

Abstract
Kelch-like protein 6 (KLHL6) is an uncharacterized gene mutated in diffuse large B-cell lymphoma (DLBCL). We report that KLHL6 assembles with CULLIN3 to form a functional CULLIN-Ring ubiquitin ligase. Mutations of KLHL6 inhibit its ligase activity by disrupting the interaction with CULLIN3. Loss of KLHL6 favors DLBCL growth and survival both in vitro and in xenograft models. We further established the mRNA decay factor Roquin2 as a substrate of KLHL6. Degradation of Roquin2 is dependent on B-cell receptor activation, and requires the integrity of the tyrosine 691 in Roquin2 that is essential for its interaction with KLHL6. A non-degradable Roquin2 (Y691F) mutant requires its RNA binding ability to phenocopy the effect of KLHL6 loss. Stabilization of Roquin2 promotes mRNA decay of the tumor suppressor and NF-κB pathway inhibitor, tumor necrosis factor-α-inducible gene 3 (TNFAIP3). Collectively, our findings uncover the tumor suppressing mechanism of KLHL6.
Introduction
B-cell cancers hijack protein ubiquitylation and degradation to promote growth and survival, as shown by the successful use of a proteasome inhibitor (bortezomib) and E3 ubiquitin ligase inhibitor (lenalinomide) in multiple myeloma (MM) and mantle cell lymphoma1. Despite the significant progress, much remains to be explored in the field of ubiquitin and molecular mechanisms of tumorigenesis.
Kelch-like protein 6 (KLHL6) is a member of the bric-a-brac/tramtrack/broad-complex (BTB) domain family of proteins with a lymphoid tissue-restricted expression pattern2,3. Whole-genome and exome sequencing have revealed cancer-associated mutations of the KLHL6 gene in B-cell malignancies, including diffuse large B-cell lymphoma (DLBCL)4–7; however, the relevance of these mutations as well as the molecular function of KLHL6 is currently unknown.
DLBCL is the most common type of lymphoid malignancies with two distinct molecular subtypes: activated B cell-like (ABC) and germinal center B cell-like (GCB) lymphoma8, 9. ABC-DLBCLs depend on hyper-activation of the inhibitor of IκB kinase (IKK) and the NF-κB transcription factor program for their proliferation and survival10, 11. This is evidenced by frequent mutations in the BCR pathway, including activating mutations of positive (CD79A/B and CARD1112, 13) and inactivating mutations of negative (TNFAIP312–15) NF-κB regulators. How KLHL6 contributes to the pathology of human DLBCL and whether it influences NF-κB activation are currently unknown.
Regulatory networks that promote cancer progression modulate gene expression at the level of mRNA stability16, 17. The RNA‐binding proteins RC3H1 and RC3H2 (from now on Roquin1 and Roquin2) promote mRNA decay via recognition of stem–loop motifs in the 3′ untranslated region (UTR) of target mRNAs18, 19. Through this recognition, Roquins recruit the CCR4-CAF1-NOT complex, leading to mRNA deadenylation and subsequent destabilization18–22. In T-cells, Roquin proteins contribute to immune homeostasis by promoting decay of Inducible T-cell Costimulator (ICOS). However, the role of Roquin in B-cell cancers has not been investigated18, 23.
Here, we demonstrate that KLHL6 is an E3 ligase for Roquin2. Cancer-associated mutations of KLHL6 inhibit its ubiquitin ligase activity and inactivation or loss of KLHL6 in ABC-DLBCL promotes cancer cell growth and survival through stabilization of Roquin2 and subsequent decay of the TNFAIP3 mRNA. This study shows how ABC-DLBCL cells hijack the ubiquitin pathway to promote their proliferation via alteration of the mRNA decay process.
