

Abstract
Protease-activatable viruses (PAV) based on adeno-associated virus have previously been generated for gene delivery to pathological sites characterized by elevated extracellular proteases. “Peptide locks”, composed of a tetra-aspartic acid motif flanked by protease cleavage sequences, were inserted into the virus capsid to inhibit virus-host cell receptor binding and transduction. In the presence of proteases, the peptide locks are cleaved off the capsid, restoring the virus’ ability to bind cells and deliver cargo. Although promising, questions remained regarding how the peptide locks prevented cell binding. In particular, it was unclear if the tetra-amino acid (4AA) motif blocks receptor binding via electrostatic repulsion or steric obstruction. To explore this question, we generated a panel of PAVs with lock designs incorporating altered 4AA motifs, each wielding various chemical properties (negative, positive, uncharged polar, and hydrophobic) and characterized the resultant PAV candidates. Notably, all mutants display reduced receptor binding and decreased transduction effciency in the absence of proteases, suggesting simple electrostatics between heparin and the D4 motif do not play an exclusive role in obstructing virus-receptor binding. Even small hydrophobic (A4) and uncharged polar (SGGS) motifs confer a reduction in heparin binding compared to the wild type. Furthermore, both uncharged polar N4 and Q4 mutants (comparable in size to the D4 and E4 motifs respectively, but lacking the negative charge) demonstrate partial ablation of heparin binding. Collectively, these results support a possible dual mechanism of PAV lock operation, where steric hindrance and electrostatics make nonredundant contributions to the disruption of virus-receptor interactions. Finally, because of high virus titer production and superior capsid stability, only the negatively charged 4AA motifs remain viable design choices for PAV construction. Future studies probing the structure–function relationship of PAVs will further expand its promise as a gene delivery vector able to target diseased tissues exhibiting elevated extracellular proteases.
Keywords: adeno-associated virus, stimulus-responsive, enzyme-responsive, gene delivery, matrix metalloproteinases, synthetic virology
Graphical Abstract
INTRODUCTION
Extracellular proteases, such as matrix metalloproteinases (MMPs), are elevated at sites of disease in various pathologies, such as cancer1 and cardiovascular diseases.2 Increased MMP activity has been implicated in disease progression and, thus, has been used as a biomarker to target therapeutics to affected tissues.2–8 For example, Ge et al. developed a polyethylenimine-gelatin gene delivery vector that upon MMP cleavage becomes activated to interact with the target cell membrane.9 More recently, Gjetting et al. synthesized lipo-nanoparticles with a lipid moiety that is susceptible to protease cleavage for delivery to diseased sites.10 Several viral vectors, including retrovirus and measles virus,11,12 have also been engineered to be protease-responsive. Thus, activation by an intrinsic biomolecular stimulus,13 such as an extracellular protease, is proving to be a promising approach for targeted therapy.14
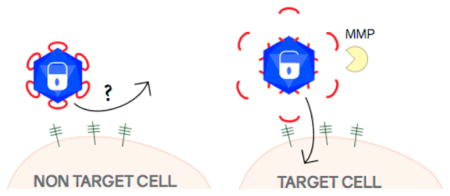
Previously, we constructed a protease-activatable virus (PAV) based on adeno-associated virus serotype 2 (AAV2) that delivers transgenes upon activation by MMPs.17 AAV is a promising delivery vector for gene therapy of various human diseases,15 including cancer.16 Our design strategy involves the insertion of a “peptide lock” into the heparan sulfate proteoglycan (HSPG) binding region in the AAV2 capsid.17 The prototype peptide lock consists of a tetra amino acid motif (4AA motif), comprising four negatively charged aspartic acids, flanked by MMP cleavage consensus sequences (Figure 1). With the peptide locks in place, the PAVs are unable to transduce cells. Then, in the presence of target MMPs, the peptide locks are cleaved off the capsid, allowing the viruses to regain their ability to bind HSPG and deliver transgenes.
Figure 1.

Protease-activatable virus (PAV) design and concept. (a) Peptide lock consists of the tetra amino acid (4AA) motif flanked by MMP cleavage consensus sequences. Scissile bonds are indicated by yellow arrows. Glycine linkers separate the MMP consensus sequences and the 4AA motif. (b) Peptide locks inserted into the AAV capsid prevent virus binding to cellular receptors. In the presence of MMPs, the MMP consensus sequences are cleaved, releasing the peptide locks from the capsid and restoring viral transduction.
