

Abstract
Prion-like proteins have the capacity to adopt multiple stable conformations, at least one of which can recruit proteins from the native conformation into the alternative fold. Although classically associated with disease, prion-like assembly has recently been proposed to organize a range of normal biochemical processes in space and time. Organisms from bacteria to mammals use prion-like mechanisms to (re)organize their proteome in response to intracellular and extracellular stimuli. Prion-like behavior is an economical means to control biochemistry and gene regulation at the systems level, and prions can act as protein-based genes to facilitate quasi-Lamarckian inheritance of induced traits. These mechanisms allow individual cells to express distinct heritable traits using the same complement of polypeptides. Understanding and controlling prion-like behavior is therefore a promising strategy to combat diverse pathologies and organize engineered biological systems.
Graphical abstract
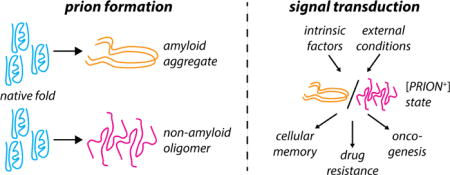
Introduction
The organization of biochemical reactions in space and time is a hallmark of life from bacteria to metazoa. Organisms have evolved diverse strategies to achieve this objective, ranging from membrane-bound and proteinaceous organelles to granules composed of both protein and nucleic acid. Subcellular structures have long been known [1,2], but one apparently ubiquitous mode of organization has only been recognized more recently: prion-like assembly of proteins. Here, we will consider the phenomenon of “prion-like assembly” to broadly encompass the self-templating of protein conformations to mediate the formation of an assembly, aggregate, or oligomer [Fig. 1]. This templating may be stochastic or regulated, and may exhibit a range of stabilities.
Figure 1.
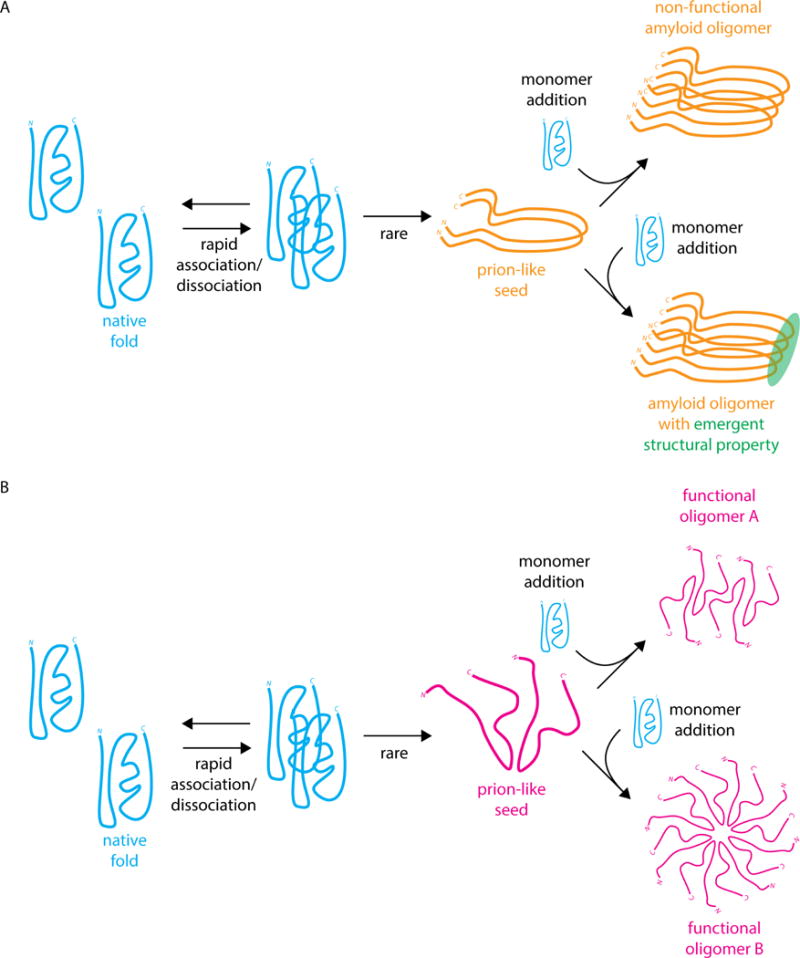
(A) Classical amyloid prion formation by nucleation of a prion-like seed followed by recruitment of monomers to the prion-like conformation to generate a cross-beta sheet amyloid oligomer [12]. The amyloid oligomer could be non-functional (top), as in the [PSI+] prion, or gain an emergent structural property (bottom), as in the HET-s prion [13] (B) Hypothetical mechanism of prion-like oligomerization leading to two distinct prion-like oligomers that maintain their molecular activity and are not amyloid in nature.
Prions are a unique class of proteins that can adopt stable alternative conformations that are capable of recruiting other proteins to adopt the alternate conformational state, constituting a structurally transmissible entity [3–5]. Prions exhibit dominant inheritance through meiosis, are seldom lost, and form a cross-beta sheet amyloid structure in the prion state [6]. Conventionally, these states are denoted as [PRION+], with the brackets denoting extranuclear inheritance and the + denoting dominance through mating. Most prions rely heavily on the activity of molecular chaperones to propagate across generations. For some this manifests in a strong dependence on the disaggregase Hsp104 [7–9] for propagation, but others rely on Hsp70 or other chaperones [10]. Infectious protein conformations have most often been associated with neurodegenerative disease, but recent discoveries across the tree of life make it clear that prion-like behavior is also a common part of normal biochemical organization and function [11]. Defined broadly, prion-like protein assembly is a potentially versatile strategy for cells to integrate diverse cellular and external inputs and respond by reorganizing key biochemical processes.
