

Abstract
Motivation
Epigenetic mechanisms are known to play a major role in breast cancer. However, the role of 5-hydroxymethylcytosine (5hmC) remains understudied. We hypothesize that 5hmC mediates redox regulation of gene expression in an aggressive subtype known as triple negative breast cancer (TNBC). To address this, our objective was to highlight genes that may be the target of this process by identifying redox-regulated, antioxidant-sensitive, gene-localized 5hmC changes associated with mRNA changes in TNBC cells.
Results
We proceeded to develop an approach to integrate novel Pvu-sequencing and RNA-sequencing data. The result of our approach to merge genome-wide, high-throughput TNBC cell line datasets to identify significant, concordant 5hmC and mRNA changes in response to antioxidant treatment produced a gene set with relevance to cancer stem cell function. Moreover, we have established a method that will be useful for continued research of 5hmC in TNBC cells and tissue samples.
Availability and implementation
Data are available at Gene Expression Omnibus (GEO) under accession number GSE103850.
1 Introduction
DNA methylation, in the form of a methyl group at the 5 position of cytosine (5mC), is arguably the most studied epigenetic modification and the role of 5mC in gene regulation in cancer is well described (Portela and Esteller, 2010). Global average loss of 5mC is observed in tumor tissues while at the same time select dense hypermethylation at CpG islands in the promoters of tumor suppressors silences their expression in cancer (Das and Singal, 2004). By comparison, less is known about the role of 5-hydroxymethylcytosine (5hmC) in cancer. 5hmC was first discovered in mammalian DNA in 1972 and in the decades that followed, it had been dismissed as a DNA oxidation event of no important functional consequence and difficulty in measuring 5hmC hindered studies (Dizdaroglu, 1998; Penn et al., 1972). The 5hmC modification was largely dismissed until in 2009 researchers reported that ten-eleven-translocation (TET) dioxygenase converts 5mC to 5hmC (Dizdaroglu, 1998; Kriaucionis and Heintz, 2009; Penn et al., 1972; Tahiliani et al., 2009). Like 5mC, global averages of 5hmC are reduced in cancer tissues (Kudo et al., 2012; Yang et al., 2013). 5hmC is also a stable modification (Hahn et al., 2013); but, opposite to 5mC, the enrichment of 5hmC at gene promoters and gene coding sequences is associated with increased gene expression (Bhattacharyya et al., 2013; Ficz et al., 2011; Khare et al., 2012; Lister et al., 2013; Mellen et al., 2012; Song et al., 2011). Recent reports (Bhattacharyya et al., 2013; Chapman et al., 2015) and our own data, described herein, provide early evidence that cancer-specific, localized 5hmC enrichment occurs at select gene coding regions.
Recent advances in high-throughput sequencing approaches have facilitated studies of 5hmC localization at sequence-level resolution. Based on a survey of the literature, examples include a study comparing a non-cancerous nevus sample and a melanoma sample, which applied an approach using 5hmC-DNA immunoprecipitation and high-throughput sequencing to discover a difference in the number of gene-containing 5hmC peaks (Lian et al., 2012). Investigating the SK-N-BE(2) neuroblastoma cell line, Mariani et al. employed the hMe-Seal method, which captures biotinylated 5hmC followed by high-throughput sequencing (Mariani et al., 2014). Another group also used this method to study 5hmC in a study of the T84 colon cancer cell line in relation to colonocytes from two patients (Chapman et al., 2015). From a study of pancreatic cancer cell lines, Bhattacharyya et al. reported redistribution of 5hmC enrichment patterns at promoter sequences using a HELP-GT technique, where researchers glycosylate, capture and sequence 5hmC-modified DNA regions (Bhattacharyya et al., 2013). Chen et al. used the TET-assisted BS-sequencing method—which combines a glycosylation step, TET enzyme treatment to convert 5mC to 5caC, bisulfite treatment and high-throughput sequencing—to measure 5hmC in tumor and matched adjacent normal kidney specimens from two clear cell renal carcinoma patients (Chen et al., 2016).