Results
KLHL6 mutations in human DLBCL abolish its catalytic function as cullin3-RING-ligase complex (CRL3)
Analysis of genomic databases of human mature B-cell cancer patients revealed mutations of the KLHL6 gene in DLBCL (http://cancergenome.nih.gov/ and4,5,6,7), chronic lymphocytic leukemia (CLL)24 and multiple myeloma (MM)25 (Fig. 1a). DLBCL cohorts displayed the highest rate of genetic mutations (Fig. 1a), which are similarly stratified amongst GCB-DLBCL, ABC-DLBCL and uncharacterized DLBCL (Fig. 1b). Most mutations in DLBCL are missense and monoallelic, with a low number in non-sense and frameshift mutations (Fig. 1c, Supplementary Table 1 and4,5,6,7). Majority of mutations clusters near and inside the BTB-domain of KLHL6 with mutational hotspots in Leucines 65 and 90 (Fig. 1c). Moreover, re-analysis of published SNP array data26 revealed infrequent deletion of the KLHL6 locus (Fig. S1a), while ~6% of DLBCL tumors displayed lower expression of KLHL6 transcript (Fig. S1b).
Figure 1. KLHL6 mutations in human DLBCL abolish its catalytic function as CRL3.
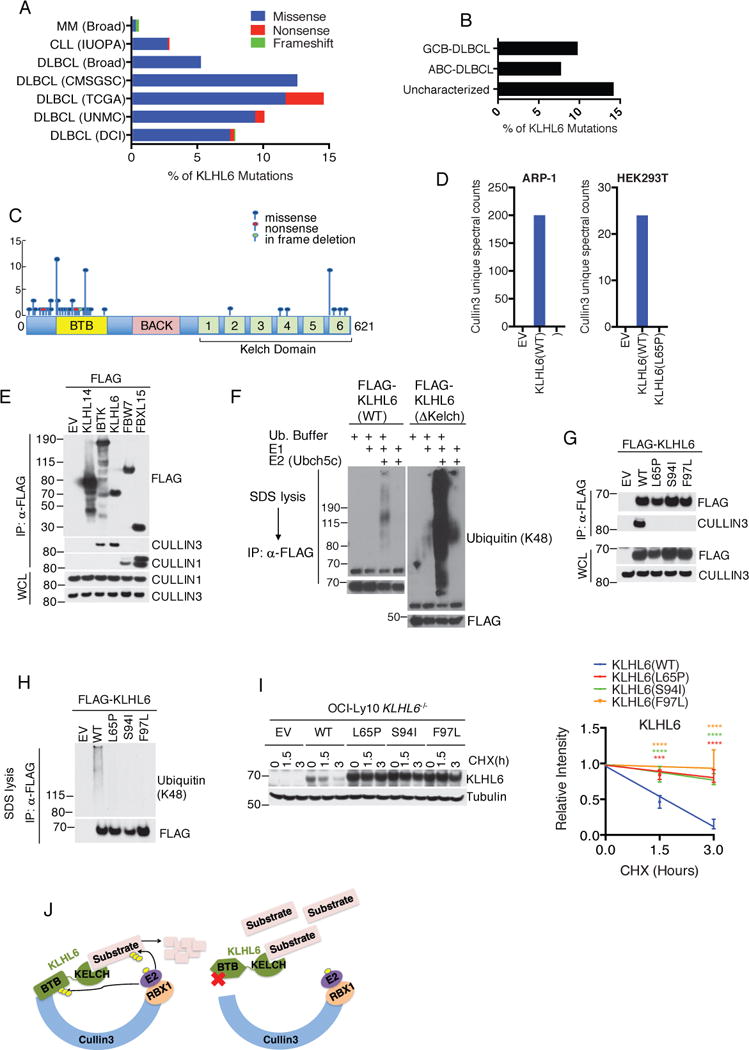
(a) Percentage of KLHL6 mutations in patients with diffuse large B-cell lymphoma (DLBCL) [University of Nebraska Medical Center (UNMC), n=140 patients, The Cancer Genome Atlas (TCGA), n=48, Canada’s Michael Smith Genome Sciences Centre (CMSGSC), n= 96, and Broad Institute (Broad), n=58, Duke Cancer Institute (DCI), n= 1001], Chronic Lymphocytic Leukemia (CLL) [Departamento de Bioquímica y Biología Molecular, Instituto Universitario de Oncología (IUOPA), n=586] and Multiple Myeloma (MM) (Broad), n=205].
(b) Percentage of KLHL6 mutations in DLBCL subtypes. Cohorts from UNMC and CMSGSC were pooled and sub-classified as Activated B-cell-like (ABC)-, Germinal Center B-cell-like (GC)- and Uncharacterized-DLBCL.
