

Abstract
The complement system is a complex, carefully regulated proteolytic cascade for which suppression of aberrant activation is of increasing clinical relevance, and inhibition of the complement alternative pathway is a subject of intense research. Here, we describe the nanobody hC3Nb1 that binds to multiple functional states of C3 with subnanomolar affinity. The nanobody causes a complete shutdown of alternative pathway activity in human and murine serum when present in concentrations comparable with that of C3, and hC3Nb1 is shown to prevent proconvertase assembly, as well as binding of the C3 substrate to C3 convertases. Our crystal structure of the C3b–hC3Nb1 complex and functional experiments demonstrate that proconvertase formation is blocked by steric hindrance between the nanobody and an Asn-linked glycan on complement factor B. In addition, hC3Nb1 is shown to prevent factor H binding to C3b, rationalizing its inhibition of factor I activity. Our results identify hC3Nb1 as a versatile, inexpensive, and powerful inhibitor of the alternative pathway in both human and murine in vitro model systems of complement activation.
Keywords: innate immunity, complement, convertase, antibody, inhibitor, structural biology, alternative pathway
Introduction
The complement system is an important effector of innate immunity and plays a crucial role in the detection, phagocytosis, and killing of invading pathogens and clearance of immune complexes. Complement is also increasingly recognized for its role in maintenance of host homeostasis through clearance of apoptotic and necrotic cells (1). Furthermore, cumulative evidence emphasizes that complement is a key player during developmental processes, including synaptic pruning where inputs inefficient in driving postsynaptic responses are eliminated in early development (2).
Activation of the complement system relies on pattern-recognition molecules recognizing pathogen-associated molecular patterns or danger-associated molecular patterns on the activator. There are three well-characterized activation pathways for complement, the classical pathway, the lectin pathway, and the alternative pathway (AP).2 All three pathways involve cleavage of the protein C3 into the 10-kDa anaphylatoxin C3a and the 175-kDa opsonin C3b (see Fig. 1A). The pathways converge via the AP into a common terminal pathway in which the proteolytic cleavage of C5 elicits a potent proinflammatory response and triggers the assembly of an intricate membrane penetrating complex capable of killing specific pathogens (3). The C3b molecules generated in any of the three pathways may attach covalently to activator hydroxyl or amino groups through their nucleophilic attack on an internal thioester accessible in nascent C3b. Upon association of C3b and factor B (FB), the proconvertase C3bB is formed, and FB is subsequently cleaved (to Bb and Ba) by factor D (FD). The resulting C3bBb complex (the AP C3 convertase) then cleaves C3, thereby enabling AP amplification to occur (see Fig. 1A). The AP also initiates continuously through slow hydrolysis of the C3 thioester, which is known as “tick-over.” The product C3(H2O) is formed in the fluid phase, but it may become recruited to an activator by a binding partner such as properdin (FP) and subsequently recruit FB, resulting in formation of another form of the AP C3 convertase: C3(H2O)Bb. Both AP C3 convertases are inherently labile and dissociate irreversibly with half-lives in the range of 2 min (4) but are stabilized 5–10-fold by FP binding (5, 6). Healthy host cells may become tagged by C3b because of C3 tick-over or through the “bystander” effect, in which C3b generated on a nearby activator attaches to a host cell. The AP is therefore tightly regulated to avoid damage on self, and convertases on host cells are quickly dissociated by negative regulators with decay acceleration activity. Cofactors also assist factor I (FI) in degrading C3b irreversibly to iC3b and C3dg, which are inactive in convertase formation (see Fig. 1A).
Figure 1.

The alternative pathway and negative stain EM analysis. A, the AP can be initiated by tick-over generated C3(H2O) or from C3b generated within the classical or lectin activation pathways. Both C3b or C3(H2O) can form the proconvertase with FB. C3b can be degraded to iC3b by FI assisted by a cofactor. The (P) signifies that the AP convertases may bind and be stabilized by properdin. B, negative stain EM 2D classes of the complex between hC3Nb1 and C3b. The TE domain adopts multiple alternative positions as compared with the C3b MG1–8 domains. Scale bar, 100 Å. C, 3D reconstruction of negative stain EM particles of the C3b–hC3Nb1 complex with the crystal structure of C3b (PDB entry 5FO7) fitted to the envelope. Excess density corresponding to the Nb is marked by asterisks.
Modulators of the immune system have a great potential in both basic research and as therapeutics. Complement overactivation caused by genetic or acquired abnormalities (mutations or autoantibodies) or tissue alterations (ischemia/reperfusion, neoplastic transformation) induces tissue damage and sustains chronic inflammation (7, 8). The C5-specific mAb eculizumab inhibits C5 cleavage and thereby functions as an overall inhibitor of the terminal pathway. This antibody has successfully been used for 10 years to treat patients suffering from the two rare diseases: atypical hemolytic uremia and paroxysmal nocturnal hemoglobinuria, proving that systemic and chronic inhibition of the complement terminal pathway can be done without severe adverse effects (9, 10). This has sparked substantial interest in developing putative complement modulating drugs targeting also the upstream classical, lectin, and alternative pathways (8, 11). In addition to their value as possible future therapeutics, such modulators are also useful tools for analysis of the molecular events in the complement cascade for both in vitro and in vivo investigations. A recent example is the use of the C3 inhibitor compstatin to challenge the idea that FP acts as a pattern-recognition molecule in the alternative pathway independent of prior C3b deposition (12).
In this study, we have developed a nanobody (Nb) toward C3. Nbs are increasingly used in structural biology, imaging, assays, biosensors, and diagnostics, and Nb selection after llama immunization is well-established (13). These single domain antibodies are derived from the VHH domain of cameloid antibodies and contain three complementarity-determining regions (CDRs) specific to each Nb that mediate antigen binding. Because of their small size (∼13 kDa) and stable format, recombinant Nbs are often soluble and may be expressed in a fast and cost-effective manner in Escherichia coli. Hence, this class of miniature antibodies is attractive for proof-of-principle development of immune modulators.
Here, we describe, to our knowledge, the first Nb-based inhibitor of a complement activation pathway. We have selected the nanobody hC3Nb1 against human C3b and show that it binds with low-nanomolar or picomolar affinity to native C3, C3b, iC3b, and the C3(H2O) mimic C3-methylamine (C3MA) and yields efficient inhibition of the alternative pathway in human and murine serum. The crystal structure of the C3b–hC3Nb1 complex was determined at 2.65 Å resolution, which allows us to rationalize the complete inhibition of AP activation and FI–mediated C3b degradation exerted by hC3Nb1. Our results, combined with the ease of producing and modifying Nbs, identifies hC3Nb1 as a versatile and powerful inhibitor of the alternative pathway that demonstrates great promise as a unique tool for studying complement C3.
Results
Nanobody selection and structural studies of its complex with C3b
To select a Nb specifically targeting human C3b, we immunized a llama with purified C3b, and from a blood sample we generated a phage display library in which the Nbs were presented as pIII fusions. We performed two rounds of phage display selection and hereafter randomly picked 96 colonies and tested these in an ELISA against immobilized C3b. The positive clones were sequenced, aligned, and clustered into families, and the corresponding Nbs were produced in bacteria as His-tagged proteins and purified to homogeneity. Based on initial C3 deposition assays, we chose to focus on one Nb termed hC3Nb1. To study the interaction between C3b and this Nb, we isolated the complex by size-exclusion chromatography (SEC), and aliquots taken from the early part of the SEC peak were analyzed by single particle negative stain EM (Fig. 1B). Using the MG domains of C3b filtered to a resolution of 80 Å as a reference model, we obtained a 3D reconstruction with an approximate resolution of 25 Å in which the MG domains, the C345c domain, the CUB, and the associated thioester (TE) domains of C3b were easily recognized (Fig. 1C). The TE domain was separated by ∼10 Å from the MG1 domain, as previously observed in small angle X-ray scattering studies of C3b and its classical pathway homolog C4b (14, 15). Additional density proximal to the MG7 domain of a docked C3b molecule (Fig. 1C) fitted the approximate size of a Nb, suggesting that this domain contributed significantly to the epitope.
To study the C3b–hC3Nb1 interaction in detail, we cocrystallized the complex, collected X-ray diffraction data extending to a maximum resolution of 2.65 Å, and solved the structure by molecular replacement. The resulting model confirmed the interaction between hC3Nb1 and the C3b MG7 domain (Fig. 2A), and clear omit electron density for unmodeled CDRs was present (Fig. 2B). Iterative manual rebuilding and refinement resulted in a final model of the C3b–hC3Nb1 complex with an Rfree value of 0.26 and excellent overall geometry (Table 1). The epitope recognized by the Nb is formed by two surface-exposed loops of the MG7 domain together with a small part of the Nt-α′ region generated upon cleavage of C3 (Fig. 2C). The C3b-interacting residues in hC3Nb1 forming the paratope are located almost exclusively in CDR2 and CDR3. The two interacting surfaces from C3b and hC3Nb1 exhibit a shape complementarity of 0.786, which is a relatively high value for intermolecular interfaces between two proteins (16). In a systematic examination of 131 antigen–nanobody complexes determined at a resolution better than 3.0 Å, we found that the mean value of surface complementarity is 0.715. However, 16 complexes had a higher complementarity than observed for the C3b–hC3Nb1 complex, indicating that the observed complementarity is high but not exceptional for a Nb–antigen complex (Fig. S1A). According to PISA (17), the interface is relatively polar and contains 12 hydrogen bonds and 2 salt bridges. The interaction between C3b and hC3Nb1 buries 1410 Å2 of surface area, which is slightly below the average value of 1636 Å2 observed in our collection of 131 high-resolution antigen–nanobody structures (Fig. S1B).