The aforementioned studies largely focused on chronicling the genome-wide distribution of 5hmC enrichment patterns in cancer and are lacking in integration of complimentary high-throughput gene transcript data to identify 5hmC modified sequences that are associated with regulation of gene mRNA expression. Nevertheless, in the current decade, researchers have gained understanding of the role of 5hmC to regulate expression of select genes to impact cell function, particularly in the context of human stem cells where epigenetic regulation of gene expression is critical for control of pluripotency and differentiation. In human pluripotent stem cells (hPSCs), self-renewal function depends on TET1 activity and 5hmC enrichment at specific gene promoters (Ficz et al., 2011; Ito et al., 2010; Yildirim et al., 2011). Fine-tuned redox signaling also regulates both hPSC self-renewal and cellular differentiation (Bigarella et al., 2014). Coulter et al. made the mechanistic connection that TET1-generated 5hmC mediates gene expression changes induced by reactive oxygen species (ROS) (Coulter et al., 2013).
In a recent study of triple negative breast cancer (TNBC), a molecular subtype of breast cancer that lacks expression of estrogen receptor-alpha (ERα), progesterone receptor (PR), or the HER2 oncogene, we observed that TNBC cells exhibit elevated levels of intracellular ROS relative to nonmalignant breast epithelial cells (Bao et al., 2017). Furthermore, treatment with select antioxidants, which reduce ROS and thus perturb the redox state of the cell, inhibits survival and self-renewal capacity of breast cancer stem cell-like cells (CSCs) (Bao et al., 2017). The relevance of identifying a means to inhibit CSCs is that they are identified in patient tumors, and breast cancer cell line cultures as the fraction of self-renewing, tumor initiating cancer cells that also give rise to drug resistance and metastatic recurrence (Dave et al., 2012; Fillmore and Kuperwasser, 2008; Liu et al., 2010; Pattabiraman and Weinberg, 2014). The molecular mechanisms relaying the effects of oncogenic redox signaling on gene expression in cancer cells are not fully understood. Drawing on potential parallels to self-renewal capacity of hPSCs outlined earlier, we hypothesize that 5hmC mediates redox-regulation of a gene expression program in TNBC that promotes the CSC phenotype.
To address this hypothesis, our objective was to highlight genes that may be the target of this process by identifying ROS-regulated, antioxidant-sensitive, gene-localized 5hmC changes associated with gene expression changes in TNBC cells. To that end, the current report details the generation and differential analysis of genome-wide, transcript level and 5hmC-enrichment datasets from TNBC cell cultures before and after treatment with an antioxidant. Transcriptome data were the result of high-throughput RNA sequencing and genome-wide 5hmC patterns were measured by an endonuclease PvuRts1I (Pvu)-digested DNA sequencing method, called Pvu-sequencing, previously established by us in (Cingolani et al. 2013). Pvu-sequencing takes advantage of the specificity for the Pvu enzyme to selectively cleave 5hmC-modified consensus sequence (5′-hmCN11-12/N9-10 G-3′) for efficient targeted sequencing of 5hmC-modified DNA. We present the approach we developed to integrate these novel datasets, to yield a set of genes where 5hmC levels and gene expression changes were coordinated and subject to regulation by antioxidant drug treatment.
2 Materials and methods
2.1 Cell line and patient tissue samples
The TNBC cell line MDA-MB-468 (from ATCC, Manassas, VA) was cultured in DMEM medium (HyClone, Logan, UT) supplemented with 10% FBS (HyClone, Logan, UT). Cell cultures were treated with vehicle control or 10 μM CAT-SKL for 24 h to derive three independent biological replicate sample sets. CAT-SKL is a recombinant antioxidant enzyme containing an 11 arginine residue cell penetrating peptide domain and an altered carboxy terminal peroxisomal targeting signal, expressed in bacteria and purified on nickel-affinity resins as described by (Giordano et al., 2012; Young et al., 2008). The activity of CAT-SKL treatment was confirmed in parallel cultures where an 80–90% decrease in ROS levels were measured using the Amplex Red Hydrogen Peroxide/Peroxidase Assay kit following the manufacturer’s instructions (Molecular Probes®, Eugene, OR). The MDA-MB-468 cell line was authenticated by short tandem repeat (STR) analysis using the PowerPlex(r) 16 system (Promega, Madison, WI). De-identified fresh-frozen breast cancer specimens and matched adjacent normal tissues were obtained from Asterand, PLC (Detroit, MI). Approval for the genomic investigation of archived and de-identified specimens was obtained from the Institutional Review Board at Wayne State University, Detroit, MI.
