

Abstract
Coiled-coil-helix-coiled-coil-helix domain–containing 10 (CHCHD10) and CHCHD2 (MNRR1) are homologous proteins with 58% sequence identity and belong to the twin CX9C family of proteins that mediate cellular stress responses. Despite the identification of several neurodegeneration-associated mutations in the CHCHD10 gene, few studies have assessed its physiological role. Here, we investigated CHCHD10's function as a regulator of oxidative phosphorylation in the mitochondria and the nucleus. We show that CHCHD10 copurifies with cytochrome c oxidase (COX) and up-regulates COX activity by serving as a scaffolding protein required for MNRR1 phosphorylation, mediated by ARG (ABL proto-oncogene 2, nonreceptor tyrosine kinase (ABL2)). The CHCHD10 gene was maximally transcribed in cultured cells at 8% oxygen, unlike MNRR1, which was maximally expressed at 4%, suggesting a fine-tuned oxygen-sensing system that adapts to the varying oxygen concentrations in the human body under physiological conditions. We show that nuclear CHCHD10 protein down-regulates the expression of genes harboring the oxygen-responsive element (ORE) in their promoters by interacting with and augmenting the activity of the largely uncharacterized transcriptional repressor CXXC finger protein 5 (CXXC5). We further show that two genetic CHCHD10 disease variants, G66V and P80L, in the mitochondria exhibit faulty interactions with MNRR1 and COX, reducing respiration and increasing reactive oxygen species (ROS), and in the nucleus abrogating transcriptional repression of ORE-containing genes. Our results reveal that CHCHD10 positively regulates mitochondrial respiration and contributes to transcriptional repression of ORE-containing genes in the nucleus, and that genetic CHCHD10 variants are impaired in these activities.
Keywords: mitochondria, mitochondrial disease, cytochrome c oxidase (Complex IV), energy metabolism, hypoxia, neurodegenerative disease, cell stress, scaffolding protein, transcriptional regulator
Introduction
Coiled-coil-helix-coiled-coil-helix domain–containing 10 (CHCHD10) is a member of the twin CX9C family of proteins. This family, of which a number of members have displayed roles in disease (1), consists of proteins that contain two pairs of cysteine residues separated usually by nine amino acids. These four cysteines form two disulfide bonds that stabilize the prototypical coiled-coil-helix-coiled-coil-helix domain (CHCHD) that traps CHCHD10 and other twin CX9C family members in the mitochondrial intermembrane space (IMS)2. Other members of this family include CHCHD4 (also called Mia40), necessary for import and proper folding of twin CX9C proteins in the IMS (2), and CHCHD3, which acts with CHCHD6 to stabilize mitochondrial cristae (3, 4). Another recently characterized member of this family is MNRR1 (mitochondrial nuclear retrograde regulator 1, also called CHCHD2), a protein that is up-regulated under hypoxic stress, and maximally so at 4% oxygen. Besides residing in the mitochondria, MNRR1 is also present in the nucleus. Mitochondrial MNRR1 interacts with cytochrome c oxidase (COX) and is required for optimal enzyme function. A stable knockdown of MNRR1 resembles a mitochondrial disease phenotype with decreased oxygen consumption, decreased cellular growth and mitochondrial membrane potential, and increased production of reactive oxygen species (ROS) (5). Nuclear MNRR1 regulates transcription of genes that harbor a conserved 13-bp sequence needed for the response to low oxygen levels that is termed the oxygen-responsive element (ORE) (6).
CHCHD10, although highly expressed in heart and skeletal muscle (7), was discovered to be mutated in a proportion of individuals suffering from several neurodegenerative diseases (8–17). The clinical effects of these mutations have been well described (18). Little is known, however, about the physiological role of CHCHD10 (7). Most studies have focused on characterizing CHCHD10's effects in mitochondria by testing the effects of pathogenic mutations in a cell culture system (8, 9, 14). These studies have suggested that CHCHD10 affects stability of mitochondrial cristae (9, 14), COX activity (7), formation of mitochondrial networks (8), stability of mitochondrial DNA, and apoptosis (14).
Here, we demonstrate that CHCHD10, like MNRR1, is a hypoxia-responsive gene whose product functions in both the mitochondria and the nucleus. In the mitochondria, CHCHD10 also interacts with COX and stimulates oxygen consumption. Furthermore, CHCHD10 regulates phosphorylation of MNRR1, thereby contributing to the role of MNRR1 in maintenance of electron transport chain function. However, unlike MNRR1, CHCHD10 in the nucleus functions as a transcriptional repressor of genes containing the ORE. To gain insight into CHCHD10's normal and pathogenic roles, we tested two disease-associated point mutations (G66V and P80L) for nuclear and mitochondrial function. We find that both mutants fail to repress transcription at the ORE. The same mutants also are defective for regulation of oxygen consumption and increase production of mitochondrial ROS. The mitochondrial defects seen with the CHCHD10 mutants result from its reduced interaction with MNRR1.
We previously showed that MNRR1 is phosphorylated by ARG (Abl2 kinase), promoting binding to and activation of COX (22). We now show that CHCHD10 acts as a scaffolding protein, assisting ARG-mediated phosphorylation of MNRR1, and that this scaffolding does not occur when CHCHD10 harbors either the G66V or the P80L replacements, leading to decreased oxygen consumption and increased ROS levels. Our findings provide new mechanistic insights into the role of CHCHD10 under physiological conditions and a potential mechanism for the pathogenic effects seen in several mutant versions.
Results
CHCHD10 is a hypoxia-sensitive gene
Given the previous observations that MNRR1 production is maximally sensitive to 4% hypoxia (6), we tested the hypoxia sensitivity of CHCHD10 at a range of physiologically encountered oxygen tensions (19) between 1 and 13%. CHCHD10 is maximally up-regulated at 8% oxygen and down-regulated at 4% oxygen in a manner opposite to MNRR1, which is maximally induced at 4% oxygen and down-regulated at 8% and higher (Fig. 1A). The up-regulation of CHCHD10 at 8% is transcriptionally mediated (Fig. 1B). To confirm these effects, we analyzed CHCHD10 levels at 7, 8, and 9% oxygen. The maximally up-regulated CHCHD10 at 8% oxygen takes place in a surprisingly narrow window (Fig. 1C). Thus, different ratios of these two proteins are present at oxygen concentrations in the range analyzed.
Figure 1.

CHCHD10 is a hypoxia-sensitive gene. A, HeLa cells were incubated for 48 h at the stated O2 levels. Whole cell lysates were separated on an SDS-PAGE gel and analyzed for CHCHD10 and MNRR1 levels. GAPDH was probed as a loading control. B, real-time PCR analysis of transcript levels of endogenous CHCHD10 at 20, 8, and 4% oxygen (n = 4; *, p < 0.05). C, as in A but not probed for MNRR1. Error bars in graphs represent 1 standard deviation from the mean of repeat determinations.
CHCHD10 is localized to both the nucleus and the mitochondria
CHCHD10 has been characterized as a mitochondrial protein in previous studies (7, 9). Because of its high degree of conservation with MNRR1 (58% identical), a bi-organellar protein localized to both the mitochondria and the nucleus (6), we examined the subcellular localization of CHCHD10. Similar to MNRR1, CHCHD10 is also present in both the mitochondria (primarily) and the nucleus as analyzed by cell fractionation (Fig. 2A) and by confocal microscopy (Fig. 2B). The mitochondrial localization of CHCHD10 is well established (7, 9, 20). In the nucleus CHCHD10 colocalizes with DAPI as suggested by a positive Pearson correlation coefficient. A recent study also showed nuclear localization (21). We further validated the nuclear signal by use of a stable knockdown of CHCHD10 in HEK293 cells (Fig. 2B, top panel, far right). In these cells there is a clear reduction in the nuclear signal (Fig. 2B, top panel) compared with WT cells also at 20% oxygen (Fig. 2B, middle panel, bar graph to right of panel), suggesting the signal does not arise from antibody cross-reactivity. At 8% oxygen CHCHD10 is increased in both the nucleus and the mitochondria (Fig. 2B).
Figure 2.

