

Abstract
Mesenchymal (MES) and proneural (PN) are two distinct glioma stem cell (GSC) populations that drive therapeutic resistance in glioblastoma (GBM). We screened a panel of 650 small molecules against patient-derived GBM cells to discover compounds targeting specific GBM subtypes. Arsenic trioxide (ATO), an FDA-approved drug that crosses the blood–brain barrier, was identified as a potent PN-specific compound in the initial screen and follow-up validation studies. Furthermore, MES and PN GSCs exhibited differential sensitivity to ATO. As ATO has been shown to activate the MAPK-interacting kinase 1 (MNK1)-eukaryotic translation initiation factor 4E (eIF4E) pathway and subsequent mRNA translation in a negative regulatory feedback manner, the mechanistic role of ATO resistance in MES GBM was explored. In GBM cells, ATO-activated translation initiation cellular events via the MNK1–eIF4E signaling axis. Furthermore, resistance to ATO in intracranial PDX tumors correlated with high eIF4E phosphorylation. Polysomal fractionation and microarray analysis of GBM cells were performed to identify ATO’s effect on mRNA translation and enrichment of anti-apoptotic mRNAs in the ATO-induced translatome was found. Additionally, it was determined that MNK inhibition sensitized MES GSCs to ATO in neurosphere and apoptosis assays. Finally, examination of the effect of ATO on patients from a phase I/II clinical trial of ATO revealed that PN GBM patients responded better to ATO than other subtypes as demonstrated by longer overall and progression-free survival.
Implications
These findings raise the possibility of a unique therapeutic approach for GBM, involving MNK1 targeting to sensitize MES GSCs to drugs like arsenic trioxide.
Introduction
Mesenchymal (MES) and proneural (PN) glioma stem cells (GSC) are the two most well-defined cancer stem cell (CSC) populations in glioblastoma (GBM), the deadliest primary malignant brain tumor (1, 2). MES and PN GSCs are tumor-initiating cells that can be found concurrently within the same tumors, and increased intratumoral heterogeneity promotes a more resistant phenotype (3, 4). Effective treatment of GBM will require the development of therapies that specifically target these distinct GSC populations.
Using a panel of ex vivo patient-derived xenograft (PDX) cell cultures, we screened a library of 650 compounds for anti-proliferative activity with the aim of identifying compounds with differential activity against GBM molecular subtypes. Arsenic trioxide (ATO) was identified as a potent inhibitor of non-MES GBM cells. We confirmed these findings in a follow-up screen of 120 compounds used at multiple doses, specifically using MES and PN GBM cells. PN GBM demonstrated increased sensitivity to ATO as well as a number of other cytotoxic agents, including temozolomide, the standard-of-care chemotherapy for GBM. MES and PN GSC neurosphere cultures also demonstrated this differential sensitivity to ATO.
ATO is an FDA-approved drug for the treatment of relapsed/refractory acute promyelocytic leukemia (APL) harboring the t(15;17) translocation and has been shown to cross the blood–brain barrier in APL patients with CNS disease (5). Furthermore, ATO has shown preclinical efficacy against GSCs through several mechanisms including activation of apoptosis and autophagy, degradation of the PML protein, and inhibition of the sonic hedgehog signaling pathway (6–8). Currently, ATO is under investigation in a phase I/II clinical trial in GBM (9, 10). Given this information, we sought to uncover the mechanisms that drive differential ATO responses in GBM.
Translation is the most energetically demanding process in the cell and is an emerging resistance mechanism in cancer (11, 12). The MAPK-interacting kinases (MNKs) regulate initiation of cap-dependent translation through phosphorylation of the mRNA-binding protein, eukaryotic translation initiation factor 4E (eIF4E; refs. 13, 14). After activation of upstream MAPK signaling through either p38 or ERK, MNK binds to the eukaryotic translation initiation factor 4G (eIF4G), facilitating phosphorylation of eIF4E and translation of eIF4E-sensitive mRNAs, many of which include potent oncogenes (15). Several stimuli can activate this signaling cascade including hypotonic stress, radiation, interferon signaling, and chemotherapy (16, 17). Activation of translation allows the cell to adapt to stressful stimuli and is a mechanism of resistance in cancer (16, 18). In GBM, MNK signaling and mRNA translation have been implicated in resistance to the alkylating agent, temozolomide, as well as radiation (19, 20). Furthermore, we previously demonstrated that MNK activation is particularly important for the maintenance of therapy-resistant MES GSCs (21).
Here, we explored the role of MNK signaling in the regulation of ATO responses in established GBM models and patient-derived MES and PN GSC lines. We found that ATO activates MNK–eIF4E in GBM cells and that, in an intracranial PDX model of GBM, MNK activation correlates with ATO resistance. Such resistance is likely mediated by MNK1, to which ATO directly binds, increasing kinase activity. Given that MNK directly regulates translational activation, we tested the effect of ATO on translation in a GBM cell line with an NF-1 mutation, a characteristic of MES GBM (22). Comparing the polysomal fraction of untreated and ATO-treated GBM cells followed by microarray and gene set enrichment analysis (GSEA), we identified an ATO-induced translatome that is enriched for anti-apoptotic mRNAs, suggesting a translationally mediated resistance mechanism to ATO in GBM. Through analysis of gene expression data from The Cancer Genome Atlas (TCGA), we explored the relationship between the MNK1 gene (MKNK1) and GSC markers in GBM. We found that MKNK1 expression correlates positively with MES GSC genes, negatively with PN GSC genes, and predicts poor survival in GBM suggesting that MNK1 is enriched in the MES phenotype and overexpression of MNK1 confers therapeutic resistance in GBM patients. Next, we analyzed data from GBM patients in a phase I/II clinical trial of Trisenox (ATO). MES GBM patients had higher eIF4E phosphorylation and were enriched for gene sets linked to the activation of translation and translation initiation. Finally, PN GBM patients had the longest overall and progression-free survival. Altogether, our findings suggest that MNK1–eIF4E signaling drives ATO resistance in GBM and suggest that MNK inhibition can sensitize MES GSCs to ATO.
Materials and Methods
Cell culture and reagents
Established GBM cell lines (U87, LN229, and LN18) were grown in DMEM (Thermo Fisher) with 10% FBS (Thermo Fisher) and gentamycin (1 mg/mL). The identity of established cell lines was authenticated by short-tandem repeat (STR) analysis (Genetica DNA Laboratories) in August 2016. PDX cell lines were grown in serum-free Neurobasal medium (Thermo Fisher) with N-2 Supplement at a 1X final concentration (Thermo Fisher), EGF (50 ng/mL), bFGF (25 ng/mL), and penicillin/streptomycin (10 U/mL). Patient-derived MES (83Mes) and PN (AC17PN) GSC lines have been previously described (2, 21). GSCs were grown in DMEM/F12 with EGF (20 ng/mL), bFGF (20 ng/mL), heparin (5 μg/mL), B27 (2%), and gentamycin (1 mg/mL) in nonadherent flasks. All cells were grown at 37 C in 5% CO2. Pharmaceutical-grade ATO (Trisenox) used for in vitro and in vivo experiments was purchased from Teva Pharmaceuticals. The MNK inhibitor, CGP57380, was purchased from Santa Cruz Biotechnology. The arsenic–biotin conjugate (As-Biotin) was purchased from Toronto Research Chemicals and has been previously described (23).
Viability assays
Cell viability for GSCs (83Mes and AC17PN) and the established GBM cell lines (U87, LN229, and LN18) was assessed using the Cell Proliferation Reagent WST-1 (Roche) according to the manufacturer’s instructions. Briefly, cells were seeded into 96-well plates at a density of 3,000 cells per well in the presence of the indicated drugs. Cells were incubated at 37 C in 5% CO2. After 5 days, the WST-1 reagent was added (10%, v/v) and cell viability was assessed using the Synergy HT plate reader and the Gen5 software (BioTek).
For ex vivo PDX GBM cells, 2,500 cells were plated per well in a 384-well tissue culture treated plate (Greiner) in 25 μL of supplemented Neurobasal medium. Twenty-five nanoliters of 1,000 drugs were added using a Labcyte Echo acoustic dispensing device and the cells were incubated for 96 hours. Cell viability was assessed using CellTiterGlo (Promega) on a Synergy 2 plate reader with Gen5 software (BioTek).
Immunoblotting
Cells were treated as indicated, washed with 1 × PBS, and harvested. Pellets were washed with 1 × PBS, snap-frozen, and lysed in phosphorylation lysis buffer (pH 7.9) containing 50 mmol/L HEPES, 150 mmol/L NaCl, 1 mmol/L MgCl2, 0.5% sodium deoxycholate, and supplemented with phosphatase and protease inhibitors (Roche). Protein concentration was quantified by Bradford assay (Bio-Rad) with the Synergy HT plate reader and Gen5 software (BioTek). Equal amounts of protein lysate were subjected to SDS-PAGE (Bio-Rad) followed by transfer to Immobilon PVDF membranes (Millipore). Membranes were blocked with 5% BSA in 1 × TBST for 1 hour. Membranes were incubated with the indicated primary antibodies overnight at 4 C, washed three times with 1 × TBST, incubated with the appropriate anti-rabbit (GE Healthcare) or anti-mouse (Bio-Rad) horseradish peroxidase (HRP)-conjugated secondary antibody for 1 hour, washed three times with 1 TBST, and analyzed using the WesternBright ECL substrate (Advansta) and autoradiography film (Denville Scientific) or the ChemiDoc MP Imaging System and Image Lab 5.0 software (Bio-Rad). Primary antibodies were purchased from Cell Signaling Technologies (phospho-eIF4E, eIF4A, MNK1, PARP), Santa Cruz Biotechnology (eIF4E, HSP90α/β), or Millipore (GAPDH). Where indicated, antibodies were removed using Restore PLUS Western Blot Stripping Buffer (Thermo Fisher) to allow for incubation with additional antibodies.
Intracranial PDX mouse study and IHC
Mouse studies were carried out in accordance with the Northwestern Institutional Animal Care and Use Committee (IACUC) and were performed at the Northwestern Developmental Therapeutics Core. Nude mice (CrTac:NCr-Foxn1nu) were purchased from Taconic Biosciences and used between the ages of 6 and 15 weeks. Tumors were established by intracranial injection of the patient-derived GBM cell line, 373811, expressing a luciferase reporter (25,000 cells/mouse). After tumor formation was con-firmed using an in vivo imaging system (IVIS), mice were randomized based on IVIS signal and weight. Mice were treated with vehicle control or Trisenox (ATO) three times per week at a dose of 5 mg/kg by intraperitoneal injection. Mice were euthanized after completion of the study (4 weeks of treatment) or 20% reduction in initial weight.