2.2 High-throughput Pvu-sequencing
DNeasy Blood and tissue kit (Qiagen, Valencia, CA) was used to extract genomic DNA from the fresh frozen tissues and MDA-MB-468 cells. RNaseA treatment was done according to the manufacturer’s instructions (4 μL of RNaseA at 100 mg/mL) to obtain RNA free genomic DNA. PvuRts1I restriction digestion of the genomic DNA was done according to manufacturer’s specifications (Active Motif, Carlsbad, CA). The digested DNA was purified using QIAquick PCR Purification Kit (Qiagen, Valencia, CA). The eluted DNA was used to prepare libraries using ThruPLEX-FD prep kit (Rubicon Genomics, Ann Arbor, MI). Sequencing was done using the Illumina HiSeq 2500 platform (San Diego, CA) high-output mode with parameters: 50 bp paired end reads, 12 libraries per lane. Sequenced reads were demultiplexed using Illumina’s CASAVA 1.8.2 software (www.illumina.com/). Quality of sequencing data was assessed using FastQC software available from Babraham Bioinformatics (www.bioinformatics.babraham.ac.uk/). Reads were aligned to the Human Genome Consortium’s reference genome hg19 (Lander et al., 2001) using Novoalign (www.novocraft.com) and reads were selected for those mapping to a unique location in the genome, resulting in approximately 20 million high quality paired-end reads (passing QC30) per sample. In an additional quality control step, the Pvu-Seq reads were filtered to retain those containing the PvuRts1I cognate sequence utilizing the PERL script provided by Sun et al. (2013). This showed that more than 75% of the reads contained the cognate sequence. We used a maximum value inference approach to deal with sample-level missing values. The genomic coordinates of sequences (>20 read counts) were used to determine overlap with the 5′UTR, 3′UTR, coding exon, intron and intergenic regions obtained from UCSC Genome Browser (assembly February 2009 GRCh37/hg19) (Meyer et al., 2012) using the BEDtools suite (Dale et al., 2011; Quinlan and Hall, 2010) and a custom R script.
Pvu-sequencing data and RNA-sequencing data (described below) sets are available at the Gene Expression Omnibus database accession number GSE103850.
2.3 High-throughput RNA-sequencing
Total RNA was isolated from MDA-MB-468 cells using the RNeasy plus kit (Qiagen, Valencia, CA). RNA sequencing libraries were prepared with the Illumina TruSeq v2 kit and sequenced using an Illumina HiSeq2500 (6 per lane, high-output mode, 50 bp, paired end reads). Sequenced reads were demultiplexed using Illumina’s CASAVA 1.8.2 software. Quality of sequencing data was assessed using FastQC software. Reads were aligned to the Human Genome Consortium’s reference genome hg19 (Lander et al., 2001) using Novoalign (www.novocraft.com) and reads were selected for those mapping to a unique location in the genome. Data were pre-processed with htseq-count (Anders et al., 2015) in order to map aligned reads to the University of California, Santa Cruz (UCSC) hg19 annotated transcripts.
2.4 Differential analysis and integration of Pvu-sequencing and RNA-sequencing datasets
Our survey of the literature revealed that many 5hmC mapping studies were focused on localization or distribution of 5hmC throughout the genome (Stroud et al., 2011; Szulwach et al., 2011; Wu et al., 2011; Yu et al., 2012). Also, studies reporting on 5hmC and gene expression associations generally relied on steady state data (Greco et al., 2016; Lin et al., 2017; Xu et al., 2011). These analyses clustered the genes into groups with different 5hmC levels and then compared the mean levels of gene expression for these groups to assess the correlation of 5hmC and gene expression between the groups (Greco et al., 2016; Lin et al., 2017). Furthermore, the gene-level association between 5hmC and gene expression was evaluated only for select genes from the group based on functional annotation of the genes as opposed to using the high-throughput data. The challenge before us was to develop an unbiased method appropriate for integrating Pvu-sequencing and RNA-sequencing data types to identify associated changes in the perturbed datasets.