CHCHD10 is localized to both the nucleus and the mitochondria. A, nuclear and mitochondrial fractions (HEK293 cells) were analyzed for levels of CHCHD10. Effectiveness of fractionation was shown using DRBP76 as a nuclear marker and NDUFS3 as a mitochondrial marker. B, representative images of CHCHD10 (red) colocalization with the nucleus (DAPI, blue; merge, purple) in HEK293 WT and CHCHD10-KD cells. The Pearson correlation coefficient values (negative in the KD cells) indicate the specificity of nuclear signal of CHCHD10 (top panel). To show effectiveness of CHCHD10 knockdown, equal amounts of lysate from HEK293 WT and CHCHD10-KD cells were probed for CHCHD10 levels (top panel, right). GAPDH was probed as loading control. Representative images of CHCHD10 (red) localization in HEK293 cells at normoxia (20% O2) (middle panel) and hypoxia (8% O2) (bottom panel). Right, relative Pearson correlation coefficient values (negative in the KD cells) indicate the specificity of nuclear signal of CHCHD10 (n = 4; *, p < 0.05). C, equal numbers of HEK293 WT and CHCHD10-KD cells were plated and grown in medium containing either glucose or galactose as the primary carbon source. Growth was assessed in real time using CellTiter 96 AQueous One Solution Cell Proliferation Assay (MTS) (n = 4; *, p < 0.05 for CHCHD10-KD as compared with WT grown in galactose; NS for glucose). D, equal numbers of HEK293 WT and CHCHD10-KD cells were plated and grown in galactose medium (n = 4 each in quadruplicate; *, p < 0.05). E, total cellular ROS was measured from WT and CHCHD10-KD HEK293 cells using CM-H2DCFDA (n = 4; *, p < 0.05). NS, not significant. Error bars in graphs represent 1 standard deviation from the mean of repeat determinations.
To gain further insight into CHCHD10's role in vivo we tested cell growth of WT versus CHCHD10 knockdown (KD) cells in both glucose and galactose media. Growth of cells with glucose as the primary carbon source yielded indistinguishable growth rates independent of CHCHD10 level. However, when galactose was used as the primary carbon source, requiring cells to make ATP primarily through mitochondria (22), CHCHD10-KD cells grew more slowly than WT cells (Fig. 2C). The decrease in growth in galactose media was also seen by counting live cell numbers (Fig. 2D). We also tested production of total cellular ROS using CM-H2DCFDA and found that CHCHD10-KD cells generate higher ROS levels (Fig. 2E). Increased ROS levels and slower growth specifically in galactose indicate defective oxidative phosphorylation.
CHCHD10 stimulates COX activity in the mitochondria
CHCHD10 has previously been characterized as a soluble protein in the mitochondrial IMS (9), and a transient knockdown of CHCHD10 decreased COX activity and ATP levels (7). Complementing these observations, we find that CHCHD10 copurifies with COX (Fig. 3A, upper left) and interacts with the intact complex in vivo (Fig. 3A, upper right). Moreover, CHCHD10 is defective for its interaction with COX in the absence of MNRR1 (Fig. 3A, lower). When we separated purified bovine heart COX on a high percentage urea-acrylamide gel that separates COX into its component subunits (23), we detected CHCHD10 immunologically, supporting its interaction with COX (Fig. 3B). The amount of CHCHD10 appears, however, to be submolar, similar to MNRR1 (Fig. 3B) (5). Interestingly, when we added increasing amounts of recombinant CHCHD10 to purified COX in vitro we observed stimulation of COX activity (Fig. 3C). We then tested the effect of CHCHD10 on COX activity in vivo, by measuring intact cellular oxygen consumption. In WT cells, overexpression of CHCHD10 increases oxygen consumption (Fig. 3D) whereas in CHCHD10-KD cells oxygen consumption is decreased to ∼60% that in WT cells. Overexpression of CHCHD10 in KD cells restores oxygen consumption. Note that the restoration is equivalent to WT cells overexpressing CHCHD10 (Fig. 3D). We then asked whether overexpression of WT-CHCHD10 can increase oxygen consumption in MNRR1-KO cells. We found that it could not (Fig. 3E), consistent with the requirement for MNRR1 for CHCHD10 to interact with COX (Fig. 3A, lower). We also examined oxygen consumption under 8% hypoxia in WT and CHCHD10-KD cells, which was increased by nearly 90% in WT cells and 40% in CHCHD10-KD cells (Fig. 3F, left). We reasoned that the increase in CHCHD10-KD cells may be because of up-regulation of CHCHD10 under hypoxia and examined protein levels in CHCHD10-KD cells at 8%. Using Western blot analysis we see that CHCHD10 levels are up-regulated in the CHCHD10-KD under hypoxia comparable to WT levels (Fig. 3F, right). A similar effect was seen on oxygen consumption. Under hypoxia, oxygen consumption in CHCHD10-KD cells increased to levels comparable to WT at 20%. Hence, at 8% hypoxia, the up-regulated CHCHD10 increases oxygen consumption rate. Taken together, the data show that CHCHD10 functions to stimulate COX activity under normoxia as well as hypoxia in the mitochondria and that the presence of MNRR1 is necessary for this stimulation.
Figure 3.

CHCHD10 regulates COX activity in the mitochondria via phosphorylation of MNRR1. A, upper, purified COX from cow liver and heart and mitochondrial fractions from HEK293 cells were probed for CHCHD10. COX2 is probed as a loading control for the tissue COX, and COX2 beads were used to immunoprecipitate endogenous COX from HEK293 cells. All samples were probed for CHCHD10. Lower, to show lack of COX binding of CHCHD10 in MNRR1-KO cells, both WT and HEK293 MNRR1-KO cells were transfected with an expression plasmid for WT-CHCHD10 (FLAG-tagged). After 48 h, COX was immunoprecipitated from mitochondrial lysates with COX4-conjugated beads and probed for CHCHD10 (FLAG). Input fractions were also probed for CHCHD10 (FLAG) and MNRR1 levels. B, purified bovine heart COX (lane 1) and recombinant His-tagged CHCHD10 (lane 2) were separated on a 9-inch-long 50% acrylamide gel containing 7.2 m urea (23). Samples were separated in parallel and one set was stained with Coomassie Brilliant Blue and the other was transferred to a membrane and probed for CHCHD10, MNRR1, and COX4. C, in vitro COX activity was assessed using a kit (Sigma) that measures change in cytochrome c absorbance. Dialyzed purified COX and cytochrome c from bovine heart were used for the assay (n = 4; *, p < 0.05). COX (10 nm) activity was measured after supplementation with the stated amounts of CHCHD10. D, HEK293 WT and CHCHD10-KD cells were transfected with either empty vector (EV) or WT-CHCHD10. After 24 h, 40,000 cells per well were plated and used for measuring oxygen consumption using the Seahorse Bioanalyzer (n = 4; *, p < 0.05; **, p < 0.01). E, HEK293 MNRR1-KO cells were transfected with either empty vector (EV) or WT-CHCHD10. After 24 h, 40,000 cells per well were plated and used for measuring oxygen consumption using the Seahorse Bioanalyzer (n = 3, NS). F, left, WT-HEK293 or CHCHD10KD-HEK293 (30,000 cells/well) were plated in a 96-well plate and incubated at 20% O2 or 8% O2 for 24 h. Oxygen consumption was analyzed using the Oxygen Consumption Rate Assay Kit (Cayman) per the manufacturer's instructions (n = 4, each in triplicate). Right, equal amounts of the cell lysates from either WT or CHCHD10-KD incubated at 20% O2 and 8% O2 were loaded on an SDS-PAGE gel and probed with anti-CHCHD10 antibody. Tubulin was probed as loading control. NS, not significant. Error bars in graphs represent 1 standard deviation from the mean of repeat determinations.
Defective mitochondrial oxygen consumption in CHCHD10-KD cells arises via defective phosphorylation of MNRR1
To investigate the mechanism of decreased oxygen consumption in CHCHD10-KD cells, we assessed whether CHCHD10's homolog MNRR1 can rescue the decreased oxygen consumption. We found that transfecting increasing amounts of MNRR1 cannot increase oxygen consumption in CHCHD10-KD cells (Fig. 4A). Because mitochondrial stimulation of respiration by MNRR1 requires its phosphorylation on Tyr-99 by Abl2 kinase/ARG (24), we examined the phosphorylation level of endogenous MNRR1. Despite elevated levels of MNRR1 in the mitochondria, MNRR1 is less phosphorylated in CHCHD10-KD than in WT cells (Fig. 4B). Furthermore, phosphorylation of overexpressed mitochondrial MNRR1 was reduced (Fig. 4C). To investigate whether ARG is responsible for reduced MNRR1 phosphorylation, we examined levels of ARG in CHCHD10-KD mitochondria and found them to be lower than WT levels (Fig. 4C).