Brains were harvested from mice and fixed in 10% buffered formalin. Brains were embedded in paraffin and sectioned in preparation for IHC. For clinical trial samples, tumors were acquired in accordance with the Northwestern Institutional Review Board (IRB). Paraffin-embedded samples were sectioned prior to IHC. For xenograft and clinical trial samples, tumors were stained with the phospho-eIF4E (Ser209) antibody from Abcam (EP2151Y) as previously described using the BOND-MAX Automated IHC/ISH Stainer and Polymer Detection System (Leica Biosystems; ref. 21). Staining intensity was assessed by a board-certified neuropathologist (CH) who was blinded to sample identity.
Gene knockdown
MNK1, MNK2, and control siRNA were purchased from Dharmacon (GE Healthcare). GBM cells were transfected with control, MNK1, MNK2, or MNK1/MNK2 siRNAs using the Lipofectamine RNAiMAX Reagent and the Opti-MEM medium (Thermo Fisher). After 24 hours transfection, cells were treated with or without ATO for the indicated times. Knockdown was validated by Western blot analysis or qRT-PCR.
Pull-down assays
Arsenic binding was assessed using a previously described streptavidin pull-down assay (24, 25). Briefly, LN229 cells or recombinant MNK1 protein were treated with the arsenic-biotin (As-Biotin) conjugate for 2 hours. Following treatment, cells were lysed using phosphorylation lysis buffer. Protein concentration was quantified by Bradford assay (Bio-Rad) with the Synergy HT plate reader and Gen5 software (BioTek Instruments). Equal amounts of lysate were incubated with streptavidin beads (Invitrogen) overnight. Alternatively, untreated or treated recombinant protein was incubated with streptavidin beads overnight. Beads were washed 3 × with phosphorylation lysis buffer and samples were subjected to SDS-PAGE and immunoblotting.
Cap-binding pull-down assay was carried out using Immobilized γ-Aminophenyl-m7GTP (C10-spacer)-Agarose beads (Jena Bioscience). Briefly, LN229 cells were treated with or without ATO at the indicated doses for 6 hours. Following treatment, cells were lysed using NP-40 lysis buffer containing 40 mmol/L HEPES, 120 mmol/L NaCl, 1 mmol/L EDTA, 10 mmol/L sodium pyrophosphate, 50 mmol/L NaF, 0.1% NP-40, 10 mmol/β-glycerophosphate, and 10% glycerol supplemented with phosphatase and protease inhibitors (Roche). Protein concentration was quantified by Bradford assay (Bio-Rad) with the Synergy HT plate reader and Gen5 software (BioTek Instruments). Equal amounts of lysate were incubated with m7GTP beads overnight. Beads were washed 3 with NP-40 buffer and samples were subjected to SDS-PAGE and immunoblotting.
In vitro kinase assay
Recombinant, active MNK1 protein (SignalChem) was used for the in vitro kinase assay. The ADP-Glo Kinase Assay (Promega) was used according to the manufacturer’s instructions. Briefly, 100 ng MNK1 was incubated with increasing concentrations of ATO at 30 C for 30 minutes in the presence of the MNK1 peptide substrate, EIF2S (SignalChem). Kinase activity was assessed by luminescence using a Synergy HT plate reader and the Gen5 software (BioTek).
qRT-PCR
To confirm siRNA knockdown, cells were snap frozen, resuspended in lysis buffer, and homogenized using the QIAshredder (Qiagen). RNA was purified using the RNeasy Mini Kit (Qiagen) according to the manufacturer’s instructions. qRT-PCR for siRNA knockdown and analysis of polysomal RNA was performed using specific primers for MKNK1, MKNK2, FZD6, or GAPDH (Thermo Fisher).
Neurosphere assay and extreme limiting dilution analysis
MES GSCs were seeded at increasing cell concentrations in round-bottom 96-well plates (Greiner Bio-One) containing the indicated drugs. After one week incubation at 37 C in 5% CO2, spheres were stained with acridine orange at 0.1 μg/mL as previously described (26). Spheres were then imaged using a Cytation 3 Cell-Imaging Multi-Mode Reader and analyzed using the Gen5 software (BioTek). Spheres 150 μm in diameter were scored positively. Extreme limiting dilution analysis (ELDA) was carried out as previously described (27) using the ELDA online software.
Apoptosis assay
MES GSCs grown as neurospheres in non-adherent flasks were treated with the indicated drugs for 48 hours and harvested for apoptosis assay. Spheres were washed with 1 × PBS and dissociated into single cells prior to staining with propidium iodide (PI) and FITC Annexin V using the BD Pharmingen FITC Annexin V Apoptosis Detection Kit I (BD Biosciences). Within 1 hour of staining, cells were analyzed by flow cytometry and FlowJo 10 for Mac.
Accession numbers
RNASeq data were made publically available through the European Nucleotide Archive (ENA). The accession number is PRJEB20631. Microarray data accession number is in process.
Additional materials and methods are listed in the Supplementary Methods.
Results
High-throughput screen identifies ATO as a potent inhibitor of PN GBM growth
We sought to identify compounds that specifically disrupt the growth of different GBM molecular subtypes. Toward this end, we analyzed the gene expression profiles of short-term GBM cultures, and subtyped 39 patient-derived GBM cell lines into the four GBM molecular subtypes: MES, PN, CL, and N (ref. 22; Fig. 1A). We then tested the effect of a panel of 650 compounds on cell viability in five patient-derived GBM cell lines as well as one human fibroblast cell line and one established GBM cell line. In our initial screen, ATO was found to be the most potent compound in non-MES cells (Fig. 1B and C; Supplementary Table S1).
Figure 1.
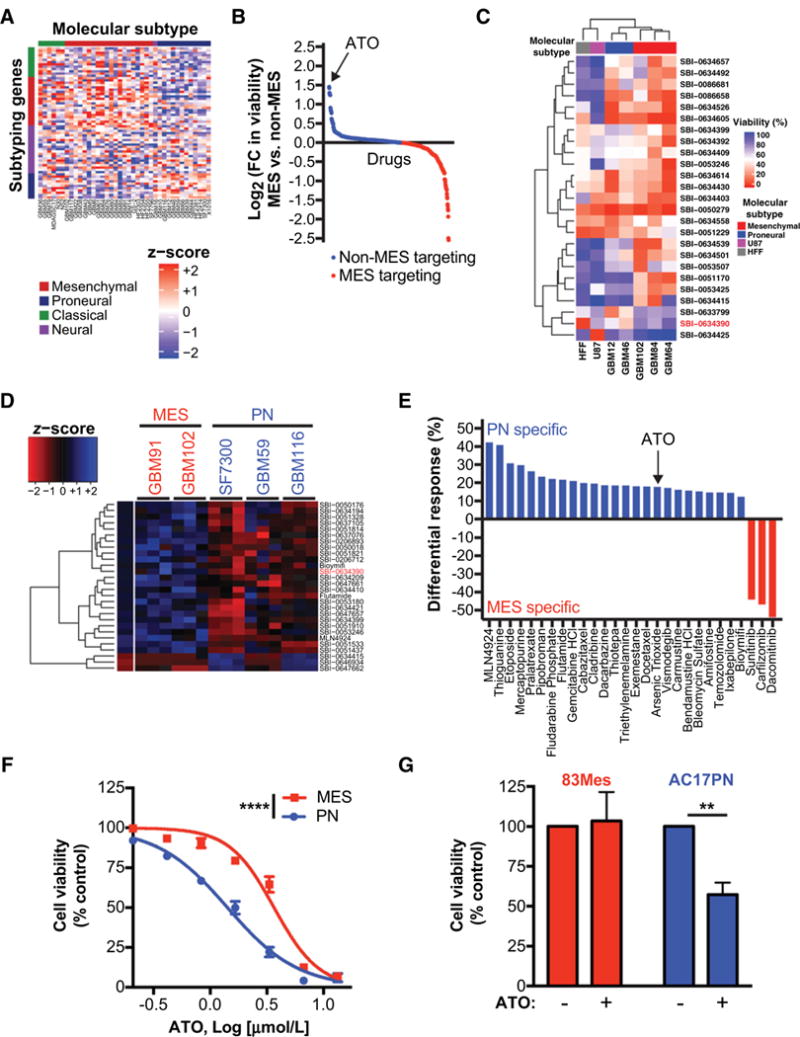
High-throughput screens identify differential ATO sensitivity in PN and MES GBM. A, RNA was extracted from orthotopically grown GBM PDX models. Transcriptomic profiling was performed using Agilent 44K array. Expression data was filtered for mouse contamination and molecular subtype analysis was performed using 66 subtype classification according to Verhaak et al. (22). B and C, Preliminary CellTiter-Glo viability screen was performed on short-term cultures of GBM PDX models, U87, and Human foreskin fibroblast (HFF) at 10 μmol/L. Only compounds with a statistically significant differential effect on cell viability are shown in the graph and heatmap. In this preliminary screen against 650 compounds, ATO (SBI-0634390; red) was identified as a small molecule with specificity towards non-MES GBM cells. D and E, ATO specificity was further validated by a secondary CellTiter-Glo viability screen, where selected 120 compounds were screened against short-term cultures of GBM PDX models at three different concentrations (0.1, 1, and 10 μmol/L). Only compounds with a statistically significant differential effect on cell viability are shown in the graph and heatmap. Averages for triplicates for each concentration are presented in the heatmap (D). Differential responses to compounds (E) show PN and MES specific compounds. F, Subtype-specific effects of ATO were further validated by performing dose-response experiments against selected MES and PN GBM ex vivo PDX models. Non-linear dose-response curve fit, F test,*****, P ≤ 0.0001. G, 83Mes and AC17PN GSCs were treated with ATO (1 μmol/L) for 5 days and cell viability was analyzed by WST-1 assay. Data represent means ± SEM of three independent experiments. Unpaired, two-tailed t test,**, P ≤ 0.01.
