

Abstract
Acquired resistance to cetuximab, an antibody that targets the epidermal growth factor receptor (EGFR), impacts clinical benefit in head and neck, and colorectal cancers (CRC). One of the mechanisms of resistance to cetuximab is the acquisition of mutations that map to the cetuximab epitope on EGFR and prevent drug binding. We find that necitumumab, another FDA approved EGFR antibody, can bind to EGFR that harbors the most common cetuximab resistance substitution, S468R (or S492R, depending on the amino acid numbering system). We determined an X-ray crystal structure to 2.8 Å resolution of the necitumumab Fab bound to an S468R variant of EGFR domain III. The arginine is accommodated in a large, pre-existing cavity in the necitumumab paratope. We predict that this paratope shape will be permissive to other epitope substitutions, and show that necitumumab binds to most cetuximab- and panitumumab-resistance EGFR variants. We find that a simple computational approach can predict with high success which EGFR epitope substitutions abrogate antibody binding. This computational method will be valuable to determine whether necitumumab will bind to EGFR as new epitope resistance variants are identified. This method could also be useful for rapid evaluation of the effect on binding of alterations in other antibody/antigen interfaces. Together these data suggest that necitumumab may be active in patients who are resistant to cetuximab or panitumumab through EGFR epitope mutation. Further, our analysis leads us to speculate that antibodies with large paratope cavities may be less susceptible to resistance due to mutations mapping to the antigen epitope.
Keywords: Antibody drug, acquired resistance, X-ray crystallography, EGFR, computational predictions
INTRODUCTION
The Epidermal Growth Factor Receptor (EGFR), one of the most intensely studied receptor tyrosine kinases (RTKs), has been implicated in many human cancers (1), and is the target of successful tyrosine kinase inhibitors (TKIs) and antibody based drugs (2, 3). Three monoclonal antibodies that bind the extracellular region of EGFR and inhibit receptor activation have been FDA approved: cetuximab that is in current use to treat colorectal and head and neck cancers (4, 5); panitumumab that is also in use to treat CRC (6); and necitumumab that recently gained approval for use in squamous non-small cell lung cancer following demonstration of overall survival benefit in this setting (7). These agents, alone or in combination with chemotherapy or radiation, lead to responses in a number of cancers (7-10). As is observed with many targeted therapies, acquired resistance to EGFR antibodies often develops and impedes the clinical benefit of these drugs (1, 11).
Resistance to targeted therapies can occur by the acquisition of mutations that create a variant of the target that abolishes drug binding (12). This mechanism is particularly common in cancer patients treated with TKIs, and more recently, acquired mutations that affect binding of therapeutic antibodies to EGFR were identified in colorectal tumors (13-17) (Supplementary Table S1). A frequent, and well-studied, case of resistance to EGFR TKIs in lung cancer is acquisition of a secondary mutation in the EGFR kinase domain, the “gatekeeper” mutation T790M (or T766M with amino acid numbering starting at the beginning of the mature polypeptide, which is used in this manuscript) (18). An appreciation for the molecular mechanism of T790M acquired resistance (19) led to development of third generation inhibitors that can circumvent this resistance mechanism (20). In this study, we investigate the molecular basis for cetuximab resistance due to EGFR mutation, and show that the EGFR antibody drug necitumumab has the potential to overcome or circumvent this type of resistance.
A cetuximab-resistant EGFR was first observed in CRC cell lines: the substitution of arginine at S468 (S492 when numbered from beginning of the signal peptide; S468/492R) (13). This same mutation was subsequently observed in tumors of CRC patients whose disease had progressed following cetuximab treatment (13, 14). The incidences of S468/492R substitution is considerable in CRC patients who received cetuximab. A retrospective analysis of plasma samples from a phase III trial (ASPECCT) comparing cetuximab and panitumumab in a large number of CRC patients reported somatic S468/492R mutation in 16 % of cases in the cetuximab arm compared to only 1 % of patients in the panitumumab arm (8, 21). These data are consistent with the observation that panitumumab can bind to, and inhibit S468/492R EGFR (13). Additional somatic mutations that map to the cetuximab epitope on EGFR have been seen in cetuximab-resistant CRC tumors: G441/465R, G441/465E, K443/467T (14, 16), or identified in CRC cell lines: S440/464L and I467/491M (14). Substitutions at G441/465 and S440/464 have also been seen in panitumumab-resistant CRC tumors (15, 17). In contrast, the K443/467T variant that binds to panitumumab has not been observed following treatment with this drug (14). Loss of effectiveness of therapeutic EGFR antibodies through epitope mutations constitutes an important resistance mechanism in CRC patients.
In this study, we evaluate the potential of necitumumab as an inhibitor of cetuximab- and/or panitumumab-resistant EGFR. We previously reported the X-ray crystal structure of the necitumumab Fab fragment bound to EGFR (22). The necitumumab epitope on EGFR is extremely similar to that of cetuximab, although the paratopes are quite different. Here we show that necitumumab binding is essentially unaffected by the S468R substitution that completely blocks cetuximab binding. We solve the X-ray crystal structure to 2.8 Å resolution of the necitumumab Fab (Fab11F8) in complex with isolated domain III from EGFR (sEGFRd3) with S468R substitution (sEGFRd3-S468R). The R468 side chain is clearly resolved, and is accommodated in a cavity in the necitumumab paratope that lies between the heavy and light chains. Interestingly, a similar structural explanation for binding of panitumumab to EGFR-S468R has been reported (23). The necitumumab paratope cavity is significantly larger than that of panitumumab, suggesting that many epitope substitutions in EGFR may be accommodated without loss of binding to necitumumab. We use computational methods to assess whether necitumumab can bind to cetuximab- and panitumumab-resistant EGFR variants, and verify the computational results with experimental binding data. We find that necitumumab can bind to most cetuximab- and panitumumab-resistant EGFR variants, and that a simple computational approach can predict whether or not an epitope substitution will abolish antibody binding. Our findings suggest that necitumumab, which was recently approved to treat squamous non-small cell lung carcinoma, may be effective in some cetuximab-resistant CRC cases.
METHODS
Stable cell lines
HIV-1-based lentiviral expression vectors (pLVX-IRES-puro/neo; Clontech) containing full length EGFR and EGFR-S468R cDNA were produced by GeneWiz (South Plainfield, NJ) and used to generate vectors to express C-terminal enhanced green fluorescent protein (eGFP) fusion of EGFR and EGFR-S468R. These plasmids were transfected into Lenti-X 293T cells with lentiviral packaging plasmids and polyethylenimine linear. Recombinant lentiviruses were collected 48 hours after transfection and used to transduce HELA (ATCC® CCL-2™, obtained in 2010) and LK2 cells (HSRRB, Japan: lot#040196, obtained in 2011). Stably transduced cells were selected with puromycin (0.5 µg/ml) or neomycin (500 µg/ml) for 96 hours. LK2 cells tested negative for mycoplasma with the MycoAlert kit from Lonza. HELA cells were not tested for mycoplasma. The length of time between cell line thawing and use in experiment did not exceed 4 weeks (two or more passages).