Figure 4.

Defective mitochondrial oxygen consumption in CHCHD10-KD cells arises from defective phosphorylation of MNRR1. A, overexpression of WT-MNRR1 fails to suppress the oxygen consumption defect of CHCHD10-KD. Upper, HEK293 WT and CHCHD10-KD cells (bars 1 and 2) are each overexpressing an empty vector (EV). CHCHD10-KD cells (D10KD) are overexpressing increasing amounts of MNRR1 (bars 3–5) or WT-CHCHD10 (bar 6). In each case, 40,000 cells per well were taken and oxygen consumption was measured 48 h after transfection (n = 4; *, p < 0.05; **, p < 0.01). Lower, equal amounts of lysate were probed for MNRR1 (shown at two exposure levels) and CHCHD10 levels. GAPDH was probed as a loading control. B, purified mitochondrial fractions from WT or CHCHD10-KD cells (D10KD) were immunoprecipitated with phosphotyrosine (pY) beads. Equal volumes of immunoprecipitate were separated on an SDS-PAGE gel and probed for MNRR1 and CHCHD10 levels. NDUFS3 was probed as a loading control and nuclear protein DRBP76 monitored fractionation. C, WT or CHCHD10-KD cells were transfected with FLAG-tagged MNRR1 plasmid. After 48 h, equal amounts of purified mitochondrial lysate were used for immunoprecipitation (IP) with FLAG beads. Equal IP volumes were separated by SDS-PAGE and probed with anti-pY and -FLAG (for MNRR1). Input fractions were probed for ARG and CHCHD10 levels; TOM20 was a loading control and nuclear protein DRBP76 monitored fractionation. D, above, CHCHD10-KD cells were transfected with either empty vector (EV) or one of the following FLAG-tagged constructs: WT-MNRR1 (WT-R1), Y99E-MNRR1 (Y99E-R1), or Y99F-MNRR1 (Y99F-R1). Y99E and Y99F represent glutamic acid (phosphomimetic) and phenylalanine (nonphosphorylatable) replacements, respectively, for WT tyrosine at position 99. After transfection (48 h), 40,000 cells per well were utilized for oxygen consumption measurements with the Seahorse Bioanalyzer (n = 4; *, p < 0.05). Below, equal amounts of lysate were probed for FLAG (MNRR1 expression) and the GAPDH loading control. E, in vitro COX activity was assessed using an assay kit (Sigma). Dialyzed purified COX and cytochrome c from bovine heart were used for the assay of COX with the addition of CHCHD10, MNRR1 (R1) bearing a phosphomimetic replacement (Y99E), and CHCHD10 plus MNRR1-Y99E (n = 4; *, p < 0.05). NS, not significant. Error bars in graphs represent 1 standard deviation from the mean of repeat determinations.
We hypothesized from the above observations that reduced ARG in mitochondria caused reduced MNRR1 phosphorylation, and thus reduced interaction with COX and decreased oxygen consumption, thereby rationalizing the inability of the overexpressed MNRR1 to suppress the oxygen consumption defect in CHCHD10-KD cells (Fig. 4A). To test this hypothesis, we overexpressed phosphomimetic MNRR1 (Y99E), which is expected to circumvent the reduced MNRR1 phosphorylation seen in CHCHD10-KD cells. We indeed observed rescue of oxygen consumption in CHCHD10-KD cells (Fig. 4D) whereas we did not see this rescue when we overexpressed WT or nonphosphorylatable (Y99F) MNRR1 at the same level. We can also see this effect in vitro using purified components. Maximum COX stimulation is seen with a combination of phosphomimetic MNRR1 and CHCHD10 as compared with each by itself (Fig. 4E).
In the nucleus CHCHD10 functions as a repressor at the ORE
The presence of CHCHD10 in both the nucleus and the mitochondria suggests that CHCHD10, like MNRR1, plays a role in both organelles. To characterize the effects of CHCHD10 in the nucleus, we used a luciferase reporter carrying the COX4I2 promoter, which harbors the 13-bp ORE (25). Unlike MNRR1 (6), overexpression of CHCHD10 inhibits transcription of the COX4I2 reporter; this inhibition requires an intact ORE (6) (Fig. 5A). We further investigated this inhibition in CHCHD10-KD cells with reporters harboring both a COX4I2 ORE (Fig. 5B) and an MNRR1 ORE (Fig. 5C). These results suggest that both COX4I2 and MNRR1 are transcriptionally up-regulated in CHCHD10-KD cells. We also examined reporter activity at 8% oxygen and found that transcription of the COX4I2 reporter is repressed at 8% oxygen to ∼60% of transcription at 20% oxygen (Fig. 5D). This is comparable to the effect seen by overexpressing WT-CHCHD10 under normoxia.
Figure 5.

CHCHD10 functions as a repressor at the ORE in the nucleus. A, upper, HEK293 cells were cotransfected with 100 ng COX4I2-luciferase WT or mutant reporter plasmid and either 200 ng of an empty vector (EV) or a WT-CHCHD10 expression plasmid and a dual luciferase assay was performed 48 h after transfection (n = 4; *, p < 0.05). Lower, equal amounts of nuclear lysate overexpressing empty vector (EV) or WT-CHCHD10 were loaded on an SDS-PAGE gel and probed with CHCHD10. DRBP76 was probed a loading control and mitochondrial protein NDUFS3 monitored fractionation. B, HEK293 or CHCHD10-KD cells were transfected with 100 ng COX4I2-luciferase WT or mutant reporter plasmid and a dual luciferase assay was performed 48 h after transfection (n = 4; *, p < 0.05). C, HEK293 or CHCHD10-KD cells were transfected with 100 ng MNRR1-luciferase reporter plasmid and assessed as in B (n = 4; *, p < 0.05). D, HEK293 cells were cotransfected with 100 ng COX4I2-luciferase WT reporter plasmid and either 200 ng empty vector (EV) or WT-CHCHD10 plasmid and incubated at 20% O2 or 8% O2. After 48 h, a dual luciferase assay was performed to measure reporter activity (n = 4; *, p < 0.05). E, HEK293 cells were transfected with a CXXC5 expression plasmid; 48 h after transfection nuclear lysate was immunoprecipitated with CXXC5 beads and probed for endogenous CHCHD10 (top). HEK293 cells were transfected with a CHCHD10 expression plasmid; after 48 h nuclear lysate was immunoprecipitated with CHCHD10 beads and probed for endogenous CXXC5 (bottom). F, HEK293 cells (WT or CXXC5 KD) transfected with a CHCHD10 expression plasmid were used for a DNA-binding assay. Anti-CHCHD10 immunoprecipitate was used to detect bound ORE-specific DNA by PCR amplification using endogenous COX4I2 ORE-specific primers. Knockdown of CXXC5 was confirmed by Western blotting; GAPDH was probed as a loading control. G, HEK293 cells were transfected with empty vector (EV) or FLAG-tagged MNRR1 expression plasmids. After 48 h, equal amounts of a nuclear-enriched lysate were immunoprecipitated with FLAG beads and probed for CHCHD10. Equal amounts of input lysate were also separated on an SDS-PAGE gel and probed for MNRR1 (FLAG) and CHCHD10 levels. Nuclear protein PCNA was used as a loading control and mitochondrial protein NDUFS3 was used to monitor fractionation. NS, not significant. Error bars in graphs represent 1 standard deviation from the mean of repeat determinations.
Transcription at the ORE was previously shown to be governed by three proteins: RBPJk, CXXC5, and MNRR1 (6). At 20% oxygen, CXXC5 interacts with RBPJk at the ORE to repress transcription, whereas at 4% MNRR1 displaces the inhibitory CXXC5 and activates RBPJk-mediated transcription at the ORE (6) (see Fig. 7B). Because CHCHD10 function requires an intact ORE, we hypothesized that CHCHD10 interacts with CXXC5 to strengthen repression at the ORE and tested this hypothesis by examining the interaction of CHCHD10 with CXXC5. We found that CHCHD10 co-immunoprecipitates with CXXC5 and vice versa (Fig. 5E). This presence of CXXC5 is essential for CHCHD10 to interact with the ORE because transient knockdown of CXXC5 prevents the interaction of CHCHD10 with the COX4I2 ORE (Fig. 5F). These results suggest that CXXC5 is necessary for CHCHD10 to function at the ORE. Furthermore, CHCHD10 does not interact with MNRR1 in the nucleus (Fig. 5G), consistent with opposed functions of CHCHD10 and MNRR1 in this cellular compartment.