To expand upon these findings, we tested a subset of these compounds in two MES and three PN GBM cell lines at three different concentrations and once again found that PN GBM cells are more sensitive to ATO than MES GBM cells (Fig. 1D and E; and Supplementary Table S2). The normalized cell viabilities after treatment with each compound were compared between MES and PN GBM cells and only 28 of the 120 tested compounds, including ATO, were found to be statistically significant at an FDR adjusted P-value < 0.05. Other cytotoxic agents were also found to be more effective in PN GBM, including temozolomide, the standard-of-care chemotherapy used for the treatment of GBM (Supplementary Fig. S1). Next, we tested ATO in a dose-response experiment and found similar results, validating our high-throughput screen (Fig. 1F). Finally, we tested the effect of ATO on patient-derived MES and PN GSC lines. We found that MES GSCs are resistant to and PN GSCs are sensitive to low-dose ATO (Fig. 1G).
As ATO treatment has been shown to activate MNK and several upstream kinases (MAPKs, MAPKKs, and MAPKKKs) leading to therapeutic resistance (28–34), we sought to determine whether this mechanism regulated ATO responses in GBM. Analysis of gene expression data from previously published patient-derived MES and PN GSC lines (2) revealed that MES GSCs overexpress genes associated with translation initiation, translation, and eIF4E upregulation (Supplementary Fig. S2). However, PN GSCs over-express hedgehog signaling genes, which are established targets of ATO in GSCs (6). Taken together, these findings suggest that MES GBM and GSCs, which overexpress the MNK1 kinase (21) and are enriched for genes that control translation, are more resistant to ATO and other cytotoxic agents than PN GBM.
MNK–eIF4E signaling regulates ATO responses in GBM
To uncover the role of MNK signaling in the regulation of ATO responses, we treated two established GBM cell lines with increasing doses of ATO and observed a dose-dependent increase in phosphorylation of the downstream MNK substrate, eIF4E (Fig. 2A). Consistent with these findings, pretreatment of GBM cells with the MNK inhibitor CGP57380 prevented ATO-induced eIF4E phosphorylation (Fig. 2B). Furthermore, MNK inhibition sensitized GBM cells to the antiproliferative effects of ATO (Fig. 2C). As experiments in cell culture do not fully recapitulate the complexity of GBM tumor biology, we used an intracranial xenograft model using a patient-derived GBM cell line orthotopically transplanted into nude mice. Mice were treated with a vehicle or a clinically-relevant dose of ATO and no decrease in body weight was observed, suggesting minimal toxicity in this setting (Fig. 2D). ATO did prolong survival in tumor-bearing mice, however, this difference failed to meet statistical significance, suggesting a resistance mechanism in these tumors (Fig. 2E). By IHC staining, the tumors in the ATO-treated animals could be separated into two equally-sized groups based on degree of eIF4E phosphorylation. Interestingly, plotting survival based on level of eIF4E phosphorylation showed a trend toward longer overall survival in the low eIF4E group (Fig. 2F). Finally, represented a different way, comparing mice in the ATO-treated cohort based on survival response showed lower eIF4E phosphorylation in the longer survivors (Fig. 2G and H). These findings suggest that MNK–eIF4E signaling may be an important feedback mechanism that negatively controls ATO responses in GBM.
Figure 2.
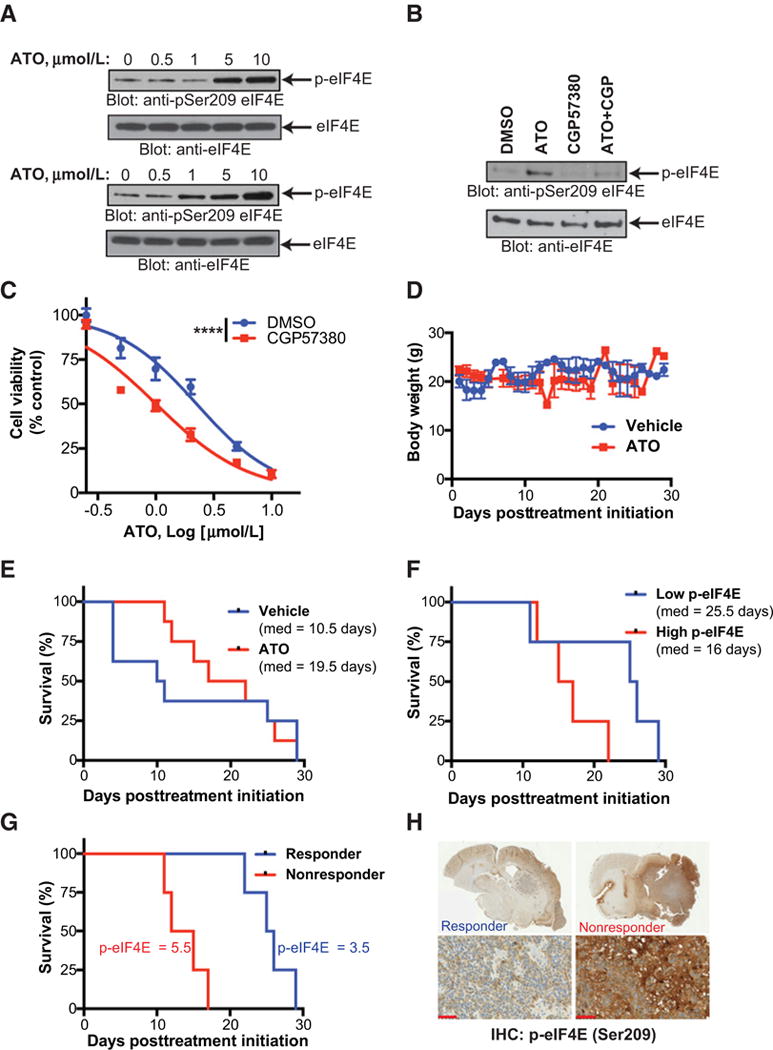
ATO activates MNK signaling in vitro and MNK activity is associated with poor response to ATO in vivo. A, U87 (top) and LN229 (bottom) cells were treated with increasing concentrations of ATO for 90 minutes. Whole cell lysates were subjected to immunoblotting with antibodies against phospho-eIF4E (Ser209) or eIF4E. B, Immunoblotting analysis as in (A). U87 cells were pretreated with CGP57380 (10 μmol/L) or DMSO for 1 hour followed by treatment with ATO (5 μmol/L) for 90 minutes. C, U87 cells were treated with increasing concentrations of ATO and CGP57380 (10 μmol/L) for 5 days and cell viability was analyzed by WST-1 assay. Data represent means ± SEM of three independent experiments. Nonlinear dose–response curve fit, F test, ****, P ≤ 0.0001. D and E, Nude mice were inoculated with a patient-derived xenograft via intracranial injection. After tumor formation, mice were randomized and treated with either vehicle control or ATO (5 mg/kg) three times per week. Body weight in vehicle control and ATO-treated mice is shown. Log-rank Mantel–Cox, P = 0.76. F, Brains were sectioned and stained with phospho-eIF4E (Ser209) antibody. Following staining, tumors were ranked blindly. Survival of mice with high (above the median) or low (below the median) phospho-eIF4E staining was compared. Log-rank Mantel–Cox, P = 0.10. G, Mice in the ATO treatment group with long survival (responders) or short survival (nonresponders) are shown. Log-rank Mantel–Cox, P = 0.007. H, Representative phospho-eIF4E staining from ATO responder and nonresponders is shown, scale bar = 50 μm.
MNK1 is required for ATO-induced eIF4E phosphorylation
Building upon our preliminary findings, we sought to identify the mechanism by which ATO activates MNK signaling. First, we used siRNA to specifically knockdown the MNK1 or MNK2 genes, alone or in combination, in two GBM cell lines. After knockdown, we treated cells with or without ATO to identify which gene is responsible for ATO-induced MNK activation. We found that MNK1, but not MNK2, is required for activation of eIF4E phosphorylation in the setting of ATO treatment in GBM cells (Fig. 3A–D). We also found that ATO activates MNK activity in LN229 cells (which express MNK1 and MNK2) but not in LN18 cells (MNK1 below detectable levels; Fig. 3E). These results support a conclusion that GBM cells depend upon MNK1 for activation of eIF4E phosphorylation after ATO treatment.
Figure 3.
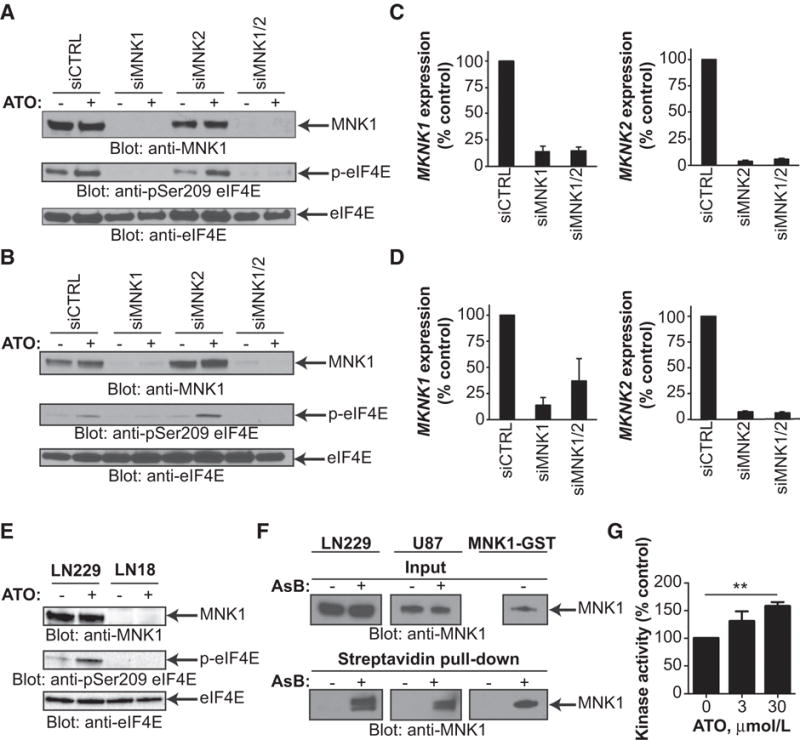
MNK1 is required for ATO-induced eIF4E phosphorylation in GBM and arsenic binds to MNK1 and activates kinase activity. A and B, U87 (A) and LN229 (B) cells were transfected with control siRNA or siRNA targeting MNK1, MNK2, or combination. Cells were treated with or without ATO (2 μmol/L) for 90 minutes. Whole cell lysates were subjected to immunoblotting with antibodies against MNK1, phospho-eIF4E (Ser209), or eIF4E. C and D, Cells from A and B were collected and RNA was extracted. Expression of MKNK1 and MKNK2 genes was determined by qRT-PCR normalizing to GAPDH. Data represent means ± SEM of two independent experiments. E, LN229 (MNK1 expressing) or LN18 (MNK1 undetectable) cells were treated with or without ATO (5 μmol/L) for 90 minutes. Whole cell lysates were subjected to SDS-PAGE followed by transfer to PVDF membranes. Membranes were incubated with antibodies against phospho-eIF4E (Ser209), eIF4E, or MNK1. F, LN229, U87, or recombinant MNK1-GST protein were treated with DMSO or the biotinylated arsenic compound, As-Biotin (AsB), for 2 hours. Whole cell lysates or recombinant protein were incubated with streptavidin beads overnight. Streptavidin pull-down and input control were subjected to immunoblotting with an antibody against MNK1. G, Recombinant MNK1-GST protein was treated with increasing concentrations of ATO for 30 minutes in the presence of the MNK1 peptide substrate, EIF2S. Kinase activity was assessed using the ADP-Glo Kinase Assay. Data represent means ± SEM of three independent experiments. Unpaired, two-tailed t test, **, P ≤ 0.01.
