

See Bernasconi (doi:10.1093/aww202) for a scientific commentary on this article.
Temporal lobe epilepsy (TLE) is associated with a high prevalence of cognitive decline. Tai et al. reveal the presence of hyperphosphorylated tau in resected temporal lobe tissue from patients with TLE. The quantity of pathological tau correlates with cognitive decline and its distribution suggests an epilepsy-related tauopathy.
Keywords: temporal lobe epilepsy, neurofibrillary tangles, tau, Alzheimer’s disease, dementia
See Bernasconi (doi:10.1093/aww202) for a scientific commentary on this article.
Temporal lobe epilepsy (TLE) is associated with a high prevalence of cognitive decline. Tai et al. reveal the presence of hyperphosphorylated tau in resected temporal lobe tissue from patients with TLE. The quantity of pathological tau correlates with cognitive decline and its distribution suggests an epilepsy-related tauopathy.
Abstract
See Bernasconi (doi:10.1093/aww202) for a scientific commentary on this article.
Temporal lobe epilepsy, the most prevalent form of chronic focal epilepsy, is associated with a high prevalence of cognitive impairment but the responsible underlying pathological mechanisms are unknown. Tau, the microtubule-associated protein, is a hallmark of several neurodegenerative diseases including Alzheimer’s disease and chronic traumatic encephalopathy. We hypothesized that hyperphosphorylated tau pathology is associated with cognitive decline in temporal lobe epilepsy and explored this through clinico-pathological study. We first performed pathological examination on tissue from 33 patients who had undergone temporal lobe resection between ages 50 and 65 years to treat drug-refractory temporal lobe epilepsy. We identified hyperphosphorylated tau protein using AT8 immunohistochemistry and compared this distribution to Braak patterns of Alzheimer’s disease and patterns of chronic traumatic encephalopathy. We quantified tau pathology using a modified tau score created specifically for analysis of temporal lobectomy tissue and the Braak staging, which was limited without extra-temporal brain areas available. Next, we correlated tau pathology with pre- and postoperative cognitive test scores and clinical risk factors including age at time of surgery, duration of epilepsy, history of secondary generalized seizures, history of head injury, handedness and side of surgery. Thirty-one of 33 cases (94%) showed hyperphosphorylated tau pathology in the form of neuropil threads and neurofibrillary tangles and pre-tangles. Braak stage analysis showed 12% of our epilepsy cohort had a Braak staging III-IV compared to an age-matched non-epilepsy control group from the literature (8%). We identified a mixture of tau pathology patterns characteristic of Alzheimer’s disease and chronic traumatic encephalopathy. We also found unusual patterns of subpial tau deposition, sparing of the hippocampus and co-localization with mossy fibre sprouting, a feature of temporal lobe epilepsy. We demonstrated that the more extensive the tau pathology, the greater the decline in verbal learning (Spearman correlation, r = −0.63), recall (r = −0.44) and graded naming test scores (r = −0.50) over 1-year post-temporal lobe resection (P < 0.05). This relationship with tau burden was also present when examining decline in verbal learning from 3 months to 1 year post-resection (r = −0.54). We found an association between modified tau score and history of secondary generalized seizures (likelihood-ratio χ2, P < 0.05) however there was no clear relationship between tau pathology and other clinical risk factors assessed. Our findings suggest an epilepsy-related tauopathy in temporal lobe epilepsy, which contributes to accelerated cognitive decline and has diagnostic and treatment implications.
Introduction
Patients with temporal lobe epilepsy (TLE) may suffer from cognitive decline and have an increased prevalence of developing dementias including Alzheimer’s disease (Høgh et al., 2002). The mechanisms contributing to cognitive decline in TLE are unknown, with few effective management options (Hermann et al., 2006).
Cross-sectional neuropsychological studies have shown that patients with chronic partial epilepsy treated with anti-epileptic drugs (AEDs) have lower cognitive scores, including verbal memory tests, compared to age-matched controls: one study shows older patients with TLE, aged 80 years and above, performed the worst even when compared to control individuals diagnosed with mild cognitive impairment (Griffith et al., 2006, 2007). An increasing number of older patients with drug-refractory TLE are being offered temporal lobe resection as treatment and age above 50 years at time of surgery has been identified as a major risk factor for subsequent memory decline (Thompson et al., 2015). Other clinical risk factors associated with cognitive decline include duration of epilepsy, seizure type, head injuries and cognitive reserve (Oyegbile et al., 2004; Helmstaedter and Elger, 2009; Black et al., 2010).
Evidence suggests an association between epilepsy and dementia as epidemiological data show increased prevalence of dementia and Alzheimer’s disease in individuals with chronic epilepsy (Gaitatzis et al., 2004; Tellez-Zenteno et al., 2005). Transgenic mice models of familial Alzheimer’s disease have been shown to suffer from recurrent seizures (Palop et al., 2007) while serial neuroimaging in chronic epilepsy, particularly TLE, shows progressive grey matter volume loss and cortical thinning, which are associated with cognitive impairment (Liu et al., 2003; Cormack et al., 2005; Lin et al., 2007; Bernhardt et al., 2009). Despite accumulating epidemiological, radiological and animal model evidence associating epilepsy with dementia, there are few studies of the pathological changes that may underlie cognitive decline in epilepsy.
Tau, a microtubule-associated protein, is a natively unfolded protein in human brains and has several roles including microtubule assembly and stabilization (Goedert and Spillantini, 2006; Ittner et al., 2010). A defining pathological feature of several neurodegenerative diseases, including Alzheimer’s disease and chronic traumatic encephalopathy (CTE) is aggregated, hyperphosphorylated tau. Hyperphosphorylated tau is a key component of neurofibrillary tangles (NFTs), which are central to the diagnosis and staging of such diseases (Braak et al., 2011; McKee et al., 2013). Pathological tau is also associated with early cognitive decline in other diseases such as Parkinson’s disease, motor neuron disease and, recently, Huntington’s disease (Wolfe, 2012; Vuono et al., 2015).
In a recent human post-mortem analysis of 138 patients with chronic, refractory epilepsy, we reported neurofibrillary tangle (hyperphosphorylated tau) pathology with age-accelerated changes within the mid-Braak stages (III/IV) compared to an age-matched non-epilepsy series (Thom et al., 2011). Interestingly, increasing Braak stages did not clearly correlate with cognitive decline measured prior to death. Traumatic brain injury, indicated primarily by histological findings of frontotemporal contusions, correlated significantly with hyperphosphorylated tau burden and suggested an underlying CTE pathological process.
Post-mortem analysis of brain tissue usually occurs some years after the last cognitive assessment. By contrast, temporal lobe resections performed for refractory epilepsy allows contemporaneous histological examination and clinical assessments. Examination of temporal lobe resection tissue in 47 patients with TLE revealed granule cell layer dispersion correlating inversely with verbal memory scores (Kandratavicius et al., 2013). Microtubule-associated proteins, MAP2 and tau, were present but with variable correlation to clinical outcome and cognitive decline with below average verbal memory scores described with increased tau expression in the CA2 region. Sheng et al. (1994) identified increased immunoreactive amyloid-β precursor protein in temporal lobe tissue from eight patients with TLE while a larger study of 101 temporal lobe specimens compared with 406 post-mortem controls showed an age-accelerated presence of senile amyloid plaques in 10% of epilepsy patients (Mackenzie and Miller, 1994).
In this study, we aimed to assess the extent, nature and significance of tau pathology in temporal lobectomy subjects aged between 50–65 years of age at time of surgery. We also compared and contrasted our findings with those reported for Alzheimer’s disease and CTE. We then aimed to investigate the relationship between tau burden and postoperative decline in cognitive test scores and to examine the effect of potential clinical risk factors in this interaction. We hypothesized that hyperphosphorylated tau pathology in the resected temporal lobe, reflective of tau pathology in the entire brain, is associated with cognitive decline in our cohort.
Materials and methods
Case selection
Post-surgical cases (n = 33) were selected from the archives of the Department of Neuropathology, National Hospital for Neurology and Neurosurgery, London, from 1995–2014. We included all patients with TLE who had undergone anterior temporal lobe resection, aged between 50 and 65 years at the time of surgery, with pathological diagnosis of hippocampal sclerosis. All cases had a primary clinical diagnosis of TLE with history of chronic, drug-refractory disease. Cases excluded were of non-hippocampal sclerosis pathology, those who developed epilepsy during a course of another neurodegenerative disease, including Alzheimer’s disease, and if consent for research was not recorded. The tissues were consented for use in research and the study was approved by the local ethics committee.
