

Abstract
Summary
The genome-wide chromosome conformation capture (Hi-C) has revealed that the eukaryotic genome can be partitioned into A and B compartments that have distinctive chromatin and transcription features. Current Principle Component Analyses (PCA)-based method for the A/B compartment prediction based on Hi-C data requires substantial CPU time and memory. We report the development of a method, CscoreTool, which enables fast and memory-efficient determination of A/B compartments at high resolution even in datasets with low sequencing depth.
Availability and implementation
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
The development of the proximity-ligation based methods for chromatin conformation capture (3 C, 4 C, 5 C and Hi-C) has greatly improved the understanding of three-dimensional chromatin organization in the eukaryotic nucleus (Gibcus and Dekker, 2013). One important feature found in mammalian Hi-C studies is that the genome is organized into A or B compartments (Lieberman-Aiden et al., 2009). Whereas the A compartment corresponds to genomic regions containing transcriptionally active and open chromatin (Lieberman-Aiden et al., 2009), the B compartment corresponds to the heterochromatin regions associated with the nuclear lamina and nucleolar (Stevens et al., 2017; van Steensel and Belmont 2017). Recent studies showed that the A/B compartment organization is independent of the topologically associated domains (TADs) (Dixon et al., 2012) and is more conserved than the organization of TADs at the single-cell level (Nora et al., 2017; Stevens et al., 2017). Therefore, understanding the A/B compartment organization is critical in deciphering 3D genome organization.
The current method for calculating A/B compartments is based on the Principal Component Analysis (PCA) of the normalized Hi-C interaction matrix (Lieberman-Aiden et al., 2009). The first eigenvector (Principal Component 1, PC1) of the correlation matrix is then defined as the compartment score, and genomic windows with positive or negative compartment scores are defined as A or B compartment, respectively. The PCA-based method has two major limitations. First, PCA is a descriptive statistical method designed for reducing dimensionality and the exact biological meaning of the compartment score is elusive. In this sense, compartment scores calculated from different Hi-C datasets may not be directly comparable. Second, PCA is slow and memory-inefficient when applied to large interaction matrix. This prohibits its application to high-resolution analysis of the compartment structure for most labs.
Here, we proposed a statistical model to infer A/B compartments from Hi-C data. The output compartment score reflects the chance of a genomic window being in the A compartment. The implemented tool, namely CscoreTool, is ∼30 times faster and more memory-efficient than the existing PCA-based method for the same resolution. CscoreTool also works at high resolution for datasets with low sequencing depth.
2 Materials and methods
We assume that each genomic window i has a chance Pi to be in the A-compartment in an individual cell. By defining C-score as , which ranges between −1 and 1, we deduce a log-likelihood function
where nij is the observed number of contacts, dij is the distance along the genome, H(dij) is the scaling factor accounting for the decrease of interactions at longer genomic distance, and Bi and Bj are the bias factors from Hi-C experiments, which could come from PCR biases or genome mappability. We performed maximum-likelihood estimation for the model parameters B, C and H using an iterative algorithm (See the Supplementary Methods for details). Unlike eigenvectors, C-scores of different samples can be directly compared because they have clear biological implications.
We implemented the algorithm with C ++ and tested the resultant tool, namely CscoreTool, on the high resolution Hi-C datasets of the GM12878, HUVEC, K562, and NHEK cell lines (Rao et al., 2014). Among PCA-based variants, we chose a widely used tool HOMER for comparison (Heinz et al., 2010). We also compared the performance to custom C ++ script using libpca, which is a C ++ PCA implementation. Mapped reads (to mm9) for Hi-C libraries of GM12878, HUVEC, K562 and NHEK cells were downloaded from the GEO database (GSE63525). Only reads with MAPQ ≥ 30 at both ends were kept, and since the analyses for each chromosome are independent, we focused our test on chromosome 1. CscoreTool was tested for 1, 5, 10, 25, 50 and 100 Kb resolutions, while HOMER was tested for 10, 25, 50 and 100 Kb resolutions with all the other parameters set as default. All tests were performed on the Memex high-performance computer of Carnegie Institution for Science. Since HOMER does not support parallelization within one chromosome, we used only one CPU (2.5 GHz) for the comparison.
3 Results
We first compared the running time and memory usage between CscoreTool and HOMER using the GM12878 dataset. At the same resolutions, CscoreTool is ∼30 times faster than HOMER (Fig. 1A). HOMER uses 40 min to 3 h for PCA analysis at 25–100 Kb resolutions but requires over 20 h for 10 Kb-resolution, and it stopped running at 5 Kb resolution because it could not handle the large matrix. In contrast, CscoreTool uses a few minutes for analyses at 25–100 Kb resolution; 35 min for 10 Kb resolution; 2 h for analyses at 5 Kb resolution; and ∼3 days for analysis at 1 Kb resolution. The time consumed can be further reduced by using parallelization. The memory usage of CscoreTool is also much less than HOMER (Fig. 1B). The 1Kb-resolution analysis by CscoreTool uses < 10 GB memory, whereas HOMER uses >60GB memory for the same dataset at the 10–100Kb resolutions that we could test. To test whether the better performance of CscoreTool is mainly because of C ++ language, we compared CscoreTool to custom PCA script using the libpca library written in C ++ (referred to as PCA_C ++ in the following). The PCA_C ++ method is >2 times faster than HOMER, but still ∼10 times slower than CscoreTool at 10 Kb resolution (Fig. 1A). PCA_C ++ uses about 20 GB at 10 Kb resolution, but stops working at 5 Kb resolution due to an error in acquiring memory. Thus, CscoreTool is much faster and more memory-efficient than PCA-based compartment analysis methods.