As arsenic compounds can directly bind to proteins or enzymes, altering their activity (24, 35), we set out to determine whether arsenic could bind MNK1. Arsenic compounds form covalent bonds with cysteine residues, and in some cases, histidine residues, leading to conformational changes that alter kinase activity or promote degradation (25, 36). Furthermore, MNK1 contains multiple cysteine residues, including four in close proximity, which may facilitate binding for arsenic. We used a previously described trivalent As-Biotin conjugate to determine whether arsenic binds MNK1 (23). We treated GBM cells or recombinant MNK1 protein with or without As-Biotin, and used streptavidin beads to pull-down biotinylated conjugates. In both cases, substantial amounts of MNK1 were found only in the As-Biotin–treated samples suggesting that the arsenic compound bound to MNK1 (Fig. 3F). Next, we used an in vitro kinase assay to determine how ATO affected MNK1 kinase activity. We found that ATO increased MNK1 kinase activity, suggesting a role for direct binding in the activation of the kinase following ATO treatment (Fig. 3G). In summary, these results show that ATO treatment in GBM leads to eIF4E phosphorylation through a MNK1-dependent mechanism that may be partially affected by direct binding of arsenic to MNK1 and direct activation of kinase activity.
ATO activates translation of anti-apoptotic mRNAs in NF1-mutant GBM
MNK1–eIF4E signaling directly regulates translation initiation leading to the synthesis of oncogenic proteins (37). Given the important role of MNK1 in ATO-mediated eIF4E phosphorylation, we sought to explore the effect of ATO treatment on mRNA translation. To explore a possible role for mRNA translation as an ATO resistance mechanism in MES GBM, we used the NF1-mutant GBM cell line, LN229, for translation analysis as mutation of the NF1 tumor suppressor drives MAPK signaling and is characteristic of the MES phenotype in GBM (22, 38).
To uncover the effect of ATO on cap-dependent translation, we employed a 5′ cap-binding assay using m7GTP-coupled beads. Treatment of LN229 cells with ATO resulted in a subsequent increase in loading of eIF4E and eIF4A, both members of the eIF4G cap-binding complex, to the 5′ cap (Fig. 4A). As cap formation is essential for translation initiation, we next sought to identify the ATO-induced translatome using polysomal fractionation. Cells were treated with or without ATO and monosomal and polysomal mRNA were separated via hypotonic lysis and polysomal fractionation (Fig. 4B). We found that ATO treatment did not significantly affect global translation profile under these conditions, as indicated by similar monosomal and polysomal profiles. To further elucidate the effects of ATO treatment on translation, we used microarray analysis of total (i.e., unfractionated) and polysomal mRNA of ATO treated and untreated GBM cells (Fig. 4C). We found that a number of coding genes were significantly increased in the polysomal as compared to total fractions (Fig. 4D). In the ATO-treated polysomes, 96 transcripts were significantly upregulated, with 51 of these transcripts found in the ATO-treated polysomes but not in the untreated polysomes (Supplementary Table S3). Of these transcripts, Frizzled Class Receptor 6 (FZD6), a master regulator of the MES phenotype in GSCs (39), was enriched in ATO treated polysomes. The upregulation of FZD6 was confirmed by qRT-PCR (Fig. 4E). Importantly, no change in FZD6 was observed in the total mRNA fractions.
Figure 4.
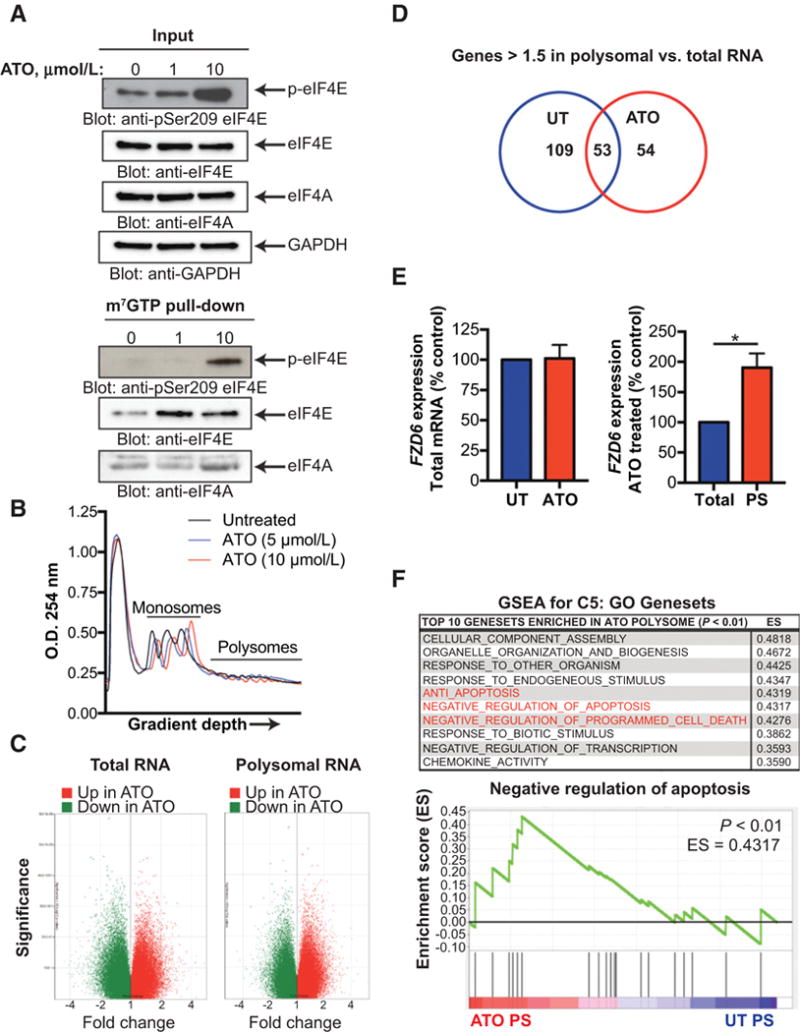
Polysomal profiling reveals ATO-induced translatome in GBM. A, LN229 cells were treated with increasing concentrations of ATO for 6 hours. Whole cell extracts were incubated with m7GTP-Agarose beads overnight. m7GTP-Agarasoe pull-down and input control were subjected to SDS-PAGE followed by immunoblotting with antibodies against phospho-eIF4E (Ser209), eIF4E, eIF4A, or GAPDH. B, LN229 cells were treated with increasing concentrations of ATO for 6 hours. Following treatment, cells were lysed with hypotonic lysis buffer, separated by a sucrose gradient (10–50%), and the O.D. 254 nm was analyzed. Graph represents O.D. as a function of gradient depth. C and D, LN229 cells were treated as in B. Total and polysomal mRNA were isolated and transcript expression was analyzed using the Clariom D microarray. Coding genes up- or downregulated after ATO treatment in polysomal and total RNA are shown. Genes >1.5 times increased in polysomal RNA as compared to total RNA for untreated and ATO treated cells are shown in the Venn diagram. E, Total and polysomal mRNA (as in B) was isolated and pooled and relative FZD6 mRNA expression was determined by qRT-PCR using GAPDH for normalization. Data represent means ± SEM of three independent experiments. Unpaired, two-tailed t test,*, P ≤ 0.05. F, Gene expression of ATO polysomes and total polysomes was analyzed by GSEA. The top 10 C5: GO gene sets enriched in ATO polysomes as compared to untreated polysomes are listed. Gene sets involved in apoptosis or cell death are highlighted in red. Enrichment plot of the negative regulation of apoptosis (GO:0043066) gene set in untreated and ATO polysomes is shown. Nominal P < 0.01.
We next sought to define the genes enriched in the ATO polysomes as compared with the untreated polysomes. Using GSEA, we found numerous gene sets significantly enriched in the ATO polysomes. Among the top ten Gene Ontology (GO) gene sets, three were involved in the inhibition of apoptosis (Fig. 4F). These findings suggest that ATO treatment activates translation of anti-apoptotic genes, preventing cell death and promoting resistance in MES GBM.
MNK1 expression correlates with MES GSC genes and predicts poor survival in GBM
GBMs are heterogeneous tumors containing a subpopulation of CSCs that mediate therapy resistance and tumor recurrence. Given that MNK1 is crucial for ATO-resistance in MES GBM, we sought to investigate whether this resistance mechanism is also conserved in MES GSCs. We analyzed GBM TCGA data and found that MKNK1 expression positively correlated with MES GSC genes, ALDH1A3 and CD44, and negatively correlated with PN GSC genes, OLIG2 and SOX2 (Fig. 5A). Similarly, ALDH1A3, negatively correlated with OLIG2 and SOX2 in this dataset (Fig. 5B). Furthermore, analysis of patient samples from different cellular compartments show that MKNK1 is differentially expressed throughout GBM tumors (Fig. 5C). The PN GSC gene, SOX2, exhibits a different pattern of expression that reflects the inverse correlation of MKNK1 and PN GSC genes (Fig. 5C). We next analyzed the effect of MKNK1 expression in GBM and other gliomas on survival. We found the higher MKNK1 expression in GBM when compared with low-grade gliomas (Fig. 5D). In low-grade gliomas, high MKNK1 expression predicts poor prognosis (Fig. 5E). Similarly, we found that high MKNK1 expression predicts poor prognosis in GBM patients (Fig. 5F). As the glioma CpG island methylator phenotype (G-CIMP) and MGMT-pro moter methylation status are among the most significant positive prognostic factors in GBM (40), we analyzed the effect of MKNK1 expression on survival in patients without these alterations. Importantly, MKNK1 expression predicted poor survival in non-G-CIMP/MGMT-promoter unmethylated GBM patients (Fig. 5G). These findings suggest that GBM patients with enrichment of MES GSC genes and MNK1 may have poorer prognosis and led us to investigate the role of MNK1 in MES GSCs.