Clinical data
Demographic data and the clinical history of each case were retrieved from clinical notes. Recorded information included the age at onset of epilepsy, duration of epilepsy, side of surgery and handedness. Information on seizure type and frequency, occurrence of status epilepticus and history of head injury was also noted when available. Any reports of cognitive decline postoperatively or development of dementia were also noted.
Cognitive test data
Temporal lobe epilepsy patients who undergo temporal lobe resections usually undergo cognitive testing during the pre-operative pathway and at 3 and 12 months postoperatively. Preoperative cognitive data were available for 27 patients (82%). A measurement for ‘change in cognition over time’ was derived from paired preoperative to 1 year postoperative test scores, and 3 month postoperative to 1 year postoperative test scores that were available for 21 patients (63%).
Intellectual level was measured using the Wechsler Adult Intelligence Scale (WAIS). All patients with pre- and postoperative data had an IQ of >69.
Memory and other cognitive measures were performed using the List Learning and Design Learning subtests from the Adult Memory and Information Processing (AMIPB) and its successor the BIRT memory and information processing battery (BIMPB) provided measures of verbal learning and recall and visual learning and recall. These measures have been shown to be sensitive to temporal lobe pathology and have been described previously (Thompson et al., 2015). The Graded Naming Test and tests of phonemic and semantic fluency provided measures of word-retrieval proficiency (Thompson et al., 2015). Test scores were converted into z-scores based on age-related norms. A negative z-score indicated a decline in memory.
Immunohistochemistry
Resected tissue was archived as formalin-fixed, paraffin-embedded tissue blocks containing temporal lobe and hippocampus for all cases and additional blocks of pes hippocampus, parahippocampal gyrus and amygdala were available for 13 more recent temporal lobe resections. Blocks were selected for immunohistochemical analysis according to anatomical position, best preserved tissue, and representative pathology (from review of archived stained sections previously prepared) (Supplementary Table 1).
AT8 (hyperphosphorylated tau) and βA4 (amyloid-β) immunohistochemistry was performed on 5-µm thick paraffin-embedded sections using the BOND-MAX automated immunostainer (Leica, Microsystems) following established laboratory protocols with primary antibody anti-AT8 (1:1200,-Innogenetics, AutogenBioclear) or anti-βA4 (1:100, DAKO). Positive (confirmed cases with Alzheimer’s disease) and negative controls were included for each staining run. Sections from selected cases were also stained with both anti-RD3 and anti-RD4 specific for 3-repeat (1:3000) and 4-repeat tau isoforms (1:4000; gift from Rohan de Silva) using automated immunostainer (A.Menarini Diagnostics Ltd). All sections were viewed using a light microscope (Olympus BX40). Selected slides were digitized using a whole slide scanner (Leica SCN400, Leica Microsystems). Images were acquired using a digital camera (Nikon Eclipse 80i) or as a snapshot from digitized slides.
Immunofluorescence
Sequential double-labelled immunofluorescent studies were performed manually over 3 days based on previously published protocols to identify specific cell populations, which co-localize with pathological tau (Thom et al., 2011). In brief, after sections were microwaved at 800 W for 12 min in the antigen unmasking solutions (H3300, Vector Labs), blocking solution containing 2.5% normal horse serum (Vector Labs) was applied. Sections were incubated in anti-AT8 primary antibody solution overnight at 4°C (1:1200). The next day, monoclonal-specific horseradish peroxidase-conjugated secondary antibodies (Vector Labs) were added for 30 min before fluorescein-conjugated antibodies diluted in tyramide-amplifying buffer (1:800; Perkin Elmer) was applied for 5 min. Sections were immersed in 0.9% hydrogen peroxidase solution for 10 min, and then the second primary antibodies were applied as described in Supplementary Table 2. The next day, sections were incubated in solutions consisting of species-specific secondary antibodies conjugated to either Alexa Fluor® 546 (Lifesciences) for 2 h at room temperature, or horseradish peroxidase and then Cy3 diluted in tyramide-amplifying buffer (1:800; Perkin Elmer) for 30 and 5 min, respectively. All sections were coverslipped in Vectashield mounting media with 4’, 6-diamidino-2-phenylindole (DAPI; Vector Labs) and washes were performed using phosphate-buffered saline (Oxoid Limited). Negative controls with omission of all or one primary or secondary antibodies were included in each run, and no false positive labelling was observed in negative controls.
Analysis of tau and amyloid pathology
We assessed tau burden semiquantitatively using two methods. Firstly, the presence and location of AT8 immunolabelling (hyperphosphorylated tau) was assessed using a ‘limited Braak staging’ according to the standards published by Braak et al. (2006). Resected temporal lobe tissue only allows for Braak staging from 0–IV (as stages V–VI require examination of extra-temporal regions which were not removed during surgery). Second, we created a ‘modified tau score’ specifically for assessing AT8 labelling (tau burden) within resected temporal lobe tissue. The modified tau score ranges from 0–6 and follows early Braak stages (0–IV) but allows greater sensitivity with a wider scoring range and did not follow the anatomical progression of the Braak staging. Table 1 gives a detailed breakdown of the modified tau scores and how they compare with Braak stages. In terms of tau burden, modified tau score 0 (no AT8 labelling) is equivalent to Braak stage 0 while modified tau score 6 (greatest tau load) is equivalent to Braak stage IV (representative images for each modified tau score are found in Supplementary Fig. 2). We noted other AT8 labelling characteristics including the cell type and location, in particular glial or neuronal and perivascular or sulcal. We also noted distributions of AT8 labelling that were consistent with CTE based on standards published by McKee et al. (2015).
Table 1.
Modified tau score description and comparison to (limited) Braak staging
| Modified tau score | Braak staging progression | ||
|---|---|---|---|
| 0 | No AT8 labelling in any region | 0 | No AT8 labelling in any region |
| 1 | Subpial band staining or rare NT staining or rare axon positive in any region | + | Few immunopositive neurons (NFTs or pre-tangles) with low densities of NTs (at ×20) with no specific pattern |
| 2 | Rare NT in > 1 region or mild NTs stain in one regions (identified at ×20 magnification). | ||
| 3 | Two of the following: mild NTs stain in >1 region; moderate NTs stain in one regions (identified on ×10 magnification) or occasional NFT noted. | I | Mild NT staining of low density localized in the trans-entorhinal region |
| 4 | Two of the following: NFT evidence; mild NTs in >2 regions or moderate threads in >1 region | II | Moderate NT staining in the superficial cortical layers and mild NT in the deep cortical layers of the entorhinal region |
| 5 | Moderate to heavy NT staining in >1 region | III | Moderate NT staining in the superficial and deep cortical layers and/or involvement of the neocortex adjoining the occipito-temporal gyrus |
| 6 | Moderate to heavy NT staining in most regions of temporal lobe | IV | Moderate NTs in middle temporal gyrus |
NFT = neurofibrillary tangle; NT = neuropil thread.
Amyloid-β immunohistochemistry (βA-4 labelling) was assessed with plaque scores of sparse, moderate or frequent, as outlined in the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) (Alafuzoff et al., 2008). Additional pathological features including type of hippocampal sclerosis (HS), mossy fibre sprouting, brain trauma, and cerebrovascular disease were also noted.
Experimental design
Pathology analysis and scoring was performed by two independent assessors (X.T. and M.T.). Cases with different scores were reassessed together and a final score was agreed upon. Neuropsychometry analysis was performed independently by P.T. and comparison with pathology examination was done only when all scores were finalized.