(c) Schematic representation of KLHL6 mutations (BTB, Broad-Complex, Tramtrack and Bric-a-brac; BACK, BTB and C-terminal Kelch plant homeodomain).
(d) Proteomic analysis of KLHL6 complex. Spectral counts for CULLIN3 are shown. EV, empty vector. This analysis was performed once in HEK293T and ARP-1 cells.
(e) Immunoblot analysis of immunoprecipitated FLAG-tagged E3 ligases in HEK293T cells. WCL, whole cell lysates. EV, empty vector.
(f) In vitro ubiquitylation reaction of immunopurified FLAG-KLHL6 and ΔKelch mutant.
(g) Immunoblot analysis of immunoprecipitated FLAG-tagged KLHL6 wild-type (WT), BTB-mutants (L65P, S94I and F97L), or empty vector (EV) in HEK293T cells.
(h) In vitro ubiquitylation reaction of immunopurified FLAG-KLHL6 wild-type (WT) and BTB-mutants (L65P, S94I and F97L).
(i) Immunoblot analysis of whole cell lysates from GFP+ OCI-Ly10 KLHL6−/− cells (clone-derived) retrovirally transduced with cDNAs encoding an empty vector (EV), KLHL6(WT) or BTB-mutants (L65P, S94I and F97L) and carrying a GFP marker (left panel). Cells were treated with cycloheximide (CHX) for the indicated times. Right panel shows quantification of KLHL6 protein levels (mean±s.d., n=3 independent experiments, two-way ANOVA, *** P value≤0.001; ****P value≤0.0001).
(j) Schematic model of Cullin3-Ring-Ligase (CRL3)-KLHL6 complex.
Unprocessed original scan of immunoblots for (e,f,g,h,i) are shown in Supplementary Fig. 8, and statistical source data and exact P values for (i) can be found in Supplementary Table 6. Unless otherwise noted, immunoblots are representative of three independent experiments.
To understand the impact of these cancer-associated mutations, we compared the protein interactome of KLHL6(WT) to that of the cancer mutant KLHL6(L65P). FLAG-KLHL6(WT) or FLAG-KLHL6(L65P) complexes were immunopurified from two cell lines (HEK293T and ARP-1) and the tryptic digestion of each protein eluate was analyzed by mass spectrometry (Supplementary Table 2). Unique spectral counts corresponding to CULLIN3 were identified in KLHL6(WT), but not in KLHL6(L65P) immunoprecipitates (Fig. 1d and Supplementary Table 2).
We validated our proteomic analysis via immunoprecipitation of KLHL6 with endogenous CULLIN3, but not CULLIN1, in HEK293T cells (Fig. 1e). By carrying out an in vitro ubiquitylation assay, we found that KLHL6 promoted self-ubiquitylation and, notably, its BTB-domain alone [KLHL6(ΔKelch)] was sufficient in catalyzing self-polyubiquitylation to a greater degree (Fig. 1f). These data suggest that KLHL6 assembles a functional CULLIN3-RING ubiquitin Ligase (CRL3)27 (Fig. 1j).
We then investigated the effect of DLBCL-associated BTB-domain mutations on KLHL6 ligase assembly and activity. All mutations tested (L65P, S94I, F97L) disrupted binding to CULLIN3 (Fig. 1g) and consequently led to a loss of self-polyubiquitylation in vitro (Fig. 1h). Correspondingly, the protein levels of BTB-domain KLHL6 mutants were remarkably high at steady state and displayed extended half-lives as compared to those of wild-type (Fig. 1i), suggesting loss of KLHL6 self-ubiquitylation affects its turnover in cells.
KLHL6 interacts with and promotes the ubiquitylation and degradation of Roquin2
Since KLHL6(L65P) is unable to promote ubiquitylation, we reasoned that it might trap (i.e. interact with, but not ubiquitylate) substrates (Fig. 1j). By ranking proteins by the number of unique spectral counts, we identified Roquin2 as a potential substrate (Fig. 2a and Supplementary Table 2).
Figure 2. KLHL6 interacts and promotes ubiquitylation and degradation of Roquin2.