Figure 5.
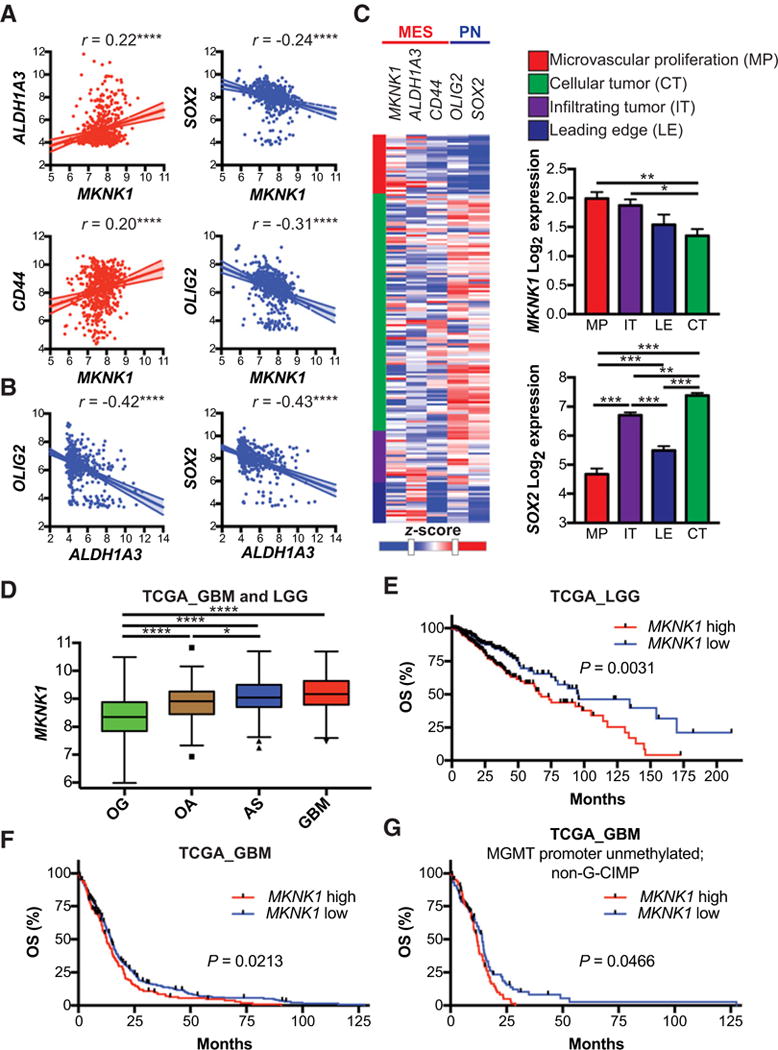
Expression of MES GSC markers correlate with MKNK1 and MKNK1 expression predicts poor survival in GBM patients. A and B, Multiple correlation analysis of MES and PN GSC markers in TCGA patients. Pearson’s correlation analysis,****, P ≤ 0.0001. C, Heatmap showing expression of MKNK1, MES GSC markers (ALDH1A3, CD44) and PN GSC markers (OLIG2, SOX2) in different tumor regions: microvascular proliferation (MP), infiltrating tumor (IT), leading edge (LE), and cellular tumor (CT). Bar graphs depict summarized data from heatmap. Comparisons between MKNK1 and SOX2 means in different tumor regions is shown with Tukey’s honest significant difference test,*, P ≤ 0.05; **, P ≤ 0.01;***, P ≤ 0.001. D, Expression of MKNK1 in GBM and low-grade gliomas: oligodendroglioma (OG), oligoastrocytoma (OA), and astrocytoma (AS). One-way ANOVA,*, P ≤ 0.05;****, ≤ 0.0001. E and F, Overall survival (OS) analysis of low-grade glioma (E), all GBM (F), and MGMT promoter unmethylated; non-G-CIMP GBM (G) patients with high and low expression of MKNK1 (MNK1). Log-rank (Mantel-Cox) test P-values are shown. Expression data downloaded from GlioVis (http://gliovis.bioinfo.cnio.es; ref. 57). Heatmap generated using the IVY Glioblastoma Project. ©2015 Allen Institute for Brain Science. Ivy Glioblastoma Atlas Project. Available from: glioblastoma.alleninstitute.org.
MNK inhibition sensitizes MES GSCs to ATO
We next examined the effect of ATO on patient-derived MES GSCs. GSCs are known to be particularly resistant to conventional therapies (2). We found that ATO treatment induces eIF4E phosphorylation in a MES GSC line (Fig. 6A). As with established GBM cells (Fig. 2), MNK inhibition blocked ATO-induced eIF4E phosphorylation in MES GSCs (Fig. 6B). Building upon our previous finding that ATO treatment activates translation of anti-apoptotic mRNAs in GBM (Fig. 4), we next tested whether MNK inhibition could sensitize MES GSCs to ATO and induce apoptosis. We found that concomitant treatment with ATO and a MNK inhibitor led to an increase in apoptosis (Fig. 6C–E). Furthermore, treatment of MES GSCs with ATO and a MNK inhibitor led to a significant decrease in neurosphere formation indicating depletion of the CSC populations (Fig. 6F). These findings confirm that ATO stimulates MNK activity in MES GSCs and suggest that combination treatment with a MNK inhibitor and ATO may be an effective strategy to deplete this CSC population.
Figure 6.
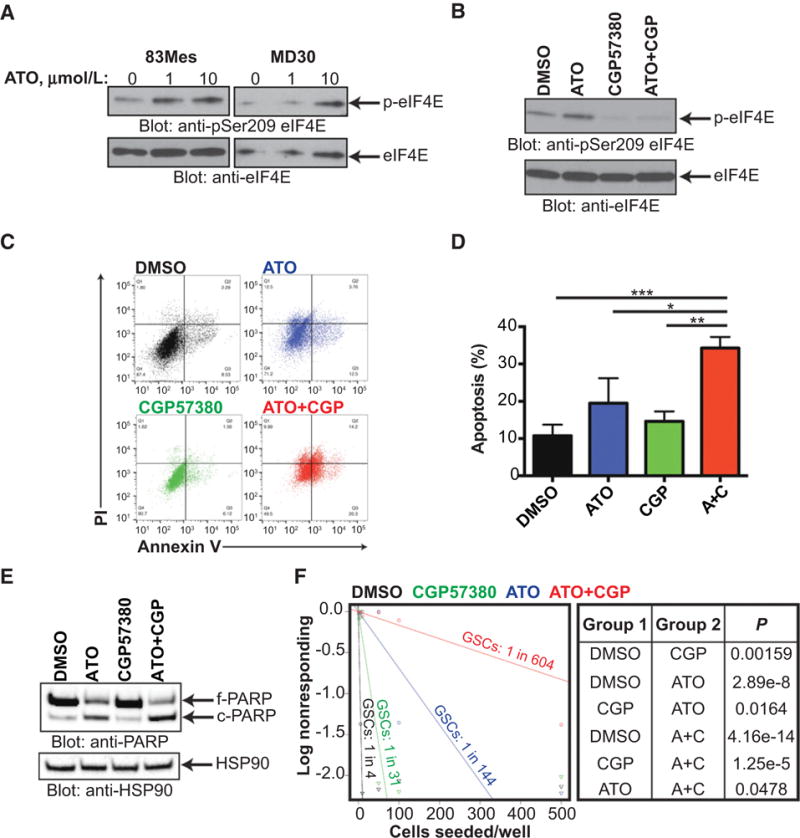
MNK inhibition sensitizes MES GSCs to ATO in neurosphere formation and apoptosis assays. A, 83Mes cells were treated with increasing concentrations of ATO for 90 minutes and whole cell lysates were subjected to immunoblotting with antibodies against phospho-eIF4E (Ser209) or eIF4E. B, 83Mes cells were pre-treated with CGP57380 (10 μmol/L) or DMSO for 1 hour followed by treatment with ATO (5 μmol/L) for 90 minutes and subjected to immunoblotting with antibodies against phospho-eIF4E (Ser209) or eIF4E. C, 83Mes cells were treated with DMSO, ATO (5 μmol/L), CGP57380 (10 μmol/L), or combination for 2 days. After treatment, apoptosis was assessed by co-staining cells with propidium iodide (PI) and Annexin V-FITC followed by flow cytometry analysis. Representative dot plots are shown. D, Annexin V positive cells from (C) were quantified to determine total apoptosis. Data represent means ± SEM of four independent experiments. Paired two tailed t test,*, P ≤ 0.05;**, P ≤ 0.01;***, P ≤ 0.001. E, 83Mes cells were treated as in (D) and whole cell lysates were subjected to immunoblotting with antibodies against PARP or HSP90. F, 83Mes cells were seeded into 96-well round-bottom plates at densities ranging from 1 to 500 cells per well in the presence of DMSO, ATO (5 μmol/L), CGP57380 (10 μmol/L), or combination. After 1 week incubation, neurospheres were stained with acridine orange and neurosphere formation was assessed and extreme limiting dilution analysis (ELDA) was performed (http://bioinf.wehi.edu.au/software/elda/; ref. 27). P-values from chi-square analysis are shown.