Figure 2.

The crystal structure of the C3b–hC3Nb1 complex. A, cartoon representation of the complex showing the Nb located close to the MG7 domain and the Nt-α′ region. B, omit 2mFo − DFc electron density contoured at 1σ around the CDR3 loop of hC3Nb1 and Arg855 of C3b. C, overview of the interaction surface between C3b and hC3Nb1 involving CDR2 and CDR3 from the Nb and MG7 and Nt-α′ of C3b. D–G, details of the interaction between hC3Nb1 and C3b. Salt bridges and hydrogen bonds are indicated by black dotted lines.
Table 1.
Data collection and refinement statistics
Rmeas = Σhkl(N/(N − 1))½Σi|Ii(hkl) − I(hkl)|/ΣhklΣiIi(hkl). CC½ is the correlation between random half-data sets. The statistics for the highest-resolution shell are shown in parentheses.
| Data collection | |
|---|---|
| Wavelength (Å) | 0.9802 |
| Resolution range (Å) | 62.88–2.65 (2.745–2.65) |
| Space group | P21212 |
| Cell dimensions: a, b, c (Å) | 255.31, 64.88, 144.72 |
| Total reflections | 2,860,846 (70,986) |
| Unique reflections | 70,861 (6900) |
| Multiplicity | 40.4 (10.3) |
| Completeness (%) | 99.84 (98.94) |
| Mean I/sigma(I) | 17.42 (0.86) |
| Wilson B-factor | 86.41 |
| Rmeas | 0.167 (4.17) |
| CC1/2 | 0.999 (0.611) |
| Refinement | |
| Rwork (%) | 23.54 (36.96) |
| Rfree (%) | 26.04 (42.30) |
| Non-hydrogen atoms (protein/glycan) | 13,132/53 |
| Bonds lengths (Å) | 0.002 |
| Angles (°) | 0.52 |
| Ramachandran favoured/outliers (%) | 96.68/0.0 |
| Clash score | 4.30 |
| Average B-factor | 113.91 |
Residues from CDR2 and CDR3 appear to be equally important for C3b recognition (Fig. 2, D–G). From CDR2 Tyr59 in combination with Thr50–Ile51–Asn52 form a pocket for the surface-exposed Phe920 in C3b MG7 domain (Fig. 2D), whereas Asn52, Arg53, and Ser54 form hydrogen bonds with the Nt-α′ region in C3b (Fig. 2E). In particular C3b Glu759 is contacted by Asn52 but also engages in an electrostatic interaction with Arg57 (Fig. 2F). The interaction of CDR3 with C3b is dominated by the insertion of hC3Nb1 Trp102 into a cavity formed by C3b Arg855, Ile921, and Leu860 with the tryptophan–arginine side chain interaction featuring a classical planar aromatic stacking (Fig. 2G). In addition, the main chain of Gly101 and Trp102 in CDR3 are engaged in hydrogen bonding with C3b, and Glu109 interacts electrostatically with the side chain of C3b Arg855, thereby promoting its stacking with hC3Nb1 Trp102. In summary, our structural analysis revealed that hC3Nb1 extensively recognizes an epitope formed primarily by two loops in the C3b MG7 domain by a combination of polar and hydrophobic interaction, with residues in the C3b Nt-α′ region also forming polar contacts with the Nb.
hC3Nb1 is a powerful inhibitor of the alternative pathway
By comparing the structure of the C3b–hC3Nb1 complex with our model for convertase–substrate recognition (18), it was inferred that hC3Nb1 prevents substrate binding to the AP C3 convertase. In addition, superposition of C3b–hC3Nb1 with the structure of the proconvertase C3bB (19) suggested that the Nb also prevents FB from binding to C3b (see below). To assess the influence of hC3Nb1 on AP activity, the deposition of C3 fragments by the AP was quantified using zymosan-coated microtiter wells. The results showed that hC3Nb1 effectively inhibits the AP in human serum. Complete inhibition was observed at hC3Nb1 concentrations corresponding approximately to an equimolar ratio between hC3Nb1 and C3 (Fig. 3A). Furthermore, the relative contributions of Nb residues suggested to contribute significantly to C3b recognition were examined by introducing the point mutations R53A, R57A, W102A, and Y112A in hC3Nb1. Of these, W102A completely abolished the ability of hC3Nb1 to inhibit the AP, whereas more modest effects were observed for the other mutations. The hC3Nb1 epitope is relatively conserved between human and murine C3 (Fig. 3B). We therefore tested whether hC3Nb1 also inhibits the AP in murine serum, and we found that hC3Nb1 was an equally effective inhibitor of the murine AP (Fig. 3C). Moreover, the effects of the various hC3Nb1 mutants were similar to those observed in human serum.
Figure 3.

The hC3Nb1 is a potent inhibitor of the alternative pathway. A, assay in human serum on a zymosan-coated AP activating surface. The horizontal axis gives the concentration of nanobodies present in the serum. The vertical axis shows the level of C3 fragment deposition with 100% deposition defined as the signal from serum without added nanobody. The dotted line indicates the final molar concentration of C3 in the assay based on a concentration of 5.3 μm in undiluted plasma (61). B, multiple sequence alignment of the hC3Nb1 epitope from human C3 and four animals of relevance for animal models for which residues marked with gray shading differ from the human sequence. Colored triangles indicate residues involved in interactions with hC3Nb1. C, assay in mouse serum on a zymosan-coated alternative pathway activating surface depicted as in A. The dotted line represents the final molar concentration of C3 in the assay assuming 4.2 μm C3 in 100% serum from C57BL6 male mice (62).
The hC3Nb1 nanobody inhibits proconvertase assembly and factor I degradation of C3b
Our structure predicts that hC3Nb1 interferes with FB binding to C3b because the glycan attached to Asn122 in the FB CCP2 domain cannot be accommodated simultaneously with the Nb (Fig. 4A). To test this putative role of FB Asn122 glycan in the inhibitory mechanism, we mutated the asparagine to alanine (N122A) in a FB molecule already carrying the convertase-stabilizing mutation D279G. We then conducted SEC analysis of samples in which the two FB variants (Asn122/Gly279 and Ala122/Gly279) were mixed with C3b or a preformed C3b–hC3Nb1 complex (Fig. 4, B–E). The Nb prevented binding of FB Asn122/Gly279 to C3b as indicated by SEC, because the major peak eluted 0.22 ml later in the presence of hC3Nb1 and did not contain FB, whereas the peak containing free FB increased significantly (Fig. 4, B and C, and Fig. S2). In contrast, when FB Ala122/Gly279 was used, the major peak eluted slightly earlier (0.06 ml; Fig. 4E) and contained FB, C3b, and hC3Nb1 (Fig. S2) and the peak for unbound FB did not increase. Hence, the mutually exclusive binding of the Nb and FB to C3b is due to spatial overlap with the Asn122 glycan. Because this residue and Ser124 are conserved in murine FB, the Asn-linked glycan is likely to be present, which would explain the hC3Nb1-mediated inhibition of AP activity in murine serum (Fig. 3C).
Figure 4.

hC3Nb1 inhibits convertase assembly and FH-mediated FI cleavage of C3b caused by spatial overlap with FH and FB. A, superposition of the C3b–hC3Nb1 and the C3bB (PDB entry 2XWB) structures shows that the N122 linked glycan of FB overlaps significantly with the Nb. B–E, SEC analysis of the AP proconvertase with and without hC3Nb1. The Nb inhibits the formation of the AP proconvertase only when the Asn122–linked glycan is present on FB. F, superposition of the C3b–hC3Nb1 structure onto the structure of C3b bound to FH CCP1–4 (PDB entry 2WII) reveals that the binding sites of FH CCP1 and the Nb overlap substantially. G, SDS-PAGE analysis of a FH-assisted FI cleavage assay showing that hC3Nb1 completely inhibits iC3b formation.