Figure 1 presents the workflow for our approach. In step 1, RNA-sequencing or Pvu-sequencing read counts from three nontreated cultures or three CAT-SKL-treated cultures were obtained from fastq files by alignment with novoalign and quantification using htseq-count or custom scripts and then averaged and analyzed with HOMER software (homer.ucsd.edu/) (Heinz et al., 2010), specifically, DESeq-1.18.0 (Anders and Huber, 2010) to obtain differential 5hmC and differential mRNA expression gene lists (P < 0.05), and their log2 fold-change values. In step 2, a min–max normalization was applied to the fold-change values for each data type. For each transcript, we computed the normalized distance [1–abs(5hmC–mRNA)] to a linear model fitted to the principal diagonal (x = y) to assess how close the 5hmC and RNA data values are at the transcript level. This distance is used to evaluate the correlation between the 5hmC and RNA values. In step 3, a high correlation filter was applied to the aforementioned distance statistic. Transcripts with a correlation statistic higher than 75% are highlighted in the results (Fig. 2). Gene ontology analysis was run using iPathwayGuide (www.advaitabio.com) (Ahsan and Drăghici, 2017; Drăghici et al., 2007; Khatri et al., 2002; Voichiţa et al., 2012) with Elim correction to identify molecular functions significantly enriched in our data.
Fig. 1.

Workflow of the integrative analysis for gene expression and 5-hydroxymethylation (5hmC). The integrative analysis aims to discover genes regulated by 5hmC in breast cancer. In step 1, we select transcripts that have both significantly different gene expression and significantly different 5hmC levels. In step 2, we apply a correlation filter where correlation denotes concordant gene expression and 5hmC (both up or both down). In step 3, we apply a high correlation filter based on a distance statistic computed to assess the relevant magnitude of change in gene expression and 5hmC. The output is a list of highly correlated/concordant genes
Fig. 2.

Correlated 5hmC and mRNA expression changes induced by antioxidant treatment of TNBC cells. (A) Heat map of 182 genes showing correlated 5hmC and mRNA changes induced by antioxidant treatment of the MDA-MB-468 TNBC cell line. The blue column reflects the correlation value (from 30 to 100%). 5hmC and mRNA levels were the average of 3 biological replicates, treated versus untreated differences (P<0.05), according to Pvu-seq and RNA-seq analyses. (B) 59 genes with the highly correlated (75–100%) 5hmC and mRNA changes increased by antioxidant treatment. (C) 63 genes with the highly correlated (75–100%) 5hmC and mRNA changes decreased by antioxidant treatment. The 6 genes listed in panel (E) are denoted by dark green points. (D) The iPathwayGuide Gene Ontology analysis tool was applied to the highly correlated gene results to identify significant molecular function associations. (E) The highly correlated genes that overlapped with genes identified in differential analysis of 5hmC levels in TNBC patient samples. Each of the genes listed are denoted by dark green points in panel (C) and showed significantly higher 5hmC levels in a patient TNBC sample relative to matched adjacent noncancer tissue. (F) Integrative Genomics Viewer (IGV) display of tiled reads region of the genome surrounding the transcription start site of the DNA and RNA binding protein gene TARDBP. Peak enrichment in the patient tumor specimen (top panel) is absent in the noncancer adjacent specimen (lower panel)
3 Results
3.1 Correlated 5hmC and mRNA expression changes induced by antioxidant treatment of TNBC cells
The result of our approach outlined earlier was a list of 182 genes showing correlated 5hmC and mRNA changes induced by antioxidant treatment (Fig. 2A). The highest correlated (>75%) subset of genes, both upregulated (Fig. 2B) and downregulated (Fig. 2C), was used in gene ontology analysis to identify over-represented molecular functions (Fig. 2D). For evidence that results from Pvu-sequencing analysis of cell lines is relevant for tumors we proceeded to analyze differential 5hmC-modified gene-associated sequences in two sets of patient-matched TNBC and adjacent noncancer tissue specimens. A table (Fig. 2E) lists genes from differential Pvu-sequencing analysis of tissue samples that overlapped with the highest correlated set of genes from cell line analysis. All of these overlapping genes were significantly lower in the noncancer relative to cancer samples and downregulated by antioxidant treatment in TNBC cell line cultures. Figure 2F displays the mapped Pvu-seq reads in patient tissue samples for TAR DNA binding protein (TARDB). TARDBP is of particular interest because it is mechanistically linked to alternative splicing of methyl-CpG-binding domain protein 2 (MBD2) (Dreumont et al., 2009; Lu et al., 2014; Sureau et al., 2001), and we recently reported on the importance of the MBD2 splice variant, MBD2_v2, for survival and generation of TNBC CSCs (Bao et al., 2017).