Statistical analysis
Statistical analysis and graphical representation was performed using SPSS for windows (IBM Corporation, version 20) or Excel 2010 (Microsoft Office). Student t-testing was used to compare Braak staging between the study cohort and age-matched population controls from a post-mortem series (Braak et al. 2013). Spearman correlation analysis was carried out with modified tau scores in relationship with age at time of surgery and onset of epilepsy as well as change in neuropsychometry scores from pre-operative to 12 months postoperative and 3 months postoperative to 12 months postoperative. Statistical analysis between modified tau scores with ANOVA testing, Pearson χ2 testing or likelihood-ratio χ2 testing were used for other clinical characteristics including duration of epilepsy, side of resection, history of head injury, history of secondary generalized seizures and handedness of each patient. Multi-linear regression was also carried out between modified tau score and clinical characteristics. Measure of Agreement (Cohen’s κ coefficient) was used to assess inter-observer agreement of modified tau scoring. For all statistical methods, a P-value of <0.05 was considered significant.
Results
Hyperphosphorylated tau (AT8 labelling) in resected temporal lobe tissue
Using immunohistochemistry for AT8 labelling, we identified hyperphosphorylated tau pathology in the form of neuropil threads, neurofibrillary tangles and pre-tangles within temporal lobe tissue (Fig. 1A and B). We applied the 6-point modified tau score and limited Braak staging for tau semiquantification and showed 31 of 33 cases (93.9%) had evidence of AT8 labelling (Fig. 2A). Twelve cases (36%) scored a modified tau score 3, the most frequent distribution observed. Two cases (6%) scored 0 (virtually no AT8 labelling), five cases (15%) scored 1, seven cases (21%) scored 2, two cases (6%) scored 4, four cases (12%) scored 5, and one case (3%) scored 6 (Fig. 2). Agreement was achieved within a 1-point margin for modified tau scoring in 91% of the cases with a moderate level of agreement between observers (Cohen’s coefficient κ = 0.54, P < 0.005).
Figure 1.
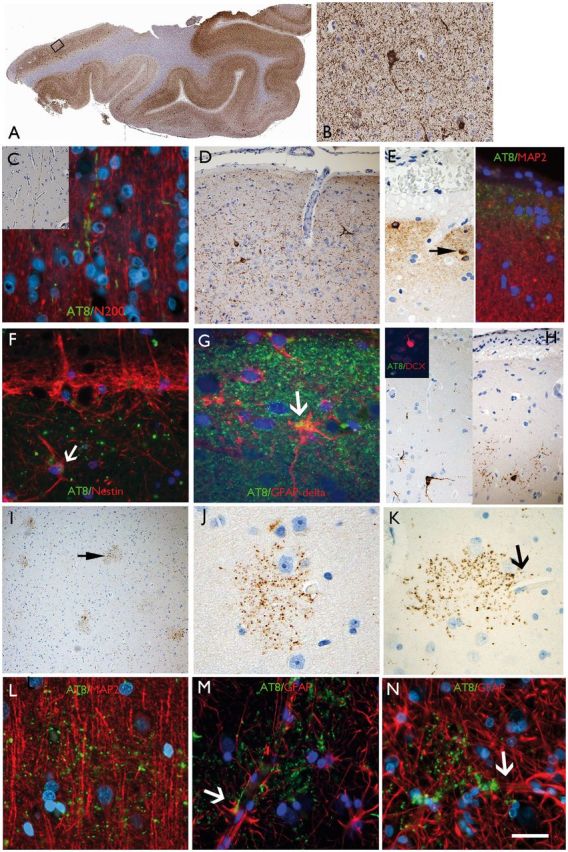
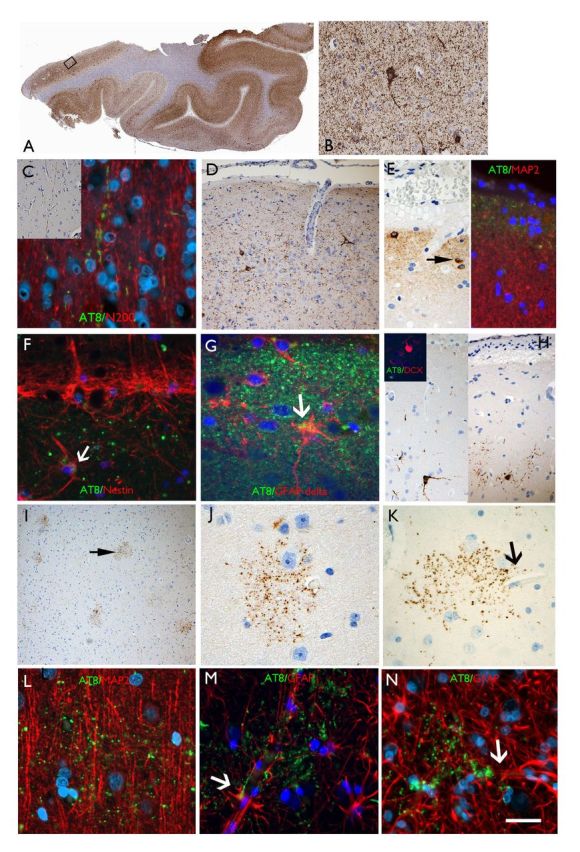
AT8 patterns in TLE/HS cases in the temporal neocortex. (A) Section of temporal lobe from case with maximal modified tau score of 6 showing dense accumulation of tau with AT8 immunohistochemistry in all gyri. The rectangle is shown at higher magnification in B, highlighting neuropil threads and neuronal labelling. (C) Axonal labelling with AT8 in the cortex (Case 24) in inset and double labelling with neurofilament (N200) showing beaded-like AT8 staining along the trajectory of radial cortical axons. (D) CTE-like pattern with focal increased AT8 in threads and small neurons in the superficial cortical layers. (E) Subpial granular band of labelling and positive neurons, reminiscent of Cajal-Retzius cells (arrow); double labelling with MAP2 (dendritic marker) on the right side of the panel, shows AT8 labelling in the compartment above dendritic MAP2 labelling supporting this more likely represents superficial subpial axonal projections. (F) Double labelling between AT8 and nestin in layer I showed no overlap with the granular, axonal-like AT8 labelling and the nestin in the subpial glia with occasional possible co-localization (arrow). (G) Layer I in a further case with AT8 showing a mainly beaded axonal staining pattern and only rare possible expression in GFAP-delta positive subpial astroglial cell. (H) Small neurons at interface of layer I and II were AT8 positive but double-labelling with doublecortin (DCX) in selected cases (inset) did not shown any AT8 positivity in these immature cell types that are known to reside in this cortical layer in the temporal lobe. (I) ‘Granular aggregates’ of tau were noted in the temporal cortex in five cases scattered in the cortex pattern but not typical of neuritic plaques of Alzheimer’s disease or glial inclusions. (J) Occasionally these aggregates surrounding cortical neurons but without definite labelling of the neuronal cell body. (K) Granular aggregates were observed in the vicinity of small capillaries (arrow). (L) The granular aggregates did not appear to co-localize with dendrites on double-labelling with MAP2 (a neuronal dendritic marker). (M and N) Labelling with GFAP confirmed that the AT8-granular aggregates were not in astroglia but through highlighting the glial foot processes along the vessels, indicated their proximity to vascular channels. Supplementary Fig. 3 contains a panel of similar immunofluorescence images captured with a non-red/green colour spectrum. Scale bars = 500 µm in A, 70 µm in I, 50 µm in B and D, 20 µm in C, E, F, G, J, K L, M and N.
Figure 2.

Quantifying tau pathology in temporal lobe tissue of epilepsy patients. Histogram showing modified tau score (described in Table 1) distribution across study cohort. Thirty-one of 33 cases showed AT8 labelling with modified tau score 3 being most common.
Comparing patients aged 50–60 years at the time of the surgery (26 cases) with age-matched population controls from a post-mortem series of 330 patients in the literature (Braak et al., 2011), we found a higher proportion of patients with Braak stages III–IV (12% versus 8% but the differences were not significant). As noted above, comparisons of Braak stage V–VI could not be performed as our cohort lacked extra-temporal tissue samples.
Distribution of hyperphosphorylated tau relative to Alzheimer’s disease and chronic traumatic encephalopathy patterns
Having found high tau phosphorylation burden within the resected temporal lobe tissue of some epilepsy cases, we next compared the distribution and patterns of tau accumulation to known tauopathies Alzheimer’s disease and CTE.
