified (26). In this study, C. difficile genotype ST37 was successfully identified with a combination of two peaks (m/z 3,242 and 3,286). ST37 is one of the most common sequence types of C. difficile found in China that has a significantly different resistance pattern than others. The MALDI-TOF MS-based rapid identification method for C. difficile ST37 is likely to have an important impact in clinical practice (6, 11, 12).
In summary, C. difficile genotype ST37 possesses several unique genotypic and phenotypic characteristics, including increased antibiotic resistance and A−B+ phenotype for the tcdA and tcdB genes; this strain currently circulates as a dominant strain in mainland China (8–10). A pilot proteomic analysis indicated that the presences of m/z 3,242 and 3,286 peaks cannot be explained by common postgenomic modifications, such as methylation. The two peaks, especially the m/z 3,242 peak, exist at a low level at the current detection sensitivity in some non-ST37 isolates. Additional studies are being done to characterize these two molecules with peaks at m/z 3,242 and 3,286 that appear to be specific for C. difficile genotype ST37.
ACKNOWLEDGMENTS
We thank medical technologists working in clinical laboratories in hospitals and centers for disease control and prevention in Hebei and Zhejiang Provinces and New York State for technical assistance.
This study was supported in part by a Natural Science Fund of Hebei Province grant (2013206450), a research contract between the Memorial Sloan Kettering Cancer Center and bioMérieux, Inc. (SK2012-0595), and an NIH/NCI P30 Cancer Center Support Grant (CA008748).
D.H.P. and M.W. are employees of bioMérieux, Inc., and bioMérieux SA, respectively. We declare that there are no conflicts of interest for the other authors.
REFERENCES
- 1.Gerding DN. 2010. Global epidemiology of Clostridium difficile infection in 2010. Infect Control Hosp Epidemiol 31(Suppl 1):S32–S34. doi: 10.1086/655998. [DOI] [PubMed] [Google Scholar]
- 2.Valiente E, Cairns MD, Wren BW. 2014. The Clostridium difficile PCR ribotype 027 lineage: a pathogen on the move. Clin Microbiol Infect 20:396–404. doi: 10.1111/1469-0691.12619. [DOI] [PubMed] [Google Scholar]
- 3.Rupnik M, Wilcox MH, Gerding DN. 2009. Clostridium difficile infection: new developments in epidemiology and pathogenesis. Nat Rev Microbiol 7:526–536. doi: 10.1038/nrmicro2164. [DOI] [PubMed] [Google Scholar]
- 4.Geric B, Rupnik M, Gerding DN, Grabnar M, Johnson S. 2004. Distribution of Clostridium difficile variant toxinotypes and strains with binary toxin genes among clinical isolates in an American hospital. J Med Microbiol 53:887–894. doi: 10.1099/jmm.0.45610-0. [DOI] [PubMed] [Google Scholar]
- 5.Kuehne SA, Cartman ST, Heap JT, Kelly ML, Cockayne A, Minton NP. 2010. The role of toxin A and toxin B in Clostridium difficile infection. Nature 467:711–713. doi: 10.1038/nature09397. [DOI] [PubMed] [Google Scholar]
- 6.Collins DA, Hawkey PM, Riley TV. 2013. Epidemiology of Clostridium difficile infection in Asia. Antimicrob Resist Infect Control 2:21. doi: 10.1186/2047-2994-2-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao Q, Wu S, Huang H, Ni Y, Chen Y, Hu Y, Yu Y. 2016. Toxin profiles, PCR ribotypes and resistance patterns of Clostridium difficile: a multicentre study in China, 2012–2013. Int J Antimicrob Agents 48:736–739. doi: 10.1016/j.ijantimicag.2016.09.009. [DOI] [PubMed] [Google Scholar]
- 8.Tian TT, Zhao JH, Yang J, Qiang CX, Li ZR, Chen J, Xu KY, Ciu QQ, Li RX. 2016. Molecular characterization of Clostridium difficile isolates from human subjects and the environment. PLoS One 11:e0151964. doi: 10.1371/journal.pone.0151964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gu SL, Chen YB, Lv T, Zhang XW, Wei ZQ, Shen P, Li LJ. 2015. Risk factors, outcomes and epidemiology associated with Clostridium difficile infection in patients with haematological malignancies in a tertiary care hospital in China. J Med Microbiol 64:209–216. doi: 10.1099/jmm.0.000028. [DOI] [PubMed] [Google Scholar]
- 10.Jin D, Luo Y, Huang C, Cai J, Ye J, Zheng Y, Wang L, Zhao P, Liu A, Fang W, Wang X, Xia S, Jiang J, Tang YW. 2017. Molecular epidemiology of Clostridium difficile infection in hospitalized patients in eastern China. J Clin Microbiol 55:801–810. doi: 10.1128/JCM.01898-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim J, Kang JO, Pai H, Choi TY. 2012. Association between PCR ribotypes and antimicrobial susceptibility among Clostridium difficile isolates from healthcare-associated infections in South Korea. Int J Antimicrob Agents 40:24–29. doi: 10.1016/j.ijantimicag.2012.03.015. [DOI] [PubMed] [Google Scholar]