3.2 Antioxidant treatment induced changes to 5hmC distribution across genomic regions
We proceeded to use the antioxidant-treated and untreated cell line Pvu-sequencing data to map the genomic distribution of 5hmC. The gene 5′-untranslated region (5′ UTR), 3′-untranslated region (3′ UTR), coding exon, and intron were defined according to the University of California, Santa Cruz (UCSC) Genome Browser (Meyer et al., 2012; Karolchik et al., 2013; Tyner et al., 2016). For each genomic region, coverage was defined as the percentage of all bases within the entire genomic element that had a sequence-read mapped to it. Results showed that in the untreated cells a higher percentage of 5hmC is localized at the 5′ UTR region, while in the antioxidant-treated cells, the percentage distribution is approximately uniform across all genomic regions (Fig. 3).
Fig. 3.

Antioxidant treatment induced changes to 5hmC distribution across genomic regions in TNBC cells. For each genomic region, coverage was defined as the percentage of all bases within the entire genomic element that had a sequence-read mapped to it. Regions were defined according to the University of California, Santa Cruz (UCSC) Genome Browser
4 Discussion
TNBC is a molecular subtype of breast cancer that accounts for approximately 15–20% of invasive breast cancer diagnoses in the United States. TNBC is defined by the lack of molecular features that are also biomarkers for use of effective targeted therapies for treatment of other breast cancer subtypes (i.e. ERα or HER2 positive). Thus, TNBC is lacking in therapy options, which contributes to why women diagnosed with TNBC have the lowest 5-year survival rates among all BC patients. The lack in understanding of what drives TNBC and how to treat it is the motivation for our research of this subtype.
The result of our approach to merge data from differential analysis of genome-wide, high-throughput TNBC cell line datasets to identify significant, concordant 5hmC and mRNA changes in response to antioxidant treatment produced a biologically relevant gene set with functional relevance for CSCs and RNA processing. Among these genes, the mRNA splicing factor TARDBP is of interest because it is mechanistically linked to expression of the alternative mRNA splicing variant MBD2_v2 (Dreumont et al., 2009; Lu et al., 2014; Sureau et al., 2001). Previously, we identified that the methyl-CpG binding domain 2 gene expression, specifically the alternative mRNA splicing variant MBD2_v2, is rapidly downregulated by CAT-SKL treatment in TNBC cells. We subsequently confirmed that downregulation of MBD2_v2 is key to the antioxidant effect of inhibiting survival and self-renewal capacity of CSCs (Bao et al., 2017). Our present analysis also identified the vimentin gene, which demonstrated a profile similar to TARDBP in antioxidant treated TNBC cells and patient specimens. The role for vimentin in defining the CSC phenotype is also established (Morel et al., 2008).
The results of our analysis of genomic region distribution before and after treatment with CAT-SKL suggest that antioxidant-sensitive, redox signaling in TNBC predominantly supports 5hmC enrichment localized to 5′UTR gene sequences, which includes gene transcription start sites. Based on embryonic stem cell (ESC) research (Huang et al., 2014), this result may be implicating TET1 in the redox regulation of TNBC genes. In ESCs, 5hmC levels at promoters and transcription start sites of expressed genes are attributed to TET1, and 5hmC in gene bodies and exon boundaries is attributed to TET2 (Huang et al., 2014). We are planning a follow-up investigation to learn whether antioxidant treatment of TNBC cells impacts TET1 function or TET1 DNA localization (antioxidant treatment did not change TET1 mRNA expression in our dataset) and thereby downregulates the 5hmC and mRNA expression for genes such as TARDBP in TNBC cells.