We have demonstrated that our vector works well in exhibiting protease-switchable transduction behavior.17,18 However, a question that remained was whether the aspartic acid residues of the 4AA motif were optimal for peptide lock operation or if other amino acids may be used as the 4AA motif. The tetra-aspartic acids were initially chosen because we hypothesized that the negatively charged amino acids inserted into the receptor binding domain of AAV2 would electrostatically repel the binding of negatively charged HSPG by the virus capsid. Effective HSPG binding by the patch of positively charged amino acid residues on the AAV2 capsid is required for successful cellular transduction.19 To address this question, we synthesized and characterized PAV mutants with different 4AA motifs to study the effect of motif chemical properties on PAV function. Our results suggest tetra-aspartic acid and tetra-glutamic acid motifs yield the best PAV properties to date.
EXPERIMENTAL METHODS
Construction of PAV Plasmids
All PAV plasmids were produced from pRC_RR, a plasmid containing AAV serotype 2 (AAV2) rep and cap genes with NgoMIV and KasI sites at position 586 of cap.17 PAV plasmids containing various peptide locks were constructed by varying the tetra-amino acid motif of the inactivating domain. Oligonucleo-tides encoding the various locks were annealed and ligated into the NgoMIV and KasI sites in the pRC_RR plasmid. PAV variant plasmids (pPAV-D4, pPAV-E4, pPAV-K4, pPAV-R4, pPAV-A4, pPAV-N4, pPAV-Q4, pPAV-SG2S) were sequence-verified.
Virus Production
PAV variants were produced through a triple plasmid cotransfection of (a) pXX6–80, a helper plasmid encoding adenoviral proteins; (b) pscGFP, self-complementary GFP transgene cassette; and (c) packaging plasmids encoding PAV capsid derivatives.17 Wild-type (wt) capsid AAV2 vector was produced through triple transfection of pXX6-80, pscGFP, and pXX2. Human Embryonic Kidney 293T cells (HEK 293T) were split 1:2 and plated on ten 15 cm plates (BD Falcon) coated with 0.001% poly-L-lysine (Sigma) 24h before transfection. Forty-8 h post-transfection, the producer cells were harvested, pelleted, and resuspended in 1× Gradient Buffer (GB: 10 mM Tris, pH 7.6, 10 mM MgCl2, 150 mM NaCl). Cells were then lysed with three freeze–thaw cycles followed by benzonase treatment (50 units/ml, Sigma) at 37 °C for 40 min. Cell lysate was centrifuged at 4 °C at 3,000g for 20 min and the supernatant was collected for iodixanol density gradient separation (Optiprep, Beckman Ultra-Clear QuickSeal Tubes). Samples were centrifuged at 18 °C at 48,000 rpm for 1h 45 min. Viruses were extracted from the 40% iodixanol layer and stored at 4 °C in 3 mL cryovials (Biotix). Viruses were purified with an anion exchange column (Pall Corporation) and concentrated with an amicon filter tube (Millipore) as published elsewhere.17
Quantification of Virus Titers
Quantitative polymerase chain reaction (qPCR) was used to determine genomic viral titers as described elsewhere.20 Briefly, viral titers were quantified using a C1000 thermal cycler (Bio-Rad) with SYBR green (Applied Biosystems) and primers against the CMV promoter (forward: 5′-TCACGGGGATTTCCAAGTCTC-3′ and reverse: 5′-AATGGGGCGGAGTTGTTACGA-3′).
Western Blotting and Silver Staining
Western blot analysis was performed to detect viral capsid proteins. Samples were heat-denatured in LDS sample buffer and sample reducing agent (NuPAGE, Life Technologies) at 75 °C for 15 min. Denatured samples were loaded and separated via electrophoresis on a 4–12% Bis-Tris gel (NuPAGE, Life Technologies), transferred onto a nitrocellulose membrane, and probed sequentially with B1 primary antibody (American Research Products) followed by horseradish peroxidase conjugated antimouse secondary antibody (American Research Products). Lumi-Light Western Blotting Substrate (Roche Applied Science) was then added to the nitrocellulose membrane and imaged with Fuji Las-4000.
Silver stain analysis was also conducted to detect viral capsid proteins as well as capsid proteolytic fragments. Viral samples were denatured in LDS and separated via electrophoresis on a 4–12% Bis-Tris gel (NuPAGE, Life Technologies). Viral capsid proteins and proteolytic fragments were detected with a silver stain kit according to manufacturer’s protocol (Life Technologies, Carlsbad, CA).
Heparin Affinity Assay
PAV mutants were tested for heparin binding ability. Viruses were added to heparin-agarose beads (Sigma H-6508) and incubated for 15 min at room temperature while continuously rotating. Samples were centrifuged for 5 min at 6000g and unbound fractions were collected. The beads were incubated sequentially with buffers containing increasing NaCl concentrations (300 mM, 500 mM, 700 mM, and 1 M) in order to elute heparin-bound viruses. All unbound and bound fractions were quantified for viral genome amounts via qPCR.