- 12.King AM, Mackin KE, Lyras D. 2015. Emergence of toxin A-negative, toxin B-positive Clostridium difficile strains: epidemiological and clinical considerations. Future Microbiol 10:1–4. doi: 10.2217/fmb.14.115. [DOI] [PubMed] [Google Scholar]
- 13.Cairns MD, Preston MD, Lawley TD, Clark TG, Stabler RA, Wren BW. 2015. Genomic epidemiology of a protracted hospital outbreak caused by a toxin A-negative Clostridium difficile sublineage PCR ribotype 017 strain in London, England. J Clin Microbiol 53:3141–3147. doi: 10.1128/JCM.00648-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shin BM, Kuak EY, Yoo SJ, Shin WC, Yoo HM. 2008. Emerging toxin A−B+ variant strain of Clostridium difficile responsible for pseudomembranous colitis at a tertiary care hospital in Korea. Diagn Microbiol Infect Dis 60:333–337. doi: 10.1016/j.diagmicrobio.2007.10.022. [DOI] [PubMed] [Google Scholar]
- 15.Arvand M, Hauri AM, Zaiss NH, Witte W, Bettge-Weller G. 2009. Clostridium difficile ribotypes 001, 017, and 027 are associated with lethal C. difficile infection in Hesse, Germany. Euro Surveill 14(45):pii=19403. doi: 10.2807/ese.14.45.19403-en. [DOI] [PubMed] [Google Scholar]
- 16.Griffiths D, Fawley W, Kachrimanidou M, Bowden R, Crook DW, Fung R, Golubchik T, Harding RM, Jeffery KJ, Jolley KA, Kirton R, Peto TE, Rees G, Stoesser N, Vaughan A, Walker AS, Young BC, Wilcox M, Dingle KE. 2010. Multilocus sequence typing of Clostridium difficile. J Clin Microbiol 48:770–778. doi: 10.1128/JCM.01796-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dierig A, Frei R, Egli A. 2015. The fast route to microbe identification: matrix assisted laser desorption/ionization-time of flight mass spectrometry (MALDI-TOF MS). Pediatr Infect Dis J 34:97–99. doi: 10.1097/INF.0000000000000601. [DOI] [PubMed] [Google Scholar]
- 18.van Veen SQ, Claas EC, Kuijper EJ. 2010. High-throughput identification of bacteria and yeast by matrix-assisted laser desorption ionization-time of flight mass spectrometry in conventional medical microbiology laboratories. J Clin Microbiol 48:900–907. doi: 10.1128/JCM.02071-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nomura F. 2015. Proteome-based bacterial identification using matrix-assisted laser desorption ionization-time of flight mass spectrometry (MALDI-TOF MS): A revolutionary shift in clinical diagnostic microbiology. Biochim Biophys Acta 1854:528–537. doi: 10.1016/j.bbapap.2014.10.022. [DOI] [PubMed] [Google Scholar]
- 20.Sauget M, Valot B, Bertrand X, Hocquet D. 2017. Can MALDI-TOF mass spectrometry reasonably type bacteria? Trends Microbiol 25:447–455. doi: 10.1016/j.tim.2016.12.006. [DOI] [PubMed] [Google Scholar]
- 21.van Belkum A, Welker M, Pincus D, Charrier JP, Girard V. 2017. Matrix-assisted laser desorption ionization time-of-flight mass spectrometry in clinical microbiology: what are the current issues? Ann Lab Med 37:475–483. doi: 10.3343/alm.2017.37.6.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Steensels D, Verhaegen J, Lagrou K. 2011. Matrix-assisted laser desorption ionization–time of flight mass spectrometry for the identification of bacteria and yeasts in a clinical microbiological laboratory: a review. Acta Clin Belg 66:267–273. [DOI] [PubMed] [Google Scholar]
- 23.McMullen AR, Wallace MA, Pincus DH, Wilkey K, Burnham CA. 2016. Evaluation of the Vitek MS matrix-assisted laser desorption ionization–time of flight mass spectrometry system for identification of clinically relevant filamentous fungi. J Clin Microbiol 54:2068–2073. doi: 10.1128/JCM.00825-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cabrolier N, Sauget M, Bertrand X, Hocquet D. 2015. Matrix-assisted laser desorption ionization–time of flight mass spectrometry identifies Pseudomonas aeruginosa high-risk clones. J Clin Microbiol 53:1395–1398. doi: 10.1128/JCM.00210-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clark CG, Kruczkiewicz P, Guan C, McCorrister SJ, Chong P, Wylie J, van Caeseele P, Tabor HA, Snarr P, Gilmour MW, Taboada EN, Westmacott GR. 2013. Evaluation of MALDI-TOF mass spectroscopy methods for determination of Escherichia coli pathotypes. J Microbiol Methods 94:180–191. doi: 10.1016/j.mimet.2013.06.020. [DOI] [PubMed] [Google Scholar]