Figure 7.
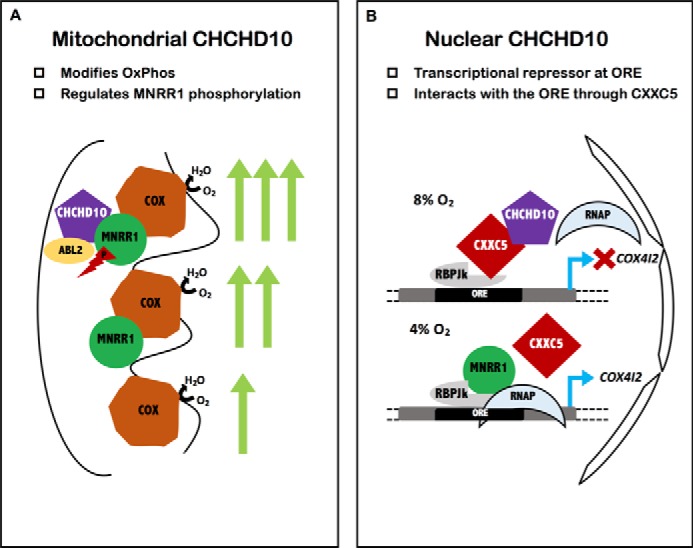
Model for CHCHD10 functioning as a bi-organellar regulator of oxidative phosphorylation. A, shows the binding of MNRR1 to COX to provide increased COX activity; the phosphorylation of MNRR1 by Abl2 kinase increases MNRR1 binding to COX (24). Mitochondrial CHCHD10 functions as a scaffold for recruitment of Abl2 kinase (ARG) to phosphorylate MNRR1. B, shows that, maximally at 8% oxygen, nuclear CHCHD10 is up-regulated and functions as an inhibitor of transcription by binding to repressor protein CXXC5 and strengthening its binding. At 4% oxygen the production of transcription stimulator MNRR1 increases, displacing CXXC5. The effect on a known ORE-containing gene, COX4I2, is indicated.
Point mutations in CHCHD10 abrogate its function in both the nucleus and the mitochondria
More than 10 different mutations in CHCHD10 have been linked with neurodegenerative diseases, and most lie within the region encoded by exon 2 (amino acids 15–87) (26). Two functionally uncharacterized mutations found in this region, G66V and P80L, were chosen for study because they have been associated with several different phenotypes including ALS (9, 12), Charcot-Marie-Tooth neuropathy type 2A (CMT2A) (27), and spinomuscular atrophy (15). In fact, the G66V mutation has been associated with a spectrum of phenotypes within the same family (28). Importantly, unlike some mutations, these have not been identified in any control population in several different studies (29), strengthening their disease association. To investigate the nuclear function of these mutants, we examined their ability to inhibit the COX4I2 reporter. We found that overexpression of these CHCHD10 mutants, unlike WT, failed to repress transcription of the COX4I2 reporter (Fig. 6A). We also examined these effects at the translational level by analyzing levels of COX4I2 and MNRR1, two ORE-regulated proteins. Both are repressed by WT-CHCHD10 but not by the mutants (Fig. 6B). To identify the mechanism for the failure to repress, we compared the interaction of WT-CHCHD10 and the point mutants with ORE inhibitor protein CXXC5. Both mutant CHCHD10 proteins were found to be defective for interaction with CXXC5 (Fig. 6C).
Figure 6.

Point mutations in CHCHD10 abrogate CHCHD10's function in the nucleus and the mitochondria. A, top, HEK293 cells were cotransfected with 100 ng COX4I2-luciferase WT or mutant reporter plasmid and either 200 ng empty vector (EV) or the different CHCHD10 expression plasmids. After 48 h, a dual luciferase assay was performed to measure reporter activity (n = 4; *, p < 0.05). Bottom, equal amounts of nuclear fractions transfected with empty vector (EV), WT-CHCHD10, or point mutants were separated on an SDS-PAGE gel and probed with CHCHD10. DRBP76 was probed as a loading control and mitochondrial protein NDUFS3 monitored fractionation. B, equal amounts of whole cell lysates transfected with empty vector (EV), or with WT or point mutants of CHCHD10, were separated on an SDS-PAGE gel and probed for MNRR1 and CHCHD10. GAPDH was used as a loading control. C, HEK293 cells were cotransfected with CXXC5 and with WT or CHCHD10 (FLAG-tagged) point mutants. After 48 h nuclear lysates were immunoprecipitated with CXXC5-conjugated beads and probed for FLAG-tagged CHCHD10. Equal amounts of input fractions were also probed for FLAG and CXXC5. D, above, WT cells were transfected with an empty vector as a control and CHCHD10-KD cells were transfected with either empty vector (EV), WT, or point mutants for CHCHD10. Oxygen consumption was measured after 24 h (n = 4; *, p < 0.05). Below, equal amounts of whole cell lysates of WT cells transfected with an empty vector and CHCHD10-KD cells transfected with empty vector (EV), WT-CHCHD10, or point mutants were separated on an SDS-PAGE gel and probed with CHCHD10. GAPDH served as a loading control. E, WT cells were transfected with an empty vector as a control and CHCHD10-KD cells were transfected with either empty vector (EV), WT, or point mutants for CHCHD10. After 24 h, 40,000 cells/well were plated on a 96-well plate and mitochondrial membrane potential was measured using TMRM (Invitrogen) per the manufacturer's protocol (n = 4, each in quadruplicate). F, HEK293 cells were transfected with an empty vector (EV), or with WT or mutant CHCHD10 expression plasmids. After 48 h, 1.5 × 105 cells were plated on a 24-well plate and mitochondrial ROS production was measured with MitoSOX Red (n = 4; *, p < 0.05). Results are shown as fluorescence relative to the empty vector sample. G, HEK293 cells were cotransfected with WT or (FLAG-tagged) CHCHD10 point mutants. After 48 h mitochondria-enriched lysates were used for immunoprecipitation (IP) of FLAG-tagged CHCHD10 and probed (IB) for MNRR1 and COX6B levels. Equal amounts of input fractions were also probed for FLAG (for CHCHD10), MNRR1, and COX6B. NS, not significant. Error bars in graphs represent 1 standard deviation from the mean of repeat determinations.
Because CHCHD10 interacts with COX in the mitochondria and promotes its activity, we examined whether either of the two mutations could rescue the oxygen consumption defect seen in CHCHD10-KD cells. We found that, unlike WT-CHCHD10, neither point mutant was able to rescue these defects, being no better than an overexpressed empty vector (Fig. 6D). We also tested the mitochondrial membrane potential in CHCHD10-KD cells and found it to be ∼50% of WT cells. Overexpression of WT-CHCHD10 can rescue this defect whereas neither point mutant was able to rescue the defective membrane potential (Fig. 6E). Moreover, each mutant increased production of mitochondrial ROS, in contrast to WT-CHCHD10, which decreased ROS production (Fig. 6F). To examine the mechanism by which this could occur, we tested for the interaction of WT or mutant CHCHD10 with MNRR1 and COX. Each mutant protein was defective for binding to MNRR1 and thereby COX (Fig. 6G). A failure to bind MNRR1 leads to reduced ARG-mediated phosphorylation of MNRR1, which then cannot bind to and stimulate COX activity. Hence, the G66V and P80L mutant CHCHD10 proteins may be defective for stimulating mitochondrial oxygen consumption, presumably leading to production of ROS.