Nuclease Protection Assay
Capsid stability of PAV mutants was analyzed with a benzonase protection assay. PAV mutants were mixed with Endo buffer (1.5 mM MgCl2, 0.5 mg/mL BSA, 50 mM Tris, pH 8.0) and incubated with benzonase nuclease (250 units/μL; Sigma) or sham buffer (50% glycerol, 50 mM Tris-HCl, pH 8.0, 20 mM NaCl, 2 mM MgCl2) at 37 °C for 30 min. Reactions were terminated with 0.5 M EDTA. Viral titers were then quantified with qPCR to calculate the percentage of protected viral genomes.
Cellular Transduction
HEK293T cells were seeded on poly-L-lysine-coated 48-well plates at 1 × 105 cells/well. PAV mutants were added to cells at a multiplicity of infection (MOI) of 500 and incubated in serum-free medium for 12 h. Complete medium containing serum was added 12 h post-transduction. Cells were harvested 48 h post-transduction for flow cytometry with FACSCanto II flow cytometer (BD Biosciences).
Proteolysis
PAV mutants were tested for MMP susceptibility. Protease activity was first calibrated with the fluorogenic substrate Mca-PLGL-Dpa-AR (Calbiochem) at 5 nM enzyme and 5 μM substrate to determine initial enzyme velocities, as previously described.17 Viruses were diluted in reaction buffer (MMP Buffer: 50 mM Tris-Cl, pH 7.4, 150 mM NaCl, 5 mM CaCl2 and 1× GB-PF68). MMP-7 (Enzo Life Sciences, Farmingdale, NY) was diluted in vehicle buffer (50 mMTris, pH 7.5, 300 mM NaCl, 5 mM CaCl2, 10 μM ZnCl2, 0.5% Brij-35, 30% glycerol) and mixed with diluted virus. Reactions were incubated at 37 °C for 12h and terminated with 50 mM EDTA. For cellular transduction, MMP-treated viruses were added to HEK293T cells and incubated for 48 h. After 48 h, cells were harvested and analyzed with flow cytometry.
RESULTS
Production of Viruses with Peptide Lock Variants
We synthesized a panel of PAV variants to test the effects of 4AA motif properties on virus production and performance. Quantitative polymerase chain reaction (qPCR) was used to quantify the titers of the PAV variants produced (Figure 2). The viruses with negatively charged 4AA motifs, PAV-D4 and PAV-E4, yield titers that are within 1-log of wild-type (wt), indicating these motifs do not adversely impact virus capsid assembly and genome packaging. PAV-A4, -N4, and -Q4, are within 1–2 log of wt. The positively charged 4AA motif mutants, PAV-K4, -R4, and the polar uncharged -SG2S display the most dramatic decreases in titers that are 2-log fold lower than wt. Therefore, the AAV capsid appears to be most tolerant to peptide locks with negatively charged 4AA motifs and poorly tolerates positively charged 4AA motifs.
Figure 2.

Titers of PAV candidates with different tetra amino acid (4AA) motifs. D4, tetra aspartic acid. E4, tetra glutamic acid. K4, tetra lysine. R4, tetra arginine. A4, tetra alanine. N4, tetra asparagine. Q4, tetra glutamine. SG2S, serine-glycine-glycine-serine. Virus titers were quantified with qPCR. Standard error of the mean (SEM) of two independent virus preparations are shown. All mutant titers are statistically different compared to wt (p < 0.001, student t test), with the exception of PAV-E4.
Western blot was performed to detect viral capsid proteins (VPs) and qualitatively assess peptide lock incorporation into the capsid (Figure 3). Because the iodixanol density gradient separation process collects assembled capsids, and these separated virus samples are the ones run on the Western blot, the bands in Figure 3 capture the VP subunit profiles in the assembled capsids. Most PAV variants display VP expression patterns similar to wt, although with a noticeable MW shift due to peptide lock insertion. These results indicate the peptide locks are successfully inserted into the VP subunits and the subunits are able to assemble into the capsid. The VP band intensities of the PAV-A4 and PAV-SG2S viruses are lighter compared to other mutants because they had lower genomic titers. Interestingly, the PAV-K4 variant displays an irregular pattern with multiple VP fragments on the Western blot, which may suggest increased susceptibility of PAV-K4 to trypsin and potentially other proteases during virus production.21 Considering the qPCR and Western blot data together, the PAV mutants demonstrate successful peptide lock insertion, capsid assembly, and genome packaging, albeit at lower levels compared to wt for most of the mutants.
Figure 3.