Antibody studies with stable cell lines
Cetuximab, necitumumab, and control human IgG were supplied by Eli Lilly and Company. HELA cells expressing EGFR-eGFP or EGFR-S468R-eGFP were fixed (4% paraformaldehyde in PBS, 20 minutes) and stained with IgG-alexa647, cetuximab-alexa647 or necitumumab-alexa647 (10µg/ml, 3 hours). Images were acquired on a Nikon-A1 confocal microscope with 20X air objective (Nikon 1.2NA) in channel mode to eliminate cross-talk using 4X line averaging. Images were analyzed using Nikon Elements and equivalent level adjustments applied across all images. For flow cytometry, suspensions of blocked HELA-EGFR-eGFP and HELA-EGFR-S468R-eGFP cells were incubated with primary antibody (nine 3-fold serial dilutions from 66.67nM) then alexa647-conjugated anti-human secondary antibody (Jackson Labs: 109-05-003). Data were acquired on the IntelliCyt iQue Flow Cytometer and analyzed in FlowJo and GraphPad Prism. Antibody-induced EGFR-eGFP degradation was assessed by flow cytometry following 24-hour incubation of HELA-EGFR-eGFP and HELA-EGFR-S468R-eGFP cells with cetuximab, necitumumab, or control IgG. Median fluorescence intensity (MFI) values were normalized to untreated controls. For receptor activation assays, subconfluent LK-EGFR and LK-EGFR-S468R were serum starved (4 hours) in the presence of 1000 nM cetuximab, necitumumab, or control IgG. Cells were stimulated for 10 minutes with epiregulin (EREG) or transforming growth factor α (TGFα), at 100 and 10 nM respectively (Peprotech). Cell lysates were subjected to Western blot analysis as described (24) using mouse Akt mAb (#2920), rabbit mAb to phosphorylated S473 of Akt (pAktS473; #4060), rabbit EGFR mAb (#4267), and rabbit mAb to phosphorylated Y1068 of EGFR (pEGFRY1068; #3777; all Cell Signaling Technology). IRDye 680 conjugated goat anti-mouse (#926-68070) and IRDye 800 conjugated goat anti-rabbit (#926-32211) (LI-COR Biosciences) were used as secondary antibodies. Signal was detected by Odyssey Infrared Imaging System (LI-COR).
Generation of sEGFR proteins and Fab fragments
The sEGFR (amino acids [aa] 1-618 of mature EGFR), and isolated EGFR domain III (sEGFRd3; aa 1–4 followed by 311–514) were produced in Sf9 cells and purified as described (22, 25). Cetuximab resistance variants were created using PCR methods and produced as for wild type proteins. FabC225 was provided by Eli Lilly and Company. Fab11F8 was generated by papain digestion of mAb using a Pierce Fab Preparation Kit and purified on a Superose 12 column (GE Healthcare) in 25 mM HEPES, 100 mM NaCl, pH 7.5 (SEC buffer).
Surface Plasmon Resonance (SPR) binding studies
SPR experiments were carried out as previously described (22, 25). Data for wild type sEGFR (or sEGFRd3) were fit to a simple one-site Langmuir binding model using Prism 6 (GraphPad Software, Inc.). Bmax values from these fits were used to normalize the equilibrium SPR responses for sEGFR variants and these fraction maximal SPR values fit with Bmax fixed to 1.0. The mean KD value was determined from the fits to at least three independent, normalized binding curves (Supplementary Table S2).
Crystallization, data collection and structure determination
A 1:2 molar ratio of sEGFRd3-S468R and Fab11F8 was purified on Superose 6 column (GE Healthcare) in SEC buffer. Fractions containing sEGFRd3-S468R/Fab11F8 complex were concentrated to 5-8 mg/ml for crystallization using the hanging drop vapor diffusion method at 20°C. Needle-like crystals were obtained from drops comprising 0.5 µl complex plus 0.5 µl reservoir solution (50 mM sodium acetate, 250 mM ammonium sulfate, 15-20% PEG3350, pH 5.0). Streak seeding was used to obtain large hexagonal plate crystals in drops containing 0.5 µl complex (6 mg/ml) and 0.5 µl reservoir solution (50 mM sodium acetate, 200 mM potassium citrate, 15-20% PEG3350, pH 6-6.3). Crystals were flash frozen in liquid nitrogen following brief exposure to reservoir solution supplemented with 12 % ethylene glycol. Data were collected at the GM/CA @ APS beamline 23-ID-D using a Pilatus 6M detector and were processed in HKL2000. Data collection statistics are summarized in Supplementary Table S3.
The sEGFRd3-S468R/Fab11F8 structure was solved by the method of molecular replacement (MR) using the program PHASER (26). Domain III and Fab11F8 from PDB ID 3B2U were used as independent search models. Model was rebuilt in COOT (27) and refined in PHENIX (28). Coordinates for the sEGFRd3-S468R/Fab11F8 have been deposited to the RCSB Protein Data Bank (6B3S).
Computational Methods
The “Calculate Mutation Binding Energy” tool within the “Design Protein” module of the Discovery Studio Client 4.1 (DSC4.1) (29) was used to estimate the change in binding free energy (ΔΔG) for binding of Fabs to wild type and mutated sEGFR. Three terms, weighted empirically, are used to compute binding free energy: a Van der Waals term, an electrostatic term, and an entropy term that accounts for side chain flexibility (30, 31). This protocol uses the CHARMM36 force field with a pair-wise Generalized Born approximation (CHARMM GBIM) (32). Altered side chains were modeled in DSC4.1 using the wild-type EGFR/Fab complexes for cetuximab (PDB:1YY9), necitumumab (PDB: 3B2V) and panitumumab (PDB: 5SX4). The calculated energy term for the wild type protein was subtracted from value for the EGFR variant to evaluate ΔΔG, ΔΔS, ΔΔEelec, and ΔΔEVdW, (Table 1 and Supplementary Table S4).
Table 1.
Changes in experimental binding and DSC4.1 computed electrostatic energy term (ΔΔEelec) for interaction of cetuximab (C225), necitumumab (11F8) and panitumumab (pani) with wild type and epitope mutated EGFR variants.
| Mutationa | KDmut/KDWT b | Experimental Bindingc | ΔΔEelec | ||||||
|---|---|---|---|---|---|---|---|---|---|
| C225 | 11F8 | pani | C225 | 11F8 | pani | C225 | 11F8 | pani | |
| S440L | >1000 | 8.8 ± 1.2 | NB | ⊗ | ● | ⊗ | 0.58 | 0.03 | 0.12 |
| G441E | NB | NB | NB | ⊗ | ⊗ | ⊗ | 4.58 | 3.80 | 0.73 |
| G441R | NB | NB | NB | ⊗ | ⊗ | ⊗ | 1.30 | −0.06 | 0.00 |
| K443T | 120 ± 20 | 2.3 ± 0.1 | ✓ | ⊗ | ● | ● | 0.80 | −0.06 | 0.21 |
| I467M | 46 ± 4 | 3.9 ± 0.4 | NB | ⊗ | ● | ⊗ | 0.09 | −0.13 | 0.08 |
| S468R | >1000 | 2.0 ± 0.2 | 3.4 | ⊗ | ● | ● | 1.64 | −0.51 | −0.08 |
| Q384A | 2.1 ± 1.1 | 13 ± 1 | – | ● | ⊗ | – | −0.04 | 0.08 | – |
| K443A | 1.3 ± 0.4 | 0.9 ± 0.1 | − | ● | ● | − | 0.67 | −0.38 | – |
| S468I | 0.7 ± 0.1 | 0.27 ± 0.03 | – | ● | ● | – | 0.28 | −0.38 | – |
| N473A | 0.9 ± 0.1 | 0.6 ± 0.1 | – | ● | ● | – | −0.02 | −0.20 | – |
|
| |||||||||
| TPd | 2 | 6 | 1 | ||||||
| TN | 6 | 2 | 4 | ||||||
| FP | 0 | 1 | 0 | ||||||
| FN | 2 | 1 | 1 | ||||||
| Accuracy | 81 % | ||||||||
| Precision | 90 % | ||||||||
The first 6 variants (bold) are associated with cetuximab and/or panitumumab resistance (Supplementary Table S1), lower 4 mutations from Li et al (22).