The idea that TET1 could play an oncogenic role in TNBC appears to be contrary to reports in the literature. Yang et al. reported that decreased TET1 expression associates with decreased genome-wide 5hmC levels and that low TET1 expression correlates with worse breast cancer patient outcomes (Yang et al., 2013). The only precursor substrate from which 5hmC can be derived is 5mC; thus, the genome-wide loss of 5mC in cancer (Tsai et al., 2015) likely underlies the genome-wide loss of 5hmC. Moreover, their analysis of TET1 mRNA expression employed a semi-quantitative RT-PCR approach and used glyceraldehyde-3-phosphate dehydrogenase (GAPDH) as a control gene. The report did not acknowledge that GAPDH is upregulated in advanced BC and high GAPDH levels are associated with worse outcomes (Wang et al., 2013). Therefore, using GAPDH expression as the normalizer may be problematic. Consistent with this idea, when we used the KM plotter database (Győrffy et al., 2013) to analyze TET1 expression in a whole-genome microarray dataset the results showed that low TET1 mRNA expression in breast cancer specimens is significantly associated with better outcomes, specifically for patients with TNBC (Fig. 4). According to the same database (Győrffy et al., 2013), analyzing a similar number of specimens, there is no association for TET1 and survival outcomes for ERα/PR-positive or HER2-positive breast cancers, and there are no associations for TET2 or TET3 with outcomes for any breast cancer subtype. For other evidence that TET1 specifically functions to promote cancer, we refer to the oncogenic role of TET1-dependent 5hmC to regulate homeobox gene expression in the development of MLL-rearranged leukemia (Huang et al., 2013). On the other hand, the potential tumor suppressor role for TET2 is underscored by frequent loss-of-function TET2 mutations in myeloid cancers (Delhommeau et al., 2009).
Fig. 4.
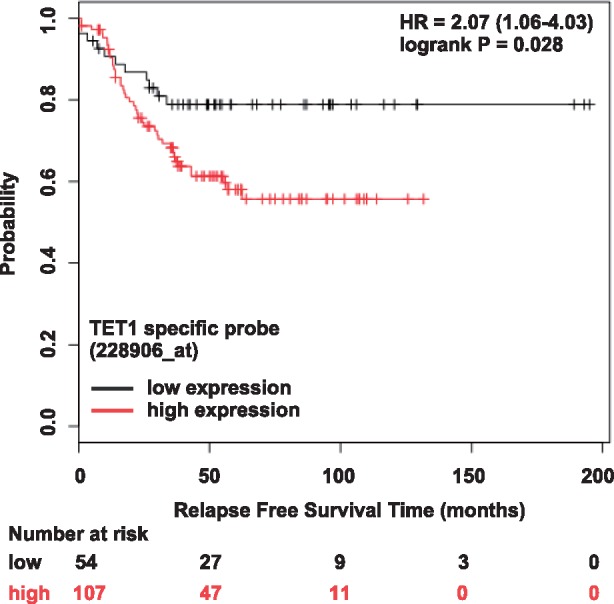
TET1 mRNA expression in TNBC. Low TET1 mRNA expression in TNBC tumor specimens (black) is associated with better patient outcomes (longer relapse free survival time). Results from a query using the on-line KM Plotter database and analysis tool (Győrffy et al., 2013)
To conclude, the work we present provides insights surrounding how oncogenic redox signaling and the epigenetics of 5hmC may converge to direct gene expression programs in TNBC. Moreover, we have established an approach for continued research of 5hmC and gene expression in breast cancer-derived cell lines and patient tissue specimens. Epigenetics plays a major role in tumor initiation and cancer progression (Feinberg et al., 2016). Insights from research surrounding the role of the well-known 5mC modification in cancer are motivating efforts to develop locus-specific diagnostic testing platforms and novel treatment strategies (Farlik et al., 2016; Guo et al., 2014). This report reinforces that it may not be far-reaching to think that with further research 5hmC could also have a bearing on diagnostics and treatment decision-making for TNBC patients.
Acknowledgement
The authors thank Dr Stanley Terlecky for supplying the CAT-SKL.
Funding
The authors thank The Believe Foundation and Joann M. Deliz and James W. Deliz for supporting this research study in memory of David Bergman. The institutional award ACS/IRG#11-053-01 (also to ABF) provided additional funding. The NIEHS Center grant P30 ES020957 to the Institute of Environmental Health Sciences and the NIH Center grant P30 CA022453 to the Karmanos Cancer Institute support the Genomics Core at Wayne State University.
Conflict of Interest: none declared.