Detection of viral capsid proteins (VPs) and peptide lock incorporation of PAV candidates via Western blot. Same volume of iodixanol-separated virus was loaded per well, except for the R4 blot in which the same number of viral genomes were loaded for wt and R4. Capsid subunits were probed with the B1 antibody, which detects a c-terminal epitope shared among all three VPs. VP1, VP2, and VP3 indicate wt capsid subunits. VP1-PAV, VP2-PAV, and VP3-PAV indicate capsid subunits with peptide lock insertions. Molecular weight ladder locations are indicated on the left side of blots. Most virus variants display only three VP bands similarly to wt capsid. PAV-K4 displays an irregular pattern of multiple bands beyond the expected three VP bands.
Capsid Stability of Viruses with Peptide Lock Variants
We next tested the stability of the formed capsids using a nuclease protection assay. A stable, well-formed capsid should effectively protect its encapsidated genome from degradation by nucleases. PAV mutants were treated with benzonase and the level of genome protection was quantified with qPCR (Figure 4). As expected, the wt control displays full genome protection, while the negative control (naked plasmid DNA) exhibits complete degradation. PAV-R4, -A4, -N4, -Q4, and -SG2S exhibit wt-like abilities to protect their genomes. Thus, although these variants are produced at lower titers than wt, the assembled capsids are stable. PAV-D4 and -E4, produced at wt-like titers, show protection levels slightly less than wt but the differences are not statistically significant. PAV-K4 has the lowest level of genome protection, which could potentially be due to protease susceptibility during capsid assembly, as suggested by Western blotting. The PAV-K4 virus appears to be the most defective virus thus far, with a low production titer, an irregular VP band pattern, and lowest capsid stability.
Figure 4.

Capsid stabilities of PAV candidates as assessed by their abilities to protect their genomes from nuclease digestion. PAV candidates were incubated with benzonase nuclease to measure capsid stability. % genome protection was quantified with qPCR. Naked plasmid DNA is used as a control to ensure nuclease activity. Error bars represent SEM of two independent experiments performed in duplicate. Asterisk indicates p < 0.05 by student t test.
Impact of Peptide Lock Variants on Heparin Binding
We conducted a heparin affinity assay to test if 4AA motif properties play a role in locking the virus. A successful peptide lock design should ablate heparin binding by the virus in absence of proteases. Our wt positive control elutes from the heparin resin at 500 mM NaCl and higher (Figure 5) as expected from previous reports.17,20 AAV serotype 4 (AAV4) serves as the negative control for this assay because it binds sialic acid instead of heparin.22 As expected, the majority of AAV4 do not bind the heparin coated beads. Most of the PAV mutants display low heparin binding, with the exception of the three polar uncharged motifs (SG2S, N4, and Q4) that appear to retain intermediate heparin binding ability. Overall, this data demonstrates negatively charged, positively charged, and hydrophobic 4AA motifs are successfully able to disrupt heparin binding by the virus.
Figure 5.

Ability of various 4AA motifs to disrupt virus-heparin binding. Viruses were eluted from heparin agarose beads by incubating with increasing NaCl concentration buffers. qPCR was used to measure virus amounts in the elutions. AAV serotype 2 (AAV2) binds to heparin and elutes off the heparin beads at 500–700 mM NaCl. AAV serotype 4 (AAV4) does not bind to heparin. Error bars represent SEM of two independent experiments.
Ability of Peptide Lock Variants to Block Cellular Transduction
We performed a transduction assay to investigate whether various peptide locks are able to prevent cellular transduction by the viruses in the absence of proteases, as desired. In comparison to wt, all of the PAV mutants demonstrate significantly lower gene delivery efficiencies (Figure 6a–c). The D4, E4, K4, R4, and A4 peptide locks are able to decrease the transduction index (TI) at least 6-fold compared to wt. N4, Q4, and SG2S are the least effective in blocking cellular transduction. N4 and Q4 decrease transduction 2-fold, while SG2S performs slightly better, yielding a 4-fold decrease. In summary, all inserted 4AA motifs with the exception of the polar uncharged motifs (N4, Q4, SG2S) appear to lock the virus effectively regardless of chemical properties.
Figure 6.

Transduction assay to test if the various 4AA motifs are able to inhibit cellular transduction by PAV candidates in the absence of proteases. Viral genomes at a multiplicity of infection (MOI) of 500 were added to HEK293T cells. (a, b) %GFP+ cells and geometric mean fluorescence intensity (gMFI) were quantified with flow cytometry and normalized to wt values. (c) Transduction index (TI) is %GFP+ cells multiplied by gMFI and is a linear indicator of viral transduction efficiency.17 Error bars represent SEM of two independent experiments conducted in duplicate. (d) Transduction inhibition as a function of heparin binding index. Lower the heparin binding index (i.e., less effective heparin binding ability), the better the inhibition of transduction in the absence of proteases. Data points for A4, SG2S, Q4, N4, and wt are plotted. The regression line is shown, with an R2 value of 0.90.