Factor H (FH) is a fluid phase AP regulator acting as a decay accelerating factor promoting dissociation of Bb from the AP C3 convertase and as cofactor for FI in the degradation of C3b to iC3b (Fig. 1A). Through recognition of host specific glycans such as sialic acid, heparin, and sulfated glycosaminoglycans, FH can distinguish self from nonself and inhibit AP amplification on host cells together with three host cell membrane-bound regulators (20). In factor H, the four N-terminal CCP domains interact with the C3b Nt-α′ region; the MG domains 1, 2, 6, and 7; and the TE domain, whereas the terminal CCPs 19–20 exclusively contact the TE domain (21–23). Both decay accelerating and cofactor activities are located within FH CCP 1–4. The binding of these FH domains to C3b is likely to confer a C3b conformation suitable for FI binding leading to the subsequent cleavage of the C3b CUB domain (24). A structural comparison of the C3b–FH CCP 1–4 complex with the C3b–hC3Nb1 complex showed that the binding sites of the Nb and FH CCP1 substantially overlap, thus predicting that hC3Nb1 prevents FH from binding to C3b (Fig. 4F). Moreover, this would imply that hC3Nb1 prevents FI-mediated C3b cleavage. We tested this hypothesis by incubation of C3b, FH, and FI in the presence and absence of 1.5 molar excess of hC3Nb1 compared with C3b and followed FI degradation of the C3b α′-chain over time by SDS-PAGE analysis (Fig. 4G). As expected, we observed fast degradation in the absence of the Nb that was complete after 2 h, whereas we did not observe any significant C3b α′-chain degradation after 8 h in the presence of hC3Nb1. This shows that hC3Nb1 is an efficient inhibitor of FH cofactor activity.
hC3Nb1 binds with picomolar affinity to multiple functional states of C3
The observation that hC3Nb1 acts as an efficient inhibitor when present in roughly stoichiometric amounts relative to C3 in AP assays prompted us to quantitate the strength of the interaction by surface plasmon resonance (SPR). We introduced a C-terminal substrate motif for the BirA enzyme in hC3Nb1, and after biotinylation of the C-terminal residue, we bound the Nb to streptavidin immobilized by amine coupling on an SPR sensor chip. This made it possible to regenerate the sensor with low pH between different analytes (C3b, native C3, iC3b, and C3MA) and competitors for hC3Nb1 (FH, FB, and variants of C3Nb1). One additional major advantage of this approach was that the Nb CDR regions were likely not to be influenced by immobilization, because they are located at the opposite end of the protein compared with the biotinylated C terminus. Both C3b (Fig. 5A) and C3 (Fig. 5B) formed a high-affinity complex with hC3Nb1 exhibiting especially low off-rates of 5.1 × 10−5 s−1 and 4.5 × 10−5 s−1, whereas the dissociation constants for the C3b–hC3Nb1 and C3–hC3Nb1 complexes were 240 and 890 pm, respectively. C3MA and iC3b also bound tightly to the hC3Nb1 (Fig. S3, A and B), but because of interaction of the sample with the reference layer, the kinetics constants could not be determined. We also performed competition assays with different hC3Nb1 mutants to compare their relative affinity for C3b. C3b was preincubated with 10-fold molar excess of the Nb mutants or the WT Nb before being injected on the hC3Nb1 surface. The W102A mutant was unable to compete with the immobilized WT Nb for binding to C3b, whereas all other mutants showed at least some binding to C3b as evidenced by their competition with immobilized hC3Nb1 (Fig. 5C). The R57A variant showed only a slight reduction in signal as compared with C3b alone, signifying that it only has limited affinity toward C3b. Both R53A and Y112A reduced the signal significantly as compared with C3b alone, although not as much as WT hC3Nb1, showing that these mutants also have reduced affinities toward C3b. Similarly, we performed competition assays with C3b in complex with either FB, FB(D279G) or FH. In these experiments we observed a clear reduction in binding of C3b to the Nb surface for all complexes as compared with C3b alone (Fig. 5D). This further confirms that the C3b–FH and C3b–FB complexes are mutually exclusive with the hC3Nb1–C3b interaction.
Figure 5.

SPR analysis of the interaction of hC3Nb1 with C3b and C3. A and B, representative SPR concentration series for C3b (A) or C3 (B) are displayed. The measured (gray) and fitted (red) SPR curves are shown for 50, 20, 10, 5, 2, 1, 0.25, and 0.1 nm for C3b or 20, 10, 5, 2.5, 1.25, 0.61, 0.32 and 0.15 nm for C3. The ka, kd, and KD values are given as their average fitted from three independent concentration series ± the standard deviation. C, competition SPR assay with 50 nm C3b preincubated with 500 nm of different hC3Nb1 mutants. D, competition SPR assay with 50 nm C3b preincubated with 500 nm FB or FH. In C the curve for hC3Nb1(W102A) is hidden below the C3b curve. RU, response units.
hC3Nb1 prevents convertase recognition of C3
The strong binding to native C3 was somewhat surprising given that the C3b Nt-α′ region, which is located far from the MG7 domain in native C3, was observed to be part of the epitope. To investigate whether the strong binding of hC3Nb1 to native C3 was due to induction of a C3(H2O)-like conformation, we incubated freshly purified C3 and the Nb in the presence of 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB) and quantitated DTNB reaction with free thiol groups appearing from cleavage of C3 thioester. In a parallel experiment, we treated C3 with methylamine, and the methylamine reaction indicated the presence of 1.00 ± 0.016 intact thioester group/C3 molecule, whereas incubation with hC3Nb1 was unable to induce any significant increase in absorbance. These findings imply that the Nb indeed binds to C3 without inducing its conversion to a C3(H2O)-like conformation.
The strong binding of the Nb to native C3 combined with our substrate–convertase model suggested that hC3Nb1 not only prevents FB from binding to C3b but may also interfere with AP activation by precluding the substrate C3 from being recognized by the C3 convertases. To address whether the mechanism of AP inhibition exerted by hC3Nb1 is solely at the level of proconvertase formation (C3b binding) or also extends to the substrate level (C3 binding), we used the C3 homolog cobra venom factor (CVF), which forms a fluid phase C3/C5 convertase together with host FB. We formed the CVFBb convertase and studied the effect of the Nb on C3 cleavage. CVFBb cleaved most of the C3 substrate after 480 min, whereas in a parallel experiment with hC3Nb1 present in 1.2-fold molar excess, only a minor fraction of C3 was cleaved (Fig. 6A). In contrast, the Nb did not interfere with C5 cleavage by CVFBb (Fig. 6B), showing that the observed inhibition of C3 cleavage is not due to hC3Nb1 binding to CVF. Thus the observed abrogation of C3 cleavage confirms that the Nb also functions as an inhibitor at the C3 substrate level in addition to preventing AP proconvertase assembly. This was further supported in a functional assay where activation of the lectin pathway supported assembly of the CP/LP C3 convertase C4b2a. The addition of hC3Nb1 significantly decreased C3 deposition, but even at the highest concentration of 700 nm, inhibition beyond 70% compared with the control nanobody was not observed (Fig. 6C). The fact that the LP inhibition by hC3Nb1 is markedly less efficient than inhibition of the AP (Fig. 3), combined with the results obtained with CVFBb (Fig. 6A), suggests that the inhibitory mechanism exerted by hC3Nb1 in the alternative pathway mainly stems from the inhibition of C3bB assembly rather than the ability of hC3Nb1 to interfere with C3 substrate binding to the C3bBb convertase.
Figure 6.
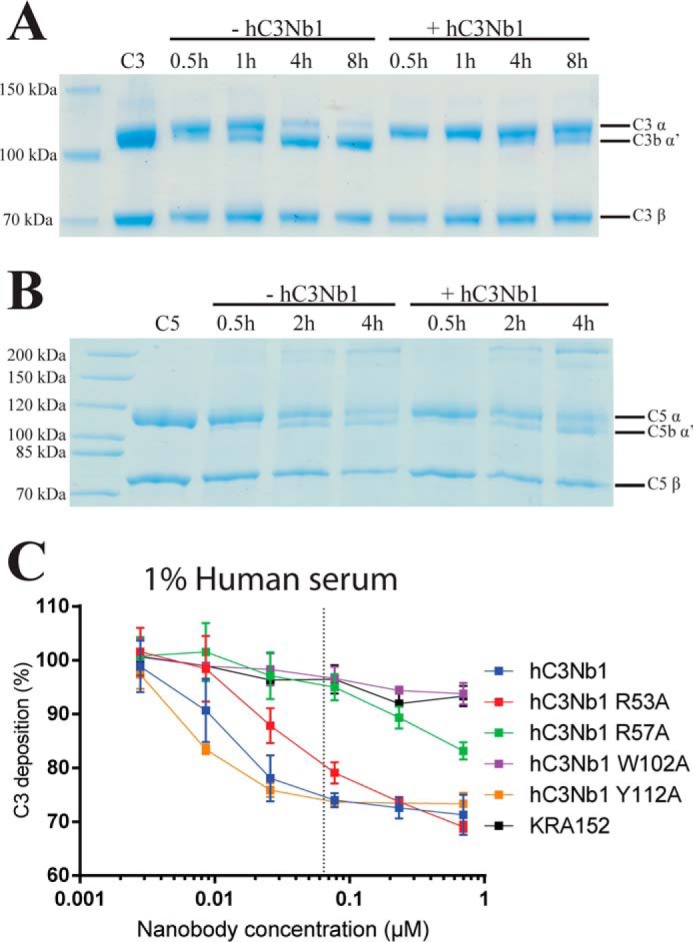
The hC3Nb1 nanobody also inhibits C3 cleavage at the substrate level. A, C3 cleavage assay with the CVFBb convertase revealing that also for this convertase not containing C3b, hC3Nb1 is an inhibitor of C3 cleavage. B, C5 cleavage assay with the CVFBb convertase demonstrating that hC3Nb1 has no effect on C5 cleavage. The bands around at and below the 200-kDa marker represents C5b not completely reduced/denatured. C, assay in 1% human serum on a mannan-coated LP activating surface. The horizontal axis gives the concentration of nanobodies added to the serum. The vertical axis shows the level of C3 fragment deposition with 100% deposition defined as the signal from serum without added nanobody. The dotted line indicates the final molar concentration of C3 in the assay based on a concentration of 5.3 μm in undiluted plasma.