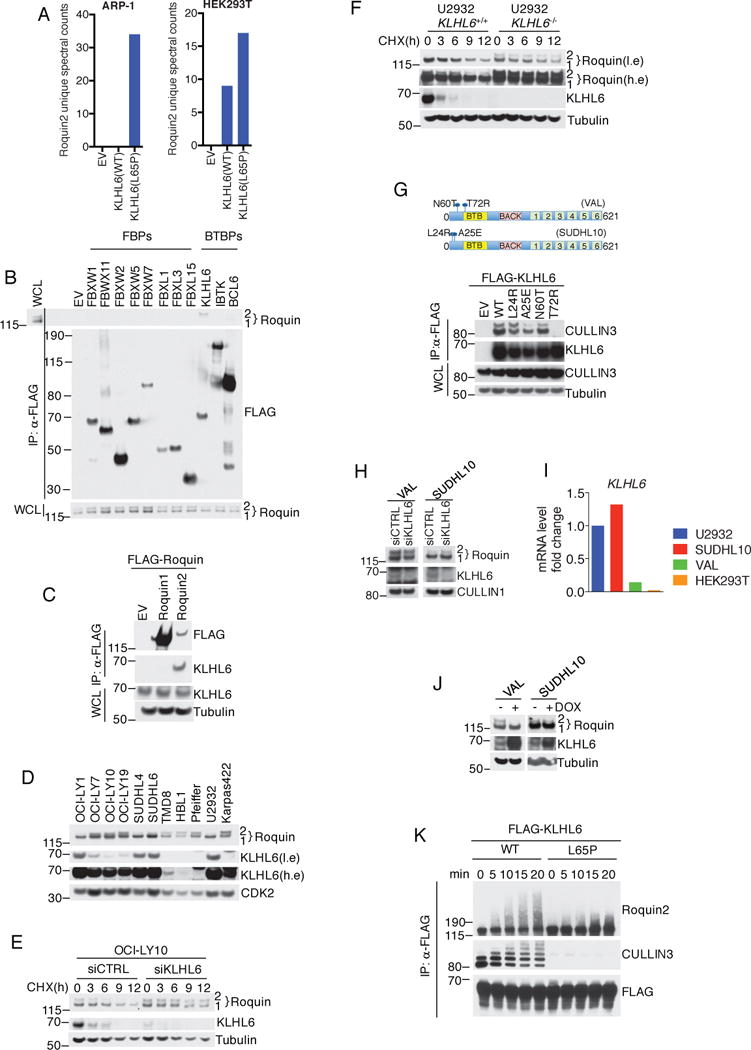
(a) Proteomic analysis of KLHL6 immunoprecipitations. Spectral counts for Roquin2 proteins are shown. EV, empty vector. The analysis was performed once in two different cell lines (HEK293T and ARP-1).
(b) Immunoblot analysis of immunoprecipitated FLAG-F-box proteins (FBPs) or BTB proteins (BTBPs) in HEK293T cells. Lane 1 shows whole cell lysates (WCL) from cells transfected with an empty vector.
(c) Immunoblot analysis of immunoprecipitated FLAG-Roquin1 or FLAG-Roquin2 in HEK293T cells.
(d) Immunoblot analysis of human DLBCL cell lysates. A low exposure (l.e.) and high exposure (h.e.) are shown. A representative blot from two independent experiments is shown.
(e) Immunoblot analysis of whole cell lysates from OCI-LY10 cells electroporated with indicated siRNAs and treated with cycloheximide (CHX). Quantification and statistical analysis is shown in Fig. S2d.
(f) KLHL6+/+ and KLHL6−/− U2932 cells(clone-derived) were processed as in (e). A low exposure (l.e.) and high exposure (h.e.) are shown. Quantification and statistical analysis is shown in Fig. S2d.
(g) Schematic representation of KLHL6 protein displaying endogenous mutations in VAL and SUDHL10 (top panel). Bottom panel shows immunoblot analysis from immunoprecipitated FLAG-tagged KLHL6 wild-type (WT), KLHL6 mutants (L24R, A25E, N60T, and T72R), or empty vector (EV) in HEK293T cells.
(h) Immunoblot analysis of whole cell lysates from VAL and SUDHL10 cells electroporated with siRNA scramble (siCTRL) or targeting KLHL6 (siKLHL6).