Considering the data gathered thus far, it appears that successful transduction inhibition by the peptide lock is correlated to the peptide lock’s ability to prevent heparin binding. To develop a rough design “rule-of-thumb” that may help us screen through different future PAV candidates, we calculated a heparin binding index for each candidate based on the results of the heparin affinity assay
The fraction of viruses eluting in the 500 mM NaCl fraction receive a greater weight of 0.6, compared to 0.4 for the 300 mM NaCl fraction, because these viruses have more wt AAV2-like heparin binding ability. Thus, a larger heparin binding index value indicates the virus retains more wt AAV2-like heparin binding ability. AAV2 and AAV4 have heparin binding indices of 0.39 and 0.06, respectively. Plotting the heparin binding index versus fraction transduction inhibition of the PAV candidates (only those with less than complete inhibition) reveals a linear trend with an R2 value of 0.9 (Figure 6d). The PAV candidates with complete transduction inhibition (D4, E4, K4, and R4) all display heparin binding indices of less than 0.2. Peptide locks that yield low heparin binding index values (i.e., effective inhibition of heparin binding) are able to achieve greater transduction inhibition. Thus, based on the results of the heparin binding assay, if a PAV candidate displays a heparin binding index of less than 0.2, we can expect it to have successful complete transduction inhibition in the absence of proteases. Moving forward, the K4, R4, A4, N4, Q4, and SG2S variants were not analyzed further because of their suboptimal production titers (all six variants), an irregular VP band pattern and unstable capsid (K4), and incomplete locking of the capsid (N4, Q4, and SG2S).
Ability of Peptide Locks to Be Cleaved by Extracellular Proteases
To determine whether the PAV-E4 variant is susceptible to proteases and also regains the ability to transduce cells after protease detection, the virus was treated with matrix metalloproteinase 7 (MMP7) and assayed for capsid proteolysis and cellular transduction ability. PAV-D4 served as the positive control in these studies since we have previously shown this variant is proteolyzed by MMP7 and displays MMP-responsive transduction behavior.17 A silver stain was performed to assess PAV-E4 susceptibility to MMP7 (Figure 7). As expected, the wt negative control shows no protease susceptibility. In contrast, the PAV-D4 positive control displays proteolytic susceptibility to MMP7, and not the sham condition. PAV-E4 demonstrates MMP susceptibility similar to PAV-D4. This data indicates the negatively charged 4AA motifs, E4 and D4, are proteolyzed similarly by MMP7.
Figure 7.

Detection of proteolyzed VP fragments of PAV-D4 and PAV-E4 in the presence of MMP7. Viruses were incubated in sham or MMP7 conditions, denatured, and then run on a 4–12% bis-tris gel. The gel was subsequently processed with the silver stain to visualize VP bands, either intact or proteolyzed. The band corresponding to the added MMP7 enzyme can be observed at the bottom of the image.
Finally, we transduced HEK293T cells with viruses, with and without MMP treatment. In the presence of MMP7, PAV-D4 and PAV-E4 display increased gene delivery efficiencies compared to the sham condition (Figure 8). The PAVs are less efficient compared to wt, which has been previously observed.17,18 Both of the tested PAVs exhibit 2-fold lower TI values in the unlocked state (i.e., MMP treatment condition) compared to wt. These findings indicate PAV-D4 and PAV-E4 are able to regain transduction abilities in the presence of MMP7, albeit at a lower-than-wt efficiency. Taken together, these data demonstrate the negatively charged 4AA motifs, D4 and E4, are the best options among our panel of variants. On the basis of viral production titers, capsid assembly, receptor binding, and protease-activatable transduction abilities, the D4 and E4 variants appear to be functionally interchangeable.
Figure 8.

Recovery of cellular transduction ability of PAV-D4 and PAV-E4 in the presence of MMP7. Viral genomes at a multiplicity of infection (MOI) of 500 were added to HEK293T cells. %GFP+ cells and geometric mean fluorescence (gMFI) were quantified with flow cytometry. Error bars represent SEM of two independent experiments conducted in duplicate.
DISCUSSION
Prior work by others have demonstrated the AAV2 capsid uses patches of positively charged amino acid residues to bind negatively charged HSPG on cell surfaces.19 HSPG binding by AAV2 is the first important step in cellular transduction.23 In our previous work aimed at building a PAV prototype,17 we hypothesized inserting negatively charged amino acids as part of the peptide lock would interfere with AAV2-HSPG interaction through an electrostatic repulsion mechanism. Indeed, our prior results show PAVs in the locked state are poorly internalized by cells, and PAV activation with MMPs dramatically enhances their cellular uptake.17 Here, we investigated whether our original hypothesis was correct or if amino acids with any chemical property may be used as the 4AA motif.