Discussion
Mitochondrial energy production is regulated to respond to extracellular changes, a key feature of cellular homeostasis that requires mitonuclear signaling to alter gene transcription (30, 31). We previously showed that MNRR1 is a component of a mitonuclear pathway whose signaling is altered under oxidative stress (5, 6, 24) and we now show that it functions in coordination with CHCHD10. CHCHD10 and MNRR1 belong to the twin CX9C family of proteins that primarily form scaffolds for the formation and the organization of the large macromolecular complexes found in mitochondria (32) such as cytochrome c oxidase, a complex composed of 14 subunits (33). MNRR1 and CHCHD10 are highly conserved (58% in humans); have a common homolog in yeast, Mix17; and appear to be the product of a gene duplication event prior to the mammalian radiation. Moreover, the presence of disulfide linkages to stabilize the CHCH domain imparts potential redox sensitivity to these proteins, which may be essential to their ability to regulate the stability of COX under altered oxygen tensions (32). The fact that MNRR1 and CHCHD10 function as a complex was also recognized in a recent analysis of an ALS-associated mutation, R15L, in CHCHD10 (20).
CHCHD10 and MNRR1 in the mitochondria bind to COX and regulate its activity. MNRR1's mitochondrial role has been characterized previously (5), including the key step of its phosphorylation by Abl2 kinase/ARG (24). In the present study, we identified the mitochondrial role of CHCHD10 to recruit ARG to phosphorylate MNRR1. This phosphorylation enhances the binding of MNRR1 to COX and increases COX activity. The above role for CHCHD10 is supported by the observations of reduced phosphorylation of MNRR1 and decreased oxygen consumption in CHCHD10-KD cells. Importantly, only a phosphomimetic version of MNRR1, but not WT-MNRR1, can rescue the respiration defect in CHCHD10-KD cells (Fig. 4D).
MNRR1 and CHCHD10 are also present in the nucleus, where they positively and negatively regulate transcription of ORE-harboring genes, respectively. Moreover, both proteins are oxygen regulated, induced maximally at two distinct oxygen tensions, 4% for MNRR1 (6) and 8% for CHCHD10 (Fig. 1). Thus, the relative effect of each on transcription of ORE-containing genes may change continuously as oxygen levels decline. Experimental oxygen tension is 20%, although this is high compared with physiological levels in vivo, which range from ∼3 to 8% in most tissues and ∼13% in lung (19, 34). One of the most critical periods of oxygen sensing is during placental development when oxygen levels switch from 2 to 3% prior to 12 weeks of gestation to 8% afterward (35). Different oxygen levels likely require the regulation of factors that respond appropriately and alter transcription. The DNA element in the CHCHD10 promoter that responds maximally at 8% oxygen is yet to be characterized. Furthermore, other cellular factors besides MNRR1 and CHCHD10 and other oxygen-responsive elements may be responsible for regulating mitochondrial function at different oxygen tensions. The twin CX9C protein CHCHD3 is also known to regulate transcription, at the BAG1 promoter (36), although its hypoxia sensitivity is yet to be tested.
Mutations in CHCHD10 have been associated with a number of neurodegenerative diseases. Several studies have characterized the effects of CHCHD10 by analyzing these mutations in skin fibroblasts from patients (9, 14, 20) or by overexpression of these mutations in WT cells (8, 37). Here we show that CHCHD10 plays a key role in the regulation of electron transport chain function in the mitochondria as well as a novel role in the nucleus as a transcriptional repressor at the ORE. We selected two CHCHD10 mutations, P80L and G66V, for detailed study. These mutations are associated with several phenotypes across different populations and are absent in control populations. In the mitochondria, each mutant decreased mitochondrial oxygen consumption and increased production of mitochondrial ROS. High ROS levels have been extensively associated with neurodegeneration and specifically are consistent with neurodegenerative phenotypes such as ALS (38); motor neuron disease, in which ROS may affect cross talk between neurons and muscles (39); and Parkinson's disease (40). Unlike WT-CHCHD10, which is necessary to recruit ARG to phosphorylate MNRR1 (Fig. 4, B and C), the mutants do not interact with MNRR1 (Fig. 6G) and they may not recruit ARG to phosphorylate MNRR1. MNRR1 phosphorylation by ARG is essential to its role in regulation of COX activity, and a mutant version of MNRR1 that has defective phosphorylation has been linked to another neurodegenerative disorder, Charcot-Marie-Tooth disease type 1A (24). Thus, even in the presence of elevated levels of MNRR1 the two mutant versions of CHCHD10 prevent phosphorylation of MNRR1, giving rise to the mitochondrial phenotype seen. A recent study also showed that CHCHD10 is localized to both the mitochondria and the nucleus and that its normal function prevents the neurotoxic mitochondrial localization of the protein TDP-43 whereas two other disease-associated mutations, R15L and S59L, promote its cytoplasmic mislocalization (21). These mutations have also been shown to accumulate excessive mitochondrial iron (37).
A feature of the pathogenic mutations not well explained thus far is their generally heterozygous appearance, suggesting they are dominant mutations, whereas in cultured cells the two mutations examined do not appear dominant. Despite being overexpressed, endogenous expression needs to be reduced to see a clear phenotype. An important caveat, however, is that the neurodegenerative diseases to which these mutations have been linked show late onset. Consequently, these experiments do not preclude a small degree of haploinsufficiency that, over time, leads to the observed pathophysiologies. Because there is evidence of population-specific effects of some of the mutations (41–43), it is also possible that other variants must be present to elicit a full phenotype, a possibility also suggested by the diversity of neurodegenerative diseases in which mutations in CHCHD10 have been found.
In summary, we have characterized the role of CHCHD10 as a key modulator of mitochondrial function via interorganellar signaling (Fig. 7). The failure of several pathogenic mutants, which no longer interact with COX, to rescue the mitochondrial effects of CHCHD10 depletion provides a potential mechanism for CHCHD10 dysfunction during the pathogenesis of neurodegenerative diseases. In addition, the altered transcriptional regulation in the nucleus of these mutants at ORE-harboring promoters provides a further pathogenic pathway. Lastly, the hypoxia sensitivity of both CHCHD10 and MNRR1 transcription places the CHCHD10-MNRR1 pair in a regulatory network that can respond to altered oxygen levels. Such altered levels may be present in different tissues, during different developmental stages, or as a result of disease, and whose lack may therefore contribute to neurodegenerative disease phenotypes with which CHCHD10 mutations have been associated.
Experimental procedures
Cell culture
Experiments were performed with HEK293 cells and HeLa (human cervical cancer) cells. The HEK293 and HeLa cells were cultured in Dulbecco's modified Eagle's medium (DMEM) (HyClone, Logan, UT) with 10% FBS (Sigma-Aldrich) plus antibiotic and antimycotic (HyClone); CHCHD10 knockdown (sc-72754, Santa Cruz Biotechnology (SCBT), Dallas, TX) cells were grown under puromycin selection. HEK293 cells stably overexpressing WT or mutant CHCHD10 were selected using G418.
The CHCHD10 shRNA is a pool of two different shRNA plasmids: hairpin sequence GATCCGTGCAAGTACTACCATGGTTTCAAGAGAACCATGGTAGTACTTGCACTTTTT and corresponding siRNA sequences (sc-72754A) (sense, GUGCAAGUACUACCAUGGUTT; antisense, ACCAUGGUAGUACUUGCACTT) and the sc-72754B hairpin sequence GATCCCAGACCACAACACCAGATTTTCAAGAGAAATCTGGTGTTGTGGTCTGTTTTT and corresponding siRNA sequences (sc-72754B) (sense, CAGACCACAACACCAGAUUTT; antisense, AAUCUGGUGUUGUGGUCUGTT). Note that all sequences are provided in 5′ → 3′ orientation.
Effector and reporter plasmids
The WT and mutant 579-bp COX4I2 promoter luciferase reporter plasmids (6) and MNRR1 reporter plasmid (5) have been described previously. The WT MNRR1 and CHCHD10 expression plasmid was cloned in the pCI-Neo vector with a C-terminal 3× FLAG epitope. The G66V and P80L mutants were cloned in the pCI-Neo vector using overlap extension PCR. The mutation was confirmed by sequencing. All the expression plasmids were purified using the EndoFree plasmid purification kit (Qiagen, Valencia, CA). CXXC5 was expressed from a pCI-Neo vector.
Transient transfection of HEK293 cells
HEK293 cells were transfected with the indicated plasmids using TransFast transfection reagent (Promega, Madison, WI) according to the manufacturer's protocol. A TransFast:DNA ratio of 3:1 in serum- and antibiotic-free medium was used. Following incubation at room temperature for ∼15 min, the cells were overlaid with the mixture. The plates were incubated for 1 h at 37 °C followed by replacement with complete medium and further incubation for the indicated time interval.