KD values for cetuximab (C225) and necitumumab (11F8) are from Supplementary Table S2 (S440L, K443T, I467M, S468R) and from (22) (Q384A, K443A, S468I, N472A). Data for panitumumab (pani) for S468R is from (23). NB – no binding from (14, 17) and Octet data. ✓ indicates binding but no affinity reported (14).
⊗ No binding (KD value more than 10-fold greater than wild type).
● WT binding (KD value within 10-fold of value for wild type).
For counting of true vs false predictions, an EGFR variant is considered an experimental “binder” if the KD value is less than 10-fold greater than the KD value for wild type (●). The computational filter is set at ΔΔE < 0. True positives (TP, binder (●) with negative computed energy) and true negatives (TN, non-binders (⊗) with positive or zero computed energy term) are highlighted in bold. Accuracy is defined as (TP + TN)/(Total number of variants) and precision is defined as TP/(TP + FP).
For molecular dynamics (MD) simulations, PDB IDs 1YY9 and 3B2V (glycans removed) where used as initial models and amino acids substitutions introduced using Molecular Operating Environment (MOE) (33). Models were transferred to Schrodinger Maestro 2015.4 (34) for protein preparation and Desmond simulation setup with OPLS3 force field and TIP3P water. Solvated complexes were parameterized with CHARMM27 force field using Desmond’s vippar protocol. NPT Molecular Dynamics used standard Desmond simulation protocol with Langevin thermostat and Particle Mesh Ewald methods. Desmond GPU simulations (200ns) were run in triplicates for Fab complexes with wild type and mutated sEGFRd3. Simulations were analyzed with VMD (35). Domain III main chain atoms were used for superposition prior to RMSD computations. RMSD computations were carried out for all CDR main chain atoms and for only the VH and VL CDR3 main chain atoms. Resulting data was analyzed and displayed with Tibco Spotfire.
RESULTS
Necitumumab binds to and inhibits the most common mutated form of EGFR (S468R) that is resistant to cetuximab
We first investigated whether necitumumab binds to cell-surface EGFR with an S468R substitution (EGFR-S468R). HELA cells expressing EGFR-eGFP or EGFR-S468R-eGFP were fixed and stained with either cetuximab or necitumumab bearing an alexa647 conjugate (Fig. 1A). Cetuximab binds robustly to the surface of cells expressing wild type EGFR but not to the cells expressing EGFR-S468R, as expected (13). Necitumumab shows strong surface staining on both EGFR-S468R-eGFP and EGFR-eGFP cells, indicating that necitumumab can bind to EGFR-S468R. Flow cytometry was used to determine median fluorescence intensity (MFI) over a range of antibody concentration (Fig. 1B). Cetuximab and necitumumab bind to cells expressing wild type EGFR with apparent KD values in the subnanomolar range. Cetuximab binding to cells expressing the S468R variant is not detectable, whereas necitumumab retains subnanomolar range binding to these cells.
Figure 1. Necitumumab binds to and inhibits EGFR harboring the S468R cetuximab resistance mutation.

A, confocal imaging of HELA cells expressing EGFR-eGFP and EGFR-S468R-eGFP stained with alexa647 labeled antibodies (left, gray scale). Right panels show an overlay of the alexa647 and GFP fluorescence. B, binding of the same antibodies to EGFR and EGFR-S468R expressing HELA cells analyzed by flow cytometry. The median fluorescence intensity (MFI) values are plotted over a concentration range from 0.01 to 67 nM of cetuximab (red), necitumumab (blue) or control IgG (black). There is negligible binding of cetuximab or necitumumab to parental HELA cells over this concentration range (Supplementary Fig. S1C). Plots are derived from three technical replicates, and are representative of data from three independent experiments. C, normalized GFP MFI signal for HELA cells expressing EGFR-eGFP or EGFR-S469R-eGFP following 48-hours pre-incubated with the indicated concentrations of cetuximab (red), necitumumab (blue), or IgG (black). MFI values are normalized to the EGFR-GFP signal with no antibody treatment. Plots are from two technical replicates and representative of data from two independent experiments. D, effect of antibody pre-treatment (1,000 nM) on EGFR activation in LK2-EGFR or LK2-EGFR-S468R cells. Cells were stimulated with EREG or TGFα (100 nM and 10 nM, respectively) and immunoblotted for total and phosphorylated EGFR and AKT (pEGFRY1068 and pAktS473).
Cetuximab and necitumumab inhibit EGFR activity through a number of mechanisms including blockade of ligand-induced EGFR activation and induction of receptor internalization and degradation (1). We asked whether necitumumab retains these inhibitory activities against EGFR-S468R. To quantify the fraction of internalized EGFR that is degraded, we used flow cytometry to measure the EGFR-eGFP signal after 48 hours of antibody exposure. For EGFR-eGFP cells, both cetuximab and necitumumab reduce the eGFP signal in a dose dependent manner, with EC50 of 3.6 and 3.9 nM, respectively (Fig. 1C). For EGFR-S468R-eGFP cells, cetuximab has no effect on eGFP levels, whereas necitumumab induces EGFR-S468R degradation with an EC50 (5.0 nM) that within two-fold that for wild type EGFR. To assess the ability of these antibodies to block EGFR activation and downstream signaling, we generated LK2 squamous cell lung carcinoma cells that stably express intact full-length EGFR (wild type or S468R). These stable LK2 cells show similar antibody binding characteristics as the HELA cells (Supplementary Fig. S1). Robust phosphorylation of EGFR, and activation of Akt, is stimulated in both LK2-EGFR and LK2-EGFR-S468R cells with EREG, a low affinity EGFR ligand, and TGFα, a high affinity EGFR ligand (Fig. 1D). Pretreatment of LK2-EGFR cells with cetuximab or necitumumab reduces EGFR phosphorylation (pEGFR) to basal levels and impairs Akt phosphorylation (pAkt) for both ligands. For the LK2-EGFR-S468R cells, cetuximab fails to inhibit EGFR activation and downstream signaling: both pEGFR and pAkt levels are the same as in the absence of antibody. Necitumumab completely inhibits EREG induced EGFR phosphorylation and significantly reduces TGFα induced pEGFR. Together, these results indicate that the EGFR-S468R substitution has only a minor impact on the ability of necitumumab to bind to and inhibit EGFR, whereas cetuximab is unable to bind to or inhibit EGFR-S468R.
To assess the impact of the S468R substitution on cetuximab and necitumumab binding quantitatively, we turned to SPR/BIAcore analysis using the soluble extracellular region of EGFR (sEGFR) and the antibody Fabs, as we have done previously (22, 25) (Fig. 2 and Supplementary Table S2). Wild type sEGFR binds to immobilized cetuximab Fab (FabC225) and necitumumab Fab (Fab11F8) with KD values of 5.5 ± 0.3 and 6.1 ± 0.6 nM, respectively. Binding of sEGFR-S468R to FabC225 is undetectable in this assay (KD >> than 4µM, the highest concentration of sEGFR tested). In stark contrast, the binding of sEGFR-S468R to Fab11F8 is 12 ± 1 nM, only two-fold weaker than the binding of the wild type protein to this Fab (Supplementary Table S2).
Figure 2. Necitumumab binds with high affinity to sEGFR harboring the cetuximab resistance mutations S468R.