Using a random peptide library approach, Perabo et al. previously found that different 7-mer peptides have differential abilities to ablate HSPG binding when inserted in the HSPG binding domain of the AAV2 capsid.24 Peptides with net positive charges were not effective in ablating binding. Conversely, peptides with net negative charges successfully ablated heparin binding. The authors suggested that peptide insertions in the heparin binding domain can affect HSPG interactions through different mechanisms that involve sterics and electrostatics.
On the basis of this prior work, it was feasible that either sterics or electrostatics could be driving the disruption of HSPG binding and cellular transduction by the 4AA motifs in the absence of MMPs. Surprisingly, almost all of the mutants, including the positively charged PAV-K4 and PAV-R4, display ablated heparin binding (Figure 5). Only the polar uncharged motifs retain marginal binding to heparin. We also observed that majority of the mutants are unable to transduce HEK293T cells in the absence of MMP, as desired (Figure 6). Again, only PAV-N4, -Q4, and -SG2S retain any appreciable level of transduction ability. Due to issues with low viral production titers and inability to effectively lock the AAV capsid, most of the PAV variants were not investigated further, leaving only PAV-D4 and PAV-E4 as viable design options. Finally, in the presence of MMP, PAV-E4 behaves similarly to PAV-D4 (Figures 7 and 8).
During the course of the study, we observed unexpected results. The AAV2 capsid does not well-tolerate the insertion of positively charged 4AA motifs in the HSPG binding domain, as PAV-K4 and PAV-R4 display the lowest viral titers (Figure 2). In particular, PAV-K4 displays an irregular Western blot pattern (Figure 3) and the capsid is unable to protect its genome effectively (Figure 4). PAV-R4, on the other hand, has the lowest titer, but the capsid that does form is able to protect its genome similarly to wt (Figure 4).
Collectively, our data suggest that both electrostatic repulsion and steric obstruction between the 4AA motif and HSPG may make nonredundant contributions to the peptide lock being able to lock the capsid successfully. Negatively and positively charged motifs, and even small hydrophobic A4 and uncharged polar SG2S motifs, are able to lock the capsid, which would suggest steric obstruction plays an important role. However, the uncharged polar N4 and Q4 motifs, which are comparable in size to the D4 and E4 motifs but lacking negative charge, yield only partial ablation of receptor binding and cellular transduction, which may suggest that steric obstruction alone is not sufficient to block receptor binding. One potential explanation could be that the polar uncharged motif-containing peptide locks, although genetically inserted into the HSPG binding domain, do not assume the correct structural configuration to effectively block HSPG binding through sterics. Determining the structures of some of these PAV variants would likely shed light on the reasons for these experimental results. Differences in the amino acid sequence of the peptide insertion may account for the differing mechanisms behind the Perabo et al. report and the PAV mutants described here.
Future work will address remaining unanswered questions about the PAV design. First, structural studies must be performed to understand why peptide locks with four negatively charged amino acid residues are well-tolerated by the AAV capsid, whereas inserting virtually an identical lock (with the only difference being four positively charged residues) hinders capsid assembly and leads to poor capsid production. Furthermore, it is presently unclear if the 4AA motif is required at all, or exactly how many amino acid residues in the motif are needed to ablate cellular transduction. Additionally, an essential and universal cellular receptor for AAV infection, named AAVR, was recently discovered.25 Whether the peptide locks interfere with AAVR binding by AAV2 remains to be determined. Lastly, creating PAVs based on other AAV serotype capsids will reveal whether the uncovered design rules are broadly applicable to AAV-based PAVs in general.
CONCLUSIONS
The overall goal of this work was to investigate the role of the 4AA motif’s chemical properties on PAV function. Interestingly, we found motifs of various properties are able to lock the AAV2 capsid. However, due to suboptimal production titers, potentially increased susceptibility to trypsin, and poor genome protection of majority of the mutants, the PAV-E4 and PAV-D4 variants appear to be the best design options so far. Continued investigation of the PAV platform may further support the translation of this promising gene delivery technology for a variety of diseases characterized by elevated levels of extracellular proteases at target tissue sites.
Acknowledgments
Funding
This material is based upon work supported by the National Science Foundation under grant 0955536, Cancer Prevention Research Institute of Texas under grant RP130455, the American Heart Association under grants 13GRNT14420044 and 15GRNT23070007, the National Institutes of Health under grant numbers R21CA187316 and R21HL126053, and a John S. Dunn Foundation award to J.S.; a National Institutes of Health Nanobiology Interdisciplinary Graduate Training Program Fellowship (T32EB009379), American Heart Association Predoctoral Fellowship (16PRE27760178), and an Alliance for Graduate Education Professoriate Fellowship from Rice University to T.M.R.; and a National Institutes of Health Cancer Nanotechnology Fellowship (T32CA196561) to M.L.H.