Real-time PCR
Total cellular RNA was extracted using an RNeasy Plus Mini Kit (Qiagen) per the manufacturer's instructions. Complementary DNA (cDNA) was generated by reverse transcriptase PCR using the ProtoScript® II First Strand cDNA Synthesis Kit (New England Biolabs, Ipswich, MA). Transcript levels were measured by real time PCR using SYBR Green on an ABI7500 system. Real-time analysis was performed by the ΔΔCT method (44). The primer sequences used were as follows: CHCHD10 (forward, CGCCTACGAGATCAGGCAGT; reverse, GGAGCTCAGACCATGGTAGTACTTG) and 18S rRNA (forward, AGTCCCTGCCCTTTGTACACA; reverse, GATCCGAGGGCCTCACTAAAC).
Hypoxia assays
Cells were maintained either at 20% oxygen (normoxia) or at 13, 9, 8, 7, 4, or 1% oxygen in a chamber infused with N2 and CO2 at 37 °C. The gas flow was controlled with PRO-OX 110 and PRO-CO2 controllers (BioSpherix, Redfield, NY) to achieve desired oxygen levels and 5% CO2. Oxygen equilibration time (3–4 h) was accounted for in the hypoxia experiments.
Luciferase reporter assays
Luciferase assays were performed using the dual-luciferase reporter assay kit (Promega) per the manufacturer's instructions. Transfection efficiency was normalized with the cotransfected pRL-SV40 Renilla luciferase expression plasmid.
DNA-binding assays
DNA-binding assays were performed as described (6) with a few modifications. HEK293 WT (scrambled) or CXXC5 KD (siRNA sc-91677 from SCBT) was transfected with a CHCHD10 expression plasmid. For sc-91677, CXXC5 siRNA is a pool of three different siRNA duplexes: sc-91677A (sense, CUCUCCCACUACUCUUCUUTT; antisense, AAGAAGAGUAGUGGGAGAGTT); sc-91677B (sense, CCAGGCCUCUUCAUUAUGATT; antisense, UCAUAAUGAAGAGGCCUGGTT); sc-91677C (sense, GCAGUUGUAGGAAUCGAAATT; antisense, UUUCGAUUCCUACAACUGCTTA). All sequences are provided in 5′ → 3′ orientation.
After 48 h proteins were cross-linked and nuclei were separated. DNA was fragmented by incubation with Sau3AI and nuclei were lysed. The antibody-matrix complex was prepared as described under “Immunoblotting and co-immunoprecipitation (IP)” using anti-CHCHD10 antibody and shaken overnight with nuclear lysate at 4 °C. Cross-linking was reversed and the precipitated DNA was purified with the QIAquick purification kit (Qiagen) and amplified with primers encompassing the segment harboring the endogenous ORE for both the CHCHD10 and IgG IP samples.
Cell proliferation assay
Cell proliferation was assessed with the CellTiter 96 AQueous One colorimetric solution kit (Promega) in a 96-well plate according to the manufacturer's instructions. Equal numbers of cells were plated and analyzed for proliferation at 24 h (∼1.5 doublings) and 48 h (∼3 doublings) by treating with CellTiter 96 cell aqueous solution and recording absorbance at 490 nm.
Cell counting assay
Live cell counts were performed by trypan blue staining on a CountessTM automated cell counter (Invitrogen). Live cells (7.5 × 104) were plated and allowed to grow for 48 h (∼3 doublings), and the number of live cells counted again.
Intact cellular oxygen consumption
Cellular oxygen consumption was measured with a Seahorse XFe24 Bioanalyzer. Cells were plated at a concentration of 40,000 per well one day prior to basal oxygen consumption measurements, performed according to the manufacturer's instructions.
For the hypoxia experiments oxygen consumption was analyzed using the Oxygen Consumption Rate Assay Kit (Cayman, Ann Arbor, MI). Cells were plated at a concentration of 30,000 per well one day prior to measurements, incubated at 20 or 8% oxygen, and measurements were performed according to the manufacturer's instructions.
Cytochrome c oxidase assay
In vitro COX activity was measured using the Cytochrome c Oxidase Assay Kit from Sigma-Aldrich as per manufacturer's instructions using bovine heart cytochrome c. The cytochrome c oxidase used was purified from bovine heart and dialyzed before being used for the assay.
ROS measurements
Total cellular ROS measurements were performed with CM-H2DCFDA (Life Technologies). Cells were distributed into 12-well plates at 105 cells per well and incubated for 24 h. Cells were then treated with 10 μm CM-H2DCFDA in serum- and antibiotic-free medium for 1 h. Cells were washed twice in PBS and analyzed for fluorescence on a Gen5 Microplate Reader (BioTek Inc, Winooski, VT). For mitochondrial ROS measurements, the cells were treated as above but with 5 μm MitoSOX Red (Life Technologies) for 30 min.
Confocal microscopy
Confocal microscopy was performed as described (24). Primary antibodies were CHCHD10 (1:50, Sigma) and COXI (1:100, Life Technologies). Secondary antibodies were goat anti-rabbit IgG Alexa Fluor 488 and goat anti-mouse IgG Alexa Fluor 596 (1:300, The Jackson Laboratory, Bar Harbor, ME). Cells were imaged with a Leica TCS S5P microscope and images were combined in Photoshop. Colocalization (overlap of the two fluorophores) and intensity (number of pixels per unit area) were quantitated using Volocity image analysis software (PerkinElmer Life Sciences). The Pearson correlation coefficient (24) in Fig. 2B is indicated on the images.
Immunoblotting and co-immunoprecipitation (IP)
Immunoblotting on a PVDF membrane was performed as described previously (5, 6). Unless specified otherwise, primary antibodies were used at a concentration of 1:500 and secondary antibodies at a concentration of 1:5000. The MNRR1 (19424–1-AP), NDUFS3 (15066–1-AP), DRBP76 (19887–1-AP), COX6B1(11425–1-AP), COX4 (11242–1-AP), GAPDH (HRP-60004), and tubulin (HRP-66031) antibodies were purchased from Proteintech (Chicago, IL). The anti-CHCHD10 (HPA003440) and anti-FLAG HRP (A8592) conjugated antibodies were purchased from Sigma-Aldrich and the COX4I2 (H00084701-M01) antibody was purchased from Abnova Corp. (Taipei City, Taiwan). The TOM20 (72610) antibody was purchased from Cell Signaling Technology (Beverly, MA), and the anti-phosphotyrosine (4G10) antibody (05–321) was purchased from EMD Millipore (Burlington, MA). Co-immunoprecipitation experiments were performed per the supplier's protocol by incubating the antibody-adsorbed beads overnight at 4 °C. For co-immunoprecipitation of FLAG-tagged proteins, Anti-FLAG M2 affinity gel (A220, Sigma-Aldrich) and anti-phosphotyrosine AC beads (sc-7020, SCBT) were used. The ARG antibody was a gift from Dr. Anthony Koleske (Yale University). The high percentage urea-acrylamide gel was run as described previously (23).
Mitochondria isolation
Mitochondria were isolated from cells with a Mitochondrial Isolation Kit (Thermo Scientific) according to the manufacturer's protocol. The nuclear fraction was obtained by low-speed centrifugation and the mitochondrial fraction was obtained after high-speed centrifugation of the nuclear supernatant. Cross-contamination between the fractions was analyzed with compartment-specific antibodies.
Statistical analysis
Statistical analyses were performed with MSTAT version 6.1.1 (N. Drinkwater, University of Wisconsin, Madison, WI). The two-sided Wilcoxon rank sum test was applied to determine statistical significance for p values. Data were considered statistically significant with p < 0.05.
Data availability
All data generated or analyzed during this study are included in this published article.
Author contributions
N. P., M. S., L. I. G., and S. A. data curation; L. I. G. and M. H. funding acquisition; N. P. and M. S. validation; N. P., M. S., L. I. G., and S. A. investigation; N. P. and M. S. writing-original draft; N. P., M. S., M. H., L. I. G., and S. A. writing-review and editing; L. I. G. and M. H. resources; M. H. methodology; L. I. G. and S. A. conceptualization; L. I. G. and S. A. supervision; L. I. G. and S. A. project administration.