Prion-like proteins as agents of disease
Prion-like behavior was first observed in the causative agent of scrapie, a neurodegenerative disease of sheep and goats [4,14]. Particles consisting only of the PrPSc protein were found to be sufficient for transmission of the disease. Infectious proteins have since been shown to be at the root of a class of transmissible encephalopathies [15]. The role of PrP in infectious human encephalopathies, including Creutzfeld-Jakob disease and Kuru, has been extensively reviewed elsewhere [16], and we will not discuss it in detail here. More recently, prion-like behavior has also been implicated in non-transmissible neurodegenerative illnesses. The amyloid-β protein in Alzheimer’s disease [17], the alpha-synuclecin protein in Parkinson’s disease [18], and the FUS protein in amylotrophic lateral sclerosis [19,20] all exhibit some degree of prion-like behavior. Moreover, iatrogenic transmission of not only Creutzfeld-Jakob disease [21], but also possibly amyloid-β aggregates, has been reported [22]. Certain mutants of the key tumor suppressor p53 also has prion-like properties [23,24]. In each of these cases, the prion state is thought to be amyloid in nature, resulting from templating natively folded proteins onto a seed with a cross-β sheet conformation.
Prion-like assembly as part of normal function
Just as foundational studies of viruses helped reveal the role of nucleic acids in heredity, so too have studies of pathogenic prions revealed prion-like behavior as a mode of information transfer that is based on protein alone. Indeed, the classical notion of prions as agents of disease has been challenged in recent years by a raft of studies implicating prion-like behavior in normal cellular function [Table 1]. The persistence of prion-like states, combined with the ability to induce their formation, presents a powerful mechanism for cells to store information for long periods in response to external stimuli, even if the individual proteins involved undergo more rapid turnover. This concept has been invoked most provocatively as an explanation for the persistence of long-term memory. Amyloidogenic behavior of the cytoplasmic polyadenylation element-binding (CPEB/Orb2) protein is critical for the formation of long-term memory in Aplysia [25], Drosophila [26], and in mice [27]. The observation that the rare Orb2A isoform seeds oligomerization implicates alternative splicing as a mechanism for the control of prion-like assembly; post-translational modifications may provide a further route for dynamic regulation of the phase-separation of CPEB4 in Xenopus and other vertebrates [28].
Table 1.
Recent descriptions of prion-like behavior in biochemical processes involved in normal cellular function. See also [10,37] for systematic approaches to identify prion-like behavior proteome-wide in S. cerevisiae.
| Biochemical process | Gene product exhibiting prion-like behavior | Species/cell line | Reference |
|---|---|---|---|
| Long-term memory formation | CPEB3 | M. musculus | [26] |
| Orb2 | D. melanogaster | [25] | |
| CPEB4 | X. laevis | [27] | |
| Antiviral signaling | MAVS | HEK293T (H. sapiens) | [28] |
| Stress granule formation | TIA-1/Sup35 | COS-7 (C. aethiops); S. cerevisiae | [30,31] |
| Transcription termination | Rho | E. coli | [32] |
| Heterokaryon incompatibility | HET-s (HET-s/HET-S) | P. anserina | [33] |
| Translation termination | SUP35 ([PSI+]) | S. cerevisiae | [34] |
| Pyrimidine biosynthesis | URE2 ([URE3+]) | S. cerevisiae | [35] |
| Chromatin remodeling | Swi1 ([SWI+]) | S. cerevisiae | [36] |
| Mating; carbon metabolism; stress response | Mot3 ([MOT3+]) | S. cerevisiae | [37] |
| Sterol biosynthesis | Mod5 ([MOD+]) | S. cerevisiae | [38] |
| Carbon metabolism | Various ([GAR+]) | S. cerevisiae | [39] |
| Prion formation | New1 ([PIN+]) | S. cerevisiae | [40] |
| Prion formation | Rnq1 ([PIN+]) | S. cerevisiae | [41] |
Prion-like organization is observed in other normal processes, including in the activation of antiviral responses mediated by the mitochondrial antiviral signaling (MAVS) protein [29]. The activated MAVS complex assembles in a prion-like fashion to form a filament on the surface of the mitochondrion [30]. This facilitates a robust switch from an inactive to an active state of the antiviral response. Another interesting case of prion-mediated subcellular organization is that of Tia1, an RNA-binding protein involved in the formation of stress granules [31] which in yeast forms a two-component prion phase in concert with the classical prion protein Sup35 to direct translation machinery to the tubulin cytoskeleton [32]. In this way, the prion-like state can control the subcellular localization of a biochemical process in fine detail in response to stress. Prion-like behavior was also recently observed in the bacterial transcriptional terminator Rho from Clostridium botulinum, the heritable aggregation of which leads to widespread transcriptional read-through [11].
These findings suggest that prion-like assembly may be an ancient and widespread mode of spatiotemporal organization, potentially predating the divergence of the bacterial and eukaryotic kingdoms. At a fundamental level, the self-templating conformational conversion inherent to prion-like ensembles creates a functional memory that can persist longer than any individual protein.
Proteins acting as genes
The heritability of the prion state makes possible the creation of heritable elements consisting only of protein, amounting to genes that are entirely divorced from nucleic acid. One of the earliest known prions of yeast, [PSI+], facilitates read-through of stop codons proteome-wide. It is thus a highly genetically compact means of precipitating large phenotypic changes [34,42], as is the bacterial Rho prion [11]. Recent studies have further shown that prion-like behavior is ubiquitous in S. cerevisiae [37,43], and that the prion state can frequently confer a fitness advantage [10].
The [GAR+] prion, which allows escape from glucose repression and converts yeast from metabolic specialists to generalists [39,44–46], the [MOT3+] prion, which modulates multicellularity [47], and the [MOD+] prion, which confers drug resistance [38], are all induced in conditions in which the [PRION+] state is adaptive. The fact that prion states can be robustly induced by environmental stimuli raises the intriguing possibility of a quasi-Lamarckian genetic element that is acquired in response to the conditions in which it confers a beneficial phenotype. The adaptive phenotype can subsequently be inherited by all mitotic and meiotic offspring due to the extranuclear localization of the prion oligomer. Furthermore, [GAR+] and [MOT3+] can be erased in unfavorable conditions (e.g. desiccation [48] and hypoxia [47], respectively). This type of reversible epigenetic behavior provides strong theoretical advantages relative to irreversible genetic mechanisms of adaptation (see below).