PN subtype is associated with improved ATO responses in GBM patients
After establishing a role for MNK signaling in ATO responses in vitro and in vivo, we sought to identify whether these results apply to a patient cohort treated with ATO. Clinical outcomes from this study have been previously reported (9, 10). Here, we specifically correlate outcomes with regard to molecular subtype. We analyzed the molecular subtype of 22 GBM patients treated with Trisenox (ATO; Fig. 7A). Interestingly, we found that enrichment of the MES gene set positively correlated with enrichment of the translation gene sets. Conversely, enrichment of the PN gene set negatively correlated with enrichment of translation gene sets (Fig. 7B and C). These results are consistent with our findings showing enrichment of translation gene sets in MES GSCs (Supplementary Fig. S2). As has been demonstrated by others (3, 4), expression of MES and PN genes exists on a spectrum with high enrichment of the MES genes predicting low expression of the PN genes. All patients were then subtyped according to MES or PN gene signatures (41) and enrichment of translation (TLN I) and translation initiation (TLN II) gene sets was compared (Fig. 7D). In both cases, tumors with high MES gene signatures were enriched for translation gene sets. Confirming our previous results (Fig. 5; Supplementary Fig. S2), these findings indicate that our MES subtype GBM clinical trial samples also exhibit higher MNK activity and translational activation.
Figure 7.
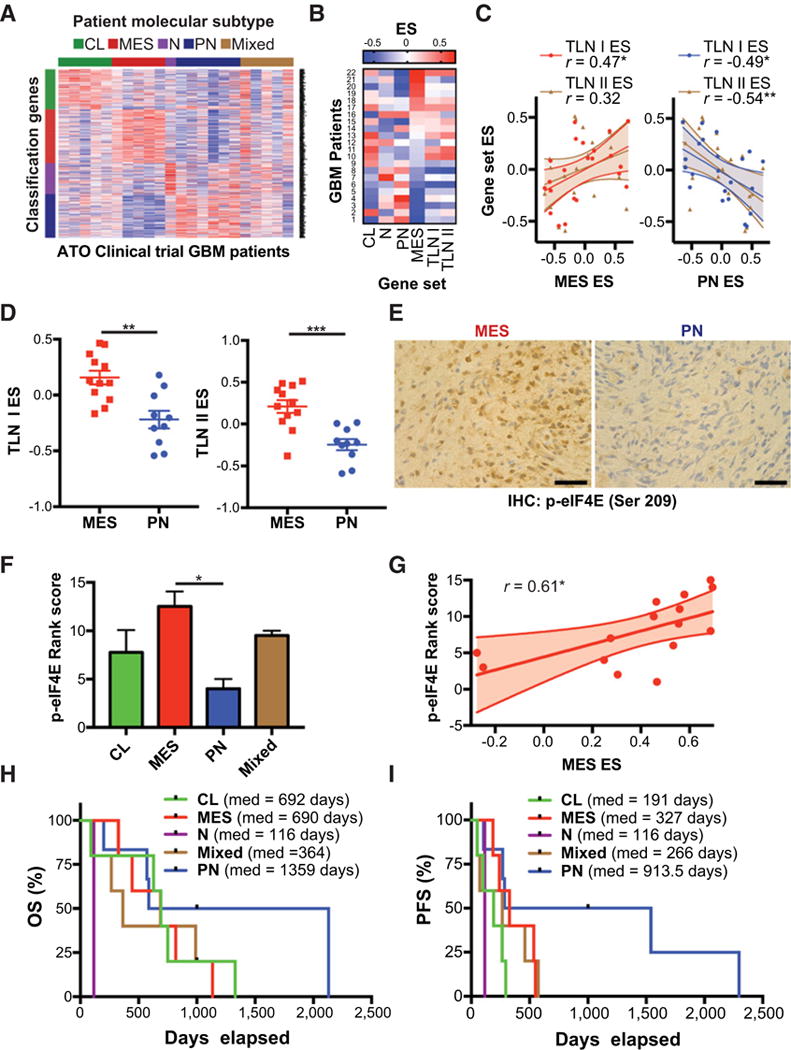
PN GBM phenotype associated with improved outcomes in ATO clinical trial. A, Tumor samples from 22 patients enrolled in a phase I/II trial of Trisenox (ATO) were analyzed by RNA-sequencing to determine molecular subtyping according to Verhaak et al. [22]. B and C, Gene set variation analysis (GSVA) of 22 patient samples. Heatmap shows GSVA enrichment scores for patients ranked according to MES ES. Pearson’s correlation analysis shows relationship between MES enrichment score (MES ES) or PN enrichment score (PN ES) and translation enrichment scores (TLN I ES, TLN II ES),*, P ≤ 0.05. D, Comparison of translation gene set enrichment scores in patients subtyped according to Carro et al. (41) (MES vs. PN). Unpaired, two-tailed t test **, P ≤ 0.01;***, P ≤ 0.001. E and F, Tumor samples were analyzed by phospho-eIF4E IHC and ranked per staining intensity. Representative phospho-eIF4E staining from MES and PN GBM patients are shown, scale bar = 50 μm. Mean staining rank score are shown for each subtype with available tumor with higher rank score corresponding with higher phospho-eIF4E intensity. One-way ANOVA,*, P ≤ 0.05. G, Correlation analysis shows relationship between MES ES and phospho-eIF4E staining rank score, *, P ≤ 0.05. H and I, Overall survival (OS) and PFS analyses for different molecular subtypes are shown.
We also found that MES subtype GBM from this cohort had significantly higher eIF4E phosphorylation than the PN subtype, suggesting that MNK is highly activated in the MES subtype (Fig. 7E and F). Furthermore, enrichment of the MES gene set positively correlated with eIF4E phosphorylation (Fig. 7G). Finally, we found that PN subtype patients responded better to ATO than other subtypes, with a median OS of 1,359 days and PFS of 913.5 days. These results implicate translation, MNK–eIF4E signaling, and the PN molecular subtype in ATO responses in GBM patients.
Discussion
GBM remains an incurable disease due, in part, to the development of therapeutic resistance driven by a subset of tumor-initiating cells, known as GSCs. When compared with PN GSCs, MES GSCs are particularly resistant to a number of different cytotoxic agents and radiation. Several mechanisms have been identified as drivers of this resistance in MES GSCs including reliance on different metabolic pathways, FOX transcription factor programs, regulatory microRNAs, and altered epigenetic states (2, 3, 39, 42). Furthermore, single-cell sequencing studies have uncovered a phenomenon in which individual GBM tumors contain clones of different molecular subtypes (i.e., intratumoral heterogeneity; ref. 4). These findings underscore the importance of appropriately characterizing GBM molecular subtypes and designing therapeutic approaches targeted to specific patient cohorts.
Here, using a high-throughput screen of 650 compounds, we identified the FDA-approved compound, ATO, as a potent inhibitor of PN GBM and GSCs, whereas MES GSCs appeared less sensitive to ATO. This can be attributed to the finding that ATO directly augments the kinase activity of MNK1 resulting in phosphorylation of eIF4E, in vitro. In line with this, we demonstrate that the MNK pathway is activated upon ATO treatment of MES GBM cells and tumor-bearing mice and this MNK activation may represent a resistance mechanism in MES GBM. We show that MNK1 is required for phosphorylation of the downstream MNK substrate and oncogene, eIF4E. Furthermore, using polysomal fractionation and gene expression profiling, we identify an ATO-induced translatome that is enriched for anti-apoptotic transcripts, suggesting a novel, translationally-driven resistance mechanism in MES GBM. Concomitantly, we show that patient-derived PN cell lines are more sensitive to ATO (and a number of other cytotoxic compounds) than MES cell lines. In line with this, we show that MES GSCs, which express elevated MNK1 and other translation initiation genes, are relatively insensitive to ATO and can be sensitized to ATO by MNK inhibition. This can be explained by the finding that ATO directly increases the kinase activity of MNK1, resulting in phosphorylation of eIF4E, in vitro. Finally, consistent with our in vitro findings, in a phase I/II clinical trial of Trisenox (ATO), PN GBM respond better to ATO as demonstrated by increased overall and progression-free survival of patients with that phenotype.
Importantly, in our study we find that ATO-induced MNK– eIF4E signaling is associated with poor survival in mice with an intracranial GBM PDX. The emergence of one group of tumor-bearing mice associated with high eIF4E phosphorylation and poor survival suggests the development of adaptive resistance to ATO. A similar phenomenon has been observed in GBM under different contexts in previous studies. For example, using single-cell phosphoproteomics to characterize adaptive resistance mechanisms in a GBM PDX model treated with an mTOR inhibitor, it was found that a subset of tumors rapidly develop therapeutic resistance via activation of several phosphorylation targets (43). Furthermore, PDX models contain heterogeneous cell populations including GSCs and differentiated cancer cells. It is conceivable that the emergence of ATO-resistant clones with high MNK activity accounts for the differential ATO responses in our system.
MNK activation and subsequent phosphorylation of eIF4E are negative prognostic factors for several cancers including malignant gliomas (44–47). In addition to regulating cap-dependent mRNA translation, eIF4E also promotes nuclear export of specific eIF4E-sensitive oncogenic mRNAs through a process that is activated by MNK-mediated eIF4E phosphorylation (48, 49). Furthermore, in addition to eIF4E, other downstream effectors may also be important in the context of ATO treatment in GBM. MNK1 and MNK2 integrate upstream signaling from p38 and ERK leading to phosphorylation of several targets (50). For example, MNK1 phosphorylates Sprouty 2 (SPRY2) at two sites (S112 and S121) stabilizing the protein and leading to its activation (51). In GBM, SPRY2 drives resistance to receptor tyrosine kinase (RTK) inhibitors (52). In addition to SPRY2, MNKs also activate the mRNA binding protein Heterogeneous Nuclear Ribonucleoprotein A1 (hnRNPA1) via activation of the p38-MNK signaling pathway in stress granules (53). The activation of hnRNPA1 promotes activation of MYC by facilitating alterative splicing of MYC-Associated Factor X (MAX; ref. 54). Conceivably, stimulation of MNK activity by ATO treatment may lead to activation of these or other downstream effectors, in addition to eIF4E. Future studies defining the role of these substrates may identify additional resistance mechanisms to ATO and other cytotoxic agents.
Arsenic species bind to proteins through coordination of cysteine and/or histidine residues in close proximity. ATO has been shown to bind to zinc fingers with at least three cysteines (i.e., C3H1 or C4 zinc fingers; ref. 36). ATO can directly bind to cysteine residues in protein kinases like hexokinase 2 (HK2) and AMP-activated protein kinase (AMPK), which results in inhibition of kinase activity (24, 35). Among the most interesting findings of our study, was the evidence for direct activation of MNK by arsenic. MNK1 contains a zinc-binding domain with four cysteine residues located in a region encompassing a helix αG domain (residues 261–290; ref. 55). Notably, this domain is unique to the MNKs and not found in other Ca2+/calmodulin-dependent protein kinases (CAMK; ref. 55). It is possible that ATO binds MNK1 to cysteines on positions C264, C268, C276, and C279. Although the role of this region is not fully understood, it may be important for substrate binding (55). Future studies will need to be conducted to uncover the role of these cysteines in MNK1 kinase activity.