Acknowledgments
We thank Dr. Anthony Koleske (Yale University) for a gift of ARG-specific antibody and Justyna Restak for help with the DNA-binding assay.
This work was supported by the Office of the Assistant Secretary of Defense for Health Affairs through the Peer Reviewed Medical Research Program under Award W81XWH-16-1-0516 and the Henry L. Brasza endowment at Wayne State University. The authors declare that they have no conflicts of interest with the contents of this article. The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the U. S. Department of Defense or the United States government.
- IMS
- intermembrane space
- ROS
- reactive oxygen species
- ORE
- oxygen-responsive element
- COX
- cytochrome c oxidase
- KD
- knockdown
- IP
- immunoprecipitation.
References
- 1. Modjtahedi N., Tokatlidis K., Dessen P., and Kroemer G. (2016) Mitochondrial proteins containing coiled-coil-helix-coiled-coil-helix (CHCH) domains in health and disease. Trends Biochem. Sci. 41, 245–260 10.1016/j.tibs.2015.12.004 [DOI] [PubMed] [Google Scholar]
- 2. Chacinska A., Pfannschmidt S., Wiedemann N., Kozjak V., Sanjuán Szklarz L. K., Schulze-Specking A., Truscott K. N., Guiard B., Meisinger C., and Pfanner N. (2004) Essential role of Mia40 in import and assembly of mitochondrial intermembrane space proteins. EMBO J. 23, 3735–3746 10.1038/sj.emboj.7600389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Darshi M., Mendiola V. L., Mackey M. R., Murphy A. N., Koller A., Perkins G. A., Ellisman M. H., and Taylor S. S. (2011) ChChd3, an inner mitochondrial membrane protein, is essential for maintaining crista integrity and mitochondrial function. J. Biol. Chem. 286, 2918–2932 10.1074/jbc.M110.171975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. An J., Shi J., He Q., Lui K., Liu Y., Huang Y., and Sheikh M. S. (2012) CHCM1/CHCHD6, novel mitochondrial protein linked to regulation of mitofilin and mitochondrial cristae morphology. J. Biol. Chem. 287, 7411–7426 10.1074/jbc.M111.277103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aras S., Bai M., Lee I., Springett R., Hüttemann M., and Grossman L. I. (2015) MNRR1 (formerly CHCHD2) is a bi-organellar regulator of mitochondrial metabolism. Mitochondrion 20, 43–51 10.1016/j.mito.2014.10.003 [DOI] [PubMed] [Google Scholar]
- 6. Aras S., Pak O., Sommer N., Finley R. Jr., Hüttemann M., Weissmann N., and Grossman L. I. (2013) Oxygen-dependent expression of cytochrome c oxidase subunit 4–2 gene expression is mediated by transcription factors RBPJ, CXXC5 and CHCHD2. Nucleic Acids Res. 41, 2255–2266 10.1093/nar/gks1454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Martherus R. S., Sluiter W., Timmer E. D., VanHerle S. J., Smeets H. J., and Ayoubi T. A. (2010) Functional annotation of heart enriched mitochondrial genes GBAS and CHCHD10 through guilt by association. Biochem. Biophys. Res. Commun. 402, 203–208 10.1016/j.bbrc.2010.09.109 [DOI] [PubMed] [Google Scholar]
- 8. Ajroud-Driss S., Fecto F., Ajroud K., Lalani I., Calvo S. E., Mootha V. K., Deng H. X., Siddique N., Tahmoush A. J., Heiman-Patterson T. D., and Siddique T. (2015) Mutation in the novel nuclear-encoded mitochondrial protein CHCHD10 in a family with autosomal dominant mitochondrial myopathy. Neurogenetics 16, 1–9 10.1007/s10048-014-0421-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bannwarth S., Ait-El-Mkadem S., Chaussenot A., Genin E. C., Lacas-Gervais S., Fragaki K., Berg-Alonso L., Kageyama Y., Serre V., Moore D. G., Verschueren A., Rouzier C., Le Ber I., Augé G., Cochaud C., et al. (2014) A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 137, 2329–2345 10.1093/brain/awu138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chaussenot A., Le Ber I., Ait-El-Mkadem S., Camuzat A., de Septenville A., Bannwarth S., Genin E. C., Serre V., Auge G., French research network on, F. T. D., Ftd, A. L. S., Brice A., Pouget J., and Paquis-Flucklinger V. (2014) Screening of CHCHD10 in a French cohort confirms the involvement of this gene in frontotemporal dementia with amyotrophic lateral sclerosis patients. Neurobiol. Aging 35, 2884.e1–2884.e4 10.1016/j.neurobiolaging.2014.07.022 [DOI] [PubMed] [Google Scholar]
- 11. Zhang M., Xi Z., Zinman L., Bruni A. C., Maletta R. G., Curcio S. A., Rainero I., Rubino E., Pinessi L., Nacmias B., Sorbi S., Galimberti D., Lang A. E., Fox S., Surace E. I., et al. (2015) Mutation analysis of CHCHD10 in different neurodegenerative diseases. Brain 138, e380 10.1093/brain/awv082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Müller K., Andersen P. M., Hübers A., Marroquin N., Volk A. E., Danzer K. M., Meitinger T., Ludolph A. C., Strom T. M., and Weishaupt J. H. (2014) Two novel mutations in conserved codons indicate that CHCHD10 is a gene associated with motor neuron disease. Brain 137, e309 10.1093/brain/awu227 [DOI] [PubMed] [Google Scholar]
- 13. Dols-Icardo O., Nebot I., Gorostidi A., Ortega-Cubero S., Hernández I., Rojas-García R., García-Redondo A., Povedano M., Lladó A., Álvarez V., Sánchez-Juan P., Pardo J., Jericó I., Vázquez-Costa J., Sevilla T., et al. (2015) Analysis of the CHCHD10 gene in patients with frontotemporal dementia and amyotrophic lateral sclerosis from Spain. Brain 138, e400 10.1093/brain/awv175 [DOI] [PubMed] [Google Scholar]
- 14. Genin E. C., Plutino M., Bannwarth S., Villa E., Cisneros-Barroso E., Roy M., Ortega-Vila B., Fragaki K., Lespinasse F., Pinero-Martos E., Augé G., Moore D., Burté F., Lacas-Gervais S., Kageyama Y., et al. (2016) CHCHD10 mutations promote loss of mitochondrial cristae junctions with impaired mitochondrial genome maintenance and inhibition of apoptosis. EMBO Mol. Med. 8, 58–72 10.15252/emmm.201505496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Penttilä S., Jokela M., Saukkonen A. M., Toivanen J., Palmio J., Lähdesmäki J., Sandell S., Shcherbii M., Auranen M., Ylikallio E., Tyynismaa H., and Udd B. (2017) CHCHD10 mutations and motor neuron disease: The distribution in Finnish patients. J Neurol. Neurosurg. Psychiatry 88, 272–277 10.1136/jnnp-2016-314154 [DOI] [PubMed] [Google Scholar]
- 16. Chio A., Mora G., Sabatelli M., Caponnetto C., Traynor B. J., Johnson J. O., Nalls M. A., Calvo A., Moglia C., Borghero G., Monsurro M. R., La Bella V., Volanti P., Simone I., Salvi F., et al. (2015) CHCH10 mutations in an Italian cohort of familial and sporadic amyotrophic lateral sclerosis patients. Neurobiol. Aging 36, 1767.e3–1767.e6 10.1016/j.neurobiolaging.2015.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kurzwelly D., Krüger S., Biskup S., and Heneka M. T. (2015) A distinct clinical phenotype in a German kindred with motor neuron disease carrying a CHCHD10 mutation. Brain 138, e376 10.1093/brain/awv014 [DOI] [PubMed] [Google Scholar]
- 18. Ait-El-Mkadem S., Chaussenot A., Bannwarth S., Rouzier C., and Paquis-Flucklinger V. (2015) CHCHD10-related disorders. In GeneReviews® (Internet) (Adam M. P., Ardinger H. H., Pagon R. A., Wallace S. E., Bean L. J. H., Stephens K., Amemiya A., eds.), University of Washington, Seattle WA: [PubMed] [Google Scholar]