Bet-hedging using switchable protein genes
Even though they arise spontaneously at low rates, prion-like states can confer adaptive phenotypes that can rapidly sweep the population in times of abrupt environmental change [45,46]. Furthermore, prion-like switches revert at much higher rates than would typically be expected for nucleic acid mutations, allowing the prion-like state to be efficiently purged from the population when it is no longer advantageous. Theoretical calculations predict that such reversible mechanisms of phenotypic switching can be favored over irreversible mutational analogs, even if the fitness benefit conferred by the prion-like state upon environmental change is modest [49,50]. For example, within such models, experimentally observed rates of switching of [GAR+] suggest that the selective advantage of reversible metabolic generalism afforded by this prion is sufficient to favor its evolutionary retention compared to a nucleic acid-based mechanism [45]. The [GAR+] state, therefore, can be thought of as a means of hedging species’ bets against dramatic environmental shifts by maintaining a small subpopulation in a state that is primed to thrive in a new environment.
Prion-like assembly from the systems biology perspective
Beyond acting as protein-based genes, the [PRION+] state can be viewed as a node that integrates diverse cellular inputs, from the proteomic state to external metabolite concentrations, and elicits heritable changes in phenotype at the single-cell level that can be highly adaptive [Fig. 2]. This idea is supported by the observation that, among the many prion-like proteins now known in S. cerevisiae, there is a striking enrichment for RNA-binding proteins and transcription factors [10,51]. These classes of proteins are uniquely suited to precipitate large phenotypic changes, as they can proximally affect the abundance of large numbers of other cellular components. Several other proteins identified as prion-like also belong to functional classes that might be expected to potentiate pleiotropic effects, such as chromatin remodelers [10,36] and DNA repair enzymes [52]. The rate of switching into the [PRION+] state can be modulated by a wide array of inputs, including missense mutations [53], alternative splicing isoforms [26], post-translational modifications [28], changes in protein abundance [54], and external conditions [38,44].
Figure 2.
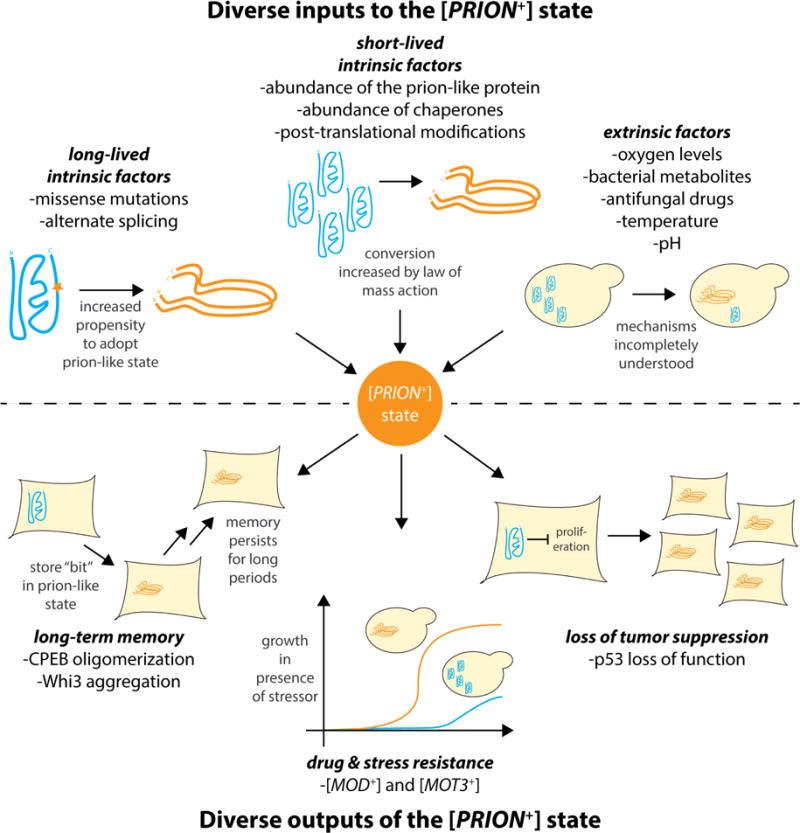
The [PRION+] state is a node that integrates diverse cell-intrinsic inputs (on short and long time scales) and cell-extrinsic stimuli, and actuates diverse phenotypic outputs. The naïve state is shown as a blue gene product; the prion-like [PRION+] state is shown as an orange gene product. Shown are selected examples of inputs and outputs of the [PRION+] state; this diagram is not exhaustive.
More broadly, prion-like assembly can be viewed as a class of phase-separation behavior. From the discovery of Cajal bodies [1] to more recent observations [55,56], phase-separated cytocondensates are implicated in a range of regulatory and metabolic processes [57,58]. These cytocondensates can be stable and self-reinforcing over many generations, as in the case of yeast prions, or subject to remodeling throughout the cell cycle, as in many subcellular phase-separated organelles. Organizing proteins in this manner can have a range of functional consequences: sequestering the activity of the prion-like protein; protecting the prion-like protein from other cellular processes; increasing the local concentration of the prion-like protein to enhance binding interactions; and enhancing the performance of prion-like enzymes and enzyme cascades. The precise kinetic effects of phase-separation on organized processes remain mysterious, and will be a fruitful area of future investigation as an increasing number of prion-like phase-separated ensembles are identified.
Avenues for control of prion-like assembly
The ubiquity of prion-like assembly both in pathology and normal function speaks to the need to understand and control prion-like behavior. The peptides responsible for prion-like behavior have been mapped in detail for amyloid prions of yeast [59–63], and prion-like amyloidogenic behavior can be conferred on otherwise non-prion-like proteins by the addition of a so-called prion domain known to be sufficient for the purpose [37,64]. Detailed investigations of the peptides and residues involved in prion-like and phase-separation behavior will continue to be valuable in understanding their sequence-structure-function relationships [65], as will emerging techniques for examining the conformations of prion-like proteins in the native context of the cellular milieu [66].