(i) Analysis of level of KLHL6 mRNA by quantitative PCR(qPCR). The value for the PCR product from U2932 cells was set as 1. A representative graph from two independent experiments is shown.
(j) Immunoblot analysis of whole cell lysates from VAL and SUDHL10 cells stably expressing KLHL6 under a doxycycline (DOX) inducible promoter with a puryomycin cassette after 12h of DOX treatment.
(k) In vitro ubiquitylation reaction of immunopurified FLAG-KLHL6 and HA-Roquin2.
Unprocessed original scan of immunoblots for (b,c,d,e,f,g,h,j,k) are shown in Supplementary Fig. 8, and source data for (i) can be found in Supplementary Table 6. Unless otherwise noted, immunoblots are representative of three independent experiments.
In agreement with our proteomic data, KLHL6 specifically co-immunoprecipitated endogenous Roquin2 in HEK293T cells (Fig. 2b). The reciprocal co-immunoprecipitation experiments confirmed Roquin2, but not Roquin1, as a KLHL6 interactor (Fig. 2c). This complex was also detectable at an endogenous level in DLBCL cells (Fig. S2a). Importantly, interaction between KLHL6 and Roquin2 required the intact Kelch domain in KLHL6 (Fig. S2b), further supporting substrate-like interaction28.
In DLBCL cell lines, abundance of KLHL6 and Roquin2 displayed an inverse correlation (Fig. 2d and S2c). To investigate whether KLHL6 controls Roquin2 protein levels, we assessed Roquin2 protein turnover upon knockdown or knockout of KLHL6 in OCI-LY10 and U2932 cells, respectively. In both cases, down-regulation of KLHL6 significantly extended the half-life of Roquin2, but not that of Roquin1 (Fig. 2e, 2f and S2d).
Gain-of-function experiments showed that re-expression of KLHL6 in cell lines with low KLHL6 expression (i.e.; HEK293T and HBL1, see Fig. 5a right panel) down-regulated Roquin2 protein levels (Fig. S2e). Moreover, BTB-domain mutants were incapable of inducing Roquin2 down-regulation (Fig. S2f). Notably, co-expression of KLHL6(WT) along with BTB-mutants still promoted Roquin2 degradation, suggesting these mutations are not dominant negative (Fig. S2g).
Figure 5. KLHL6 is a BCR/NF-κB target gene that links Roquin2 degradation to BCR signaling.
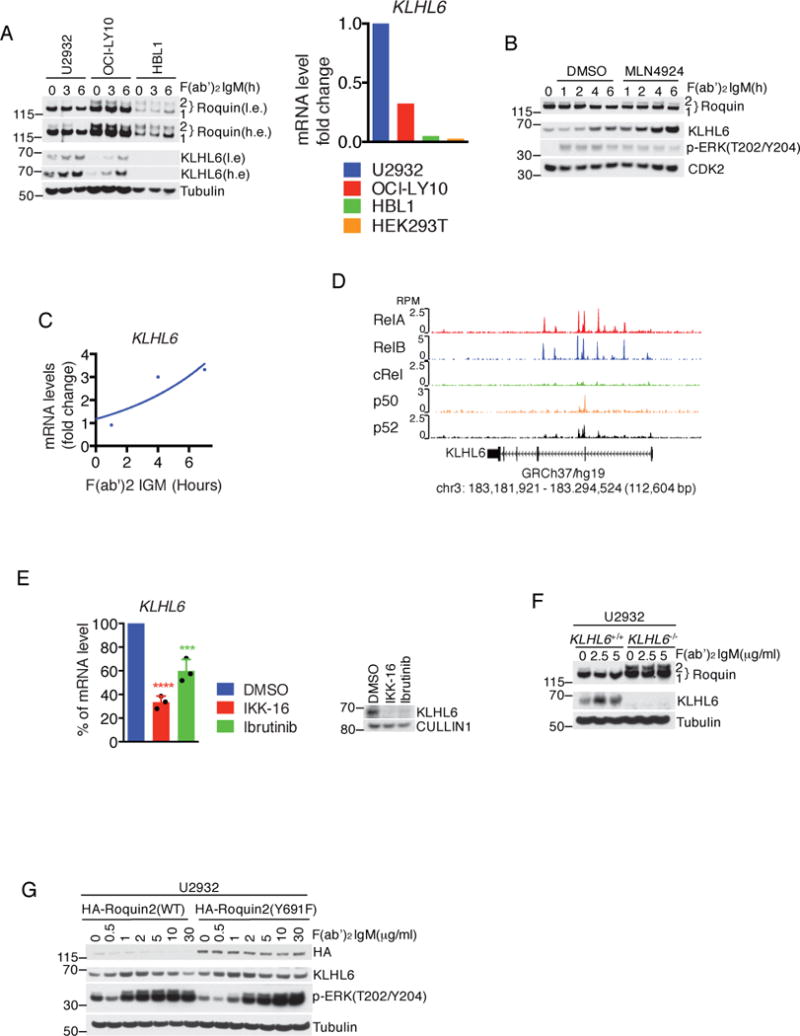
(a) Immunoblot analysis of whole cell lysates from OCI-LY10, U2932, and HBL1 cells stimulated with 10 μg/ml F(ab′)2-IgM for 3 and 6 hours (left panel). A low exposure (l.e.) and high exposure (h.e.) are shown. Right panel shows level of KLHL6 mRNA analyzed by qPCR. The value for the PCR product from U2932 was set as 1. A representative graph from two independent experiments is shown.
