

Abstract
Individuals with elevated lipid levels are at risk for developing cardiovascular disease as well as cancer. Sterol regulatory element–binding protein transcription factors (SREBPs) are inducers of lipid synthesis. Elevated SREBPs levels are linked to cell proliferation and metastasis. Using biochemical and mouse models of cancer, Zhao et al. have discovered that nuclear SREBP-1a–dependent transcription is activated by pyruvate kinase M2 in cancer cells, which promotes tumor growth. Targeting the lipogenesis pathway may therefore be a promising avenue for cancer treatment.
Keywords: cancer, tumor cell biology, dyslipidemia, cholesterol metabolism, cell proliferation, sterol regulatory element-binding protein, tumorigenesis
Introduction
The SREBP2 transcription factors (SREBP-1a, SREBP-1c, and SREBP-2) are key regulators of de novo lipid synthesis. They reside in the endoplasmic reticulum, but under conditions of low intracellular cholesterol make their way to the nucleus (Fig. 1). Once there, they direct lipid gene transcription through binding to specific sterol response elements (SREs) (1). When cholesterol levels are high, SREBPs are retained in the ER as part of a complex that includes sterol cleavage activating protein (SCAP). When cholesterol becomes depleted, the sterol-sensing domain of SCAP undergoes a conformational change that allows for its interaction with COPII transport proteins. SREBP–SCAP complexes then translocate to the Golgi (1), whereby they are cleaved by the site-1 (S1P) and site-2 proteases (S2P), generating soluble N-terminal SREBPs transcription factors (known as nBPs), which transport to the nucleus to induce transcription of genes involved in lipogenesis. Mice expressing a dominant gain-of-function SCAP allele are severely hyperlipidemic, whereas mice ablated for liver SCAP have reduced lipid levels in response to a high-fat diet (2, 3). Thus, the cholesterol-dependent SREBP pathway is critically important for regulating lipid homeostasis.
Figure 1.
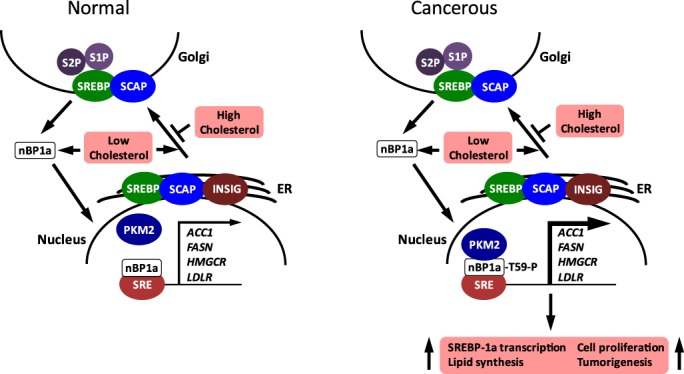
The model shows the effects of PKM2 on the cholesterol-mediated control of lipid synthesis gene expression in normal and cancerous cells. In normal cells with low cholesterol (left), the S1P/S2P proteases hydrolyze SREBP in the Golgi membrane to release the soluble transcription factor nBP1a; nBP1a enters the nucleus and binds to SREs to activate transcription of lipid synthesis genes. In this case, PKM2 is not associated with nBP1a. High cholesterol blocks the transcriptional activation of SRE-containing genes by inhibiting the vesicular trafficking of SREBP–SCAP from the ER to the Golgi (via COPII vesicles not shown). In cancerous cells with low cholesterol (right), the formation of the PKM2–nBP1a complex, with nBP1a being phosphorylated at threonine (T)-59, stimulates further (indicated by the boldface arrow) the transcription of SRE-containing lipid synthesis genes leading to lipid accumulation, cell proliferation, and tumorigenesis.
Studies have also demonstrated a strong relationship between increased lipid syntheses and accelerated metastatic potential in cancer. Many types of cancer cells have elevated SREBPs activities and lipid synthesis, which possibly supports increased cell proliferation (4). Murine cancer models have shown that pharmacological inhibitors of SREBPs activities, in addition to reducing hyperlipidemia, also reduce cell proliferation and tumor progression (5, 6). Thus, the role of SREBPs and misregulated lipid metabolism in promoting cancer progression is increasingly becoming clear.