Our findings suggest that ATO represents an effective therapeutic option for GBM in some contexts, particularly through the depletion of PN GSCs. Furthermore, our study suggests a rationale for ATO when combined with MNK inhibitors in MES GBM. Novel MNK inhibitors are currently under development, which may have clinical applications (55). Furthermore, preliminary success for the treatment of GBM has been achieved by targeting other components of the translational machinery (37, 56). For example, Ribavirin, an eIF4E targeting compound that is FDA-approved for the treatment of hepatitis C, disrupts GSCs and synergizes with chemotherapy and radiation in a GBM mouse model (56). Our study corroborates the notion of MNK-related translational activation as an attractive target for GBM and, in particular, for depletion of therapy-resistant MES GSCs.
To our knowledge, this is the first analysis of clinical trial data assessing ATO responses in different GBM molecular subtypes. Although we do not measure MNK1 protein levels directly in patient samples, our findings provide evidence that high MNK activity triggers mRNA translation of target genes that correlate with poor clinical responses to ATO. Our findings will need to be replicated in larger patient cohorts using the most recent classifications for GBM that include IDH mutation status, an important prognostic factor (40). However, our observation that MKNK1 expression correlates with poor survival in non-G-CIMP/ MGMT promoter unmethylated patients suggests that IDH mutations may not play an important role in this scenario. Our findings provide valuable insight by linking in vitro and in vivo data to a meaningful clinical scenario. As GBM is a complex, heterogeneous disease that can be difficult to model precisely with cell culture and PDX models, it is necessary to confirm preclinical research findings in patients. Our study utilizes an unbiased approach to identify ATO as a PN subtype-specific compound in patient-derived cell lines. Furthermore, we characterize MNK1–eIF4E signaling as an ATO resistance mechanism in MES GBM. Thus, our data supports the use of ATO in PN GBM patients and a combination of ATO and MNK inhibition in MES GBM.
In summary, we provide evidence implicating MNK signaling as a resistance mechanism to ATO in GBM and, in particular, MES GSCs. Further research may uncover additional ATO resistance mechanisms and present opportunities for novel combinatorial approaches. The development of treatment strategies designed to specifically target MES or PN GSCs is an attractive approach to disrupt these tumor-initiating cells and prevent tumor recurrence.
Supplementary Material
Acknowledgments
This work was supported by the NIH Grants CA121192, CA77816, and CA155566, U01CA168397 and by grant I01CX000916 from the Department of Veterans Affairs. J.B. Bell was supported in part by the MSTP NIH training grant T32 GM008152. E.M. Beauchamp and H.D. Dhruv were supported in part by The Ben and Catherine Ivy Foundation.
Scientists at Northwestern acknowledge the Northwestern Pathology Core Facility, Northwestern Flow Cytometry Core Facility, NUSeq Core, and the Northwestern Developmental Therapeutics Core for their support. Scientists at the Sanford Burnham Prebys Medical Discovery Institute acknowledge the Animal Facility and Tumor Analysis Core. Scientists at TGen acknowledge the Sequencing Core Facility.
Footnotes
Note: Supplementary data for this article are available at Molecular Cancer Research Online (http://mcr.aacrjournals.org/).
Disclosure of Potential Conflicts of Interest
P. Kumthekar is a medical advisory board member for Vivacitas; and is a consultant/advisory board member at Abbvie. J. Raizer is an Executive Medical Director at Celldex; reports receiving commercial research grants from Astra-Zeneca and Genentech; and is a consultant/advisory board member for Zio-Pharm. No potential conflicts of interest were disclosed by the other authors.
Authors’ Contributions
Conception and design: J.B. Bell, F. Eckerdt, H.D. Dhruv, S. Goldman, C.D. James, J. Raizer, L.C. Platanias
Development of methodology: J.B. Bell, F. Eckerdt, H.D. Dhruv, S. Kim, E.M. Beauchamp, A.P. Mazar
Acquisition of data (provided animals, acquired and managed patients, provided facilities, etc.): J.B. Bell, F. Eckerdt, H.D. Dhruv, D. Finlay, S. Peng, S. Kim, B. Kroczynska, K. Alley, J. Clymer, A.P. Mazar, J. Raizer
Analysis and interpretation of data (e.g., statistical analysis, biostatistics, computational analysis): J.B. Bell, F. Eckerdt, H.D. Dhruv, D. Finlay, S. Peng, S. Kim, B. Kroczynska, E.M. Beauchamp, K. Alley, C. Horbinski, K. Vuori, L.C. Platanias
Writing, review, and/or revision of the manuscript: J.B. Bell, F. Eckerdt, H.D. Dhruv, S. Kim, S.-Y. Cheng, P. Kumthekar, J. Raizer, L.C. Platanias
Administrative, technical, or material support (i.e., reporting or organizing data, constructing databases): C.D. James, I. Nakano, C. Horbinski
Study supervision: A.P. Mazar, M.E. Berens, L.C. Platanias
References
- 1.Nakano I. Stem cell signature in glioblastoma: therapeutic development for a moving target. J Neurosurg. 2015;122:324–30. doi: 10.3171/2014.9.JNS132253. [DOI] [PubMed] [Google Scholar]
- 2.Mao P, Joshi K, Li J, Kim SH, Li P, Santana-Santos L, et al. Mesenchymal glioma stem cells are maintained by activated glycolytic metabolism involving aldehyde dehydrogenase 1A3. Proc Natl Acad Sci U S A. 2013;110:8644–9. doi: 10.1073/pnas.1221478110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Segerman A, Niklasson M, Haglund C, Bergstrom T, Jarvius M, Xie Y, et al. Clonal variation in drug and radiation response among glioma-initiating cells is linked to proneural-mesenchymal transition. Cell Rep. 2016;17:2994–3009. doi: 10.1016/j.celrep.2016.11.056. [DOI] [PubMed] [Google Scholar]
- 4.Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Au WY, Tam S, Fong BM, Kwong YL. Determinants of cerebrospinal fluid arsenic concentration in patients with acute promyelocytic leukemia on oral arsenic trioxide therapy. Blood. 2008;112:3587–90. doi: 10.1182/blood-2008-06-161000. [DOI] [PubMed] [Google Scholar]
- 6.Ding D, Lim KS, Eberhart CG. Arsenic trioxide inhibits Hedgehog, Notch and stem cell properties in glioblastoma neurospheres. Acta Neuropathol Commun. 2014;2:31. doi: 10.1186/2051-5960-2-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Iwanami A, Gini B, Zanca C, Matsutani T, Assuncao A, Nael A, et al. PML mediates glioblastoma resistance to mammalian target of rapamycin (mTOR)-targeted therapies. Proc Natl Acad Sci U S A. 2013;110:4339–44. doi: 10.1073/pnas.1217602110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sun H, Zhang S. Arsenic trioxide regulates the apoptosis of glioma cell and glioma stem cell via down-regulation of stem cell marker Sox2. Biochem Biophys Res Commun. 2011;410:692–7. doi: 10.1016/j.bbrc.2011.06.060. [DOI] [PubMed] [Google Scholar]
- 9.Grimm SA, Marymont M, Chandler JP, Muro K, Newman SB, Levy RM, et al. Phase I study of arsenic trioxide and temozolomide in combination with radiation therapy in patients with malignant gliomas. J Neurooncol. 2012;110:237–43. doi: 10.1007/s11060-012-0957-6. [DOI] [PubMed] [Google Scholar]
- 10.Kumthekar P, Grimm S, Chandler J, Mehta M, Marymont M, Levy R, et al. A phase II trial of arsenic trioxide and temozolomide in combination with radiation therapy for patients with malignant gliomas. J Neurooncol. 2017;133:589–94. doi: 10.1007/s11060-017-2469-x. [DOI] [PubMed] [Google Scholar]
- 11.Siddiqui N, Sonenberg N. Signalling to eIF4E in cancer. Biochem Soc Trans. 2015;43:763–72. doi: 10.1042/BST20150126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Topisirovic I, Sonenberg N. mRNA translation and energy metabolism in cancer: the role of the MAPK and mTORC1 pathways. Cold Spring Harb Symp Quant Biol. 2011;76:355–67. doi: 10.1101/sqb.2011.76.010785. [DOI] [PubMed] [Google Scholar]
- 13.Fukunaga R, Hunter T. MNK1, a new MAP kinase-activated protein kinase, isolated by a novel expression screening method for identifying protein kinase substrates. EMBO J. 1997;16:1921–33. doi: 10.1093/emboj/16.8.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Joshi S, Platanias LC. Mnk kinase pathway: cellular functions and biological outcomes. World J Biol Chem. 2014;5:321–33. doi: 10.4331/wjbc.v5.i3.321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hinnebusch AG, Ivanov IP, Sonenberg N. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science. 2016;352:1413–6. doi: 10.1126/science.aad9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Spriggs KA, Bushell M, Willis AE. Translational regulation of gene expression during conditions of cell stress. Mol Cell. 2010;40:228–37. doi: 10.1016/j.molcel.2010.09.028. [DOI] [PubMed] [Google Scholar]
- 17.Mehrotra S, Sharma B, Joshi S, Kroczynska B, Majchrzak B, Stein BL, et al. Essential role for the Mnk pathway in the inhibitory effects of type I interferons on myeloproliferative neoplasm (MPN) precursors. J Biol Chem. 2013;288:23814–22. doi: 10.1074/jbc.M113.476192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chio II, Jafarnejad SM, Ponz-Sarvise M, Park Y, Rivera K, Palm W, et al. NRF2 promotes tumor maintenance by modulating mRNA translation in pancreatic cancer. Cell. 2016;166:963–76. doi: 10.1016/j.cell.2016.06.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wahba A, Rath BH, Bisht K, Camphausen K, Tofilon PJ. Polysome profiling links translational control to the radioresponse of glioblastoma stem-like cells. Cancer Res. 2016;76:3078–87. doi: 10.1158/0008-5472.CAN-15-3050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grzmil M, Seebacher J, Hess D, Behe M, Schibli R, Moncayo G, et al. Inhibition of MNK pathways enhances cancer cell response to chemotherapy with temozolomide and targeted radionuclide therapy. Cell Signal. 2016;28:1412–21. doi: 10.1016/j.cellsig.2016.06.005. [DOI] [PubMed] [Google Scholar]