(b) Immunoblot analysis of whole cell lysates from U2932 cells treated with 10 μg/ml of F(ab′)2-IgM for the indicated times. Where indicated, cells were pre-treated with 5μM MLN4924 for 1 hour.
(c) Analysis of level of KLHL6 mRNA by qPCR in U2932 cells treated with 10 μg/ml of F(ab′)2-IgM for the indicated times. A representative graph from two independent experiments is shown. The value for PCR product without treatment was set as 1.
(d) Visualization of ChIP-seq peaks using the University of California Santa Cruz (UCSD) Genome browser (GEO Series accession GSE55105). RPM, reads per million mapped.
(e) Same as in (c) except that U2932 cells were treated with DMSO, 10μM of IKK inhibitor (IKK-16) or 5μM of BTK inhibitor (Ibrutinib) for 6 hours. The value for PCR product present without treatment (DMSO) was set as 100% (mean±s.d., n=3 independent experiments, one-way ANOVA, *** P value≤0.001; ****P value≤0.0001). The right panel shows immunoblot analysis of whole cell lysates for the indicated proteins.
(f) Immunoblot analysis of whole cell lysates from U2932 KLHL6+/+ and KLHL6−/− (clone-derived) cells treated with increasing concentrations of F(ab′)2-IgM for 6 hours.
(g) Immunoblot analysis of whole cell lysates from U2932 cells stably expressing HA-Roquin2(WT) or HA-Roquin2(Y691F) treated with F(ab′)2-IgM for 6 hours.
Unprocessed original scan of immunoblots for (a,b,e,f,g) are shown in Supplementary Fig. 8, and source data for (a,c) and statistical source data and exact P values for (e) can be found in Supplementary Table 6. Unless otherwise noted, immunoblots are representative of three independent experiments.
Next, we utilized B-cell lymphoma cell lines harboring endogenous KLHL6 mutations. VAL cells harbor two BTB-mutations: N60T and T72R (Fig. 2g). Binding analysis revealed that only the KLHL6(T72R) mutant lost interaction with CULLIN3 (Fig. 2g), while KLHL6(N60T) mutant did not. Knockdown of KLHL6 in VAL cells did not result in Roquin2 accumulation (Fig. 2h), whereas re-expression of KLHL6 induced Roquin2 downregulation (Fig. 2j). In VAL cells, expression of KLHL6 at the mRNA level was comparatively low (Fig. 2i). This suggests that VAL cells display one KLHL6 allele inactivated by a mutation in the BTB-domain and an additional down-regulation of KLHL6 mRNA. In contrast, mutations of KLHL6 in SUDHL10 cells had no impact on KLHL6 function (Fig. 2g, 2h 2i, and 2j).