Temporal neocortex and pole
We characterized tau distribution in 24 cases in whom sufficient AT8 was present and found a mixture of pathological patterns (Supplementary Table 3). Ten cases had a ‘Braak-like’ pattern of tau pathology with greater AT8 labelling within the transentorhinal region and entorhinal region compared to temporal neocortex. In eight cases, we identified ‘CTE-like’ tau patterns, based on the identification of one or more of the following: axon labelling in white matter, cortex, layer I or hippocampus in single long or groups of axon (Figs 1C and 3N); patches of cortical AT8 labelling restricted to a single region as the middle temporal gyrus or temporal pole (Fig. 1D), greater tau labelling in neocortical samples compared to entorhinal cortex or more prominent tau accumulation in superficial (layers I–III) than deeper cortex (layers IV–VI) (Supplementary Table 3) (McKee et al., 2013). None of the cases showed the typical sulcal or well-defined perivascular neuronal tau pathology or obvious astrocytic or sub-pial tangles, considered pathognomonic of CTE.
Figure 3.
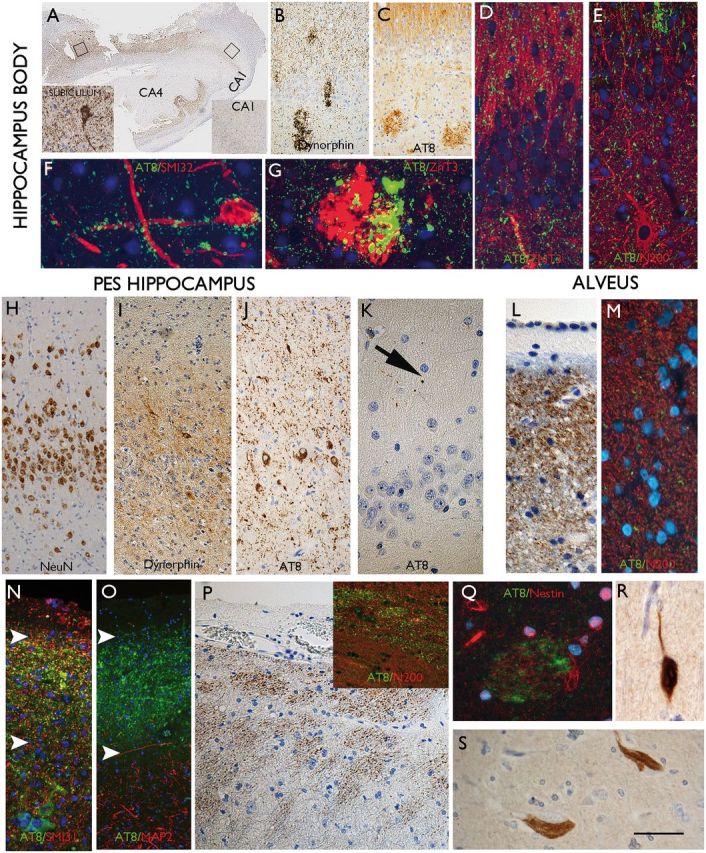
AT8 patterns in TLE/HS in the hippocampus, pes and amygdala. (A) Hippocampus body from case with highest tau load in the cortex. The hippocampus, which showed typical features of longstanding hippocampal sclerosis and gliosis with neuronal loss in CA1 and CA4. More AT8 labelling was seen in the subiculum compared to the CA1 subfield (inserts). (B) In another case of HS, mossy fibre sprouting was show with dynorphin staining and granular aggregates noted within the CA4 region and in C. A similar pattern was observed with AT8. (D) Double labelling of AT8 with ZnT3 and (E) neurofilament (N200), confirmed some overlap of labelling in the molecular layer of the dentate gyrus. (F) AT8-positive granules were observed in close proximity, surrounding a CA4 neuron and its processes and in (G) overlap of ZnT3 and AT8 was noted in the CA4 region corresponding to mossy fibre end terminals. (H) Pes hippocampus from one case showed marked dispersion of the granule cells on NeuN compared to the body also with a greater degree of mossy fibre sprouting on dynorphin stain as shown in I. (J) Abundant AT8 labelling in the granule cells and axons in the molecular layer was noted compared to very little staining in the adjacent CA1 subfield (not shown) and (K) the hippocampus body of the same cases, where only rare AT8-positive threads were noted. (L) An axonal-like pattern of labelling was noted in the alveus beneath the ependymal of the lateral ventricle and confirmed with double-labelling with N200 neurofilament marker (M) showing association of AT8-positive grains with axons. (N) In the subpial surface of the pes, a band of AT8 was noted in some case (between arrows), comparable to the subpial band in the cortex; this again had an axonal appearance with localization with non-phosphorylate neurofilament (SMI3 shown in N), but not dendritic marker (MAP2 shown in O), which labelled the processes in underlying neurons only. (P) Bundles of axons were also noted in the peri-ventricular region of the pes hippocampus white matter. (Q) Occasional fibre bundles were noted in the amygdala with AT8, which were weakly nestin-positive. (R) Labelling of neurons with 3R and (S) 4R tau isoforms was confirmed. Supplementary Fig. 3 contains a panel of similar immunofluorescence images captured with a non-red/green colour spectrum. Scale bars: A = 500 µm; B and C = 200 µm; H, I and J = 75 µm; D, E, K, N, 0, P = 50 µm; F, G, Q, R, S = 25 µm.
We also identified unusual patterns of AT8 labelling, not recognized as typical for either Alzheimer’s disease or CTE. A characteristic ‘subpial’ band of AT8 labelling was observed in 18 of the cases (Fig. 1E) with an axonal-like pattern. This band was seen in cases with both ‘Braak-like’ and ‘CTE-like’ patterns of tau pathology as well as other cases where it was the only positive AT8 labelling observed. In six of these cases there was also an impression of intense labelling of Cajal-Retzius cells in layer I (Fig. 1E) but an impression of subpial astroglial labelling, as reported in CTE and epilepsy post-mortem studies (Thom et al., 2011; McKee et al., 2013), was lacking. Double labelling with AT8 and reelin (Cajal Retzius cells) or delta-GFAP and nestin (subpial astroglial markers) did not confirm definite overlap between cell bodies or processes in this compartment (Fig. 1F and G). Prominent labelling of small neurons at the interface of layer I/II was noted in some cases (Fig. 1H); as we have previously identified populations of immature DCX-positive neurons in the superficial cortical layers of the mesial temporal lobe in temporal lobe epilepsy/HS, which have also been reported in other species (Liu et al., 2008; Xiong et al., 2008; Srikandarajah et al., 2009; Zhang et al., 2009) we explored if these subsets were vulnerable to tau accumulation, but this was not supported by double labelling (Fig. 1H, inset).
In five cases cortical granular aggregates of tau formed small ‘burst-like patterns’ of tau-positive grains. These were noted in the neocortex of cases of low tau score (Fig. 1I–N) in some cases present primarily in superficial layers. These granular aggregates did not have the typical morphology of neuritic or astroglial plaques, in that they lacked a central core or nucleus, were not specifically related to neurons (Fig. 1J) but were more frequently noted close to small capillaries (Fig. 1K), supported with double labelling for neuronal, glial and vascular markers (Fig. 1L–N). Furthermore, in all these five cases no plaques were seen with beta-amyloid. The precise cellular compartmentalization of these tau granular aggregates remains uncertain but they may represent a unique finding in epilepsy not previously reported in CTE or Alzheimer’s disease.
Hippocampus, pes hippocampus and amygdala
All patients in this group had HS and in many mossy fibre sprouting was confirmed in the routine diagnostic work-up by Timms stain or immunohistochemistry (for dynorphin or ZnT3) as evidence of epilepsy-associated hippocampal network reorganization (Thom, 2014). Relative sparing of AT8 labelling within the hippocampal subfields, particularly CA1, was a striking finding in tau-positive cases in as previously reported in post-mortem series of HS in epilepsy (Thom et al., 2011). Only nine of the 33 cases had AT8 labelling within hippocampal regions. Of these nine cases, AT8 labelling was more prominent in the subiculum while CA1, CA2, CA3 and CA4 showed proportionally much lower levels of AT8, with labelling in only four cases (Fig. 3A). This is out of step for the normal sequence of AT8 accumulation in Alzheimer’s disease, where CA1 is typically involved earlier than the subiculum (Braak et al., 2011). In addition, early involvement of granule cells was noted in seven cases (Supplementary Table 3), which are typically involved late in Alzheimer’s disease. In one case prominent labelling by AT8 of radial processes through the molecular layer of the dentate gyrus, reminiscent of the pattern of mossy fibre sprouting (Fig. 3B–E), was seen with compact aggregates in the CA4 region (Fig. 3C). There was evidence AT8 localized with ZnT3 aggregates (Fig. 3G) and around neurofilament-positive neurons in CA4 (Fig. 3F), supporting tau aggregation in the mossy fibre axons and terminals. Additional prominent staining was noted in the hippocampal body included labelling of axons in the alveus (Fig. 3 3I–M), horizontal neurons in CA1 and axons in parahippocampal gyrus white matter.