Discussion
Development of modulators of the immune system with the aim of preventing pathogenesis is a rapidly increasing topic in biomedicine, and many different principles have been described for the control of the complement system. Monoclonal antibodies, fragments of cofactors, RNA aptamers, small molecules, pathogen complement evasion proteins, directed evolution of protease inhibitor scaffolds, and siRNAs are examples of molecule classes that have been evaluated as complement suppressors (8). Considering that the nanobody technology has been known for two decades and is relatively accessible (25), it is surprising that there are, to our knowledge, no other reports describing a Nb-based inhibitor of complement activation. Here, we present such an inhibitor, the hC3Nb1 nanobody, which efficiently abrogates AP activity by interfering with both convertase formation and C3 substrate recognition, leading to complete inhibition when present in stoichiometric levels as compared with C3. We have earlier proposed a general model, based on the C5–CVF complex, of how substrates are recognized by the Bb bearing C3b in the AP C3 and C5 convertases (18). This model, combined with structural information, offers an explanation for how the C3-specific peptide compstatin (26), the C3b-specific S77 antibody (27), the ectodomain of complement receptor Ig (28), the C5-specific antibody eculizumab (29), and, in this study, hC3Nb1 interfere with substrate binding to the AP C3/C5 convertases. In addition to emphasizing how important prevention of C3bB assembly is for the inhibitory effect of hC3Nb1, the weaker inhibition observed in our C3 deposition assay with the CP/LP C3 convertase could imply that the recognition of the C3 MG7 domain is not recognized in the same manner by the C4b2a and the C3bBb convertases. A potential difference between the influence of hC3Nb1 on the activity of the two C3 convertases is reminiscent of the much higher doses of compstatin that is required to inhibit the CP C3 convertase as compared with the C3 AP convertase (30).
It is intriguing that hC3Nb1 shows tight binding not only to C3b but also to iC3b, C3MA, and native C3. Whereas the recognition of iC3b was predictable as the epitope remains intact after FI degradation, this is not true for C3MA and native C3 because the Nt-α′ region is part of the epitope in the C3b complex. Crystal structures of human and bovine native C3 (31, 32) showed that the uncleaved Nt-α region is far from the MG7 domain in native C3, and when C3a is liberated by convertase cleavage of C3, the newly formed Nt-α′ region undergoes a spectacular relocation. In nascent C3b it presumably rattles through a narrow opening between the MG2, MG3, and MG6 domains to end up at its final position next to the MG7 domain in C3b (33). When native C3 ticks over to C3(H2O) or is treated with methylamine, the relocation of the Nt-α region to the position of Nt-α′ in C3b would require the anaphylatoxin domain—encompassing C3a in native C3 and C3(H2O)—to pass through this narrow opening, which seems unlikely. Cross-linking and subsequent MS analysis of C3(H2O) indicated that the anaphylatoxin domain is within cross-linking distance of the MG3, MG7, and MG8 domains (34), and especially the MG3 cross-link is difficult to reconcile with a C3b-like position of the Nt-α region in C3(H2O). Overall, the very tight binding to C3b, iC3b, C3MA, and native C3 suggests that the Nt-α′ contribution to the C3b epitope is not major, which is consistent with the fact that the interaction is centered around two aromatic residues from the Nb CDR3 and the C3b MG7 domain.
The ability of hC3Nb1 to bind to all tested forms of C3 containing the MG7 domains and the ability to completely shut down AP C3 cleavage suggests that it is functionally similar to compstatin, which likewise binds to multiple forms of C3 and prevents C3 cleavage (reviewed in Ref. 35). Like hC3Nb1, the most efficient compstatin derivatives bind C3b with dissociation constants below 10−9 m, but compstatin to our knowledge does not interfere with FB binding and FI degradation. The hC3Nb1 also shares many characteristics with the mAb S77 binding C3b, iC3b, C3c, and hydrolyzed C3 with KD values of 1–2 nm, although S77 does not bind to native C3 (27). The two anti-C3 molecules also both inhibit FB binding to C3b, FH binding to C3b and subsequent FI degradation (27). A comparison of the structures of the C3b complexes with hC3Nb1 and S77 also reveals a striking similarity, because both antibodies rely mainly on recognition of the exposed end of the C3b MG7 domain. Their epitopes overlap significantly and both antibodies recognize the C3b MG7 loop 916–921 encompassing Phe920. This similarity can be further extended to C5 and eculizumab, which has its entire epitope located in the same exposed end of the MG7 domain (29). The S77 light chain in addition interacts electrostatically with Arg573 in the C3b MG6 domain, and the loop holding this arginine is also close to hC3Nb1 in the Nb–C3b complex. An interesting possibility is that this loop may become part of the epitope when hC3Nb1 binds to native C3.
Compared with existing C3 inhibitors, hC3Nb1 is easy to produce as a recombinant protein; it can be tagged for immobilization, the W102A variant is available as a negative control, and like other nanobodies it may be fused with itself or other proteins to confer multivalent binding and a highly specific in vivo distribution. We and others have also shown that Nbs may be engineered to contain free cysteines that are reactive with fluorescent dyes, thereby allowing their use in applications like flow cytometry, in vivo imaging, and fluorescence microscopy (36, 37). The site-specific introduction of a biotin molecule used here for SPR immobilization also opens for a wealth of applications based on the biotin–streptavidin interaction.
Both C3 and its proteolytic fragments C3b, iC3b, C3dg, and C3a are involved in a plethora of protein– interactions with proteases, receptors, and regulators (reviewed in Ref. 38). An efficient convertase inhibitor like hC3Nb1 may be used for therapeutic treatment of conditions in which the alternative pathway or the downstream terminal pathway are linked to pathogenesis. However, a complete, systemic, and chronic shutdown of the AP may have undesirable adverse effects in terms of an increased risk of infections and autoimmune diseases (35). The recently uncovered cross-talk between C3 fragments and metabolism (reviewed in Ref. 39) further complicates chronic therapeutic shutdown of the amplification pathway. Nevertheless, a wealth of AP inhibitors for possible clinical applications have been described including small-molecule FD inhibitors, FB antibodies, the compstatin peptide and its derivatives, C3b antibodies, and recombinant cofactors derived from FH and complement receptor 1 that promote C3b degradation (40). The glomerular basement membrane in the kidney lacks membrane-bound complement regulators and is therefore particularly susceptible in the case of genetic deficiencies and autoantibodies conferring dysregulation of the AP, leading to C3 glomerulopathy, for which overall blockade of the AP could be a therapeutic strategy (41).
With respect to a future in vivo evaluation of hC3Nb1, there are several challenges that must be considered. To minimize an immune response during prolonged administration, hC3Nb1 may have to be “humanized” (42). Another aspect is that Nbs in general have a short in vivo circulation time of <1 h. However, because the plasma concentration of C3 in mice is 6 μm and hC3Nb1 binds with a KD of 900 pm to human C3, hC3Nb1 may circulate bound to C3 for much longer time than a free Nb. This is supported by the finding that C3-binding compstatin derivatives with affinities comparable with that of hC3Nb1 are cleared with a half-life of 9–12 h (43). Circulation time may furthermore be substantially prolonged by fusion of hC3Nb1 to a second molecule with specificity for serum albumin (44). This would also favor recycling in early endosomes because of interaction of serum albumin with the neonatal Fc receptor. In contrast, fusion of hC3Nb1 to IgG Fc would entail a risk of generating large complexes containing two bound molecules of C3, which could deposit in organs similar to immune complex–mediated diseases (45). Our nanobody is a superb inhibitor of proconvertase assembly but also prevents FH binding to C3b and FI degradation, which is an important endogenous mechanism for AP regulation, because the iC3b product cannot serve as a scaffold for convertase formation. Structural similarity between the C3b regulator complexes (46) suggests that hC3Nb1 inhibition of regulator binding to C3b may not be limited to FH. Hence, if hC3Nb1 is used in vivo it must be carefully addressed whether the inhibitory properties of hC3Nb1 outweighs the risk of C3b accumulation that may trigger excessive AP activation if administration of the nanobody is halted. In addition, C3b and iC3b are ligands for at least five complement receptors differentially expressed on immune cells (reviewed in Ref. 3), suggesting that inhibition of iC3b formation may affect phagocytosis of complement opsonized activators.