- 19. Carreau A., El Hafny-Rahbi B., Matejuk A., Grillon C., and Kieda C. (2011) Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell. Mol. Med. 15, 1239–1253 10.1111/j.1582-4934.2011.01258.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Straub I. R., Janer A., Weraarpachai W., Zinman L., Robertson J., Rogaeva E., and Shoubridge E. A. (2018) Loss of CHCHD10-CHCHD2 complexes required for respiration underlies the pathogenicity of a CHCHD10 mutation in ALS. Hum. Mol. Genet. 27, 178–189 10.1093/hmg/ddx393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Woo J. A., Liu T., Trotter C., Fang C. C., De Narvaez E., LePochat P., Maslar D., Bukhari A., Zhao X., Deonarine A., Westerheide S. D., and Kang D. E. (2017) Loss of function CHCHD10 mutations in cytoplasmic TDP-43 accumulation and synaptic integrity. Nat. Commun. 8, 15558 10.1038/ncomms15558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Robinson B. H., Petrova-Benedict R., Buncic J. R., and Wallace D. C. (1992) Nonviability of cells with oxidative defects in galactose medium: A screening test for affected patient fibroblasts. Biochem. Med. Metab. Biol. 48, 122–126 10.1016/0885-4505(92)90056-5 [DOI] [PubMed] [Google Scholar]
- 23. Kadenbach B., Jarausch J., Hartmann R., and Merle P. (1983) Separation of mammalian cytochrome c oxidase into 13 polypeptides by a sodium dodecyl sulfate-gel electrophoretic procedure. Anal. Biochem. 129, 517–521 10.1016/0003-2697(83)90586-9 [DOI] [PubMed] [Google Scholar]
- 24. Aras S., Arrabi H., Purandare N., Hüttemann M., Kamholz J., Züchner S., and Grossman L. I. (2017) Abl2 kinase phosphorylates bi-organellar regulator MNRR1 in mitochondria, stimulating respiration. Biochim. Biophys. Acta 1864, 440–448 10.1016/j.bbamcr.2016.11.029 [DOI] [PubMed] [Google Scholar]
- 25. Hüttemann M., Lee I., Liu J., and Grossman L. I. (2007) Transcription of mammalian cytochrome c oxidase subunit IV-2 is controlled by a novel conserved oxygen responsive element. FEBS J. 274, 5737–5748 10.1111/j.1742-4658.2007.06093.x [DOI] [PubMed] [Google Scholar]
- 26. Ronchi D., Riboldi G., Del Bo R., Ticozzi N., Scarlato M., Galimberti D., Corti S., Silani V., Bresolin N., and Comi G. P. (2015) CHCHD10 mutations in Italian patients with sporadic amyotrophic lateral sclerosis. Brain 138, e372 10.1093/brain/awu384 [DOI] [PubMed] [Google Scholar]
- 27. Auranen M., Ylikallio E., Shcherbii M., Paetau A., Kiuru-Enari S., Toppila J. P., and Tyynismaa H. (2015) CHCHD10 variant p. (Gly66Val) causes axonal Charcot-Marie-Tooth disease. Neurol. Genet. 1, e1 10.1212/NXG.0000000000000003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pasanen P., Myllykangas L., Pöyhönen M., Kiuru-Enari S., Tienari P. J., Laaksovirta H., Toppila J., Ylikallio E., Tyynismaa H., and Auranen M. (2016) Intrafamilial clinical variability in individuals carrying the CHCHD10 mutation Gly66Val. Acta Neurol. Scand. 133, 361–366 10.1111/ane.12470 [DOI] [PubMed] [Google Scholar]
- 29. Marroquin N., Stranz S., Müller K., Wieland T., Ruf W. P., Brockmann S. J., Danzer K. M., Borck G., Hübers A., Weydt P., Meitinger T., Strom T. M., Rosenbohm A., Ludolph A. C., and Weishaupt J. H. (2016) Screening for CHCHD10 mutations in a large cohort of sporadic ALS patients: No evidence for pathogenicity of the p.P34S variant. Brain 139, e8 10.1093/brain/awv218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Quirós P. M., Mottis A., and Auwerx J. (2016) Mitonuclear communication in homeostasis and stress. Nat. Rev. Mol. Cell Biol. 17, 213–226 10.1038/nrm.2016.23 [DOI] [PubMed] [Google Scholar]
- 31. Kotiadis V. N., Duchen M. R., and Osellame L. D. (2014) Mitochondrial quality control and communications with the nucleus are important in maintaining mitochondrial function and cell health. Biochim. Biophys. Acta 1840, 1254–1265 10.1016/j.bbagen.2013.10.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cavallaro G. (2010) Genome-wide analysis of eukaryotic twin CX9C proteins. Mol. Biosyst. 6, 2459–2470 10.1039/c0mb00058b [DOI] [PubMed] [Google Scholar]
- 33. Balsa E., Marco R., Perales-Clemente E., Szklarczyk R., Calvo E., Landázuri M. O., and Enríquez J. A. (2012) NDUFA4 is a subunit of complex IV of the mammalian electron transport chain. Cell Metab. 16, 378–386 10.1016/j.cmet.2012.07.015 [DOI] [PubMed] [Google Scholar]
- 34. McKeown S. R. (2014) Defining normoxia, physoxia and hypoxia in tumours—implications for treatment response. Br. J. Radiol. 87, 20130676 10.1259/bjr.20130676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rodesch F., Simon P., Donner C., and Jauniaux E. (1992) Oxygen measurements in endometrial and trophoblastic tissues during early pregnancy. Obstet. Gynecol. 80, 283–285 [PubMed] [Google Scholar]
- 36. Liu H., Li Y., Li Y., Liu B., Wu H., Wang J., Wang Y., Wang M., Tang S. C., Zhou Q., and Chen J. (2012) Cloning and functional analysis of FLJ20420: A novel transcription factor for the BAG-1 promoter. PLoS One 7, e34832 10.1371/journal.pone.0034832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Burstein S. R., Valsecchi F., Kawamata H., Bourens M., Zeng R., Zuberi A., Milner T. A., Cloonan S. M., Lutz C., Barrientos A., and Manfredi G. (2018) In vitro and in vivo studies of the ALS-FTLD protein CHCHD10 reveal novel mitochondrial topology and protein interactions. Hum. Mol. Genet. 27, 160–177 10.1093/hmg/ddx397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lin M. T., and Beal M. F. (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 443, 787–795 10.1038/nature05292 [DOI] [PubMed] [Google Scholar]
- 39. Jackson M. J. (2016) Reactive oxygen species in sarcopenia: Should we focus on excess oxidative damage or defective redox signalling? Mol. Aspects Med. 50, 33–40 10.1016/j.mam.2016.05.002 [DOI] [PubMed] [Google Scholar]
- 40. Zuo L., and Motherwell M. S. (2013) The impact of reactive oxygen species and genetic mitochondrial mutations in Parkinson's disease. Gene 532, 18–23 10.1016/j.gene.2013.07.085 [DOI] [PubMed] [Google Scholar]
- 41. Li X. L., Shu S., Li X. G., Liu Q., Liu F., Cui B., Liu M. S., Peng B., Cui L. Y., and Zhang X. (2016) CHCHD10 is not a frequent causative gene in Chinese ALS patients. Amyotroph. Lateral Scler. Frontotemporal Degener. 17, 458–460 10.3109/21678421.2016.1170151 [DOI] [PubMed] [Google Scholar]
- 42. Zhou Q., Chen Y., Wei Q., Cao B., Wu Y., Zhao B., Ou R., Yang J., Chen X., Hadano S., and Shang H. F. (2017) Mutation screening of the CHCHD10 gene in Chinese patients with amyotrophic lateral sclerosis. Mol. Neurobiol. 54, 3189–3194 10.1007/s12035-016-9888-0 [DOI] [PubMed] [Google Scholar]
- 43. Wong C. H., Topp S., Gkazi A. S., Troakes C., Miller J. W., de Majo M., Kirby J., Shaw P. J., Morrison K. E., de Belleroche J., Vance C. A., Al-Chalabi A., Al-Sarraj S., Shaw C. E., and Smith B. N. (2015) The CHCHD10 P34S variant is not associated with ALS in a UK cohort of familial and sporadic patients. Neurobiol. Aging 36, 2908.e17–e18 10.1016/j.neurobiolaging.2015.07.014 [DOI] [PubMed] [Google Scholar]
- 44. Livak K. J., and Schmittgen T. D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(-ΔΔ C(T)) method. Methods 25, 402–408 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.