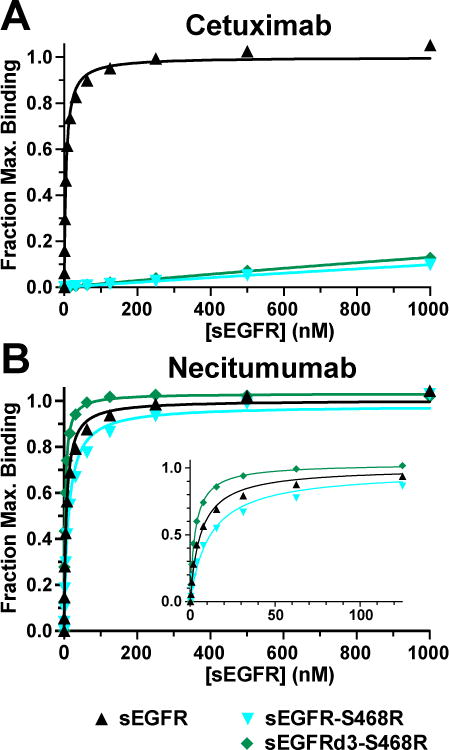
A and B, SPR analysis of sEGFR, sEGFR-S468R and sEGFRd3-S468R binding to immobilized cetuximab (A) and necitumumab (B) Fabs. Normalized equilibrium SPR response plotted as a function of protein concentration were fit to a simple one-site Langmuir binding equation. Data are representative of at least three independent measurements. Mean KD values with standard deviations are reported in Supplementary Table S2.
Structural basis for binding of sEGFR-S468R to necitumumab
To understand at a molecular level how necitumumab is able to bind with high affinity to EGFR that has an arginine at amino acid 468, we determined the X-ray crystal structure of the necitumumab Fab (Fab11F8) bound to isolated domain III of EGFR with an S468R substitution (sEGFRd3-S468R). The structure was solved to 2.8 Å resolution using MR methods, with domain III and the Fab from the sEGFRd3/Fab11F8 complex as search models. Clear electron density could be seen for R468 in initial MR phased maps (Supplementary Fig. S2). Data collection and refinement statistics are shown in Supplementary Table S3.
The overall structure of sEGFRd3-S468R/Fab11F8 is extremely similar to the structure of wildtype sEGFRd3 in complex with this same Fab (RMSD overall of 0.57 Å; Fig. 3A). The R468 side chain lies in a pre-existing cavity between the necitumumab light and heavy chains (Fig 3B and Supplementary Fig. S3A). The arginine side chain makes van der Waals contacts with hydrophobic side chains that line the paratope cavity (Fig. 3B). By contrast, the S468 of wild type EGFR interacts only with the side chains at the mouth of the cavity near CDRL3 L96 (Supplementary Fig. S3B). The R468 guanidinium group is within hydrogen bonding distance of the main chain carbonyl of CDRH3 G100A, and lies in a region of relatively high electronegative potential (Fig. 4A). Water molecules in three of the four complexes in the asymmetric unit, suggest that water mediated interactions also stabilize the arginine in the necitumumab paratope pocket.
Figure 3. Structure of the sEGFRd3-S468R/Fab11F8 complex.
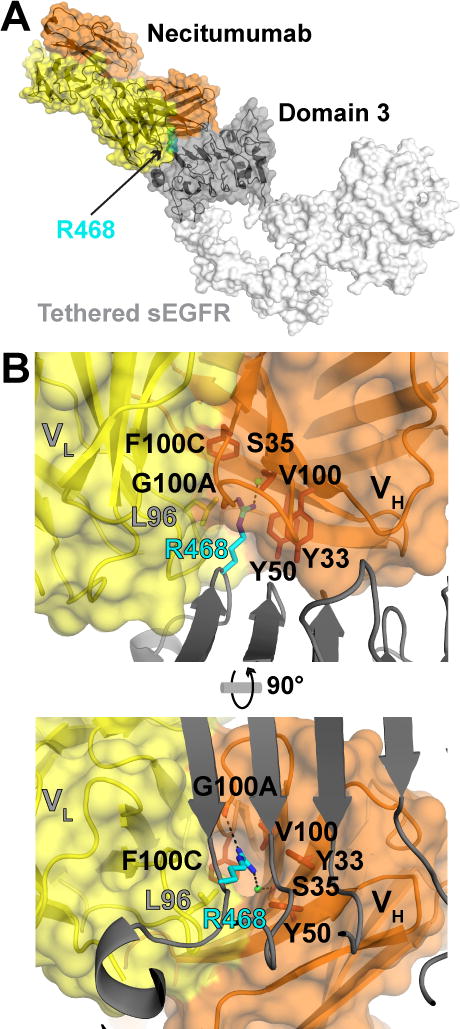
A, a transparent surface representation plus cartoon of the sEGFRd3-S468R/Fab11F8 structure with antibody orange (VH) and yellow (VL) and domain III dark gray. The R468 side chain is in sphere representation colored cyan. Domains I, II and IV that are not present in the structure are in white (from PDB 1YY9). B, orthogonal views of the necitumumab paratope, with VH and VL colored as in A, and domain III in gray cartoon. R468 (cyan) sits in a deep hydrophobic cavity between the VH and VL domains. Amino acids lining the cavity are shown in stick representation. Presumed direct and water mediated hydrogen bonds with R468 are indicated with dashed lines.
Figure 4. Comparison of necitumumab and cetuximab paratopes.

A, surface representations of necitumumab and cetuximab (PDB 1YY9) paratopes in the same orientation as in Fig. 3B (right). The electrostatic potential, calculated using the adaptive Poisson-Boltzmann solver (APBS) (49), from −5 kT (red) to +5 kT (blue) is projected on to the surface. R468 (left) and S468 (right) are in cyan stick representation. B, the paratope surfaces are colored to highlight the position of the CDRs. VL CDRs are yellow, CDR H1 pale orange, CDR H2, orange and CDR H3 brown. Side chains from the CDR H3 of cetuximab that interact with side chains in the VL CDRs are show.
The conformation of the Fab11F8 paratope when bound to sEGFRd3-S468R is very similar to that seen in the wild type complex (Supplementary Fig. S4A), with no significant difference in buried surface area (approximately 915 Å2 on domain III occluded from solvent in both cases) or in the shape complementarity parameter (0.68) (36). The cavity in the sEGFRd3/Fab11F8 complex (Fig. 4B and Supplementary Fig. S3B) is more than large enough (≈ 260 Å3) to accommodate the arginine side chain without any structural rearrangement. The relative orientation of the Fab with respect to domain III varies for the four molecules in the asymmetric unit (Supplementary Fig. S4B). This “wobble” in the docking of Fab to domain III is not seen for the eight copies of sEGFRd3/Fab11F8 in PDB ID 3B2U (Supplementary Fig. S4C). This plasticity in how R468 is accommodated may contribute to the 2-fold reduction in the KD value for binding of Fab11F8 to sEGFR-S468R compared to binding to wild type sEGFR.
Comparison of the binding of cetuximab, necitumumab and panitumumab to EGFR
X-ray crystal structures have been reported for EGFR bound to the Fabs from all three FDA-approved EGFR antibodies; cetuximab, panitumumab and necitumumab (22, 23, 25). The structure of the Fab from panitumumab has also been reported bound to an EGFR S468R variant (23). We asked whether comparison of these structures might shed some light on why CRC patients treated with cetuximab show a higher frequency of epitope resistance mutations compared to those treated with panitumumab (8, 21), and on what frequency of resistance mutations one might expect for necitumumab.
Unlike necitumumab, the cetuximab paratope does not contain a cavity between the heavy and light chains (Fig. 4B). This is due primarily to the arrangement of the VH CDRs, in particular CDRH3 (Fig. 4B). The conformation of the cetuximab CDRH3 places the Y100A side chain between the VL and VH domains creating a relatively flat paratope with no space to accommodate R468. There are several direct side chain interactions between CDRH3 and the CDRs of the VL subunit that would presumably make structural rearrangement energetically unfavorable (Fig. 4B). No polar interactions between the CDRs of the VL and VH subunits are observed in necitumumab, and CDRH3, which has a glycine at position 100A, and lies further from VL, creating the paratope cavity.