These insights have raised the possibility of designing compounds to enhance or inhibit prion-like behavior. Recently, researchers have disrupted the binding of amyloid-β to PrPC, an interaction implicated in Alzheimer’s disease, with small molecule inhibitors [67] and used an engineered peptide to induce amyloid formation and cause loss of function of the VEGFR2 protein [68]. A biotinylated isoxazole compound can also precipitate the formation of amyloid-like fibrils [69,70], and an inhibitor of p53 aggregation has also been shown to disrupt the prion-like loss of tumor suppression discussed above [71]. Since amyloid-β, for instance, exists in a range of oligomeric states with differential toxicity [72], it is important to consider the oligomeric state targeted by a therapeutic, in addition to tissue tropism and other conventional considerations, when designing anti-amyloid therapies [73]. Considering the array of processes we now know to be dependent on prion-like behaviors, interventions to promote or prevent the formation of prion-like states hold promise in the treatment of many pathologies. The prion domain of Sup35 has even been employed in vitro to organize engineered processes [74,75].
Future directions
A wealth of structural, mechanistic, and evolutionary questions remain with respect to the function of prion-like assemblies. From a structural perspective, it is apparent that the conventional amyloid prions represent an important but not all-encompassing class of possible self-templating protein conformations. Of interest in this regard are prion-like states that mediate a gain-of-function phenotype due to oligomerization, rather than a loss of function due to sequestration in amyloid. Frequently, these non-amyloid prions are not enriched in Asn and Gln residues, in contrast to classical amyloid prion domains [10]. Structural determination for prion-like assemblies remains challenging, as they often adopt heterogeneous conformations, but cryoelectron microscopy and NMR approaches offer a promising route to access their conformational ensembles [66,76].
Mechanistically, a loss of protein function due to amyloid sequestration is only one possible explanation in cases when a prion-like state leads to an emergent gain of function by the assembled aggregate, fiber, or other oligomer. Often, a combination of molecular biological and biochemical experiments, polymer physical analyses of prion-like phase-separated assemblies, and detailed kinetic models of organized protein complexes will be required to tease apart the mechanisms by which prion-like organization can enhance the function of proteins and protein ensembles.
The evolutionary history of prion-like assembly likewise remains enigmatic, as the intrinsically disordered proteins often responsible for prion-like behavior often exhibit poor sequence conservation over evolutionary time, despite frequently maintaining their disordered nature. New methods of sequence alignment and analysis will help to address this problem [77–80], which is key in understanding the diversity in and relationships between prion-like proteins. In any event, the discovery of prion-like behavior in organisms ranging from bacteria to mammals suggests that this mode of organization arose near the root of the tree of life [81].
In the same way that the discovery of pre-mRNA splicing revealed enormous hidden information content in genomic DNA [82], the discovery of prion-like alternate conformations revealed that many proteins have the potential to dramatically reshape cellular biochemistry by reorganizing the existing complement of proteins. In another parallel with splicing [83], prion-like behavior, while present in bacteria, seems to have reached its fullest elaboration in eukarya. The organization of biochemistry in space and time, the key attribute of living processes, must be increasingly sophisticated in organisms of increasing complexity, but we observe that the number of genes does not scale according to this intuition [84]. In this light, a form of organization that is self-templating presents an attractive organization strategy because of its compactness and reversibility: a biochemical system can be reshaped repeatedly on time scales both short (less than one cell cycle) and long (hundreds of generations) using a single set of genes and indeed of polypeptides. We fully expect that a broad spectrum of prion-like assemblies remain to be discovered, and that their functional consequences will be even broader than those currently known.
Highlights.
Prion-like behavior can be defined as self-templating of a protein conformation to form an “infectious” aggregate or oligomer
Prion-like behavior has been observed across multiple domains of life
Prion-like behavior contributes to both normal and pathological processes in biology
The prion-like state can act as a bistable node that integrates diverse inputs and actuates diverse outputs
Controlling protein organization using prion-like behavior is a new paradigm in biochemical regulation at the single-cell level
Acknowledgments
We thank David Garcia, Zach Harvey, Thomas Lozanoski, and Leah Sibener for critical review of the manuscript. CMJ was supported by the National Institutes of Health (NIH-1F32-GM125162 to CMJ). DFJ was supported by the National Institutes of Health (NIH-DP2-GM119140 to DFJ), the National Science Foundation (NSF-MCB116762 to DFJ), a Searle Scholar Award (14-SSP-210 to DFJ), a Kimmel Scholar Award (SFK-15-154 to DFJ), and a Science and Engineering Fellowship from the David and Lucile Packard Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cajal S. Un sencillo metodo de coloracion selectiva del reticulo protoplasmico y sus efectos en los diversos organos nerviosos de vertebrados e invertebrados. Trab Lab Investig Biol Univ Madr. 1903;2:129–221. [Google Scholar]