References
- Ahsan S., Drăghici S. (2017) Identifying significantly impacted pathways and putative mechanisms with iPathwayGuide. Curr Protoc Bioinformatics, 57, 7–15. [DOI] [PubMed] [Google Scholar]
- Anders S., Huber W. (2010) Differential expression analysis for sequence count data. Genome Biol., 11, R106.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S. et al. (2015) HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics, 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bao B. et al. (2017) Treating triple negative breast cancer cells with erlotinib plus a select antioxidant overcomes drug resistance by targeting cancer cell heterogeneity. Sci. Rep., 7, 44125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S. et al. (2013) Genome-wide hydroxymethylation tested using the HELP-GT assay shows redistribution in cancer. Nucl. Acids Res., 41, e157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigarella C.L. et al. (2014) Stem cells and the impact of ROS signaling. Development, 141, 4206–4218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman C.G. et al. (2015) TET-catalyzed 5-hydroxymethylcytosine regulates gene expression in differentiating colonocytes and colon cancer. Sci. Rep., 5, 17568.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen K. et al. (2016) Loss of 5-hydroxymethylcytosine is linked to gene body hypermethylation in kidney cancer. Cell Res., 26, 103.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani P. et al. (2013) Intronic Non-CG DNA hydroxymethylation and alternative mRNA splicing in honey bees. BMC Genomics, 14, 666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulter J.B. et al. (2013) Hydroquinone increases 5-hydroxymethylcytosine formation through ten eleven translocation 1 (TET1) 5-methylcytosine dioxygenase. J. Biol. Chem., 288, 28792–28800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale R.K. et al. (2011) Pybedtools: a flexible Python library for manipulating genomic datasets and annotations. Bioinformatics, 27, 3423–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das P.M., Singal R. (2004) DNA methylation and cancer. J. Clin. Oncol., 22, 4632–4642. [DOI] [PubMed] [Google Scholar]
- Dave B. et al. (2012) Epithelial-mesenchymal transition, cancer stem cells and treatment resistance. Breast Cancer Res., 14, 202.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delhommeau F. et al. (2009) Mutation in TET2 in myeloid cancers. N. Engl. J. Med., 360, 2289–2301. [DOI] [PubMed] [Google Scholar]
- Dizdaroglu M. (1998) Facts about the artifacts in the measurement of oxidative DNA base damage by gas chromatography-mass spectrometry. Free Radical Res., 29, 551–563. [DOI] [PubMed] [Google Scholar]
- Drăghici S. et al. (2007) A systems biology approach for pathway level analysis. Genome Res., 17, 1537–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreumont N. et al. (2009) Antagonistic factors control the unproductive splicing of SC35 terminal intron. Nucl. Acids Res., 38, 1353–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farlik M. et al. (2016) DNA methylation dynamics of human hematopoietic stem cell differentiation. Cell Stem Cell, 19, 808–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg A.P. et al. (2016) Epigenetic modulators, modifiers and mediators in cancer aetiology and progression. Nat. Rev. Genetics., 17, 284–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ficz G. et al. (2011) Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature, 473, 398–402. [DOI] [PubMed] [Google Scholar]
- Fillmore C.M., Kuperwasser C. (2008) Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res., 10, R25.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano C.R. et al. (2012) A targeted enzyme approach to sensitization of tyrosine kinase inhibitor-resistant breast cancer cells. Exp. Cell Res., 318, 2014–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco C.M. et al. (2016) DNA hydroxymethylation controls cardiomyocyte gene expression in development and hypertrophy. Nat. Commun., 7, 12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X.E. et al. (2014) Targeting tumor suppressor networks for cancer therapeutics. Curr. Drug Targets, 15, 2–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Győrffy B. et al. (2013) Online survival analysis software to assess the prognostic value of biomarkers using transcriptomic data in non-small-cell lung cancer. PLoS One, 8, e82241.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn M.A. et al. (2013) Dynamics of 5-hydroxymethylcytosine and chromatin marks in Mammalian neurogenesis. Cell Rep., 3, 291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S. et al. (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell, 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H. et al. (2013) TET1 plays an essential oncogenic role in MLL-rearranged leukemia. Proc. Natl. Acad. Sci., 110, 11994–11999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y. et al. (2014) Distinct roles of the methylcytosine oxidases Tet1 and Tet2 in mouse embryonic stem cells. Proc. Natl. Acad. Sci., 111, 1361–1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S. et al. (2010) Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature, 466, 1129–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D. et al. (2013) The UCSC genome browser database: 2014 update. Nucl. Acids Res., 42, D764–D770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khare T. et al. (2012) 5-hmC in the brain is abundant in synaptic genes and shows differences at the exon-intron boundary. Nat. Struct. Mol. Biol., 19, 1037–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatri P. et al. (2002) Profiling gene expression using Onto-Express. Genomics, 79, 266–270. [DOI] [PubMed] [Google Scholar]