In the pes hippocampal specimens, similar tau patterns were noted. In one case with more severe granule cell dispersion (Fig. 3H) and mossy fibre sprouting (Fig. 3I) in the pes compared to the hippocampal body, there was a striking increase in the AT8 labelling in the pes granule cell layer (Fig. 3J) compared to other subfields and the hippocampal body (Fig. 3K). Other observations were axonal-like labelling along the subpial surface of the pes (Fig. 3N and O) and the periventricular white matter (Fig. 3P). The amygdala region was not available in all cases and fragmented making anatomical orientation difficult; occasional findings were AT8 labelling of axonal fibres and bundles (Fig. 3Q). Supplementary Table 3 provides a summary of our classification of tau patterns.
Biochemical characterization of tau in epilepsy cohort
We further characterized tau composition by immunostaining for TDP-43 inclusions, 3R- and 4R- isoforms in four cases with more abundant AT8 labelling. Neurons and tangles were both 3R and 4R positive (Fig. 3R and S) indicating mixed 3R:4R tau isoform accumulation and was negative for TDP-43 immunoreactive inclusions. The absence of immunoreactive TDP-43 inclusions contrasts with previous reports of Alzheimer’s disease (Amador-Ortiz et al., 2007) but is consistent with epilepsy HS (Lee et al., 2008).
Absence of amyloid-β positive plaques in majority of cases
Amyloid-β positive plaques were absent in 28 cases (85%) with a ‘sparse’ plaque score identified in three cases (10%), ‘moderate’ plaque score in one case (3.0%) and ‘frequent’ plaque score in one case (3%).
The case with frequent plaques had a modified tau score of 6 and the moderate plaque case had a modified tau score of 3. The three cases with occasional plaques had a modified tau scores of 1–2. There was no statistically significant correlation between amyloid-β plaque appearance and the modified tau score (P > 0.05).
Modified tau score in relation to age and cognitive testing
A weak positive correlation was seen between modified tau score and age at time of surgery and between modified tau score and age at onset of epilepsy of the study cohort (Spearman correlation r = 0.39 and r = 0.21, respectively) (Fig. 4). No statistically significant correlations were observed between the modified tau score and preoperative neuropsychometric scores (correlation coefficient range −0.25 < r < 0.13).
Figure 4.
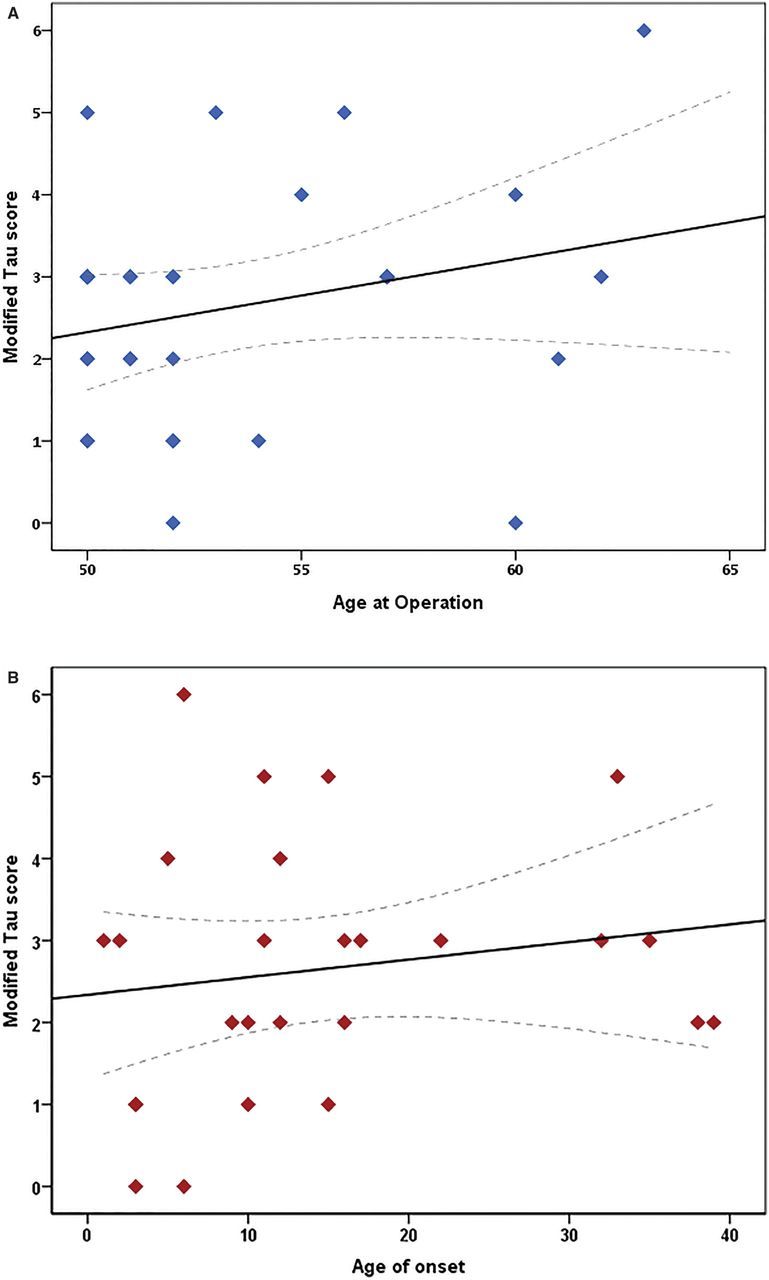
The effect of (A) age at time of surgery and (B) age of onset of epilepsy on the modified tau score in the epilepsy cohort. Scatter plot graphs with linear correlation curve fitted. Spearman correlation shows a weak relationship between both clinical factors (r = 0.39 and r = 0.21, respectively) and are not statistically significant (P > 0.05). Individual points may overlap. Interrupted line indicates 95% confidence interval for the mean modified tau score for the given age.
To further explore cognitive phenotype, we determined postoperative cognitive decline by comparing neuropsychometric scores at different time points. Neuropsychometric data were condensed into memory learning and recall components (verbal and visual) and naming ability. We first compared the scores at 1 year post operation with preoperative scores. We observed a significant decline in verbal learning and recall at 1 year (mean z-score drop −0.67 and −0.58), compared to slight improvement on the visual memory indices at 1 year (P < 0.01) (Table 2).
Table 2.
Comparing the change in neuropsychometry scores for verbal and visual memory and graded naming test between cases of different modified tau scores
|
|
Mean drop (z-score) for each Modified tau score group |
Spearman correlation significance test | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Number of cases with data available | Mean drop (z-score) | 0 | 1 | 2 | 3 | 4 | 5 | 6 | ||
| Verbal Learning | 21 | −0.67 | 0.81 | −0.25 | −0.44 | −1.20 | −0.75 | −2.63 | −2.13 | **P < 0.01 |
| Verbal Recall | 20 | −0.58 | 0.80 | −0.70 | −0.40 | −0.48 | −0.30 | −4.40 | −1.20 | *P < 0.05 |
| Visual Learning | 20 | 0.34 | 0.72 | −0.25 | 0.25 | 0.36 | 0.81 | 0.33 | 0.52 | P > 0.05 |
| Visual Recall | 20 | 0.40 | 0.25 | 1.38 | 0.83 | 0.13 | 0.50 | 0.00 | 0.00 | P > 0.05 |
When examining change in cognitive test scores over 1 year (pre-temporal lobe resection to 1 year post operation), modified tau scores had a strong negative correlation with change in verbal learning (Spearman correlation r = −0.63, *P < 0.05), a moderate negative correlation with change in verbal recall (Spearman correlation r = −0.44, *P < 0.05) and a moderate negative correlation with graded naming test scores (Spearman correlation r = −0.50, P < 0.05) (Fig. 5). No statistically significant associations were observed between the modified tau score and postoperative changes in visual memory (P > 0.05) (Table 2).