- 2.van Leeuwenhoek A. Observations On Animalcula Seen In Rain, Well, Sea And Snow-Water; As Also In Pepper-Water. :1675. [Google Scholar]
- 3○.Patino MM, Liu J-J, Glover JR, Lindquist S. Support for the Prion Hypothesis for Inheritance of a Phenotypic Trait in Yeast. Science. 1996;273:622–626. doi: 10.1126/science.273.5275.622. This manuscript convincingly demonstrates that the [PSI+] state of S. cerevisiae is conferred by a prion form of the Sup35 protein. [DOI] [PubMed] [Google Scholar]
- 4○.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136–144. doi: 10.1126/science.6801762. Prusiner shows that the causative agent of scrapie, long observed to have unusual properties, is caused by an infectious protein, or prion. [DOI] [PubMed] [Google Scholar]
- 5○.Wickner RB. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science. 1994;264:566–569. doi: 10.1126/science.7909170. In this foundational study, Wickner shows that the [URE3+] state in S. cerevisiae is conferred by a prion form of the Ure2 protein, and speculates that [PSI+] might likewise be prion-like in nature. [DOI] [PubMed] [Google Scholar]
- 6.Si K. Prions: What Are They Good For? Annu Rev Cell Dev Biol. 2015;31:149–169. doi: 10.1146/annurev-cellbio-100913-013409. [DOI] [PubMed] [Google Scholar]
- 7○.Chernoff YO, Lindquist SL, Ono B, Inge-Vechtomov SG, Liebman SW. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+] Science. 1995;268:880–884. doi: 10.1126/science.7754373. Chernoff and colleagues demonstrate that the Hsp104 disaggregase is required to propagate the [PSI+] prion in S. cerevisae. [DOI] [PubMed] [Google Scholar]
- 8.Moriyama H, Edskes HK, Wickner RB. [URE3] Prion Propagation in Saccharomyces cerevisiae: Requirement for Chaperone Hsp104 and Curing by Overexpressed Chaperone Ydj1p. Mol Cell Biol. 2000;20:8916–8922. doi: 10.1128/mcb.20.23.8916-8922.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shorter J, Lindquist S. Hsp104, Hsp70 and Hsp40 interplay regulates formation, growth and elimination of Sup35 prions. EMBO J. 2008;27:2712–2724. doi: 10.1038/emboj.2008.194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10○.Chakrabortee S, Byers JS, Jones S, Garcia DM, Bhullar B, Chang A, She R, Lee L, Fremin B, Lindquist S, et al. Intrinsically Disordered Proteins Drive Emergence and Inheritance of Biological Traits. Cell. 2016;167:369–381. doi: 10.1016/j.cell.2016.09.017. e12. The authors use an unbiased screen to assess the prion-like character of all the proteins of S. cerevisiae. 46 new prion-like proteins are described, many of which are not classically amyloid in nature. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11○.Yuan AH, Hochschild A. A bacterial global regulator forms a prion. Science. 2017;355:198–201. doi: 10.1126/science.aai7776. The authors demonstrate that the Clostridium botulinum Rho transcription termination factor exhibits prion-like behavior, the first bona fide prion discovered in bacteria. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collins SR, Douglass A, Vale RD, Weissman JS. Mechanism of Prion Propagation: Amyloid Growth Occurs by Monomer Addition. PLOS Biol. 2004;2:e321. doi: 10.1371/journal.pbio.0020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daskalov A, Gantner M, Wälti MA, Schmidlin T, Chi CN, Wasmer C, Schütz A, Ceschin J, Clavé C, Cescau S, et al. Contribution of Specific Residues of the β-Solenoid Fold to HET-s Prion Function, Amyloid Structure and Stability. PLOS Pathog. 2014;10:e1004158. doi: 10.1371/journal.ppat.1004158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alper T, Cramp WA, Haig DA, Clarke MC. Does the Agent of Scrapie Replicate without Nucleic Acid ? Nature. 1967;214:764–766. doi: 10.1038/214764a0. [DOI] [PubMed] [Google Scholar]
- 15.Prusiner SB. Prions. Proc Natl Acad Sci. 1998;95:13363–13383. doi: 10.1073/pnas.95.23.13363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Collinge J. Prion diseases of humans and animals: their causes and molecular basis. Annu Rev Neurosci. 2001;24:519–550. doi: 10.1146/annurev.neuro.24.1.519. [DOI] [PubMed] [Google Scholar]
- 17.Nussbaum JM, Schilling S, Cynis H, Silva A, Swanson E, Wangsanut T, Tayler K, Wiltgen B, Hatami A, Rönicke R, et al. Prion-like behaviour and tau-dependent cytotoxicity of pyroglutamylated amyloid-β. Nature. 2012;485:651–655. doi: 10.1038/nature11060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iljina M, Garcia GA, Horrocks MH, Tosatto L, Choi ML, Ganzinger KA, Abramov AY, Gandhi S, Wood NW, Cremades N, et al. Kinetic model of the aggregation of alpha-synuclein provides insights into prion-like spreading. Proc Natl Acad Sci. 2016;113:E1206–E1215. doi: 10.1073/pnas.1524128113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ciryam P, Lambert-Smith IA, Bean DM, Freer R, Cid F, Tartaglia GG, Saunders DN, Wilson MR, Oliver SG, Morimoto RI, et al. Spinal motor neuron protein supersaturation patterns are associated with inclusion body formation in ALS. Proc Natl Acad Sci. 2017;114:E3935–E3943. doi: 10.1073/pnas.1613854114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20○.Patel A, Lee HO, Jawerth L, Maharana S, Jahnel M, Hein MY, Stoynov S, Mahamid J, Saha S, Franzmann TM, et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell. 2015;162:1066–1077. doi: 10.1016/j.cell.2015.07.047. Patel et al. show that a mutation associated with ALS accelerates liquid-to-solid phase transitions of the FUS protein. [DOI] [PubMed] [Google Scholar]