Fig. 1.
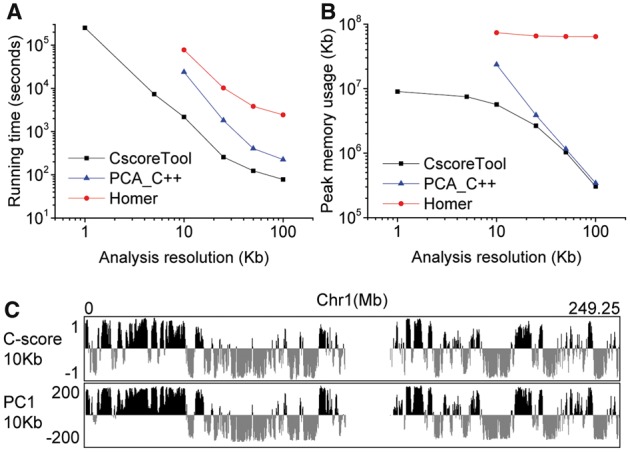
(A and B) Comparison of running time and memory usage for CscoreTool, HOMER, and PCA method written in C ++. (C) Whole-chromosome view of C-score by CscoreTool and PC1 by HOMER calculated at 10 Kb resolution
We then compared the results of CscoreTool to PCA-based methods using the GM12878 dataset. As different PCA implementations give identical results, we used PC1 from HOMER to represent PCA methods. 10 Kb resolution was used, which is the best resolution that PCA-based methods could reach. Whole-chromosome view showed that the patterns between different methods are in general similar (Fig. 1C and Supplementary Table S1), indicating that both methods capture the large-scale structure of A/B compartments. Finer scale comparison reveals some differences between the methods (Supplementary Fig. S1A-B and Table S1). For example, HOMER predicted a long region as the A-compartment (Supplementary Fig. S1A), whereas CscoreTool showed that a stretch of chromatin within this region belonged to the B-compartment. Since DNase I sensitivity is associated with chromatin in the A-compartment (Lieberman-Aiden et al., 2009), we analyzed the ENCODE DNase I data (Consortium, 2012) in these cells and found that the B-compartment predicted by CscoreTool had low DNase I sensitivity, indicating that our method correctly predicted this region as the B-compartment (Supplementary Fig. S1A). We also found small A-compartments with DNase I peaks that were missed by HOMER (Supplementary Fig. S1B). Similar regions are found for other cell types (Supplementary Figs S1C and D). More generally, we separated chromosome 1 into 4 types of regions for the GM12878 cell: common A-compartment by both methods; CscoreTool-specific A-compartment (classified as B-compartment by HOMER); HOMER-specific A-compartment (classified as B-compartment by CscoreTool); and common B-compartment (Supplementary Table S1). We then calculated the coverage by DNase I hotspots on these four types of regions, and found that the CscoreTool-specific A-compartment has similar DNase I hotspot coverage as the common A-compartment; while the HOMER-specific A-compartment has similar DNase I hotspot coverage as the common B-compartments. These results show that CscoreTool can more accurately detect the A- and B-compartments than PCA-based methods.
Rao et al. (2014) used clustering method to detect sub-compartments at 100Kb resolution for the GM12878 cell line. Although CscoreTool currently does not support sub-compartment inference, we combined their A1 and A2 into the A-compartment; B1–B4 into the B-compartment, respectively, and compared their result with ours. We found that the CscoreTool-specific A-compartment also has much higher DNase I hotspot coverage than the clustering-method-specific A-compartment, supporting that CscoreTool is better at detecting the A- and B-compartments than the clustering method.
The high-resolution GM12878 dataset we used here has 3.51 G mapped non-redundant reads (Rao et al., 2014), which requires substantial amount of sequencing that is cost-prohibitive for many labs. Therefore, we created two smaller datasets by randomly selecting 10% and 1% of from the 3.51 G mapped reads. The 10% dataset corresponds to 351 M mapped non-redundant reads, which is common for most Hi-C datasets. The 1% dataset corresponds to 35.1 M mapped non-redundant reads, which is common for low-cell-number Hi-C data such as those in early embryo development (Du et al., 2017; Ke et al., 2017). CscoreTool gave very consistent results among all sequencing depth (Supplementary Fig. S2) on large scale. In contrast, HOMER showed more inconsistency between different depths (Supplementary Fig. S2). Taken together, we show that CscoreTool can perform accurate high-resolution compartment analysis at both high and low sequencing depth with similar accuracy and it requires less time and computer memory than the commonly used PCA-based methods.
Funding
This work has been supported by the National Institutes of Health (R01GM106023 to Y.Z.).
Conflict of Interest: none declared.
Supplementary Material
References
- Consortium E.P. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature, 489, 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon J.R. et al. (2012) Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature, 485, 376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z. et al. (2017) Allelic reprogramming of 3D chromatin architecture during early mammalian development. Nature, 547, 232–235. [DOI] [PubMed] [Google Scholar]
- Gibcus J.H., Dekker J. (2013) The hierarchy of the 3D genome. Mol. Cell, 49, 773–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S. et al. (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell, 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke Y. et al. (2017) 3D chromatin structures of mature gametes and structural reprogramming during mammalian embryogenesis. Cell, 170, 367–381.e320. [DOI] [PubMed] [Google Scholar]
- Lieberman-Aiden E. et al. (2009) Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science, 326, 289–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nora E.P. et al. (2017) Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell, 169, 930–944. e922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao S.S. et al. (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens T.J. et al. (2017) 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature, 544, 59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B., Belmont A.S. (2017) Lamina-associated domains: links with chromosome architecture, heterochromatin, and gene repression. Cell, 169, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.