Figure 5.

Correlation between modified tau score and change in verbal memory domains and graded naming test scores. Decline in cognitive scores from preoperative assessment to 1 year post-temporal lobe resection shows a significant negative relationship with increasing tau burden for verbal memory domains and graded naming (Spearman coefficient r = −0.66, *P < 0.01, for verbal learning; r = −0.44, *P < 0.05, for verbal recall and r = −0.50, *P < 0.05, for graded naming test). Interrupted line indicates 95% confidence interval for the mean cognitive score for the given modified tau score.
We also examined the change in cognitive test scores from 3 months post-temporal lobe resection to 1 year post operation. Modified tau scores had a moderate negative correlation with change in verbal learning during that period (Spearman correlation r = −0.54, P < 0.05). The correlations with verbal recall (r = −0.06), visual memory (r = −0.11) and visual recall (r = −0.12) were not significant.
Formal long-term neuropsychometric follow-up was not performed for all cases of this study. We can report that the case with the highest tau burden of the study was diagnosed with Alzheimer’s dementia 9 years after temporal lobe resection. This patient had no evidence of dementia at pre-operative assessment and their temporal lobe resection tissue showed tau pathology with distributions typical to the cohort including hippocampal sparing.
Cognitive decline in relation to secondary generalized seizures and other clinical factors of epilepsy
History of secondary generalized seizures was identified in 19 of 26 cases (73%). There was a significant association between modified tau score and history of secondary generalized seizures (likelihood-ratio χ2, P < 0.05) (Supplementary Table 4). Other clinical factors including age at time of surgery (mean 53.6 years), age at onset of epilepsy (mean 14.7 years), duration of epilepsy (mean 39.5 years), side of temporal lobe resection (59.1% of cases were right sided temporal resection), history of head injury and handedness (66.7% of cases were right handed) did not have a clear relationship to modified tau score (P > 0.05). There was a greater decline in verbal memory scores over 1 year with left-sided temporal lobe resection (mean z-score drop in verbal learning, −0.97 versus −0.49, P > 0.05) compared to right-sided resections, but this was not statistically significant. Subsequent stepwise multiple regression analysis did not identify further clinical indicators as a significant independent variable associated with memory decline (verbal or visual domains) or graded naming ability.
Discussion
We confirm and extend previous findings that tau pathology is present in patients with TLE and establish a key involvement by showing a strong correlation between tau pathology and postoperative cognitive decline in a cohort of older patients with epilepsy.
Tau hyperphosphorylation is an important post-translational pathological step causing an inability to interact with microtubules (Bramblett et al., 1993) with imbalances of different tau isoforms contributing to variation across neurodegenerative tauopathies (Spillantini and Goedert, 2013). Epilepsy is not classically thought of as a neurodegenerative disease, however, we identified mixed characteristics of tau pathology similar to both Alzheimer’s disease and CTE, two neurodegenerative diseases previously linked to epilepsy (Amatniek et al., 2006; McKee et al., 2015).
Double labelling identified neuronal tau, mainly within axons, and no tau within glial cell populations, which is consistent with Alzheimer’s disease. The absence of consistent amyloid-β in this study however does not accord with typical Alzheimer’s disease and a similar absence of amyloid-β was noted in the post-mortem study by Thom et al. (2011). Based on these findings, we propose the underlying tau-related neurodegeneration in patients with TLE should not be classified as a typical Alzheimer’s disease process and other ‘tau-predominant pathologies’ should be considered.
We identified several cases with CTE-like patterns of tau distribution, but the lack of perivascular and sulcal tau foci, a prominent feature of early CTE, argues against classical CTE pathology in this epilepsy cohort. CTE tau pathology may reflect the angle of acceleration of head trauma, for example boxers experience angular acceleration injuries from trauma originating below the chin causing a predisposition to basal ganglia pathology with more prominent parkinsonism, while American football players sustain linear acceleration head injuries from in-game tackles leading to pathology that predominates fronto-temporal regions (Gavett et al., 2011). During seizures, we hypothesize two potential head injury mechanisms: falls as a result of seizures leading to direct head trauma and small repetitive trauma of the cerebrum against the skull vault during head jerking movements of secondarily generalized seizures. The latter mechanism may explain the prominent subpial location of tau pathology observed in this study. We argue that tau pathology in our study is not classical, but shares some overlap with CTE, which may be due to the nature of the repetitive head injury and intrinsic factors of epilepsy.
Another hypothesis is that epileptic ictal and interictal activity may be involved in the formation of tau pathology that is related to this specific cohort of patients with chronic TLE. We identified unusual patterns of tau pathology that include the frequent finding of a subpial band of tau pathology in almost all cases (in both Braak-like and CTE-like cases), relative sparing of tau pathology within the hippocampus but involvement of granule cells and mossy fibre pathways. In some cases, the subpial band was the only positive tau labelling and may represent the earliest site of deposition of hyperphosphorylated tau. These patterns do not conform with the stereotypical accumulation of tau (or its propagation) in the mesial temporal lobe as described in the Braak staging of Alzheimer’s disease (Braak et al., 2006; Braak and Del Tredici, 2015). In TLE/HS (as in this study cohort) there is early onset of seizures with neuronal loss and resultant alterations of circuitry and hippocampal networks (Haneef et al., 2014). With the current favoured ‘prion-like’ hypothesis of neuron-to-neuron dissemination of tau along long axonal pathways, this could be one explanation for the observed differences (Supplementary Fig. 1). There is also experimental evidence that tau is released by synaptic activity (Pooler et al., 2013; Lewis and Dickson, 2016) and it is plausible that epileptic activity in these subjects may have influenced tau accumulation patterns, particularly in the excitable granule cell layer. The Alzheimer’s disease literature suggests that tau accumulation primarily effects phylogenetically recent and less mature neurons (Braak and Del Tredici, 2015); in this TLE series, however, we did not confirm evidence for involvement of DCX-positive or reelin-positive cortical neurons. The relative lack of hippocampal involvement also distinguishes TLE/HS cases from PART (primary age-related tauopathies), which are confined to the hippocampus (Duyckaerts et al., 2015). Other unique findings in our series included cortical granular aggregates and subpial axonal bands. Possible explanations are that these features in TLE represent early patterns of tau-accumulation with ageing or related to trauma. However what seems more likely is that this represents an epilepsy-specific early ‘axonal-tauopathy’, as a consequence of seizures, ictal/interictal activity, neuronal loss and the reorganized temporal lobe axonal networks (Supplementary Fig. 1); this ‘model’ invites further investigation to explore mechanisms of tau acquisition.
Interestingly, our study did not find a correlation between tau pathology burden and pre-operative cognitive test scores. Preoperative scores represent a ‘snapshot’ of cognition affected by several factors including co-morbidities, educational level and AED medication, which is likely not to be sufficiently sensitive to be compared with tau burden. Longitudinal data afforded by the postoperative change in cognitive scores reduce contaminating influence of confounding factors such as AED medication, which is usually unaltered over the first postoperative year. Tau burden correlated with a drop in cognitive test scores, particularly verbal learning, from pre-temporal lobe resection to 1 year post operation. We also examined cognitive changes from 3 months post-temporal lobe resection to 1 year post-operation to mitigate the direct effects of the surgical resection and showed similar correlation with verbal learning scores, but poorer correlation with the other memory domains. This difference may be explained by a ‘cognitive effect’ of the actual surgery, which is excluded in the 9-month analysis or may reflect the shorter follow-up period being less sensitive in detecting cognitive changes. Longer term follow-up of cognitive decline in this cohort would be important to explore this.
Despite the atypical patterns of temporal lobe hyperphosphorylated tau in our TLE cohort and differences between cognitive decline when including surgery in the analysis, there is nevertheless evidence for clinical impact. This impact is demonstrated by the correlation between overall tau pathology burden and cognitive decline, particularly verbal learning, post-temporal lobe resection. Our study supports a neurodegenerative role for tau and advances the findings of a previous post-mortem epilepsy study, which did not find a linear correlation with higher Braak stages and pre-mortem cognitive scores but reported over 70% of Braak stage III or greater patients had progressive cognitive decline leading up to death (Thom et al., 2011).