The panitumumab paratope is similar to that of necitumumab, with a cavity between the VH and VL domains that can accommodate R468 (23). The position and conformation of the panitumumab CDRH3 are similar to that of necitumumab, with no polar interactions between the VL and VH CDRs (Supplementary Fig. S3C). Importantly, however, the panitumumab paratope cavity is considerably smaller than that observed in necitumumab (≈ 100 Å3 for panitumumab compared to ≈ 260 Å3 for necitumumab; Supplementary Fig. S3A). The depth of the panitumumab cavity is limited by VH D95 side chain, which interacts with R468. Necitumumab has a valine at this position, resulting in a wider and deeper pocket that is incompletely filled by R468 (Supplementary Fig. S3B). These observations led us to ask whether the large necitumumab paratope cavity can accommodate other EGFR variants associated with cetuximab and/or panitumumab resistance.
Antibody binding to other mutated EGFR associated with cetuximab and/or panitumumab resistance
Seven CRC resistance mutations that map to the cetuximab epitope on EGFR have been identified in tumors and cell lines (Supplementary Table S1), and it is probable that additional mutations will emerge. Where CRC patients develop resistance to standard therapy through new epitope mutations, the ability to predict whether necitumumab will retain EGFR binding, and therefore be a candidate for additional therapy, may be a valuable tool for clinicians. Computational approaches have been developed, and validated against biochemical data, to predict the effectiveness of TKIs against activating mutations in anaplastic lymphoma kinase (ALK) in neuroblastoma (37). We sought a similar approach to predict, and validate, the effect of CRC resistance mutations on antibody binding.
(i) The experimental test set
The test set includes antibody resistance variants (S440L, G441E, G441R, K443T, I467M and S468R) and additional epitope variants (Q384A, K443A, S468I and N473A; Table 1 and Supplementary Fig. S5A). S440L, K443T and I467M were generated in sEGFR and their binding evaluated by SPR. All three variants show loss of affinity for cetuximab (Fig. 5B and Supplementary Table S2). The S440L variant shows no detectable binding to FabC225 (KD value >> 4 µM), and the I467M and K443T variants show ~50- and 100-fold weaker KD values, respectively. All three of these variants retain necitumumab binding (Fig. 5A and Supplementary Table S2). The sEGFR-K443T has just 2-fold weaker affinity compared to wild type (KD value of 13 ± 1 nM), similar to sEGFR-S468R, whereas for I467M and S440L binding is 4- and 9-fold weaker (KD values of 24 ± 3 and 54 ± 7 nM), respectively. For these cetuximab resistance sEGFR variants, binding of necitumumab is 10 to > 500-fold stronger than the binding of cetuximab. The G441 variants of sEGFR could not be generated, but these variants could be make in the context of an EGFR-Fc fusion. We used bio-layer interferometry based assay (Octet/FortéBIO) to show that G441E and G441R abolish binding to necitumumab, cetuximab and panitumumab. We did not include R427C due the complication of introducing an unpaired cysteine. Data from our previous study using SPR to measure binding to the necitumumab and cetuximab Fab (22) were added to the test set to include some substitutions that retain cetuximab binding, and additional data for panitumumab were added from published studies (14, 17, 23).
Figure 5. Experimental and computational evaluation of the effect of epitope substitutions on cetuximab and necitumumab binding to EGFR.
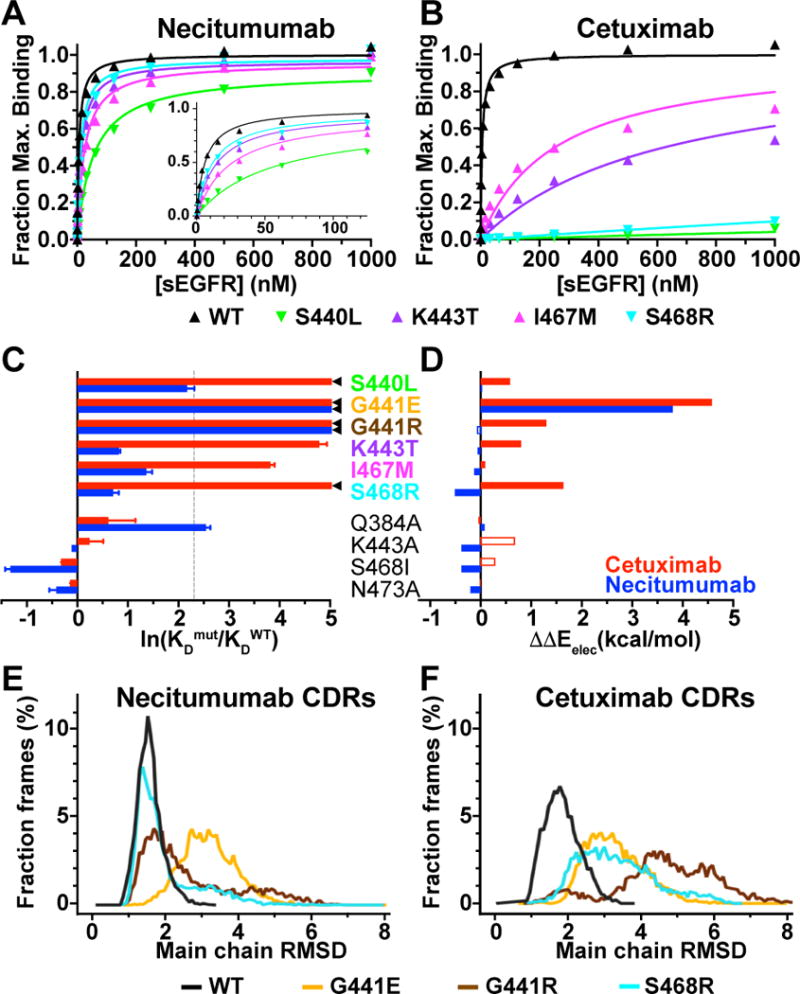
A and B, SPR binding of indicated sEGFR variants to necitumumab (A) and cetuximab (B) Fabs, analyzed as described in the legend to Fig. 2. Mean KD values are in Supplementary Table S2. C, experimental change in binding due to the indicated substitution in sEGFR for cetuximab (red) and necitumumab (blue), plotted as the natural log of the ratio of the KD value for mutated EGFR to the KD value of WT; ln(KDmut/KDWT). KDmut/KDWT values from Table 1. ◄ indicates ln(KDmut/KDWT) >> 5. Dotted line is at KDmut/KDWT = 10. D, computed change in electrostatic binding energy for each mutated EGFR relative to WT; ΔΔEelec = ΔEelecmut - ΔEelecWT colored as in C. Open bars indicated incorrect predictions (see text and Table 1). E, dynamics of necitumumab CDRs during three independent 200 ns MD simulations when bound to WT (black), G441E (yellow), G441R (brown) and S468R (cyan) EGFR. Structures from 6000 frames were aligned to frame 1 using main chain of domain III. Binned (0.07 Å) RMSD values for main chain CDR atoms, expressed as a percentage of the total number of frames, are plotted as a function of RMSD. Mean (median) of the RMSD cumulative distributions are; WT: 1.54 (1.5) Å, S468R: 1.96 (1.64) Å, G441R: 2.64 (2.17) Å, and G441E: 3.19 (3.14) Å. Differences in mean for WT and G441 substitutions are significant (P<0.001). F, the same analysis as in E for the complexes with cetuximab. Mean (median) values; WT: 1.86 (1.81) Å, S468R: 3.33 (3.19) Å, G441R: 4.71 (4.73) Å, and G441E: 3.31 (3.21) Å. Differences for WT is significant (P<0.001).