In conclusion, we have described the first nanobody-based inhibitor of the complement AP pathway and presented an elaborate analysis of its mechanism of inhibition through a combination of functional and structural studies. The hC3Nb1 is a potent AP inhibitor that can be easily prepared at a low cost for in vitro studies. The demonstrated ability to efficiently inhibit AP activity in murine serum suggests that hC3Nb1 may be further developed as a powerful inhibitor applicable in murine disease models.
Experimental procedures
Nanobody production
One llama (Lama glama) was immunized four times with a total of 500 μg of human C3b. Peripheral blood lymphocytes from a blood sample were isolated. Total RNA was extracted using RNase Plus mini kit (Qiagen), and cDNA was generated using Superscript III first-strand kit (Invitrogen) with random hexamer primers. The coding regions of the Nbs were amplified by PCR, inserted into a phagemid vector where the Nbs are fused to an E-tag and the pIII coat protein. VCSM13 helper phage was utilized for generating the final M13 phage display Nb library. For selection of C3b binding Nbs, one well in a microtiter plate was coated with 1 μg of C3b in 100 μl of PBS and after 12 h blocked with PBS supplemented with 2% (w/v) BSA. M13 phages (3 × 1012) in PBS were added and allowed to bind to the coated C3b for 1 h before 15 wash steps with PBS containing 0.1% (v/v) Tween 20. Phages were eluted using low pH by adding 0.2 m glycine (pH 2.2) for 15 min. The enriched library in the eluate was amplified and used in a second round of phage display, but this time only 0.1 μg of C3b was coated in the well, and 3 × 1012 M13 phages were used. After two rounds of phage display selection, single colonies were picked and grown in LB medium in a 96-well plate format for 6 h before Nb expression were induced with 0.8 mm isopropyl β-d-1-thiogalactopyranoside overnight at 30 °C. The 96-well plate was centrifuged, and 50 μl of the supernatant were transferred to a C3b-coated ELISA plate prepared by coating with 0.1 μg C3b/well followed by blocking with PBS with 0.1% (v/v) Tween 20 and 2% (w/v) BSA. After the addition of supernatant, the C3b-coated ELISA plate was incubated for 1 h and then washed six times in PBS with 0.1% (v/v) Tween 20 before anti-E-tag–HRP antibody (Bethyl) was added at a 1:10.000 dilution. The plate was incubated for 1 h, washed, and developed with 3,3′,5,5′-tetramethylbenzidine. The reaction was quenched with 1 m HCl, and the absorbance at 450 nm was measured. Phagemids from positive clones were isolated and sequenced, and Nbs were cloned into the pET22b(+) (Novagen) for bacterial expression.
C3, C3b, iC3b, C3MA, FB, and hC3Nb1
Human native C3 was purified from outdated plasma pools. All steps were conducted at 4 °C except for the phenyl-Sepharose. The first steps including PEG precipitation and DEAE ion exchange chromatography were conducted as described (47). To the DEAE pool 400 mm Na2SO4 was added, and the sample was applied to a phenyl-Sepharose HP column (GE Healthcare) operated at room temperature and equilibrated in 400 mm Na2SO4, 20 mm HEPES (pH 7.5). The column was washed with 200 mm Na2SO4, 20 mm HEPES (pH 7.5) and eluted with a linear gradient from 200 mm Na2SO4, 20 mm HEPES (pH 7.5) to 150 mm NaCl, 20 mm HEPES (pH 7.5). Human C3 eluted in the flowthrough and the wash step, whereas C5 eluted in the linear gradient. C3 was dialyzed against 20 mm HEPES (pH 7.5), 50 mm NaCl overnight prior to loading on a Source 15Q (GE Healthcare) equilibrated in 20 mm HEPES (pH 7.5), 25 mm NaCl. C3 was eluted by a linear gradient from 25 to 275 mm NaCl and dialyzed against 20 mm MES (pH 6.0), 50 mm NaCl overnight. A final polishing step was performed using a Source 15S (GE Healthcare) equilibrated in 20 mm MES (pH 6.0), 50 mm NaCl. C3 was eluted by a linear gradient from 50 to 400 mm NaCl. C3b, iC3b, and C3MA were prepared as described (48) except that the Cys1010 thiol from the thioester was reacted with iodoacetamide to prevent intermolecular disulfide formation. To express FB D279 S/A (active site serine mutated to alanine) not bearing an Asn-linked glycan at position 122, site-directed mutagenesis was performed using the QuikChange Lightning kit (Agilent Technologies) and the primers 5′-catcactcacagggtagta-3′ and 5′-ctccctactacgctgtgag-3′. FB D279G, FB D279G S/A, and D279G N122A S/A were purified from mammalian cell culture supernatants as described (48). The hC3Nb1 nanobody and its variants were expressed in the E. coli LOBSTR strain (49). Cells harboring the recombinant plasmid were grown at 37 °C in 2× yeast tryptone medium. Protein expression was induced at a A600 value of ∼0.6 with 0.2 mm isopropyl β-d-1-thiogalactopyranoside, and growth was continued at 18 °C overnight. All purification was performed at 4 °C. The cells pellets were resuspended in 50 mm Tris (pH 8.5), 500 mm NaCl, 20 mm imidazole, and 1 mm PMSF. The cells were lysed by sonication, and the cell debris was removed by centrifugation. The supernatant was loaded on a 1-ml Protino nickel–nitrilotriacetic acid–agarose column (Macherey–Nagel). The column was washed twice in 10 ml of 50 mm Tris (pH 8.5), 500 mm NaCl, 20 mm imidazole, and the protein was eluted in 5 ml of 50 mm Tris (pH 8.5), 500 mm NaCl, 350 mm imidazole. The eluate was dialyzed against 20 mm sodium acetate (pH 5.5), 25 mm NaCl overnight. The dialyzed protein was applied to a 1-ml Source 15S (GE Healthcare) equilibrated in 20 mm sodium acetate (pH 5.5), 25 mm NaCl. The protein was eluted by a 30-ml linear gradient from 30 to 330 mm NaCl. A final polishing step was performed by size exclusion on a 24-ml Superdex 75 (GE Healthcare) equilibrated in 20 mm HEPES (pH 7.5), 150 mm NaCl.
Negative stain EM
C3b was added a 2-fold molar excess of hC3Nb1 and incubated for 5 min at 4 °C. The complex was purified by SEC on a 24-ml Superdex 200 Increase (GE Healthcare) equilibrated in 20 mm HEPES (pH 7.5), 150 mm NaCl. Drops from the early fractions of the peak were adsorbed to glow-discharged carbon-coated copper grids, washed with deionized water, and stained with 2% (w/v) uranyl formate. Images were acquired with a FEI Tecnai G2 Spirit transmission electron microscope at 120 kV, a nominal magnification of 67.000×, and a defocus ranging from −0.7 to −1.7 μm. Automated image acquisition was performed using Leginon (50), and particles were picked with DoG picker (51) in the Appion framework (52). 2D classification was performed in RELION (53). The 2D classes corresponding to single particles were used for 3D classification. MG1–8 of C3b was filtered to 80 Å using EMAN (54) and used as an initial map for 3D classification in RELION. The 3D reconstruction was further refined in RELION using the best class from 3D classification filtered to 60 Å as the initial model.
Crystal structure determination
Crystals of the C3b–hC3Nb1 complex were grown at 19 °C in sitting drops made by mixing the complex at 6.5 mg/ml in a 1:1 ratio with reservoir solution containing 112 mm sodium citrate (pH 8), 28 mm sodium citrate (pH 6.0), 12.25% (w/v) PEG 2000 monomethyl ether, 70 mm Imidazole pH 7, 60 mm Ammonium acetate, 70 mm Tris (pH 8.5), and 13.5% (v/v) 2-methyl-2,4-pentanediol. The crystals were soaked in reservoir solution supplemented with 5% (v/v) glycerol prior to flash cooling in liquid nitrogen. The data were collected at PETRA III P13 at 100 K and processed with XDS (55). The structure was determined using the coordinates of the C3b (PDB entry 5FO7) with the C345c, CUB, and TE domains removed for molecular replacement in Phaser (56). The remaining C3b domains were placed by superposition, whereas the nanobody from the PDB entry 5IMK with the CDRs removed was manually placed in Coot (57). The resulting model was refined with rigid body refinement in Phenix.refine (58). In an iterative manner the structure was manually rebuilt in Coot and refined with Phenix.refine using positional refinement, grouped B-factors, and TLS groups. In the last round of refinement, individual B-factor refinement was used instead of grouped B-factors. Prior to the final refinement, the structure was fitted to the electron density map using iMDFF as described (59). The final structure displayed superior stereochemistry according to MolProbity (60) considering the resolution of the diffraction data. The intermolecular interface was analyzed with PyMOL 1.8.6 (63) and the CCP4 programs PISA (17) and SC (16). The figures were prepared with PyMOL. The set of high-resolution nanobody–antigen complexes was created by extracting entries containing at least one nanobody determined at a resolution better than 3.0 Å and manually inspecting these in PyMOL. Complexes in which the antigen was a peptide or formed very few contacts to the nanobody were discarded.