- 26.Reil M, Erhard M, Kuijper EJ, Kist M, Zaiss H, Witte W, Gruber H, Borgmann S. 2011. Recognition of Clostridium difficile PCR-ribotypes 001, 027 and 126/078 using an extended MALDI-TOF MS system. Eur J Clin Microbiol Infect Dis 30:1431–1436. doi: 10.1007/s10096-011-1238-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang B, Jin D, Zhang J, Sun JY, Wang X, Stiles J, Xu X, Kamboj M, Babady NE, Tang YW. 2014. Real-time cellular analysis coupled with a specimen enrichment accurately detects and quantifies Clostridium difficile toxins in stool. J Clin Microbiol 52:1105–1111. doi: 10.1128/JCM.02601-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lemee L, Dhalluin A, Testelin S, Mattrat MA, Maillard K, Lemeland JF, Pons JL. 2004. Multiplex PCR targeting tpi (triose phosphate isomerase), tcdA (toxin A), and tcdB (toxin B) genes for toxigenic culture of Clostridium difficile. J Clin Microbiol 42:5710–5714. doi: 10.1128/JCM.42.12.5710-5714.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qin J, Dai Y, Ma X, Wang Y, Gao Q, Lu H, Li T, Meng H, Liu Q, Li M. 2017. Nosocomial transmission of Clostridium difficile genotype ST81 in a general teaching hospital in China traced by whole genome sequencing. Sci Rep 7:9627. doi: 10.1038/s41598-017-09878-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheng JW, Xiao M, Kudinha T, Kong F, Xu ZP, Sun LY, Zhang L, Fan X, Xie XL, Xu YC. 2016. Molecular epidemiology and antimicrobial susceptibility of Clostridium difficile isolates from a university teaching hospital in China. Front Microbiol 7:1621. doi: 10.3389/fmicb.2016.01621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sauget M, Nicolas-Chanoine MH, Cabrolier N, Bertrand X, Hocquet D. 2014. Matrix-assisted laser desorption ionization–time of flight mass spectrometry assigns Escherichia coli to the phylogroups A, B1, B2 and D. Int J Med Microbiol 304:977–983. doi: 10.1016/j.ijmm.2014.06.004. [DOI] [PubMed] [Google Scholar]
- 32.Nakamura A, Komatsu M, Kondo A, Ohno Y, Kohno H, Nakamura F, Matsuo S, Ohnuma K, Hatano N, Kawano S. 2015. Rapid detection of B2-ST131 clonal group of extended-spectrum beta-lactamase-producing Escherichia coli by matrix-assisted laser desorption ionization–time-of-flight mass spectrometry: discovery of a peculiar amino acid substitution in B2-ST131 clonal group. Diagn Microbiol Infect Dis 83:237–244. doi: 10.1016/j.diagmicrobio.2015.06.024. [DOI] [PubMed] [Google Scholar]
- 33.Lu JJ, Tsai FJ, Ho CM, Liu YC, Chen CJ. 2012. Peptide biomarker discovery for identification of methicillin-resistant and vancomycin-intermediate Staphylococcus aureus strains by MALDI-TOF. Anal Chem 84:5685–5692. doi: 10.1021/ac300855z. [DOI] [PubMed] [Google Scholar]
- 34.Mather CA, Werth BJ, Sivagnanam S, SenGupta DJ, Butler-Wu SM. 2016. Rapid detection of vancomycin-intermediate Staphylococcus aureus by matrix-assisted laser desorption ionization–time of flight mass spectrometry. J Clin Microbiol 54:883–890. doi: 10.1128/JCM.02428-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lartigue MF, Kostrzewa M, Salloum M, Haguenoer E, Hery-Arnaud G, Domelier AS, Stumpf S, Quentin R. 2011. Rapid detection of “highly virulent” group B Streptococcus ST-17 and emerging ST-1 clones by MALDI-TOF mass spectrometry. J Microbiol Methods 86:262–265. doi: 10.1016/j.mimet.2011.05.017. [DOI] [PubMed] [Google Scholar]
- 36.Williamson YM, Moura H, Woolfitt AR, Pirkle JL, Barr JR, Carvalho MG, Ades EP, Carlone GM, Sampson JS. 2008. Differentiation of Streptococcus pneumoniae conjunctivitis outbreak isolates by matrix-assisted laser desorption ionization–time of flight mass spectrometry. Appl Environ Microbiol 74:5891–5897. doi: 10.1128/AEM.00791-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kuhns M, Zautner AE, Rabsch W, Zimmermann O, Weig M, Bader O, Gross U. 2012. Rapid discrimination of Salmonella enterica serovar Typhi from other serovars by MALDI-TOF mass spectrometry. PLoS One 7:e40004. doi: 10.1371/journal.pone.0040004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mencacci A, Monari C, Leli C, Merlini L, De Carolis E, Vella A, Cacioni M, Buzi S, Nardelli E, Bistoni F, Sanguinetti M, Vecchiarelli A. 2013. Typing of nosocomial outbreaks of Acinetobacter baumannii by use of matrix-assisted laser desorption ionization–time of flight mass spectrometry. J Clin Microbiol 51:603–606. doi: 10.1128/JCM.01811-12. [DOI] [PMC free article] [PubMed] [Google Scholar]