- 21.Swerdlow AJ, Higgins CD, Adlard P, Jones ME, Preece MA. Creutzfeldt-Jakob disease in United Kingdom patients treated with human pituitary growth hormone. Neurology. 2003;61:783–791. doi: 10.1212/01.wnl.0000084000.27403.15. [DOI] [PubMed] [Google Scholar]
- 22.Jaunmuktane Z, Mead S, Ellis M, Wadsworth JDF, Nicoll AJ, Kenny J, Launchbury F, Linehan J, Richard-Loendt A, Walker AS, et al. Evidence for human transmission of amyloid-β pathology and cerebral amyloid angiopathy. Nature. 2015;525:247–250. doi: 10.1038/nature15369. [DOI] [PubMed] [Google Scholar]
- 23.Bom APDA, Rangel LP, Costa DCF, de Oliveira GAP, Sanches D, Braga CA, Gava LM, Ramos CHI, Cepeda AOT, Stumbo AC, et al. Mutant p53 Aggregates into Prion-like Amyloid Oligomers Fibrils IMPLICATIONS FOR CANCER. J Biol Chem. 2012;287:28152–28162. doi: 10.1074/jbc.M112.340638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rangel LP, Costa DCF, Vieira TCRG, Silva JL. The aggregation of mutant p53 produces prion-like properties in cancer. Prion. 2014;8:75–84. doi: 10.4161/pri.27776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Si K, Choi Y-B, White-Grindley E, Majumdar A, Kandel ER. Aplysia CPEB can form prion-like multimers in sensory neurons that contribute to long-term facilitation. Cell. 2010;140:421–435. doi: 10.1016/j.cell.2010.01.008. [DOI] [PubMed] [Google Scholar]
- 26○.Majumdar A, Cesario WC, White-Grindley E, Jiang H, Ren F, Khan M “Repon”, Li L, Choi EML, Kannan K, Guo F, et al. Critical Role of Amyloid-like Oligomers of Drosophila Orb2 in the Persistence of Memory. Cell. 2012;148:515–529. doi: 10.1016/j.cell.2012.01.004. This study demonstrates not only that CPEB/Orb2 forms amyloid-like fibers to create long-term memory in Drosophila, but also that the rare Orb2A splice variant is likely responsible for seeding these assemblies. [DOI] [PubMed] [Google Scholar]
- 27.Fioriti L, Myers C, Huang Y-Y, Li X, Stephan JS, Trifilieff P, Colnaghi L, Kosmidis S, Drisaldi B, Pavlopoulos E, et al. The Persistence of Hippocampal-Based Memory Requires Protein Synthesis Mediated by the Prion-like Protein CPEB3. Neuron. 2015;86:1433–1448. doi: 10.1016/j.neuron.2015.05.021. [DOI] [PubMed] [Google Scholar]
- 28.Guillén-Boixet J, Buzon V, Salvatella X, Méndez R. CPEB4 is regulated during cell cycle by ERK2/Cdk1-mediated phosphorylation and its assembly into liquid-like droplets. eLife. 2016;5:e19298. doi: 10.7554/eLife.19298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29○.Hou F, Sun L, Zheng H, Skaug B, Jiang Q-X, Chen ZJ. MAVS Forms Functional Prion-like Aggregates to Activate and Propagate Antiviral Innate Immune Response. Cell. 2011;146:448–461. doi: 10.1016/j.cell.2011.06.041. Hou and colleagues demonstrate that the mitochondrial MAVS protein forms prion-like fibrillary structures that precipitate an antiviral immune response. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xu H, He X, Zheng H, Huang LJ, Hou F, Yu Z, de la Cruz MJ, Borkowski B, Zhang X, Chen ZJ, et al. Structural basis for the prion-like MAVS filaments in antiviral innate immunity. eLife. 2014;3:e01489. doi: 10.7554/eLife.01489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gilks N, Kedersha N, Ayodele M, Shen L, Stoecklin G, Dember LM, Anderson P. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol Biol Cell. 2004;15:5383–5398. doi: 10.1091/mbc.E04-08-0715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li X, Rayman JB, Kandel ER, Derkatch IL. Functional role of Tia1/Pub1 and Sup35 prion domains: directing protein synthesis machinery to the tubulin cytoskeleton. Mol Cell. 2014;55:305–318. doi: 10.1016/j.molcel.2014.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coustou V, Deleu C, Saupe S, Begueret J. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc Natl Acad Sci. 1997;94:9773–9778. doi: 10.1073/pnas.94.18.9773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34○.Cox B. Heredity - Abstract of article: Ψ, A cytoplasmic suppressor of super-suppressor in yeast. Heredity. 1965;20:505–521. This manuscript describes for the first time the [PSI+] omnipotent suppressor and its unusual pattern of inheritance through meiosis. [Google Scholar]
- 35.Lacroute F. Non-Mendelian Mutation Allowing Ureidosuccinic Acid Uptake in Yeast. J Bacteriol. 1971;106:519–522. doi: 10.1128/jb.106.2.519-522.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Du Z, Park K-W, Yu H, Fan Q, Li L. Newly identified prion linked to the chromatin-remodeling factor Swi1 in Saccharomyces cerevisiae. Nat Genet. 2008;40:460–465. doi: 10.1038/ng.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37○.Alberti S, Halfmann R, King O, Kapila A, Lindquist S. A Systematic Survey Identifies Prions and Illuminates Sequence Features of Prionogenic Proteins. Cell. 2009;137:146–158. doi: 10.1016/j.cell.2009.02.044. This study develops and verifies a computational approach to identify prion-like proteins from primary amino acid sequence, and describes the [MOT3+] prion of S. cerevisiae. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Suzuki G, Shimazu N, Tanaka M. A Yeast Prion, Mod5, Promotes Acquired Drug Resistance and Cell Survival Under Environmental Stress. Science. 2012;336:355–359. doi: 10.1126/science.1219491. [DOI] [PubMed] [Google Scholar]
- 39.Brown JCS, Lindquist S. A heritable switch in carbon source utilization driven by an unusual yeast prion. Genes Dev. 2009;23:2320–2332. doi: 10.1101/gad.1839109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Derkatch IL, Bradley ME, Hong JY, Liebman SW. Prions Affect the Appearance of Other Prions: The Story of [PIN+] Cell. 2001;106:171–182. doi: 10.1016/s0092-8674(01)00427-5. [DOI] [PubMed] [Google Scholar]