SREBPs are known to be post-translationally modified, but how these modifications regulate SREBP function is incompletely understood (7, 8). SREBPs are phosphorylated, and this either activates or inhibits their activities, depending on the particular residue phosphorylated (8). Although several kinases have been shown to regulate SREBP function directly or indirectly, a great deal of work needs to be done to comprehensively catalogue the effects of different phosphorylation events on lipid gene expression and lipid synthesis. Even more important is to determine how these events affect cancer cell proliferation and tumor progression.
Using elegant biochemical and xenograft murine models, Zhao et al. (9) have made the important discovery that a pyruvate kinase M2 (PKM2) activates nuclear SREBP-1a (nBP1a)–dependent transcription, leading to cell proliferation and increased tumor progression. Regulation is initiated through the direct interaction of PKM2 with nBP1a through a PKM2-binding motif. The interaction is highly specific, as PKM2 does not interact with nuclear SREBP-1c/SREBP-2. This role in transcriptional activation of lipid synthesis is highly novel, as PKM2 is recognized as the enzyme catalyzing the last step in glycolysis, the transphosphorylation of phosphoenolpyruvate to ADP, which generates ATP.
Zhao et al. (9) further show that PKM2–nBP1a complex formation results in increased nBP1a stability and Thr-59 phosphorylation. By using an nBP1a mutant (T59A) lacking the ability to be phosphorylated, they demonstrated the importance and fundamental nature of nBP1a Thr-59 phosphorylation by recapitulating these results in lung, colorectal, and breast cancer cell lines. In cell culture experiments, the authors expressed the nBP1a T59A allele in HepG2 cells lacking SREBP1 and found that it was unable to stimulate transcription of several nBP1a-dependent lipid genes, including FASN (fatty acid synthase), ACC1 (acetyl-CoA carboxylase), and HMGCR (HMG-CoA reductase). The T59A mutant also failed to stimulate nBP1a-dependent lipid accumulation. In contrast, an nBP1a T27A mutant allele remained fully functional when expressed in HepG2 SREBP1-null cells. Finally, the authors generated a PKM2–nBP1a complex-blocking peptide, which when administered to cancer cells inhibited nBP1a transcription and lipid accumulation.
Moving on to in vivo studies, they showed that their cell culture data could be recapitulated in xenograft models. Nude mice were injected with SREBP-1a null HepG2 cells overexpressing PKM2 or the WT or mutated nBP1a. Those cells overexpressing PKM2 gave rise to tumors similar in size to the empty vector control, whereas mice expressing WT nBP1a had larger tumors, while those expressing the Thr59A mutant had smaller tumors.
Furthermore, the authors examined tumors from 90 individuals with hepatocellular carcinoma. The authors examined each individual tumor and surrounding normal tissue in order to analyze the relationship between Thr-59 phosphorylation and prognosis. They discovered that tumors from these patients had increased nBP1a Thr-59 phosphorylation when compared with the degree seen in surrounding normal tissue. Higher nBP1a Thr-59 phosphorylation correlated with poor clinical outcomes.
Thus, Zhao et al. (9) have clearly shown that PKM2–nBP1a complex formation and subsequent Thr-59 phosphorylation stimulates nBP1a-dependent transcription and lipid accumulation, which are critical events necessary for cell proliferation and tumorigenesis (Fig. 1). Furthermore, they have presented strong evidence that Thr-59 hyperphosphorylation correlates with reduced survival rate.
The work presented by Zhao et al. (9) brings to the forefront the importance of the relationship between lipid synthesis and cancer progression. The question now is: How can we exploit this knowledge for the development of anti-cancer therapies? Understanding the molecular basis for driving PKM2-nBP1a association and subsequent activation of nBP1a should be elucidated. In addition, it will be important to uncover whether PKM2 directly phosphorylates Thr-59 on nBP1a, or whether it stimulates the association of another kinase(s), although the authors demonstrated that PKM2 could phosphorylate nBP1a Thr-59 in vitro. It will also be crucial to determine whether the PKM2–nBP1a complex is found in normal cells, as this would suggest that additional regulatory events are necessary to promote Thr-59 phosphorylation and thus cell proliferation.