(ii) Rapid computational evaluation of Binding Energy
The ability of fast computational methods to accurately calculate changes in binding free energies of antibody-antigen complexes was recently surveyed against a database of over 1,000 paratope mutations (38, 39). One of the better performing packages, Discovery Studio (29), performed well for mutations that enhance and that weaken binding. We asked whether this method, which computes three empirically-weighted energy terms, can accurately predict effects of mutations in EGFR on binding to necitumumab and cetuximab. The differences in the total and individual computed energy terms (ΔΔG, ΔΔS, ΔΔEelec, and ΔΔEVdW) for each variant compared to wild type are reported in Supplementary Table S4, alongside the experimental binding observations. The numerical correlation between calculated and experimental changes in binding energy is quite poor (Fig. 5C/D and Supplementary Table S4), which is not unexpected for this type of rapid computational approach (38). There is however predictive power in these data if the EGFR variants are partitioned into two groups. Those that have KD values within 10-fold of wild type are considered binders (●), whereas the rest are classed as non-binders (⊗). This classification can be extended to include effects of the epitope substitutions on binding to panitumumab (Table 1, pani). ΔΔEelec shows substantial predictive power to distinguish between these two groups. All of the cetuximab resistance mutations have positive ΔΔEelec for binding to cetuximab (Fig. 5D and Table 1), so are accurately classified as non-binders. K443T, I467M and S468R have negative ΔΔEelec for necitumumab binding, accurately predicting that these variants bind to necitumumab. For panitumumab, S440L, G441E, G441R and I467M are accurately predicted to be non-binders with positive ΔΔEelec for panitumumab binding. Importantly, ΔΔEelec accurately predicts that panitumumab should bind to the S468R variant (ΔΔEelec = −0.08), consistent with the reported that this variant binds panitumumab with just a 3.4-fold weaker affinity compared to wild type EGFR (23).
S440L is a false negative for necitumumab binding, although the parameters for this variant are very close to the cutoff for both the calculated and experimental data. K443T is also wrongly classified as a non-binder (false negative) for interaction with panitumumab. It should be noted that this is the only case where we assign positive experimental binding based on the binding to cells expressing this EGFR variant (14), rather than using an experimental KD value. G441R is a false positive for necitumumab binding, one of only five examples where ΔΔEelec fails to correctly classify a mutation as binder or non-binder (highlighted with the open bars in Fig. 5D and gray text in Table 1). A possible explanation for this is that the side or main chain conformation for the G441R variant is poorly modelled in DSC4.1. It is of note, however, that for S468R the DSC4.1 modeled side chain conformations are within 1.0 Å (RMSD) of the conformations observed in crystal structures (Supplementary Fig. S5B).
Overall, the ΔΔEelec filter successfully predicts nine true positives and twelve true negatives (Table 1). The ΔΔEelec < 0 filter on this small mutational dataset has 81% accuracy and 90% precision. It is of note that, for the ten cases with the most significant loss of experimental binding (marked with a ◄ in Fig. 5C for necitumumab and cetuximab, and noted NB in Table 1 for panitumumab), nine are accurately predicted to be non-binders with mostly large, positive values for ΔΔEelec.
(iii) Molecular dynamics (MD) simulations
The second computational approach utilizes MD simulation to assess the dynamics of the antibody CDRs when bound to wild type and mutated sEGFRd3. For this more computationally-intense analysis, we chose to focus on the two positions most commonly associated with resistance, S468 and G441. Three, 200 nanosecond simulations were run, and the fluctuations in the necitumumab and cetuximab CDR conformations analyzed for each complex (WT, G441E, G441R and S468R). The cumulative RMSD distribution for all six CDRs of necitumumab and cetuximab are shown in Fig. 5E/F. A low RMSD fluctuation is seen for necitumumab bound to wild type and to S468R EGFR (mean values of 1.54 Å and 1.96 Å respectively), indicating conformational rigidity of the CDRs in these cases. The CDR RMSD distribution for G441E and G441R are significantly broader with mean values of 2.64 and 3.19 Å respectively. The CDR RMSD distribution for the cetuximab/wild type EGFR complex has a mean value of 1.86 Å, consistent with a stable complex. All three cetuximab complexes with EGFR variants have broad RMSD distributions with larger mean values (3.33, 4.71 and 3.31 Å for S468R, G441R and G441E respectively). The mean CDR RMSD is less than 2.0 Å for experimentally stable complexes, whereas it is more than 2.5 Å for experimentally unstable complexes. Additionally, the non-stable interactions have wider and more variable CDR RMSD distributions than those of the stable ones. Like ΔΔEelec < 0, the mean of CDR RMSD distributions seems to be a reasonable classifier to filter epitope mutations that abrogate the binding to antibody from those that do not. Considering only CDR H3 and L3, there is a less distinct differentiation between the stable and unstable complexes (Supplementary Fig. S6B/C), due in part to the fact that main chain CDR3s of high affinity antibodies are structurally rigid (40). The trend remains apparent that stable complexes (necitumumab and cetuximab bound to wild type, and necitumumab bound to S468R) have less dynamic CDR3s than observed for the unstable complexes.
DISCUSSION
Our findings show that necitumumab binds to all reported cetuximab and panitumumab resistance mutations, with the exception of those at position G441. Necitumumab may, therefore, be active in cases where progression following cetuximab (or panitumumab) therapy was due to acquisition of resistance mutations in the EGFR antibody epitope. To date, approaches to overcome or circumvent resistance due to loss of antibody binding have focused on the use of mixtures of antibodies with non-overlapping epitopes, such as Sym004 and MM-151 (41, 42). These antibody cocktails have been shown to effectively block EGFR activation in cells and are in Phase I clinical trials (43, 44). These cocktails, like panitumumab, can block activation of EGFR-S468R, and the Phase I data suggests a clinical benefit to re-challenge in cases with this cetuximab resistance mutation (45, 46). The precise epitopes for the antibodies in the Sym004 and MM-151 cocktails have not been disclosed. It is, therefore, impossible to predict therapeutic outcome of these cocktails for other epitope mutations. In this study, we demonstrate that a simple computational approach has the potential to assess whether necitumumab is capable of binding to the mutant EGFR variants that emerge during treatment with cetuximab and/or panitumumab.
We find that, with the available experimental data on EGFR epitope substitution, the ΔΔEelec term calculated in Discovery Studio (29) is a robust classifier (filter) of the impact of epitope mutations on antibody binding. Further, this electrostatic energy filter is clearly superior to any of the other DSC4.1 energy terms. This is consistent with earlier studies that optimized electrostatics-based classifiers to filter-out CDR changes that abolish the binding of T-cell receptors to peptide-MHC complexes (47). The fact that ΔΔEelec but not ΔΔG is a reasonable estimator of the impact of an epitope mutation on the change in binding affinity of antibodies indicates that the energy target function within DSC4.1 requires further re-optimization to prospectively predict the impact of an epitope mutation on the affinity of an antibody. Optimization of the protocol to model side chain conformations could also minimize the number of FN and FP predictions that unfavorably impact the performance of the ΔΔEelec < 0 filter. This simple structure-based filter could also be further developed as a rapid classifier of epitope and/or paratope mutations that abolish the formation of other antibody/antigen interfaces. Availability of such a tool could have a profound impact on structure-based epitope mapping and/or structure-based optimization of antibody/antigen interactions.