Assay for alternative pathway activity on a zymosan surface
Zymosan-coated 96-well Maxisorp plates (Nunc) were prepared by adding 100 μl of 20 μg/ml zymosan (Sigma, catalog no. Z4250) diluted in coating buffer (50 mm sodium carbonate buffer, pH 9.6) to each well and incubating overnight at room temperature. The wells were emptied and blocked by the addition of 200 μl of 1 mg/ml human serum albumin diluted in TBS (10 mm Tris, 145 mm NaCl, pH 7.4). After 1 h of incubation at room temperature, unbound HSA were removed by washing the wells thrice in TBS/Tween (10 mm Tris, 145 mm NaCl, 0.05% (v/v) Tween 20, pH 7.4). The samples that were added to zymosan-coated wells were prepared by mixing normal human serum and nanobodies in VBS/EGTA/Mg2+ buffer (5 mm barbital, pH 7.4, 145 mm NaCl, 10 mm EGTA, 5 mm MgCl2), which blocks the classical and lectin pathways but allows C3 deposition through the alternative pathway. Serum was diluted to a final concentration of 15% (v/v), whereas nanobodies (hC3Nb1, hC3Nb1 R53A, hC3Nb1 R57A, hC3Nb1 W101A, hC3Nb1 Y112A, and KRA152) were added to final concentrations of 5.0, 2.5, 1.25, 0.63, 0.32, 0.16, 0.08, or 0.04 μm. The experiments also included a sample without Nb, wells to which only buffer was added, and a sample diluted in VBS/EGTA/Mg2+ with 10 mm EDTA as a positive control for inhibition. All samples were incubated for 1 h at room temperature to allow formation of Nb-C3 complexes. Then 100 μl of sample was added to the plate in duplicates, and the plate was incubated for 1.5 h at 37 °C. The wells were washed thrice with TBS/Tween, and deposited C3 was detected using 0.5 μg/ml biotinylated rabbit anti-C3d (Dako, Denmark, catalog no. A0063) diluted in TBS/Tween. After 2 h of incubation at room temperature, the wells were washed thrice in TBS/Tween, and 100 μl of 1 μg/ml europium-labeled streptavidin (PerkinElmer; catalog no. 1244-360) in TBS/Tween with 25 μm EDTA was added to each well. Unbound europium-labeled streptavidin was washed away with TBS/Tween after 1 h of incubation at room temperature. 200 μl of enhancement solution (Ampliquon Laboratory Reagents, Denmark, catalog no. Q99800) was added to each well, and the fluorescence was measured as time-resolved fluorescence using a Victor X5 plate reader (PerkinElmer). The background signal from the buffer was subtracted from all samples, and the signal from the sample without added Nb was defined as 100%. The assays for alternative pathway in murine serum were performed analogously to the assay in human serum. The differences were the use of a final concentration of 5% (v/v) serum from C57Bl6 male mice and the use of anti-mouse C3 antibody (Cederlane, catalog no. CL7503NA) for detection of deposited C3 fragments.
Assay for lectin pathway activity
Microtiter wells (FlouroNunc, Nunc) were coated with 1 μg of mannan (M7504, Sigma) in 100 μl of coating buffer. Residual protein-binding sites were blocked by the addition of 200 μl of 0.1% HSA in TBS. After a washing step using TBS containing 5 mm CaCl2 and 0.05% Tween 20, the wells received serum (from a donor with relatively high concentration of mannan-binding lectin) diluted in 4 mm barbital (pH 7.5), 145 mm NaCl, 2 mm CaCl2, and 1 mm MgCl2. The serum was used at a 1% final concentration with nanobody added to final concentrations of 700, 230, 78, 26, 8.6, or 2.8 nm. This was followed by incubation in the wells for 1.5 h at 37 °C. The subsequent deposition of C3 fragments onto the mannan surface was conducted and analyzed as described above for the alternative pathway assay for C3 deposition.
SEC assay for proconvertase assembly
C3b in complex with hC3Nb1 or C3b alone was mixed with 1.2-fold molar excess of either FB D279G S/A or FB D279G S/A N122A and incubated for 30 min at 4 °C in 20 mm HEPES (pH 7.5), 150 mm NaCl, 3 mm MgCl2. The samples were injected on a 24-ml Superdex 200 increase column equilibrated in 20 mm HEPES (pH 7.5), 150 mm NaCl, and 3 mm MgCl2 operated at 4 °C.
FH-mediated FI cleavage assay
C3b was incubated with 1.2-fold molar excess of hC3Nb1 for 5 min at 4 °C in 20 mm HEPES (pH 7.5), 150 mm NaCl. FH (Complement Tech) and FI was added in a 1/500 and 1/100 mass ratio to C3b, respectively. The reaction mixture was incubated at 37 °C, and samples for SDS-PAGE analysis were taken after 1, 2, 4, and 8 h.
DTNB thiol assay
To a solution containing 10.75 μm freshly purified C3 was added 100 mm Tris (pH 8.5), 1.25 mm DTNB, and either 100 mm methylamine (pH 8.5), 12 μm hC3Nb1 or reaction buffer. The three reactions were incubated for 2 h on ice after which the absorbance at 412 nm was measured. The experiment was performed twice.
CVFBb cleavage of C3 and C5
FB D279G was incubated with 1.2 molar excess of CVF and 10% (w/w) FD was added to the proconvertase, and the reaction was incubated for 15 min at room temperature followed by a 10-min incubation at 4 °C. A 100-fold molar excess of native C3 compared with FB was added to the CVFBb, the reaction was placed at 4 °C, and samples for SDS-PAGE analysis were taken after 0.5, 1, 4, and 8 h. In the C5 cleavage assay, a 100-fold molar excess of either C5 or C5 and hC3Nb1 was added instead, and samples for SDS-PAGE analysis were taken after 0.5, 2, and 4 h of incubation at 4 °C.
SPR analysis
All SPR experiments were performed on a Biacore T200 instrument in a buffer containing 20 mm HEPES (pH 7.5), 150 mm NaCl, 3 mm MgCl2. Streptavidin was immobilized on a CMD500M chip (XanTec Bioanalytics) to 100 response units. The hC3Nb1 was immobilized on the streptavidin surface through a biotinylated C-terminal Avi-tag. Native C3, C3MA, C3b, and iC3b were injected in concentrations ranging from 50 to 0.1 nm. The surface was regenerated by injecting 100 mm glycine (pH 2.7). The data were fitted with the BiaCore T200 evaluation software using a 1:1 binding model without bulk effect. The kinetics experiments were performed in triplicate. Competition assays were performed using FB (Complement Tech), recombinant FB D279G S/A, FH (Complement Tech), hC3Nb1, or hC3Nb1 mutants at 10-fold molar excess to a final concentration of 50 nm C3b. The complexes were incubated on ice for 60 min before being injected on the surface. All experiments were performed in triplicate.
Author contributions
R. K. J., R. P., J. K. J., K. R. A., S. T., N. S. L., and G. R. A. data curation; R. K. J., J. K. J., S. T., N. S. L., and G. R. A. formal analysis; R. K. J., S. T., and G. R. A. validation; R. K. J., R. P., T. A. F. G., K. R. A., and N. S. L. investigation; R. K. J. visualization; R. K. J., R. P., T. A. F. G., K. R. A., S. T., N. S. L., and G. R. A. methodology; R. K. J., R. P., T. A. F. G., J. K. J., K. R. A., S. T., N. S. L., and G. R. A. writing-original draft; T. A. F. G., K. R. A., and N. S. L. resources; J. K. J., S. T., N. S. L., and G. R. A. supervision; S. T., N. S. L., and G. R. A. funding acquisition; N. S. L. and G. R. A. conceptualization; G. R. A. project administration; G. R. A. writing-review and editing.
Supplementary Material
Acknowledgments
We thank the staff at P13 and P14 at Petra III for help during data collection.
This work was supported by funds from the Lundbeck Foundation BRAINSTRUC Centre (R155-2015-2666), Danscatt, the Novo Nordisk Foundation, and the Danish Research Council for Nature and Universe (to G. R. A.); from the Lundbeck Foundation (to N. S. L. and K. R. A.); and from the Danish Council for Independent Research, Medical Sciences (to R. P. and S. T.). The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S3.
The atomic coordinates and structure factors (code 6EHG) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- AP
- alternative pathway
- C3MA
- methylamine treated complement C3
- CCP
- complement control protein
- CDR
- complementarity-determining region
- CP
- classical pathway
- CUB
- complement C1r/C1s Uegf Bmp1
- CVF
- cobra venom factor
- DNTB
- 5, 5′-dithiobis-(2-nitrobenzoic acid)
- FB
- factor B
- FD
- factor D
- FH
- factor H
- FI
- factor I
- FP
- properdin
- LP
- lectin pathway
- MG
- macroglobulin
- Nb
- nanobody
- PDB
- Protein Data Bank
- SEC
- size-exclusion chromatography
- SPR
- surface plasmon resonance
- TE
- thioester
- DTNB
- 5,5′-dithiobis-(2-nitrobenzoic acid).