- 41.Sondheimer N, Lindquist S. Rnq1: an epigenetic modifier of protein function in yeast. Mol Cell. 2000;5:163–172. doi: 10.1016/s1097-2765(00)80412-8. [DOI] [PubMed] [Google Scholar]
- 42.Baudin-Baillieu A, Legendre R, Kuchly C, Hatin I, Demais S, Mestdagh C, Gautheret D, Namy O. Genome-wide Translational Changes Induced by the Prion [PSI+] Cell Rep. 2014;8:439–448. doi: 10.1016/j.celrep.2014.06.036. [DOI] [PubMed] [Google Scholar]
- 43.Halfmann R, Jarosz DF, Jones SK, Chang A, Lancaster AK, Lindquist S. Prions are a common mechanism for phenotypic inheritance in wild yeasts. Nature. 2012;482:363–368. doi: 10.1038/nature10875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Garcia DM, Dietrich D, Clardy J, Jarosz DF. A common bacterial metabolite elicits prion-based bypass of glucose repression. eLife. 2016;5:e17978. doi: 10.7554/eLife.17978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jarosz DF, Lancaster AK, Brown JCS, Lindquist S. An Evolutionarily Conserved Prion-like Element Converts Wild Fungi from Metabolic Specialists to Generalists. Cell. 2014;158:1072–1082. doi: 10.1016/j.cell.2014.07.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46○.Jarosz DF, Brown JCS, Walker GA, Datta MS, Ung WL, Lancaster AK, Rotem A, Chang A, Newby GA, Weitz DA, et al. Cross-Kingdom Chemical Communication Drives a Heritable, Mutually Beneficial Prion-Based Transformation of Metabolism. Cell. 2014;158:1083–1093. doi: 10.1016/j.cell.2014.07.025. The authors show that the [GAR+] prion element is found in diverse fungal species, and present an evolutionary theoretic model arguing that the reversibility of the prion-like state creates a metabolic bet-hedging mechanism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47○.Holmes DL, Lancaster AK, Lindquist S, Halfmann R. Heritable Remodeling of Yeast Multicellularity by an Environmentally Responsive Prion. Cell. 2013;153:153–165. doi: 10.1016/j.cell.2013.02.026. This study describes the [MOT3+] prion of S. cerevisiae, which controls multicellularity in response to environmental stimuli. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tapia H, Koshland DE. Trehalose Is a Versatile and Long-Lived Chaperone for Desiccation Tolerance. Curr Biol. 2014;24:2758–2766. doi: 10.1016/j.cub.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 49○.King OD, Masel J. The evolution of bet-hedging adaptations to rare scenarios. Theor Popul Biol. 2007;72:560–575. doi: 10.1016/j.tpb.2007.08.006. The theoretical framework presented in this analysis provides a basis for understanding the tradeoffs between reversible (e.g. prion-like) and irreversible (e.g. nucleic acid-based) modes of heredity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lancaster AK, Masel J. The Evolution of Reversible Switches in the Presence of Irreversible Mimics. Evolution. 2009;63:2350–2362. doi: 10.1111/j.1558-5646.2009.00729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.She R, Chakravarty AK, Layton CJ, Chircus LM, Andreasson JOL, Damaraju N, McMahon PL, Buenrostro JD, Jarosz DF, Greenleaf WJ. Comprehensive and quantitative mapping of RNA–protein interactions across a transcribed eukaryotic genome. Proc Natl Acad Sci. 2017 doi: 10.1073/pnas.1618370114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Byers JS, Garcia DM, Jarosz DF. Epigenetic regulation of genome integrity by a prion-based mechanism. bioRxiv. 2017 doi: 10.1101/152512. [DOI] [Google Scholar]
- 53.Doel SM, McCready SJ, Nierras CR, Cox BS. The dominant PNM2- mutation which eliminates the psi factor of Saccharomyces cerevisiae is the result of a missense mutation in the SUP35 gene. Genetics. 1994;137:659–670. doi: 10.1093/genetics/137.3.659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chernoff YO, Derkach IL, Inge-Vechtomov SG. Multicopy SUP35 gene induces de-novo appearance of psi-like factors in the yeast Saccharomyces cerevisiae. Curr Genet. 1993;24:268–270. doi: 10.1007/BF00351802. [DOI] [PubMed] [Google Scholar]
- 55.Chan CY, Zhao H, Pugh RJ, Pedley AM, French J, Jones SA, Zhuang X, Jinnah H, Huang TJ, Benkovic SJ. Purinosome formation as a function of the cell cycle. Proc Natl Acad Sci. 2015;112:1368–1373. doi: 10.1073/pnas.1423009112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.French JB, Jones SA, Deng H, Pedley AM, Kim D, Chan CY, Hu H, Pugh RJ, Zhao H, Zhang Y, et al. Spatial colocalization and functional link of purinosomes with mitochondria. Science. 2016;351:733–737. doi: 10.1126/science.aac6054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Banani SF, Lee HO, Hyman AA, Rosen MK. Biomolecular condensates: organizers of cellular biochemistry. Nat Rev Mol Cell Biol. 2017;18:285–298. doi: 10.1038/nrm.2017.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wu H, Fuxreiter M. The Structure and Dynamics of Higher-Order Assemblies: Amyloids, Signalosomes, and Granules. Cell. 2016;165:1055–1066. doi: 10.1016/j.cell.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.DePace AH, Weissman JS. Origins and kinetic consequences of diversity in Sup35 yeast prion fibers. Nat Struct Mol Biol. 2002;9:389–396. doi: 10.1038/nsb786. [DOI] [PubMed] [Google Scholar]
- 60.DePace AH, Santoso A, Hillner P, Weissman JS. A Critical Role for Amino-Terminal Glutamine/Asparagine Repeats in the Formation and Propagation of a Yeast Prion. Cell. 1998;93:1241–1252. doi: 10.1016/s0092-8674(00)81467-1. [DOI] [PubMed] [Google Scholar]
- 61○.Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature. 2004;428:323–328. doi: 10.1038/nature02392. In this study and the subsequent 2006 manuscript, Tanaka et al. elegantly demonstrate the connection between the physical properties of Sup35 amyloid fibrils and the phenotype exerted by the [PSI+] prion state. [DOI] [PubMed] [Google Scholar]