Comparison of the X-ray crystal structures of EGFR bound to the Fab fragments from cetuximab (25), panitumumab (23) and necitumumab (22) provides a molecular understanding for why the S468R EGFR variant cannot bind to cetuximab, but can bind to both panitumumab and necitumumab. Of note, the presence of a pocket between the heavy and light chains accommodates the larger arginine side chain in complexes with panitumumab (23) and necitumumab (this study). The cavity in necitumumab is more than 2.5-fold larger than that observed in panitumumab (Supplementary Fig. S3). We speculate that the large necitumumab cavity can accommodate a wider range of epitope substitutions, and verify experimentally and computationally that this is the case for the set of alterations considered in this study. Necitumumab binds all resistance variants except those at position G441. This includes the S440L panitumumab resistance variant, as well as the I467M mutations observed in cetuximab-resistant cells lines that fails to engage panitumumab (14). We propose that the presence of a cavity in the paratope of an antibody could correlate with less frequent onset of acquired mutations that block antibody binding. Indeed, this could be why the S468R mutation has been observed with greater frequency in the tumors of patients treated with cetuximab compared to those treated with panitumumab, which has a paratope cavity (21, 23). By extension, it would seem reasonable to speculate that the incidence of this type of resistance might be even lower if CRC patients were treated with necitumumab. There have been limited Phase II studies of necitumumab in CRC (48). It remains to be seen whether these trials will advance, and whether or not resistance through epitope mutation will emerge. We find that significant cavities in the center of the antibody paratope are relatively common in reported structures of therapeutic antibodies bound to their antigens. It remains to be seen whether the presence of such cavities will correlate with low incidence of resistance through epitope mutation in other systems.
Supplementary Material
Acknowledgments
We thank Mark Lemmon and members of the Ferguson and Lemmon laboratories for helpful advice and discussion. We thank the staff at the GM/CA @ APS at Argonne National Laboratories for help in data collection and Steve Stayrook for help in data analysis. We thank Dr. Nick Loizos, Dr. Alan C. Rigby and Dr. Gregory D. Plowman (Eli Lilly & Company) for enabling this work and their scientific input.
Financial Information: This work was supported in part by the National Cancer Institute (CA112552 and CA198164 to K.M. Ferguson). A. Bagchi was supported in part by NIGMS T32 GM008275. Financial support to all other authors was provided by Eli Lilly & Company.
Grant Support
This work was supported by NIH R01s CA112552 and CA198164 (KMF), and Eli Lilly and Company (JNH, SWE, MV, MT, MDI, AF, YS, RDN). AB was supported in part by a NIH T32-GM008275. GM/CA @ APS is funded by NCI (ACB-12002) and NIGMS (AGM-12006). APS is a DOE User Facility (DE-AC02-06CH11357).
References
- 1.Arteaga CL, Engelman JA. ERBB receptors: from oncogene discovery to basic science to mechanism-based cancer therapeutics. Cancer Cell. 2014;25:282–303. doi: 10.1016/j.ccr.2014.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roskoski R., Jr The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol Res. 2014;79:34–74. doi: 10.1016/j.phrs.2013.11.002. [DOI] [PubMed] [Google Scholar]
- 3.Veronese ML, O’Dwyer PJ. Monoclonal antibodies in the treatment of colorectal cancer. Eur J Cancer. 2004;40:1292–301. doi: 10.1016/j.ejca.2004.02.014. [DOI] [PubMed] [Google Scholar]
- 4.Lee MS, Kopetz S. Current and Future Approaches to Target the Epidermal Growth Factor Receptor and Its Downstream Signaling in Metastatic Colorectal Cancer. Clin Colorectal Cancer. 2015;14:203–18. doi: 10.1016/j.clcc.2015.05.006. [DOI] [PubMed] [Google Scholar]
- 5.Okines A, Cunningham D, Chau I. Targeting the human EGFR family in esophagogastric cancer. Nat Rev Clin Oncol. 2011;8:492–503. doi: 10.1038/nrclinonc.2011.45. [DOI] [PubMed] [Google Scholar]
- 6.Wu M, Rivkin A, Pham T. Panitumumab: human monoclonal antibody against epidermal growth factor receptors for the treatment of metastatic colorectal cancer. Clin Ther. 2008;30:14–30. doi: 10.1016/j.clinthera.2008.01.014. [DOI] [PubMed] [Google Scholar]
- 7.Thatcher N, Hirsch FR, Luft AV, Szczesna A, Ciuleanu TE, Dediu M, et al. Necitumumab plus gemcitabine and cisplatin versus gemcitabine and cisplatin alone as first-line therapy in patients with stage IV squamous non-small-cell lung cancer (SQUIRE): an open-label, randomised, controlled phase 3 trial. Lancet Oncol. 2015;16:763–74. doi: 10.1016/S1470-2045(15)00021-2. [DOI] [PubMed] [Google Scholar]
- 8.Price TJ, Peeters M, Kim TW, Li J, Cascinu S, Ruff P, et al. Panitumumab versus cetuximab in patients with chemotherapy-refractory wild-type KRAS exon 2 metastatic colorectal cancer (ASPECCT): a randomised, multicentre, open-label, non-inferiority phase 3 study. Lancet Oncol. 2014;15:569–79. doi: 10.1016/S1470-2045(14)70118-4. [DOI] [PubMed] [Google Scholar]
- 9.Bokemeyer C, Van Cutsem E, Rougier P, Ciardiello F, Heeger S, Schlichting M, et al. Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer. 2012;48:1466–75. doi: 10.1016/j.ejca.2012.02.057. [DOI] [PubMed] [Google Scholar]
- 10.Douillard JY, Siena S, Cassidy J, Tabernero J, Burkes R, Barugel M, et al. Randomized, phase III trial of panitumumab with infusional fluorouracil, leucovorin, and oxaliplatin (FOLFOX4) versus FOLFOX4 alone as first-line treatment in patients with previously untreated metastatic colorectal cancer: the PRIME study. J Clin Oncol. 2010;28:4697–705. doi: 10.1200/JCO.2009.27.4860. [DOI] [PubMed] [Google Scholar]
- 11.Misale S, Di Nicolantonio F, Sartore-Bianchi A, Siena S, Bardelli A. Resistance to anti-EGFR therapy in colorectal cancer: from heterogeneity to convergent evolution. Cancer Discov. 2014;4:1269–80. doi: 10.1158/2159-8290.CD-14-0462. [DOI] [PubMed] [Google Scholar]
- 12.Redmond KL, Papafili A, Lawler M, Van Schaeybroeck S. Overcoming Resistance to Targeted Therapies in Cancer. Semin Oncol. 2015;42:896–908. doi: 10.1053/j.seminoncol.2015.09.028. [DOI] [PubMed] [Google Scholar]
- 13.Montagut C, Dalmases A, Bellosillo B, Crespo M, Pairet S, Iglesias M, et al. Identification of a mutation in the extracellular domain of the Epidermal Growth Factor Receptor conferring cetuximab resistance in colorectal cancer. Nat Med. 2012;18:221–3. doi: 10.1038/nm.2609. [DOI] [PubMed] [Google Scholar]
- 14.Arena S, Bellosillo B, Siravegna G, Martinez A, Canadas I, Lazzari L, et al. Emergence of Multiple EGFR Extracellular Mutations during Cetuximab Treatment in Colorectal Cancer. Clin Cancer Res. 2015;21:2157–66. doi: 10.1158/1078-0432.CCR-14-2821. [DOI] [PubMed] [Google Scholar]