Age is an important factor for tau deposition (Braak et al., 2013), and thus we studied a cohort of between 50 and 65 years at time of surgery, which is much younger than the expected typical age at onset in sporadic Alzheimer’s disease. A post-mortem TLE study found consistent tau pathology in quantities higher than expected for their age (Thom et al., 2011). We observed tau pathology in almost all cases with a weak positive correlation between age at time of surgery and tau burden suggesting age is an important, but not the only, factor in tau accumulation. History of secondary generalized seizures was the only clinical factor which showed statistically significant association with tau burden and this would support the hypothesis of abnormal ictal activity contributing to an epilepsy-associated tauopathy. We feel, however, that a correlation between frequency of generalzed seizures and tau burden would offer a more robust analysis and better reflect the literature on poor cognitive outcomes in epilepsy (Elger et al., 2004). We did not identify a clear relationship between other clinical characteristics analysed including age at onset of epilepsy and duration of epilepsy with hyperphosphorylated tau burden. We observed a greater mean drop in verbal memory with left-sided temporal lobe resections which, despite not being statistically significant, is consistent with the literature (Thompson et al., 2015). The lack of clear relationship with left-sided resection and verbal memory decline may be explained by atypical language dominance and cross-dominance that is documented to occur is more frequent in epilepsy patients (Springer et al., 1999; Dijkstra and Ferrier, 2013) or due to the low number of left-sided resections in this study. Reliable clinical risk factors influencing hyperphosphorylated tau deposition would be required to develop preventative strategies.
The diagnosis of Alzheimer’s disease 9 years post-temporal lobe resection in the patient with the highest tau burden illustrates the possible long-term effect of tau pathology on cognitive decline. Despite the heavy pathological tau burden at the time of surgery, this patient had preserved Mini-Mental State Examination (MMSE) for 6 years post-operation before an appreciable decline and a (Pittsburg compound B) PiB-PET scan, which showed amyloid deposition within average age-controlled range during that time. Long-term follow-up of all cases would be needed to fully investigate the effect of tau pathology.
The importance of our findings is the demonstration of tau pathology in the brains of elderly patients with refractory TLE undergoing surgery and the correlation with postoperative cognitive decline. Temporal lobectomy is not widely offered to older patients with refractory TLE, and only in recent times are we increasingly focussing on ‘physiological’ age rather than actual age when considering candidates for surgery. Analysis of this older population with pathological correlation with ‘real-time’ neuropsychometry assessment for cognitive decline partly explains the low number of cases analysed but is a unique aspect of this study. Surgery in the elderly brain already compromised by tau pathology might accelerate the decline with the little compensatory reserve capacity. Our findings suggest that epilepsy should be added to the increasing spectrum of disorders in which tau pathology may not be the characteristic feature, however is reflective of underlying neurodegeneration and cognitive decline, including Parkinson’s disease, motor neuron disease and, recently, Huntington’s disease (Wolfe, 2012; Vuono et al., 2015).
Our study is limited in that we have examined only temporal lobe tissue, which was resected during surgery and did not have the entire brain for pathological analysis. We created a new scoring system for tau pathology within the temporal lobe, which was useful for the limited tissue available. For Braak staging comparison, similarly limited by available resected tissue, we used control data from large published post-mortem series from a normal ageing, non-epilepsy population (Braak et al., 2011); however, this may be confounded if the tau pattern in TLE does not follow the Braak distribution. We also acknowledge that we have not excluded the effect of progressive neuronal loss (resulting from seizures) as a factor of cognitive decline in our cohort. Clinical data, such as frequency of generalized seizures and head injury, were not consistently available and a prospective approach to collecting this information may prove effective. Furthermore, investigation of the effect of the MAPT haplotypes and APOE status on rate of cognitive decline would be useful to explore potential genetic factors.
In summary, we show a pathological role for the involvement of tau in clinical expression of cognitive decline in TLE. We describe unusual patterns of tau pathology, which may be related to CTE or represent an epilepsy-specific tauopathy. Further studies are needed to elucidate the exact mechanism underlying tau pathology, which may lead to new approaches for diagnosing and treating cognitive decline in temporal lobe epilepsy.
Supplementary Material
Acknowledgement
We are grateful to Derek Marsdon in neuropathology for his help.
Funding
This work is supported by the Medical Research Council (grant MR/JO127OX/1). This work was undertaken at UCLH/UCL who received a proportion of funding from the Department of Health’s NIHR Biomedical Research Centres funding scheme. Z.M. received funding from the European Union Seventh Framework Programme (FP7/2007-2013) under grant agreement EPITARGET, #602102. The Epilepsy Society Brain and Tissue Bank at UCL was funded by the Epilepsy Society.
Supplementary material
Supplementary material is available at Brain online.
Glossary
Abbreviations
- CTE
chronic traumatic encephalopathy
- HS
hippocampal sclerosis
- TLE
temporal lobe epilepsy
References
- Amador-Ortiz C, Lin W-L, Ahmed Z, Personett D, Davies P, Duara R, et al. TDP-43 immunoreactivity in hippocampal sclerosis and Alzheimer’s disease [Internet]. Ann Neurol 2007; 61: 435–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amatniek JC, Hauser WA, DelCastillo-Castaneda C, Jacobs DM, Marder K, Bell K, et al. Incidence and predictors of seizures in patients with Alzheimer’s disease [Internet]. Epilepsia 2006; 47: 867–72. [DOI] [PubMed] [Google Scholar]
- Bernhardt BC, Worsley KJ, Kim H, Evans AC, Bernasconi A, Bernasconi N. Longitudinal and cross-sectional analysis of atrophy in pharmacoresistant temporal lobe epilepsy [Internet]. Neurology 2009; 72: 1747–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black LC, Schefft BK, Howe SR, Szaflarski JP, Yeh H, Privitera MD. The effect of seizures on working memory and executive functioning performance [Internet]. Epilepsy Behav 2010; 17: 412–9. [DOI] [PubMed] [Google Scholar]
- Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. [Internet]. Acta Neuropathol 2006; 112: 389–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Thal DR, Ghebremedhin E, Del Tredici K. Stages of the pathologic process in Alzheimer disease: age categories from 1 to 100 years [Internet]. J Neuropathol Exp Neurol 2011; 70: 960–9. [DOI] [PubMed] [Google Scholar]
- Braak H, Del Tredici K. The preclinical phase of the pathological process underlying sporadic Alzheimer’s disease [Internet]. Brain 2015; 138: 2814–33. [DOI] [PubMed] [Google Scholar]
- Braak H, Zetterberg H, Del Tredici K, Blennow K. Intraneuronal tau aggregation precedes diffuse plaque deposition, but amyloid-β changes occur before increases of tau in cerebrospinal fluid [Internet]. Acta Neuropathol 2013; 126: 631–41. [DOI] [PubMed] [Google Scholar]
- Bramblett GT, Goedert M, Jakes R, Merrick SE, Trojanowski JQ, Lee VM. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding [Internet]. Neuron 1993; 10: 1089–99. [DOI] [PubMed] [Google Scholar]
- Cormack F, Gadian DG, Vargha-Khadem F, Cross JH, Connelly A, Baldeweg T. Extra-hippocampal grey matter density abnormalities in paediatric mesial temporal sclerosis [Internet]. Neuroimage 2005; 27: 635–43. [DOI] [PubMed] [Google Scholar]