- 21.Bell JB, Eckerdt FD, Alley K, Magnusson LP, Hussain H, Bi Y, et al. MNK inhibition disrupts mesenchymal glioma stem cells and prolongs survival in a mouse model of glioblastoma. Mol Cancer Res. 2016;14:984–93. doi: 10.1158/1541-7786.MCR-16-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang X, Yang F, Shim JY, Kirk KL, Anderson DE, Chen X. Identification of arsenic-binding proteins in human breast cancer cells. Cancer Lett. 2007;255:95–106. doi: 10.1016/j.canlet.2007.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Beauchamp EM, Kosciuczuk EM, Serrano R, Nanavati D, Swindell EP, Viollet B, et al. Direct binding of arsenic trioxide to AMPK and generation of inhibitory effects on acute myeloid leukemia precursors. Mol Cancer Ther. 2015;14:202–12. doi: 10.1158/1535-7163.MCT-14-0665-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang XW, Yan XJ, Zhou ZR, Yang FF, Wu ZY, Sun HB, et al. Arsenic trioxide controls the fate of the PML-RARalpha oncoprotein by directly binding PML. Science. 2010;328:240–3. doi: 10.1126/science.1183424. [DOI] [PubMed] [Google Scholar]
- 26.Eckerdt F, Alvarez A, Bell J, Arvanitis C, Iqbal A, Arslan AD, et al. A simple, low-cost staining method for rapid-throughput analysis of tumor spheroids. Biotechniques. 2016;60:43–6. doi: 10.2144/000114372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hu Y, Smyth GK. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J Immunol Methods. 2009;347:70–8. doi: 10.1016/j.jim.2009.06.008. [DOI] [PubMed] [Google Scholar]
- 28.McNeer JL, Goussetis DJ, Sassano A, Dolniak B, Kroczynska B, Glaser H, et al. Arsenic trioxide-dependent activation of thousand-and-one amino acid kinase 2 and transforming growth factor-beta-activated kinase 1. Mol Pharmacol. 2010;77:828–35. doi: 10.1124/mol.109.061507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goussetis DJ, Platanias LC. Arsenic trioxide and the phosphoinositide 3-kinase/akt pathway in chronic lymphocytic leukemia. Clin Cancer Res. 2010;16:4311–2. doi: 10.1158/1078-0432.CCR-10-1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dolniak B, Katsoulidis E, Carayol N, Altman JK, Redig AJ, Tallman MS, et al. Regulation of arsenic trioxide-induced cellular responses by Mnk1 and Mnk2. J Biol Chem. 2008;283:12034–42. doi: 10.1074/jbc.M708816200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoon P, Giafis N, Smith J, Mears H, Katsoulidis E, Sassano A, et al. Activation of mammalian target of rapamycin and the p70 S6 kinase by arsenic trioxide in BCR-ABL-expressing cells. Mol Cancer Ther. 2006;5:2815–23. doi: 10.1158/1535-7163.MCT-06-0263. [DOI] [PubMed] [Google Scholar]
- 32.Kannan-Thulasiraman P, Katsoulidis E, Tallman MS, Arthur JS, Platanias LC. Activation of the mitogen- and stress-activated kinase 1 by arsenic trioxide. J Biol Chem. 2006;281:22446–52. doi: 10.1074/jbc.M603111200. [DOI] [PubMed] [Google Scholar]
- 33.Giafis N, Katsoulidis E, Sassano A, Tallman MS, Higgins LS, Nebreda AR, et al. Role of the p38 mitogen-activated protein kinase pathway in the generation of arsenic trioxide-dependent cellular responses. Cancer Res. 2006;66:6763–71. doi: 10.1158/0008-5472.CAN-05-3699. [DOI] [PubMed] [Google Scholar]
- 34.Verma A, Mohindru M, Deb DK, Sassano A, Kambhampati S, Ravandi F, et al. Activation of Rac1 and the p38 mitogen-activated protein kinase pathway in response to arsenic trioxide. J Biol Chem. 2002;277:44988–95. doi: 10.1074/jbc.M207176200. [DOI] [PubMed] [Google Scholar]
- 35.Zhang HN, Yang L, Ling JY, Czajkowsky DM, Wang JF, Zhang XW, et al. Systematic identification of arsenic-binding proteins reveals that hexoki-nase-2 is inhibited by arsenic. Proc Natl Acad Sci U S A. 2015;112:15084–9. doi: 10.1073/pnas.1521316112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou X, Sun X, Mobarak C, Gandolfi AJ, Burchiel SW, Hudson LG, et al. Differential binding of monomethylarsonous acid compared to arsenite and arsenic trioxide with zinc finger peptides and proteins. Chem Res Toxicol. 2014;27:690–8. doi: 10.1021/tx500022j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Grzmil M, Huber RM, Hess D, Frank S, Hynx D, Moncayo G, et al. MNK1 pathway activity maintains protein synthesis in rapalog-treated gliomas. J Clin Invest. 2014;124:742–54. doi: 10.1172/JCI70198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Malone CF, Fromm JA, Maertens O, DeRaedt T, Ingraham R, Cichowski K. Defining key signaling nodes and therapeutic biomarkers in NF1-mutant cancers. Cancer Discov. 2014;4:1062–73. doi: 10.1158/2159-8290.CD-14-0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huang T, Alvarez AA, Pangeni RP, Horbinski CM, Lu S, Kim SH, et al. A regulatory circuit of miR-125b/miR-20b and Wnt signalling controls glioblastoma phenotypes through FZD6-modulated pathways. Nat Commun. 2016;7:12885. doi: 10.1038/ncomms12885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathol. 2016;131:803–20. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- 41.Carro MS, Lim WK, Alvarez MJ, Bollo RJ, Zhao X, Snyder EY, et al. The transcriptional network for mesenchymal transformation of brain tumours. Nature. 2010;463:318–25. doi: 10.1038/nature08712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheng P, Wang J, Waghmare I, Sartini S, Coviello V, Zhang Z, et al. FOXD1-ALDH1A3 signaling is a determinant for the self-renewal and tumorigenicity of mesenchymal glioma stem cells. Cancer Res. 2016;76:7219–30. doi: 10.1158/0008-5472.CAN-15-2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wei W, Shin YS, Xue M, Matsutani T, Masui K, Yang H, et al. Single-cell phosphoproteomics resolves adaptive signaling dynamics and informs targeted combination therapy in glioblastoma. Cancer Cell. 2016;29:563–73. doi: 10.1016/j.ccell.2016.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fan W, Wang W, Mao X, Chu S, Feng J, Xiao D, et al. Elevated levels of p-Mnk1, p-eIF4E and p-p70S6K proteins are associated with tumor recurrence and poor prognosis in astrocytomas. J Neurooncol. 2017;131:485–93. doi: 10.1007/s11060-016-2327-2. [DOI] [PubMed] [Google Scholar]
- 45.Zheng J, Li J, Xu L, Xie G, Wen Q, Luo J, et al. Phosphorylated Mnk1 and eIF4E are associated with lymph node metastasis and poor prognosis of nasopharyngeal carcinoma. PLoS One. 2014;9:e89220. doi: 10.1371/journal.pone.0089220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Millican-Slater RA, Sayers CD, Hanby AM, Hughes TA. Expression of phosphorylated eIF4E-binding protein 1, but not of eIF4E itself, predicts survival in male breast cancer. Br J Cancer. 2016;115:339–45. doi: 10.1038/bjc.2016.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Martinez-Saez E, Peg V, Ortega-Aznar A, Martinez-Ricarte F, Camacho J, Hernandez-Losa J, et al. peIF4E as an independent prognostic factor and a potential therapeutic target in diffuse infiltrating astrocytomas. Cancer Med. 2016;5:2501–12. doi: 10.1002/cam4.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Topisirovic I, Siddiqui N, Lapointe VL, Trost M, Thibault P, Bangeranye C, et al. Molecular dissection of the eukaryotic initiation factor 4E (eIF4E) export-competent RNP. EMBO J. 2009;28:1087–98. doi: 10.1038/emboj.2009.53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Phillips A, Blaydes JP. MNK1 and EIF4E are downstream effectors of MEKs in the regulation of the nuclear export of HDM2 mRNA. Oncogene. 2008;27:1645–9. doi: 10.1038/sj.onc.1210785. [DOI] [PubMed] [Google Scholar]
- 50.Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75:50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DaSilva J, Xu L, Kim HJ, Miller WT, Bar-Sagi D. Regulation of sprouty stability by Mnk1-dependent phosphorylation. Mol Cell Biol. 2006;26:1898–907. doi: 10.1128/MCB.26.5.1898-1907.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Walsh AM, Kapoor GS, Buonato JM, Mathew LK, Bi Y, Davuluri RV, et al. Sprouty2 drives drug resistance and proliferation in glioblastoma. Mol Cancer Res. 2015;13:1227–37. doi: 10.1158/1541-7786.MCR-14-0183-T. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guil S, Long JC, Caceres JF. hnRNP A1 relocalization to the stress granules reflects a role in the stress response. Mol Cell Biol. 2006;26:5744–58. doi: 10.1128/MCB.00224-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Babic I, Anderson ES, Tanaka K, Guo D, Masui K, Li B, et al. EGFR mutation-induced alternative splicing of Max contributes to growth of glycolytic tumors in brain cancer. Cell Metab. 2013;17:1000–8. doi: 10.1016/j.cmet.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Dreas A, Mikulski M, Milik M, Fabritius CH, Brzozka K, Rzymski T. Mitogen-activated protein kinase (MAPK) interacting kinases 1 and 2 (MNK1 and MNK2) as targets for cancer therapy: recent progress in the development of MNK inhibitors. Curr Med Chem. 2017;24:3025–53. doi: 10.2174/0929867324666170203123427. [DOI] [PubMed] [Google Scholar]
- 56.Volpin F, Casaos J, Sesen J, Mangraviti A, Choi J, Gorelick N, et al. Use of an anti-viral drug, Ribavirin, as an anti-glioblastoma therapeutic. Oncogene. 2017;36:3037–47. doi: 10.1038/onc.2016.457. [DOI] [PubMed] [Google Scholar]
- 57.Bowman RL, Wang Q, Carro A, Verhaak RG, Squatrito M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol. 2017;19:139–41. doi: 10.1093/neuonc/now247. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.