Lastly, to explore whether KLHL6 directly controls Roquin2 ubiquitylation in vitro, we incubated immunopurified KLHL6-Roquin2 complex with a ubiquitylation mix. High-molecular weight species of Roquin2 were detected with the KLHL6(WT) complex, but not with KLHL6(L65P) (Fig. 2k). Correspondingly, ubiquitylation of Roquin2 was abolished in KLHL6−/− cell lines in vivo (Fig. S2h).
KLHL6 functions as a tumor suppressor in ABC-DLBCL by regulating its growth and survival
Although mutations of KLHL6 occur at a similar rate in GCB- and ABC-DLBCL (Fig. 1B), low KLHL6 expression correlated with a significantly poorer survival in ABC-DLBCL patients (Fig. 3a) as previously reported29, 30.
Figure 3. KLHL6 functions as a tumor suppressor in ABC-DLBCL by regulating its growth and survival.
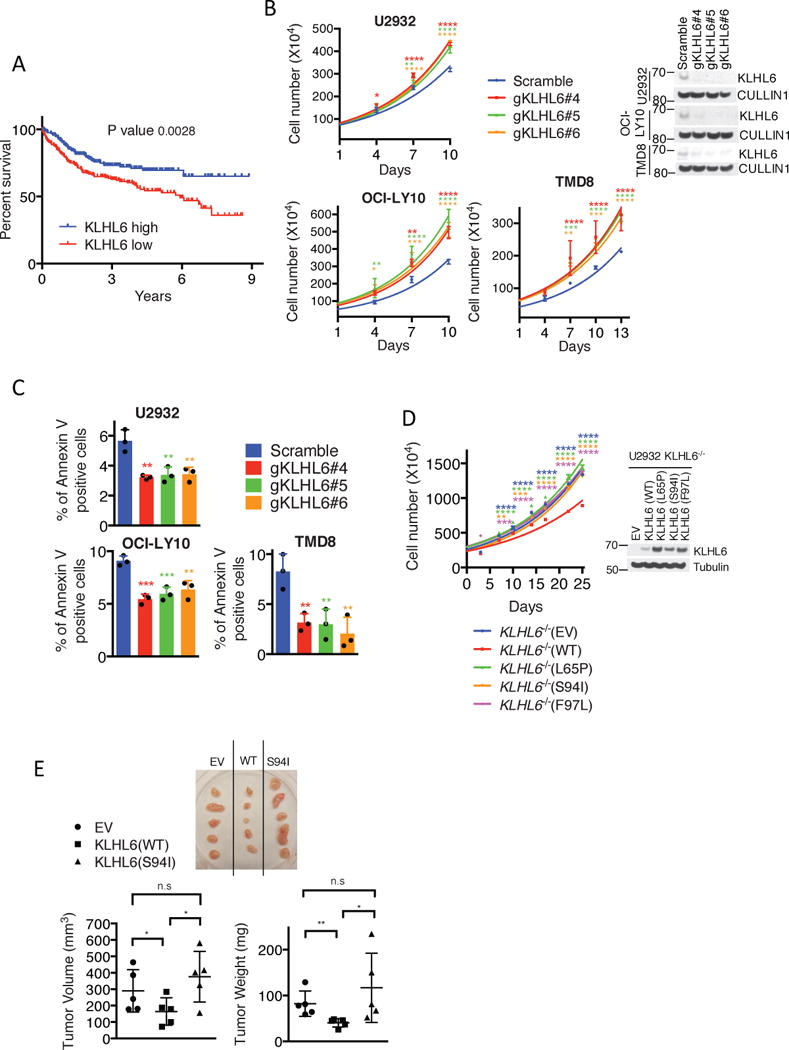
(a) Kaplan–Meier analysis based on gene expression data for ABC-DLBCL tumors (GSE10846, GSE34171 and GSE3131239-41) is shown (n=367 patients). Censored subjects are indicated on the Kaplan-Meier cure as tick mark. Statistical analysis was performed using the Log-rank (Mantel-Cox), two-sided test, 95% confidence interval.
(b) Cell counts of U2932-, OCI-LY10- and TMD8-Cas9 cells expressing the indicated gRNAs and carrying a puromycin cassette (mean±s.d., n=3 independent experiments, two-way ANOVA, *P value≤0.05; **P value≤0.01; *** P value≤0.001; ****P value≤0.0001) (left panel). Cells were grown in media containing 1μg/ml (U2932- and OCI-LY10) or 4μg/ml (TMD8) of F(ab′)2-IgM. Right panel shows immunoblot analysis of whole cell lysates.