- Kriaucionis S., Heintz N. (2009) The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science, 324, 929–930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudo Y. et al. (2012) Loss of 5-hydroxymethylcytosine is accompanied with malignant cellular transformation. Cancer Sci., 103, 670–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander E.S. et al. (2001) Initial sequencing and analysis of the human genome. Nature, 409, 860–921. [DOI] [PubMed] [Google Scholar]
- Lian C.G. et al. (2012) Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma. Cell, 150, 1135–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin I.-H. et al. (2017) Correlated 5-hydroxymethylcytosine (5hmC) and gene expression profiles underpin gene and organ-specific epigenetic regulation in adult mouse brain and liver. PLoS One, 12, e0170779.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R. et al. (2013) Global epigenomic reconfiguration during mammalian brain development. Science, 341, 1237905.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H. et al. (2010) Cancer stem cells from human breast tumors are involved in spontaneous metastases in orthotopic mouse models. Proc. Natl. Acad. Sci., 107, 18115–18120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y. et al. (2014) Alternative splicing of MBD2 supports self-renewal in human pluripotent stem cells. Cell Stem Cell, 15, 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mariani C.J. et al. (2014) TET1-mediated hydroxymethylation facilitates hypoxic gene induction in neuroblastoma. Cell Rep., 7, 1343–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellen M. et al. (2012) MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell, 151, 1417–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer L.R. et al. (2012) The UCSC Genome Browser database: extensions and updates 2013. Nucl. Acids Res., 41, D64–D69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morel A.-P. et al. (2008) Generation of breast cancer stem cells through epithelial-mesenchymal transition. PLoS One, 3, e2888.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattabiraman D.R., Weinberg R.A. (2014) Tackling the cancer stem cells—what challenges do they pose? Nat. Rev. Drug Discovery, 13, 497.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Penn N. et al. (1972) The presence of 5-hydroxymethylcytosine in animal deoxyribonucleic acid. Biochem. J., 126, 781–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portela A., Esteller M. (2010) Epigenetic modifications and human disease. Nat. Biotechnol., 28, 1057–1068. [DOI] [PubMed] [Google Scholar]
- Quinlan A.R., Hall I.M. (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C.-X. et al. (2011) Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat. Biotechnol., 29, 68–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud H. et al. (2011) 5-Hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol., 12, R54.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Z. et al. (2013) High-resolution enzymatic mapping of genomic 5-hydroxymethylcytosine in mouse embryonic stem cells. Cell Rep., 3, 567–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sureau A. et al. (2001) SC35 autoregulates its expression by promoting splicing events that destabilize its mRNAs. EMBO J., 20, 1785–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulwach K.E. et al. (2011) 5-hmC-mediated epigenetic dynamics during postnatal neurodevelopment and aging. Nat. Neurosci., 14, 1607–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M. et al. (2009) Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science, 324, 930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai K.-W. et al. (2015) Reduction of global 5-hydroxymethylcytosine is a poor prognostic factor in breast cancer patients, especially for an er/pr-negative subtype. Breast Cancer Res. Treatment, 153, 219–234. [DOI] [PubMed] [Google Scholar]
- Tyner C. et al. (2016) The UCSC Genome Browser database: 2017 update. Nucl. Acids Res., 45, D626–D634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voichiţa C. et al. (2012). Incorporating gene significance in the impact analysis of signaling pathways. In: 2012 11th International Conference on Machine Learning and Applications (ICMLA), vol. 1, pp. 126–131. IEEE, Boca Raton, FL, USA.
- Wang D. et al. (2013) The expression of glyceraldehyde-3-phosphate dehydrogenase associated cell cycle (GACC) genes correlates with cancer stage and poor survival in patients with solid tumors. PLoS One, 8, e61262.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H. et al. (2011) Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev., 25, 679–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y. et al. (2011) Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Mol. Cell, 42, 451–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H. et al. (2013) Tumor development is associated with decrease of TET gene expression and 5-methylcytosine hydroxylation. Oncogene, 32, 663–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yildirim O. et al. (2011) Mbd3/NURD complex regulates expression of 5-hydroxymethylcytosine marked genes in embryonic stem cells. Cell, 147, 1498–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young C.N. et al. (2008) Reactive oxygen species in tumor necrosis factor-α-activated primary human keratinocytes: implications for psoriasis and inflammatory skin disease. J. Investigative Dermatol., 128, 2606–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M. et al. (2012) Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell, 149, 1368–1380. [DOI] [PMC free article] [PubMed] [Google Scholar]