These very important studies lay the groundwork for furthering our understanding of the link between nBP1a Thr-59 phosphorylation, which leads to lipid accumulation, and cancer progression and tumorigenesis. These studies have also clearly made an argument that establishes PKM2 as a central player regulating lipid-dependent cell proliferation. This novel regulation of lipid synthesis adds another tier to the complexity of our understanding of cell proliferation and cancer progression.
Acknowledgment
I am grateful for the financial support of Genesis Biotechnology Group.
The author declares that he has no conflicts of interest with the contents of the article.
- SREBP
- sterol regulatory element–binding protein transcription factor
- nBP1a
- nuclear sterol response element–binding protein-1a
- PKM2
- pyruvate kinase 2
- SRE
- sterol response element
- SCAP
- sterol cleavage activating protein
- S1P
- site-1 protease
- S2P
- site-2 protease.
References
- 1. Brown M. S., Ye J., Rawson R. B., and Goldstein J. L. (2000) Regulated intramembrane proteolysis: a control mechanism conserved from bacteria to humans. Cell 100, 391–398 10.1016/S0092-8674(00)80675-3 [DOI] [PubMed] [Google Scholar]
- 2. Korn B. S., Shimomura I., Bashmakov Y., Hammer R. E., Horton J. D., Goldstein J. L., and Brown M. S. (1998) Blunted feedback suppression of SREBP processing by dietary cholesterol in transgenic mice expressing sterol-resistant SCAP(D443N). J. Clin. Invest. 102, 2050–2060 10.1172/JCI5341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Matsuda M., Korn B. S., Hammer R. E., Moon Y. A., Komuro R., Horton J. D., Goldstein J. L., Brown M. S., and Shimomura I. (2001) SREBP cleavage-activating protein (SCAP) is required for increased lipid synthesis in liver induced by cholesterol deprivation and insulin elevation. Genes Dev. 15, 1206–1216 10.1101/gad.891301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ray U., and Roy S. S. (2018) Aberrant lipid metabolism in cancer cells - the role of oncolipidactivated signaling. FEBS J. 285, 432–443 10.1111/febs.14281 [DOI] [PubMed] [Google Scholar]
- 5. Kamisuki S., Mao Q., Abu-Elheiga L., Gu Z., Kugimiya A., Kwon Y., Shinohara T., Kawazoe Y., Sato S., Asakura K., Choo H. Y., Sakai J., Wakil S. J., and Uesugi M. (2009) A small molecule that blocks fat synthesis by inhibiting the activation of SREBP. Chem. Biol. 16, 882–892 10.1016/j.chembiol.2009.07.007 [DOI] [PubMed] [Google Scholar]
- 6. Tang J. J., Li J. G., Qi W., Qiu W. W., Li P. S., Li B. L., and Song B. L. (2011) Inhibition of SREBP by a small molecule, betulin, improves hyperlipidemia and insulin resistance and reduces atherosclerotic plaques. Cell Metab. 13, 44–56 10.1016/j.cmet.2010.12.004 [DOI] [PubMed] [Google Scholar]
- 7. Raghow R., Yellaturu C., Deng X., Park E. A., and Elam M. B. (2008) SREBPs: the crossroads of physiological and pathological lipid homeostasis. Trends Endocrinol. Metab. 19, 65–73 10.1016/j.tem.2007.10.009 [DOI] [PubMed] [Google Scholar]
- 8. Lewis C. A., Griffiths B., Santos C. R., Pende M., and Schulze A. (2011) Regulation of the SREBP transcription factors by mTORC1. Biochem. Soc. Trans. 39, 495–499 10.1042/BST0390495 [DOI] [PubMed] [Google Scholar]
- 9. Zhao X., Zhao L., Yang H., Li J., Min X., Yang F., Liu J., and Huang G. (2018) Pyruvate kinase M2 interacts with nuclear sterol regulatory element–binding protein 1a and thereby activates lipogenesis and cell proliferation in hepatocellular carcinoma. J. Biol. Chem. 293, 6623–6634 10.1074/jbc.RA117.000100 [DOI] [PMC free article] [PubMed] [Google Scholar]