References
- 1. Merle N. S., Noe R., Halbwachs-Mecarelli L., Fremeaux-Bacchi V., and Roumenina L. T. (2015) Complement system part II: role in immunity. Front. Immunol. 6, 257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stephan A. H., Barres B. A., and Stevens B. (2012) The complement system: an unexpected role in synaptic pruning during development and disease. Annu. Rev. Neurosci. 35, 369–389 10.1146/annurev-neuro-061010-113810 [DOI] [PubMed] [Google Scholar]
- 3. Bajic G., Degn S. E., Thiel S., and Andersen G. R. (2015) Complement activation, regulation, and molecular basis for complement-related diseases. EMBO J. 34, 2735–2757 10.15252/embj.201591881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Fishelson Z., and Müller-Eberhard H. J. (1982) C3 convertase of human complement: enhanced formation and stability of the enzyme generated with nickel instead of magnesium. J. Immunol. 129, 2603–2607 [PubMed] [Google Scholar]
- 5. Bexborn F., Andersson P. O., Chen H., Nilsson B., and Ekdahl K. N. (2008) The tick-over theory revisited: formation and regulation of the soluble alternative complement C3 convertase (C3 (H 2 O) Bb). Mol. Immunol. 45, 2370–2379 10.1016/j.molimm.2007.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Fearon D. T., and Austen K. F. (1975) Properdin: binding to C3b and stabilization of the C3b-dependent C3 convertase. J. Exp. Med. 142, 856–863 10.1084/jem.142.4.856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ricklin D., Reis E. S., and Lambris J. D. (2016) Complement in disease: a defence system turning offensive. Nat. Rev. Nephrol. 12, 383–401 10.1038/nrneph.2016.70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Morgan B. P., and Harris C. L. (2015) Complement, a target for therapy in inflammatory and degenerative diseases. Nat. Rev. Drug. Discov. 14, 857–877 10.1038/nrd4657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hill A., DeZern A. E., Kinoshita T., and Brodsky R. A. (2017) Paroxysmal nocturnal haemoglobinuria. Nat. Rev. Dis. Primers 3, 17028 10.1038/nrdp.2017.28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zuber J., Fakhouri F., Roumenina L. T., Loirat C., Frémeaux-Bacchi V., and French Study Group for aHUS/C3G (2012) Use of eculizumab for atypical haemolytic uraemic syndrome and C3 glomerulopathies. Nat. Rev. Nephrol. 8, 643–657 10.1038/nrrheum.2012.182,10.1038/nrneph.2012.214 [DOI] [PubMed] [Google Scholar]
- 11. Melis J. P., Strumane K., Ruuls S. R., Beurskens F. J., Schuurman J., and Parren P. W. (2015) Complement in therapy and disease: Regulating the complement system with antibody-based therapeutics. Mol. Immunol. 67, 117–130 10.1016/j.molimm.2015.01.028 [DOI] [PubMed] [Google Scholar]
- 12. Harboe M., Johnson C., Nymo S., Ekholt K., Schjalm C., Lindstad J. K., Pharo A., Hellerud B. C., Nilsson Ekdahl K., Mollnes T. E., and Nilsson P. H. (2017) Properdin binding to complement activating surfaces depends on initial C3b deposition. Proc. Natl. Acad. Sci. U.S.A. 114, E534–E539 10.1073/pnas.1612385114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Könning D., Zielonka S., Grzeschik J., Empting M., Valldorf B., Krah S., Schröter C., Sellmann C., Hock B., and Kolmar H. (2017) Camelid and shark single domain antibodies: structural features and therapeutic potential. Curr. Opin. Struct. Biol. 45, 10–16 10.1016/j.sbi.2016.10.019 [DOI] [PubMed] [Google Scholar]
- 14. Rodriguez E., Nan R., Li K., Gor J., and Perkins S. J. (2015) A revised mechanism for the activation of complement C3 to C3b: a molecular explanation of a disease-associated polymorphism. J. Biol. Chem. 290, 2334–2350 10.1074/jbc.M114.605691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mortensen S., Kidmose R. T., Petersen S. V., Szilágyi Á., Prohászka Z., and Andersen G. R. (2015) Structural basis for the function of complement component C4 within the classical and lectin pathways of complement. J. Immunol. 194, 5488–5496 10.4049/jimmunol.1500087 [DOI] [PubMed] [Google Scholar]
- 16. Lawrence M. C., and Colman P. M. (1993) Shape complementarity at protein/protein interfaces. J. Mol. Biol. 234, 946–950 10.1006/jmbi.1993.1648 [DOI] [PubMed] [Google Scholar]
- 17. Krissinel E., and Henrick K. (2007) Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 10.1016/j.jmb.2007.05.022 [DOI] [PubMed] [Google Scholar]
- 18. Laursen N. S., Andersen K. R., Braren I., Spillner E., Sottrup-Jensen L., and Andersen G. R. (2011) Substrate recognition by complement convertases revealed in the C5-cobra venom factor complex. EMBO J. 30, 606–616 10.1038/emboj.2010.341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Forneris F., Ricklin D., Wu J., Tzekou A., Wallace R. S., Lambris J. D., and Gros P. (2010) Structures of C3b in complex with factors B and D give insight into complement convertase formation. Science 330, 1816–1820 10.1126/science.1195821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Meri S., and Pangburn M. K. (1994) Regulation of alternative pathway complement activation by glycosaminoglycans: specificity of the polyanion binding site on factor H. Biochem. Biophys. Res. Commun. 198, 52–59 10.1006/bbrc.1994.1008 [DOI] [PubMed] [Google Scholar]
- 21. Morgan H. P., Schmidt C. Q., Guariento M., Blaum B. S., Gillespie D., Herbert A. P., Kavanagh D., Mertens H. D., Svergun D. I., Johansson C. M., Uhrín D., Barlow P. N., and Hannan J. P. (2011) Structural basis for engagement by complement factor H of C3b on a self surface. Nat. Struct. Mol. Biol. 18, 463–470 10.1038/nsmb.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kajander T., Lehtinen M. J., Hyvärinen S., Bhattacharjee A., Leung E., Isenman D. E., Meri S., Goldman A., and Jokiranta T. S. (2011) Dual interaction of factor H with C3d and glycosaminoglycans in host-nonhost discrimination by complement. Proc. Natl. Acad. Sci. U.S.A. 108, 2897–2902 10.1073/pnas.1017087108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schmidt C. Q., Herbert A. P., Kavanagh D., Gandy C., Fenton C. J., Blaum B. S., Lyon M., Uhrín D., and Barlow P. N. (2008) A new map of glycosaminoglycan and C3b binding sites on factor H. J. Immunol. 181, 2610–2619 10.4049/jimmunol.181.4.2610 [DOI] [PubMed] [Google Scholar]
- 24. Roversi P., Johnson S., Caesar J. J., McLean F., Leath K. J., Tsiftsoglou S. A., Morgan B. P., Harris C. L., Sim R. B., and Lea S. M. (2011) Structural basis for complement factor I control and its disease-associated sequence polymorphisms. Proc. Natl. Acad. Sci. U.S.A. 108, 12839–12844 10.1073/pnas.1102167108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Muyldermans S. (2013) Nanobodies: natural single-domain antibodies. Annu. Rev. Biochem. 82, 775–797 10.1146/annurev-biochem-063011-092449 [DOI] [PubMed] [Google Scholar]
- 26. Janssen B. J., Halff E. F., Lambris J. D., and Gros P. (2007) Structure of compstatin in complex with complement component C3c reveals a new mechanism of complement inhibition. J. Biol. Chem. 282, 29241–29247 10.1074/jbc.M704587200 [DOI] [PubMed] [Google Scholar]
- 27. Katschke K. J. Jr, Stawicki S., Yin J., Steffek M., Xi H., Sturgeon L., Hass P. E., Loyet K. M., Deforge L., Wu Y., van Lookeren Campagne M., and Wiesmann C. (2009) Structural and functional analysis of a C3b-specific antibody that selectively inhibits the alternative pathway of complement. J. Biol. Chem. 284, 10473–10479 10.1074/jbc.M809106200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wiesmann C., Katschke K. J., Yin J., Helmy K. Y., Steffek M., Fairbrother W. J., McCallum S. A., Embuscado L., DeForge L., Hass P. E., and van Lookeren Campagne M. (2006) Structure of C3b in complex with CRIg gives insights into regulation of complement activation. Nature 444, 217–220 10.1038/nature05263 [DOI] [PubMed] [Google Scholar]
- 29. Schatz-Jakobsen J. A., Zhang Y., Johnson K., Neill A., Sheridan D., and Andersen G. R. (2016) Structural basis for eculizumab-mediated inhibition of the complement terminal pathway. J. Immunol. 197, 337–344 10.4049/jimmunol.1600280 [DOI] [PubMed] [Google Scholar]
- 30. Nilsson B., Larsson R., Hong J., Elgue G., Ekdahl K. N., Sahu A., and Lambris J. D. (1998) Compstatin inhibits complement and cellular activation in whole blood in two models of extracorporeal circulation. Blood 92, 1661–1667 [PubMed] [Google Scholar]