The authors thank Joanna Yang for assistance with the table of contents graphic illustration. The authors acknowledge the University of North Carolina at Chapel Hill Gene Therapy Center Vector Core for providing us with pXX2, pXX6-80, and scAAV2-CMV-GFP.
ABBREVIATIONS
- AAV
adeno-associated virus
- CMV
cytomegalovirus
- GFP
green fluorescent protein
- HSPG
heparan sulfate proteoglycan
- HEK
human embryonic kidney cells
- MMP
matrix-metalloproteinase
- MOI
multiplicity of infection
- qPCR
quantitative polymerase chain reaction
- PAV
protease-activatable virus
- PAV-D4
protease-activatable virus with tetra-aspartic acid motif
- PAV-E4
protease-activatable virus with tetra-glutamic acid motif
- PAV-K4
protease-activatable virus with tetra-lysine motif
- PAV-R4
protease-activatable virus with tetra-arginine motif
- PAV-A4
protease-activatable virus with tetra-alanine motif
- PAV-N4
protease-activatable virus with tetra-asparagine motif
- PAV-Q4
protease-activatable virus with tetra-glutamine motif
- PAV-SG2S
protease-activatable virus with serine-glycine-glycine-serine motif
- ssDNA
single-stranded DNA
- VP
viral protein
- WT
wild-type
Footnotes
Author Contributions
T.M.R. planned and conducted experiments, and collected and analyzed data. J.J. and M.L.H. planned and conducted experiments and analyzed data. J.S. analyzed data and directed the work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Notes
The authors declare no competing financial interest.
References
- 1.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2(3):161–74. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 2.Thomas CV, Coker ML, Zellner JL, Handy JR, Crumbley AJ, III, Spinale FG. Increased matrix metalloproteinase activity and selective upregulation in LV myocardium from patients with endstage dilated cardiomyopathy. Circulation. 1998;97(17):1708–15. doi: 10.1161/01.cir.97.17.1708. [DOI] [PubMed] [Google Scholar]
- 3.Gialeli C, Theocharis AD, Karamanos NK. Roles of matrix metalloproteinases in cancer progression and their pharmacological targeting. FEBS J. 2011;278(1):16–27. doi: 10.1111/j.1742-4658.2010.07919.x. [DOI] [PubMed] [Google Scholar]
- 4.Hadler-Olsen E, Winberg JO, Uhlin-Hansen L. Matrix metalloproteinases in cancer: their value as diagnostic and prognostic markers and therapeutic targets. Tumor Biol. 2013;34(4):2041–51. doi: 10.1007/s13277-013-0842-8. [DOI] [PubMed] [Google Scholar]
- 5.Roy R, Yang J, Moses MA. Matrix metalloproteinases as novel biomarkers and potential therapeutic targets in human cancer. J Clin Oncol. 2009;27(31):5287–97. doi: 10.1200/JCO.2009.23.5556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Szarvas T, vom Dorp F, Ergun S, Rubben H. Matrix metalloproteinases and their clinical relevance in urinary bladder cancer. Nat Rev Urol. 2011;8(5):241–54. doi: 10.1038/nrurol.2011.44. [DOI] [PubMed] [Google Scholar]
- 7.Schulz R. Intracellular targets of matrix metalloproteinase-2 in cardiac disease: rationale and therapeutic approaches. Annu Rev Pharmacol Toxicol. 2007;47:211–42. doi: 10.1146/annurev.pharmtox.47.120505.105230. [DOI] [PubMed] [Google Scholar]
- 8.Wang W, Schulze CJ, Suarez-Pinzon WL, Dyck JR, Sawicki G, Schulz R. Intracellular action of matrix metalloproteinase-2 accounts for acute myocardial ischemia and reperfusion injury. Circulation. 2002;106(12):1543–1549. doi: 10.1161/01.cir.0000028818.33488.7b. [DOI] [PubMed] [Google Scholar]
- 9.Ge J, Min SH, Kim DH, Lee DC, Park KC, Yeom YI. Selective gene delivery to cancer cells secreting matrix metal-loproteinases using a gelatin/polyethylenimine/DNA complex. Bio-technol Bioprocess Eng. 2012;17(1):160–167. [Google Scholar]
- 10.Gjetting T, Jolck RI, Andresen TL. Effective nanoparticle-based gene delivery by a protease triggered charge switch. Adv Healthcare Mater. 2014;3(7):1107–18. doi: 10.1002/adhm.201300503. [DOI] [PubMed] [Google Scholar]