- Dijkstra KK, Ferrier CH. Patterns and predictors of atypical language representation in epilepsy [Internet]. J Neurol Neurosurg Psychiatry 2013; 84: 379–85. [DOI] [PubMed] [Google Scholar]
- Duyckaerts C, Braak H, Brion J-P, Buée L, Del Tredici K, Goedert M, et al. PART is part of Alzheimer disease [Internet]. Acta Neuropathol 2015; 129: 749–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elger CE, Helmstaedter C, Kurthen M. Chronic epilepsy and cognition. [Internet]. Lancet Neurol 2004; 3: 663–72. [DOI] [PubMed] [Google Scholar]
- Gaitatzis A, Carroll K, Majeed A, W Sander J. The epidemiology of the comorbidity of epilepsy in the general population [Internet]. Epilepsia 2004; 45: 1613–22. [DOI] [PubMed] [Google Scholar]
- Gavett BE, Stern RA, McKee AC. Chronic traumatic encephalopathy: a potential late effect of sport-related concussive and subconcussive head trauma [Internet]. Clin Sports Med 2011; 30: 179–88, xi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goedert M, Spillantini MG. A century of Alzheimer’s disease [Internet]. Science 2006; 314: 777–81. [DOI] [PubMed] [Google Scholar]
- Griffith HR, Martin RC, Bambara JK, Faught E, Vogtle LK, Marson DC. Cognitive functioning over 3 years in community dwelling older adults with chronic partial epilepsy [Internet]. Epilepsy Res 2007; 74: 91–6. [DOI] [PubMed] [Google Scholar]
- Griffith HR, Martin RC, Bambara JK, Marson DC, Faught E. Older adults with epilepsy demonstrate cognitive impairments compared with patients with amnestic mild cognitive impairment [Internet]. Epilepsy Behav 2006; 8: 161–8. [DOI] [PubMed] [Google Scholar]
- Haneef Z, Lenartowicz A, Yeh HJ, Levin HS, Engel J, Stern JM. Functional connectivity of hippocampal networks in temporal lobe epilepsy [Internet]. Epilepsia 2014; 55: 137–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helmstaedter C, Elger CE. Chronic temporal lobe epilepsy: a neurodevelopmental or progressively dementing disease? [Internet]. Brain 2009; 132: 2822–30. [DOI] [PubMed] [Google Scholar]
- Hermann BP, Seidenberg M, Dow C, Jones J, Rutecki P, Bhattacharya A, et al. Cognitive prognosis in chronic temporal lobe epilepsy [Internet]. Ann Neurol 2006; 60: 80–7. [DOI] [PubMed] [Google Scholar]
- Høgh P, Smith SJ, Scahill RI, Chan D, Harvey RJ, Fox NC, et al. Epilepsy presenting as AD: neuroimaging, electroclinical features, and response to treatment [Internet]. Neurology 2002; 58: 298–301. [DOI] [PubMed] [Google Scholar]
- Ittner LM, Ke YD, Delerue F, Bi M, Gladbach A, van Eersel J, et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models [Internet]. Cell 2010; 142: 387–97. [DOI] [PubMed] [Google Scholar]
- Kandratavicius L, Monteiro MR, Hallak JE, Carlotti CG, Assirati JA, Leite JP. Microtubule-associated proteins in mesial temporal lobe epilepsy with and without psychiatric comorbidities and their relation with granular cell layer dispersion [Internet]. Biomed Res Int 2013; 2013: 960126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EB, Lee VM-Y, Trojanowski JQ, Neumann M. TDP-43 immunoreactivity in anoxic, ischemic and neoplastic lesions of the central nervous system [Internet]. Acta Neuropathol 2008; 115: 305–11. [DOI] [PubMed] [Google Scholar]
- Lewis J, Dickson DW. Propagation of tau pathology: hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies [Internet]. Acta Neuropathol 2016; 131: 27–48. [DOI] [PubMed] [Google Scholar]
- Lin JJ, Salamon N, Lee AD, Dutton RA, Geaga JA, Hayashi KM, et al. Reduced neocortical thickness and complexity mapped in mesial temporal lobe epilepsy with hippocampal sclerosis [Internet]. Cereb Cortex 2007; 17: 2007–18. [DOI] [PubMed] [Google Scholar]
- Liu YWJ, Curtis MA, Gibbons HM, Mee EW, Bergin PS, Teoh HH, et al. Doublecortin expression in the normal and epileptic adult human brain [Internet]. Eur J Neurosci 2008; 28: 2254–65. [DOI] [PubMed] [Google Scholar]
- Liu RSN, Lemieux L, Bell GS, Hammers A, Sisodiya SM, Bartlett PA, et al. Progressive neocortical damage in epilepsy. Ann Neurol 2003; 53: 312–324. [DOI] [PubMed] [Google Scholar]
- Mackenzie IR, Miller LA. Senile plaques in temporal lobe epilepsy [Internet]. Acta Neuropathol 1994; 87: 504–10. [DOI] [PubMed] [Google Scholar]
- McKee AC, Stein TD, Kiernan PT, Alvarez VE. The neuropathology of chronic traumatic encephalopathy [Internet]. Brain Pathol 2015; 25: 350–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH, et al. The spectrum of disease in chronic traumatic encephalopathy [Internet]. Brain 2013; 136: 43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oyegbile TO, Dow C, Jones J, Bell B, Rutecki P, Sheth R, et al. The nature and course of neuropsychological morbidity in chronic temporal lobe epilepsy [Internet]. Neurology 2004; 62: 1736–42. [DOI] [PubMed] [Google Scholar]
- Palop JJ, Chin J, Roberson ED, Wang J, Thwin MT, Bien-Ly N, et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease [Internet]. Neuron 2007; 55: 697–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pooler AM, Phillips EC, Lau DHW, Noble W, Hanger DP. Physiological release of endogenous tau is stimulated by neuronal activity [Internet]. EMBO Rep 2013; 14: 389–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng JG, Boop FA, Mrak RE, Griffin WST. Increased Neuronal β-Amyloid Precursor Protein Expression in Human Temporal Lobe Epilepsy: Association with Interleukin-1α Immunoreactivity. J Neurochem. 1994; 63: 1872–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Goedert M. Tau pathology and neurodegeneration [Internet]. Lancet Neurol 2013; 12: 609–22. [DOI] [PubMed] [Google Scholar]
- Springer JA, Binder JR, Hammeke TA, Swanson SJ, Frost JA, Bellgowan PS, et al. Language dominance in neurologically normal and epilepsy subjects: a functional MRI study [Internet]. Brain 1999; 122 (Pt 1): 2033–46. [DOI] [PubMed] [Google Scholar]
- Srikandarajah N, Martinian L, Sisodiya SM, Squier W, Blumcke I, Aronica E, et al. Doublecortin expression in focal cortical dysplasia in epilepsy [Internet]. Epilepsia 2009; 50: 2619–28. [DOI] [PubMed] [Google Scholar]
- Tellez-Zenteno JF, Matijevic S, Wiebe S. Somatic comorbidity of epilepsy in the general population in Canada [Internet]. Epilepsia 2005; 46: 1955–62. [DOI] [PubMed] [Google Scholar]
- Thom M, Liu JYW, Thompson P, Phadke R, Narkiewicz M, Martinian L, et al. Neurofibrillary tangle pathology and Braak staging in chronic epilepsy in relation to traumatic brain injury and hippocampal sclerosis: a post-mortem study [Internet]. Brain 2011; 134: 2969–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thom M. Review: hippocampal sclerosis in epilepsy: a neuropathology review [Internet]. Neuropathol Appl Neurobiol 2014; 40: 520–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson PJ, Baxendale SA, McEvoy AW, Duncan JS. Cognitive outcomes of temporal lobe epilepsy surgery in older patients [Internet]. Seizure 2015; 29: 41–5. [DOI] [PubMed] [Google Scholar]
- Vuono R, Winder-Rhodes S, de Silva R, Cisbani G, Drouin-Ouellet J, Spillantini MG, et al. The role of tau in the pathological process and clinical expression of Huntington’s disease [Internet]. Brain 2015; 138: 1907–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfe MS. The role of tau in neurodegenerative diseases and its potential as a therapeutic target [Internet]. Scientifica (Cairo) 2012; 2012: 796024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong K, Luo D-W, Patrylo PR, Luo X-G, Struble RG, Clough RW, et al. Doublecortin-expressing cells are present in layer II across the adult guinea pig cerebral cortex: partial colocalization with mature interneuron markers [Internet]. Exp Neurol 2008; 211: 271–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X-M, Cai Y, Chu Y, Chen E-Y, Feng J-C, Luo X-G, et al. Doublecortin-expressing cells persist in the associative cerebral cortex and amygdala in aged nonhuman primates [Internet]. Front Neuroanat 2009; 3: 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.