- 31. Janssen B. J., Huizinga E. G., Raaijmakers H. C., Roos A., Daha M. R., Nilsson-Ekdahl K., Nilsson B., and Gros P. (2005) Structures of complement component C3 provide insights into the function and evolution of immunity. Nature 437, 505–511 10.1038/nature04005 [DOI] [PubMed] [Google Scholar]
- 32. Fredslund F., Jenner L., Husted L. B., Nyborg J., Andersen G. R., and Sottrup-Jensen L. (2006) The structure of bovine complement component 3 reveals the basis for thioester function. J. Mol. Biol. 361, 115–127 10.1016/j.jmb.2006.06.009 [DOI] [PubMed] [Google Scholar]
- 33. Janssen B. J., Christodoulidou A., McCarthy A., Lambris J. D., and Gros P. (2006) Structure of C3b reveals conformational changes that underlie complement activity. Nature 444, 213–216 10.1038/nature05172 [DOI] [PubMed] [Google Scholar]
- 34. Chen Z. A., Pellarin R., Fischer L., Sali A., Nilges M., Barlow P. N., and Rappsilber J. (2016) Structure of complement C3(H2O) revealed by quantitative cross-linking/mass spectrometry and modeling. Mol. Cell. Proteomics 15, 2730–2743 10.1074/mcp.M115.056473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Mastellos D. C., Yancopoulou D., Kokkinos P., Huber-Lang M., Hajishengallis G., Biglarnia A. R., Lupu F., Nilsson B., Risitano A. M., Ricklin D., and Lambris J. D. (2015) Compstatin: a C3-targeted complement inhibitor reaching its prime for bedside intervention. Eur J. Clin. Invest. 45, 423–440 10.1111/eci.12419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pleiner T., Bates M., Trakhanov S., Lee C. T., Schliep J. E., Chug H., Böhning M., Stark H., Urlaub H., and Görlich D. (2015) Nanobodies: site-specific labeling for super-resolution imaging, rapid epitope-mapping and native protein complex isolation. Elife 4, e11349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hansen S. B., Laursen N. S., Andersen G. R., and Andersen K. R. (2017) Introducing site-specific cysteines into nanobodies for mercury labelling allows de novo phasing of their crystal structures. Acta Crystallogr. D Struct. Biol. 73, 804–813 10.1107/S2059798317013171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ricklin D., Reis E. S., Mastellos D. C., Gros P., and Lambris J. D. (2016) Complement component C3: The “Swiss Army Knife” of innate immunity and host defense. Immunol. Rev. 274, 33–58 10.1111/imr.12500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kolev M., and Kemper C. (2017) Keeping it all going: complement meets metabolism. Front. Immunol. 8, 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ruseva M. M., Ramaglia V., Morgan B. P., and Harris C. L. (2015) An anticomplement agent that homes to the damaged brain and promotes recovery after traumatic brain injury in mice. Proc. Natl. Acad. Sci. U.S.A. 112, 14319–14324 10.1073/pnas.1513698112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mastellos D. C., Reis E. S., Ricklin D., Smith R. J., and Lambris J. D. (2017) Complement C3-targeted therapy: replacing long-held assertions with evidence-based discovery. Trends Immunol. 38, 383–394 10.1016/j.it.2017.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vincke C., Loris R., Saerens D., Martinez-Rodriguez S., Muyldermans S., and Conrath K. (2009) General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J. Biol. Chem. 284, 3273–3284 10.1074/jbc.M806889200 [DOI] [PubMed] [Google Scholar]
- 43. Qu H., Ricklin D., Bai H., Chen H., Reis E. S., Maciejewski M., Tzekou A., DeAngelis R. A., Resuello R. R., Lupu F., Barlow P. N., and Lambris J. D. (2013) New analogs of the clinical complement inhibitor compstatin with subnanomolar affinity and enhanced pharmacokinetic properties. Immunobiology 218, 496–505 10.1016/j.imbio.2012.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tijink B. M., Laeremans T., Budde M., Stigter-van Walsum M., Dreier T., de Haard H. J., Leemans C. R., and van Dongen G. A. (2008) Improved tumor targeting of anti-epidermal growth factor receptor nanobodies through albumin binding: taking advantage of modular Nanobody technology. Mol. Cancer Ther. 7, 2288–2297 10.1158/1535-7163.MCT-07-2384 [DOI] [PubMed] [Google Scholar]
- 45. Krishna M., and Nadler S. G. (2016) Immunogenicity to biotherapeutics: the role of anti-drug immune complexes. Front. Immunol. 7, 21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Forneris F., Wu J., Xue X., Ricklin D., Lin Z., Sfyroera G., Tzekou A., Volokhina E., Granneman J. C., Hauhart R., Bertram P., Liszewski M. K., Atkinson J. P., Lambris J. D., and Gros P. (2016) Regulators of complement activity mediate inhibitory mechanisms through a common C3b-binding mode. EMBO J. 35, 1133–1149 10.15252/embj.201593673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sottrup-Jensen L., and Andersen G. R. (2014) Purification of human complement protein C5. Methods Mol. Biol. 1100, 93–102 10.1007/978-1-62703-724-2_7 [DOI] [PubMed] [Google Scholar]
- 48. Pedersen D. V., Roumenina L., Jensen R. K., Gadeberg T. A., Marinozzi C., Picard C., Rybkine T., Thiel S., Sørensen U. B., Stover C., Fremeaux-Bacchi V., and Andersen G. R. (2017) Functional and structural insight into properdin control of complement alternative pathway amplification. EMBO J. 36, 1084–1099 10.15252/embj.201696173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Andersen K. R., Leksa N. C., and Schwartz T. U. (2013) Optimized E. coli expression strain LOBSTR eliminates common contaminants from His-tag purification. Proteins 81, 1857–1861 10.1002/prot.24364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Suloway C., Pulokas J., Fellmann D., Cheng A., Guerra F., Quispe J., Stagg S., Potter C. S., and Carragher B. (2005) Automated molecular microscopy: the new Leginon system. J. Struct. Biol. 151, 41–60 10.1016/j.jsb.2005.03.010 [DOI] [PubMed] [Google Scholar]
- 51. Voss N. R., Yoshioka C. K., Radermacher M., Potter C. S., and Carragher B. (2009) DoG Picker and TiltPicker: software tools to facilitate particle selection in single particle electron microscopy. J. Struct. Biol. 166, 205–213 10.1016/j.jsb.2009.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lander G. C., Stagg S. M., Voss N. R., Cheng A., Fellmann D., Pulokas J., Yoshioka C., Irving C., Mulder A., Lau P. W., Lyumkis D., Potter C. S., and Carragher B. (2009) Appion: an integrated, database-driven pipeline to facilitate EM image processing. J. Struct. Biol. 166, 95–102 10.1016/j.jsb.2009.01.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Scheres S. H. (2012) RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 180, 519–530 10.1016/j.jsb.2012.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Ludtke S. J., Baldwin P. R., and Chiu W. (1999) EMAN: semiautomated software for high-resolution single-particle reconstructions. J. Struct. Biol. 128, 82–97 10.1006/jsbi.1999.4174 [DOI] [PubMed] [Google Scholar]
- 55. Kabsch W. (2010) Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 66, 133–144 10.1107/S0907444909047374,10.1107/S0108767310004551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Emsley P., Lohkamp B., Scott W. G., and Cowtan K. (2010) Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 10.1107/S0907444912001308,10.1107/S0108767312007234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Croll T. I., and Andersen G. R. (2016) Re-evaluation of low-resolution crystal structures via interactive molecular-dynamics flexible fitting (iMDFF): a case study in complement C4. Acta Crystallogr. D Struct Biol. 72, 1006–1016 10.1107/S2059798316012201 [DOI] [PubMed] [Google Scholar]
- 60. Chen V. B., Arendall W. B. 3rd, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Engström G., Hedblad B., Berglund G., Janzon L., and Lindgärde F. (2007) Plasma levels of complement C3 is associated with development of hypertension: a longitudinal cohort study. J. Hum. Hypertens. 21, 276–282 10.1038/sj.jhh.1002129 [DOI] [PubMed] [Google Scholar]
- 62. Kotimaa J., Klar-Mohammad N., Gueler F., Schilders G., Jansen A., Rutjes H., Daha M. R., and van Kooten C. (2016) Sex matters: systemic complement activity of female C57BL/6J and BALB/cJ mice is limited by serum terminal pathway components. Mol. Immunol. 76, 13–21 10.1016/j.molimm.2016.06.004 [DOI] [PubMed] [Google Scholar]
- 63. Schroedinger, LLC (2015) The PyMOL Molecular Graphics System, Version 1.8
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.