- 15.Siravegna G, Mussolin B, Buscarino M, Corti G, Cassingena A, Crisafulli G, et al. Clonal evolution and resistance to EGFR blockade in the blood of colorectal cancer patients. Nat Med. 2015;21:795–801. doi: 10.1038/nm.3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bertotti A, Papp E, Jones S, Adleff V, Anagnostou V, Lupo B, et al. The genomic landscape of response to EGFR blockade in colorectal cancer. Nature. 2015;526:263–7. doi: 10.1038/nature14969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Braig F, Marz M, Schieferdecker A, Schulte A, Voigt M, Stein A, et al. Epidermal growth factor receptor mutation mediates cross-resistance to panitumumab and cetuximab in gastrointestinal cancer. Oncotarget. 2015;6:12035–47. doi: 10.18632/oncotarget.3574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oxnard GR, Arcila ME, Chmielecki J, Ladanyi M, Miller VA, Pao W. New strategies in overcoming acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in lung cancer. Clin Cancer Res. 2011;17:5530–7. doi: 10.1158/1078-0432.CCR-10-2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, et al. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci USA. 2008;105:2070–5. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jia Y, Yun CH, Park E, Ercan D, Manuia M, Juarez J, et al. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature. 2016;534:129–32. doi: 10.1038/nature17960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Price TJ, Newhall K, Peeters M, Kim TW, Li J, Cascinu S, et al. Prevalence and outcomes of patients (pts) with EGFR S492R ectodomain mutations in ASPECCT: Panitumumab (pmab) vs cetuximab (cmab) in pts with chemorefractory wild-type KRAS exon 2 metastatic colorectal cancer (mCRC) J Clin Oncol (Meeting Abstracts) 2015;33:e14623. [Google Scholar]
- 22.Li S, Kussie P, Ferguson KM. Structural basis for EGF receptor inhibition by the therapeutic antibody IMC-11F8. Structure. 2008;16:216–27. doi: 10.1016/j.str.2007.11.009. [DOI] [PubMed] [Google Scholar]
- 23.Sickmier EA, Kurzeja RJ, Michelsen K, Vazir M, Yang E, Tasker AS. The Panitumumab EGFR Complex Reveals a Binding Mechanism That Overcomes Cetuximab Induced Resistance. PLoS One. 2016;11:e0163366. doi: 10.1371/journal.pone.0163366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forest A, Amatulli M, Ludwig DL, Damoci CB, Wang Y, Burns CA, et al. Intrinsic Resistance to Cixutumumab Is Conferred by Distinct Isoforms of the Insulin Receptor. Mol Cancer Res. 2015;13:1615–26. doi: 10.1158/1541-7786.MCR-15-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li S, Schmitz KR, Jeffrey PD, Wiltzius JJ, Kussie P, Ferguson KM. Structural basis for inhibition of the epidermal growth factor receptor by cetuximab. Cancer Cell. 2005;7:301–11. doi: 10.1016/j.ccr.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 26.CCP4. The CCP4 Suite: Programs for Protein Crystallization. Acta Crystallogr. 1994;D60:760–3. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 27.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr. 2010;D66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. 2010;D66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.BIOVIA. Discovery Studio Modeling Environment, DSC4.1. San Diego: Dassault Systèmes; 2016. [Google Scholar]
- 30.Spassov VZ, Yan L. pH-selective mutagenesis of protein-protein interfaces: in silico design of therapeutic antibodies with prolonged half-life. Proteins. 2013;81:704–14. doi: 10.1002/prot.24230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Spassov VZ, Yan L. A fast and accurate computational approach to protein ionization. Protein Sci. 2008;17:1955–70. doi: 10.1110/ps.036335.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Brooks BR, Brooks CL, 3rd, Mackerell AD, Jr, Nilsson L, Petrella RJ, Roux B, et al. CHARMM: the biomolecular simulation program. J Comput Chem. 2009;30:1545–614. doi: 10.1002/jcc.21287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Molecular Operating Environment (MOE) Chemical Computing Group ULC, 1010 Sherbooke St West, Suite #910, Montreal, QC, Canada, H3A 2R7. 2017 2013.08. [Google Scholar]
- 34.Schrödinger. Release 2015–4: Maestro, version 10.4. Schrödinger, LLC; New York, NY: 2015. [Google Scholar]
- 35.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14:33–8. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 36.Lawrence MC, Colman PM. Shape complementarity at protein/protein interfaces. J Mol Biol. 1993;234:946–50. doi: 10.1006/jmbi.1993.1648. [DOI] [PubMed] [Google Scholar]
- 37.Bresler SC, Weiser DA, Huwe PJ, Park JH, Krytska K, Ryles H, et al. ALK mutations confer differential oncogenic activation and sensitivity to ALK inhibition therapy in neuroblastoma. Cancer Cell. 2014;26:682–94. doi: 10.1016/j.ccell.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sirin S, Apgar JR, Bennett EM, Keating AE. AB-Bind: Antibody binding mutational database for computational affinity predictions. Protein Sci. 2016;25:393–409. doi: 10.1002/pro.2829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pires DE, Ascher DB. mCSM-AB: a web server for predicting antibody-antigen affinity changes upon mutation with graph-based signatures. Nucleic Acids Res. 2016;44:W469–73. doi: 10.1093/nar/gkw458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Haidar JN, Zhu W, Lypowy J, Pierce BG, Bari A, Persaud K, et al. Backbone flexibility of CDR3 and immune recognition of antigens. J Mol Biol. 2014;426:1583–99. doi: 10.1016/j.jmb.2013.12.024. [DOI] [PubMed] [Google Scholar]
- 41.Pedersen MW, Jacobsen HJ, Koefoed K, Hey A, Pyke C, Haurum JS, et al. Sym004: a novel synergistic anti-epidermal growth factor receptor antibody mixture with superior anticancer efficacy. Cancer Res. 2010;70:588–97. doi: 10.1158/0008-5472.CAN-09-1417. [DOI] [PubMed] [Google Scholar]
- 42.Kearns JD, Bukhalid R, Sevecka M, Tan G, Gerami-Moayed N, Werner SL, et al. Enhanced Targeting of the EGFR Network with MM-151, an Oligoclonal Anti-EGFR Antibody Therapeutic. Mol Cancer Ther. 2015;14:1625–36. doi: 10.1158/1535-7163.MCT-14-0772. [DOI] [PubMed] [Google Scholar]
- 43.Dienstmann R, Patnaik A, Garcia-Carbonero R, Cervantes A, Benavent M, Rosello S, et al. Safety and Activity of the First-in-Class Sym004 Anti-EGFR Antibody Mixture in Patients with Refractory Colorectal Cancer. Cancer Discov. 2015;5:598–609. doi: 10.1158/2159-8290.CD-14-1432. [DOI] [PubMed] [Google Scholar]
- 44.Harb WA, Adjei AA, Beeram M, Pipas JM, Chen S, Valencia A, et al. Final results of a first-in-human study evaluating the safety, pharmacology and initial efficacy of MM-151, an oligoclonal anti-EGFR antibody in patients with refractory solid tumors. J Clin Oncol (Meeting Abstracts) 2016;34:2518. [Google Scholar]
- 45.Arena S, Siravegna G, Mussolin B, Kearns JD, Wolf BB, Misale S, et al. MM-151 overcomes acquired resistance to cetuximab and panitumumab in colorectal cancers harboring EGFR extracellular domain mutations. Sci Transl Med. 2016;8:324ra14. doi: 10.1126/scitranslmed.aad5640. [DOI] [PubMed] [Google Scholar]
- 46.Sanchez-Martin FJ, Bellosillo B, Gelabert-Baldrich M, Dalmases A, Canadas I, Vidal J, et al. The First-in-class Anti-EGFR Antibody Mixture Sym004 Overcomes Cetuximab Resistance Mediated by EGFR Extracellular Domain Mutations in Colorectal Cancer. Clin Cancer Res. 2016;22:3260–7. doi: 10.1158/1078-0432.CCR-15-2400. [DOI] [PubMed] [Google Scholar]
- 47.Haidar JN, Pierce B, Yu Y, Tong W, Li M, Weng Z. Structure-based design of a T-cell receptor leads to nearly 100-fold improvement in binding affinity for pepMHC. Proteins. 2009;74:948–60. doi: 10.1002/prot.22203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Elez E, Hendlisz A, Delaunoit T, Sastre J, Cervantes A, Varea R, et al. Phase II study of necitumumab plus modified FOLFOX6 as first-line treatment in patients with locally advanced or metastatic colorectal cancer. Br J Cancer. 2016;114:372–80. doi: 10.1038/bjc.2015.480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–41. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.