- 11.Hartl I, Schneider RM, Sun Y, Medvedovska J, Chadwick MP, Russell SJ, Cichutek K, Buchholz CJ. Library-based selection of retroviruses selectively spreading through matrix metalloprotease-positive cells. Gene Ther. 2005;12(11):918–26. doi: 10.1038/sj.gt.3302467. [DOI] [PubMed] [Google Scholar]
- 12.Springfeld C, von Messling V, Frenzke M, Ungerechts G, Buchholz CJ, Cattaneo R. Oncolytic efficacy and enhanced safety of measles virus activated by tumor-secreted matrix metalloproteinases. Cancer Res. 2006;66(15):7694–7700. doi: 10.1158/0008-5472.CAN-06-0538. [DOI] [PubMed] [Google Scholar]
- 13.Evans AC, Thadani NN, Suh J. Biocomputing nanoplatforms as therapeutics and diagnostics. J Controlled Release. 2016 doi: 10.1016/j.jconrel.2016.01.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Szecsi J, Drury R, Josserand V, Grange MP, Boson B, Hartl I, Schneider R, Buchholz CJ, Coll JL, Russell SJ, Cosset FL, Verhoeyen E, et al. Targeted retroviral vectors displaying a cleavage site-engineered hemagglutinin (HA) through HA-protease interactions. Mol Ther. 2006;14(5):735–44. doi: 10.1016/j.ymthe.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 15.Kotterman MA, Schaffer DV. Engineering adeno-associated viruses for clinical gene therapy. Nat Rev Genet. 2014;15(7):445–51. doi: 10.1038/nrg3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Santiago-Ortiz JL, Schaffer DV. Adeno-associated virus (AAV) vectors in cancer gene therapy. J Controlled Release. 2016 doi: 10.1016/j.jconrel.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Judd J, Ho ML, Tiwari A, Gomez EJ, Dempsey C, Van Vliet K, Igoshin OA, Silberg JJ, Agbandje-McKenna M, Suh J. Tunable Protease-Activatable Virus Nanonodes. ACS Nano. 2014;8(5):4740–4746. doi: 10.1021/nn500550q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ho ML, Judd J, Kuypers BE, Yamagami M, Wong FF, Suh J. Efficiency of Protease-Activatable Virus Nanonodes Tuned Through Incorporation of Wild-Type Capsid Subunits. Cell Mol Bioeng. 2014;7(3):334–343. [Google Scholar]
- 19.Opie SR, Warrington KH, Jr, Agbandje-McKenna M, Zolotukhin S, Muzyczka N. Identification of amino acid residues in the capsid proteins of adeno-associated virus type 2 that contribute to heparan sulfate proteoglycan binding. Journal of virology. 2003;77(12):6995–7006. doi: 10.1128/JVI.77.12.6995-7006.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ho ML, Adler BA, Torre ML, Silberg JJ, Suh J. SCHEMA Computational Design of Virus Capsid Chimeras: Calibrating How Genome Packaging, Protection, and Transduction Correlate with Calculated Structural Disruption. ACS Synth Biol. 2013;2(12):724–733. doi: 10.1021/sb400076r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waley SG, Watson J. The action of trypsin on polylysine. Biochem J. 1953;55(2):328–37. doi: 10.1042/bj0550328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kaludov N, Brown KE, Walters RW, Zabner J, Chiorini JA. Adeno-associated virus serotype 4 (AAV4) and AAV5 both require sialic acid binding for hemagglutination and efficient transduction but differ in sialic acid linkage specificity. Journal of virology. 2001;75(15):6884–93. doi: 10.1128/JVI.75.15.6884-6893.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Summerford C, Samulski RJ. Membrane-associated heparan sulfate proteoglycan is a receptor for adeno-associated virus type 2 virions. J Virol. 1998;72(2):1438–1445. doi: 10.1128/jvi.72.2.1438-1445.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perabo L, Goldnau D, White K, Endell J, Boucas J, Humme S, Work LM, Janicki H, Hallek M, Baker AH, Buning H. Heparan sulfate proteoglycan binding properties of adeno-associated virus retargeting mutants and consequences for their in vivo tropism. J Virol. 2006;80(14):7265–9. doi: 10.1128/JVI.00076-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pillay S, Meyer NL, Puschnik AS, Davulcu O, Diep J, Ishikawa Y, Jae LT, Wosen JE, Nagamine CM, Chapman MS, Carette JE. An essential receptor for adeno-associated virus infection. Nature. 2016;530(7588):108–12. doi: 10.1038/nature16465. [DOI] [PMC free article] [PubMed] [Google Scholar]