- 62○.Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature. 2006;442:585–589. doi: 10.1038/nature04922. See comment on Tanaka et al. 2004. [DOI] [PubMed] [Google Scholar]
- 63.Tessier PM, Lindquist S. Prion recognition elements govern nucleation, strain specificity and species barriers. Nature. 2007;447:556–561. doi: 10.1038/nature05848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li L, Lindquist S. Creating a Protein-Based Element of Inheritance. Science. 2000;287:661–664. doi: 10.1126/science.287.5453.661. [DOI] [PubMed] [Google Scholar]
- 65.Simon JR, Carroll NJ, Rubinstein M, Chilkoti A, López GP. Programming molecular self-assembly of intrinsically disordered proteins containing sequences of low complexity. Nat Chem. 2017 doi: 10.1038/nchem.2715. advance online publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66○.Frederick KK, Michaelis VK, Corzilius B, Ong T-C, Jacavone AC, Griffin RG, Lindquist S. Sensitivity-Enhanced NMR Reveals Alterations in Protein Structure by Cellular Milieus. Cell. 2015;163:620–628. doi: 10.1016/j.cell.2015.09.024. The authors use sophisticated labeling and detection methods to assess the conformation of Sup35 fibrils in the context of the cellular milieu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Risse E, Nicoll AJ, Taylor WA, Wright D, Badoni M, Yang X, Farrow MA, Collinge J. Identification of a compound which disrupts binding of amyloid-beta to the prion protein using a novel fluorescence-based assay. J Biol Chem. 2015 doi: 10.1074/jbc.M115.637124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gallardo R, Ramakers M, Smet FD, Claes F, Khodaparast L, Khodaparast L, Couceiro JR, Langenberg T, Siemons M, Nyström S, et al. De novo design of a biologically active amyloid. Science. 2016;354:aah4949. doi: 10.1126/science.aah4949. [DOI] [PubMed] [Google Scholar]
- 69.Kato M, Han TW, Xie S, Shi K, Du X, Wu LC, Mirzaei H, Goldsmith EJ, Longgood J, Pei J, et al. Cell-free formation of RNA granules: low complexity sequence domains form dynamic fibers within hydrogels. Cell. 2012;149:753–767. doi: 10.1016/j.cell.2012.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Han TW, Kato M, Xie S, Wu LC, Mirzaei H, Pei J, Chen M, Xie Y, Allen J, Xiao G, et al. Cell-free formation of RNA granules: bound RNAs identify features and components of cellular assemblies. Cell. 2012;149:768–779. doi: 10.1016/j.cell.2012.04.016. [DOI] [PubMed] [Google Scholar]
- 71.Soragni A, Janzen DM, Johnson LM, Lindgren AG, Thai-Quynh Nguyen A, Tiourin E, Soriaga AB, Lu J, Jiang L, Faull KF, et al. A Designed Inhibitor of p53 Aggregation Rescues p53 Tumor Suppression in Ovarian Carcinomas. Cancer Cell. 2016;29:90–103. doi: 10.1016/j.ccell.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Benilova I, Karran E, De Strooper B. The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat Neurosci. 2012;15:349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- 73.Sevigny J, Chiao P, Bussière T, Weinreb PH, Williams L, Maier M, Dunstan R, Salloway S, Chen T, Ling Y, et al. The antibody aducanumab reduces Aβ plaques in Alzheimer’s disease. Nature. 2016;537:50–56. doi: 10.1038/nature19323. [DOI] [PubMed] [Google Scholar]
- 74.Men D, Guo Y-C, Zhang Z-P, Wei H, Zhou Y-F, Cui Z-Q, Liang X-S, Li K, Leng Y, You X-Y, et al. Seeding-Induced Self-assembling Protein Nanowires Dramatically Increase the Sensitivity of Immunoassays. Nano Lett. 2009;9:2246–2250. doi: 10.1021/nl9003464. [DOI] [PubMed] [Google Scholar]
- 75.Men D, Zhou J, Li W, Leng Y, Chen X, Tao S, Zhang X-E. Fluorescent Protein Nanowire-Mediated Protein Microarrays for Multiplexed and Highly Sensitive Pathogen Detection. ACS Appl Mater Interfaces. 2016;8:17472–17477. doi: 10.1021/acsami.6b04786. [DOI] [PubMed] [Google Scholar]
- 76.Vázquez-Fernández E, Vos MR, Afanasyev P, Cebey L, Sevillano AM, Vidal E, Rosa I, Renault L, Ramos A, Peters PJ, et al. The Structural Architecture of an Infectious Mammalian Prion Using Electron Cryomicroscopy. PLOS Pathog. 2016;12:e1005835. doi: 10.1371/journal.ppat.1005835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Capra JA, Singh M. Predicting functionally important residues from sequence conservation. Bioinformatics. 2007;23:1875–1882. doi: 10.1093/bioinformatics/btm270. [DOI] [PubMed] [Google Scholar]
- 78.Chen JW, Romero P, Uversky VN, Dunker AK. Conservation of Intrinsic Disorder in Protein Domains and Families: I. A Database of Conserved Predicted Disordered Regions. J Proteome Res. 2006;5:879–887. doi: 10.1021/pr060048x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Toth-Petroczy A, Palmedo P, Ingraham J, Hopf TA, Berger B, Sander C, Marks DS. Structured States of Disordered Proteins from Genomic Sequences. Cell. 2016;167:158–170.e12. doi: 10.1016/j.cell.2016.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Varadi M, Guharoy M, Zsolyomi F, Tompa P. DisCons: a novel tool to quantify and classify evolutionary conservation of intrinsic protein disorder. BMC Bioinformatics. 2015;16:153. doi: 10.1186/s12859-015-0592-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Riek R, Eisenberg DS. The activities of amyloids from a structural perspective. Nature. 2016;539:227–235. doi: 10.1038/nature20416. [DOI] [PubMed] [Google Scholar]
- 82.Sharp PA. Split genes and RNA splicing. Cell. 1994;77:805–815. doi: 10.1016/0092-8674(94)90130-9. [DOI] [PubMed] [Google Scholar]
- 83.Woodson SA. Ironing out the kinks: splicing and translation in bacteria. Genes Dev. 1998;12:1243–1247. doi: 10.1101/gad.12.9.1243. [DOI] [PubMed] [Google Scholar]
- 84.Cooper GM. The Complexity of Eukaryotic Genomes. 2000 [Google Scholar]