(c) Apoptosis analysis of U2932-, OCI-LY10-, and TMD8-Cas9 cells expressing the indicated gRNAs and carrying a GFP marker. Cells were grown as in (b). Apoptosis was quantified on GFP+ and Annexin V+ cells (mean±s.d., n=3 independent experiments, one-way ANOVA, **P value≤0.01; *** P value≤0.001).
(d) Left panel shows cell counts of GFP-sorted U2932 KLHL6−/− (clone-derived) cells expressing empty vector (EV), KLHL6(WT) or BTB-mutants (L65P, S94I and F97L) and carrying a GFP marker (mean±s.d., n=3 independent experiments, two-way ANOVA, **P value≤0.01; *** P value≤0.001; ****P value≤0.0001). Right panel shows the immunoblot analysis of whole cell lysates.
(e) Xenograft experiments with GFP-sorted U2932 KLHL6−/− cells expressing an empty vector (EV), KLHL6(WT) or KLHL6(S94I) and carrying a GFP marker. Top panel shows the tumors at the experimental endpoint. Tumor volume (mean±s.d., n=5 mice per group, one-way ANOVA, *P value≤0.05; n.s, not significant) and tumor weight (mean±s.d., n=5 mice per group, one-tailed t-test, *P value ≤0.05; **P value ≤0.01; n.s, not significant) are shown in the bottom left and right, respectively.
Unprocessed original scan of immunoblots for (b,d) are shown in Supplementary Fig. 8, and statistical source data and exact P values for (a,b,c,d,e) can be found in Supplementary Table 6. Unless otherwise noted, immunoblots are representative of three independent experiments.
To assess the biological effect of KLHL6 loss in ABC-DLBCL lines, we infected Cas9-expressing U2932, OCI-LY10 and TMD8 cells with lentiviruses encoding three different gRNAs targeting the KLHL6 gene locus. Ablation of KLHL6 resulted in an increase in cellular proliferation and a decrease in apoptosis (Fig. 3b and 3c). This effect was confirmed in 3D cultures as measured by a larger number and size of colonies (Fig. S3a and S3b). To rule out the possible off-target effects of gRNAs, we utilized shRNA-mediated knockdown of KLHL6 in U2932 and OCI-LY10 and observed similar results (Fig. S3c–f).
To investigate the role of cancer mutations in cell growth, we re-expressed KLHL6(WT) or KLHL6 BTB-domain mutants or an empty vector (EV) in a clonally-derived U2932 KLHL6−/− cell line (Fig. 3d). While re-expression of KLHL6(WT) decreased cell proliferation, expression of KLHL6 BTB-domain mutants phenocopied loss of KLHL6, confirming these mutations as loss of function. In xenograft models, expression of KLHL6(WT) decreased tumor burden while expression of KLHL6(S94I) displayed a similar tumor burden compared to KLHL6−/− cells (EV) (Fig. 3e).
A non-degradable Roquin2 mutant phenocopies loss of KLHL6
We mapped the KLHL6 binding motif in Roquin2. By performing mutagenesis experiments, we identified a region in Roquin2 between amino acids 640 and 700 as necessary for interaction with KLHL6 (Fig. 4a and 4b). More refined deletions narrowed the interaction domain between amino acids 690 and 695 (Fig. 4c). Alanine scanning mutagenesis of the individual residues surrounding the 691-704 region revealed that a conserved tyrosine residue, in position 691, was required for KLHL6-Roquin2 interaction (Fig. 4d). In vitro binding assays confirmed that a Roquin2 peptide from 686 to 700 directly interacts with KLHL6 (Fig. S4a–d). Mutations of tyrosine 691 into alanine or phenylalanine impaired the ability of Roquin2 to co-immunoprecipitate with KLHL6 in vivo and in vitro (Fig. 4e and S4c), suggesting that the integrity of the tyrosine hydroxyl group is critical for KLHL6 interaction.
Figure 4. A non-degradable Roquin2 mutant phenocopies loss of KLHL6.
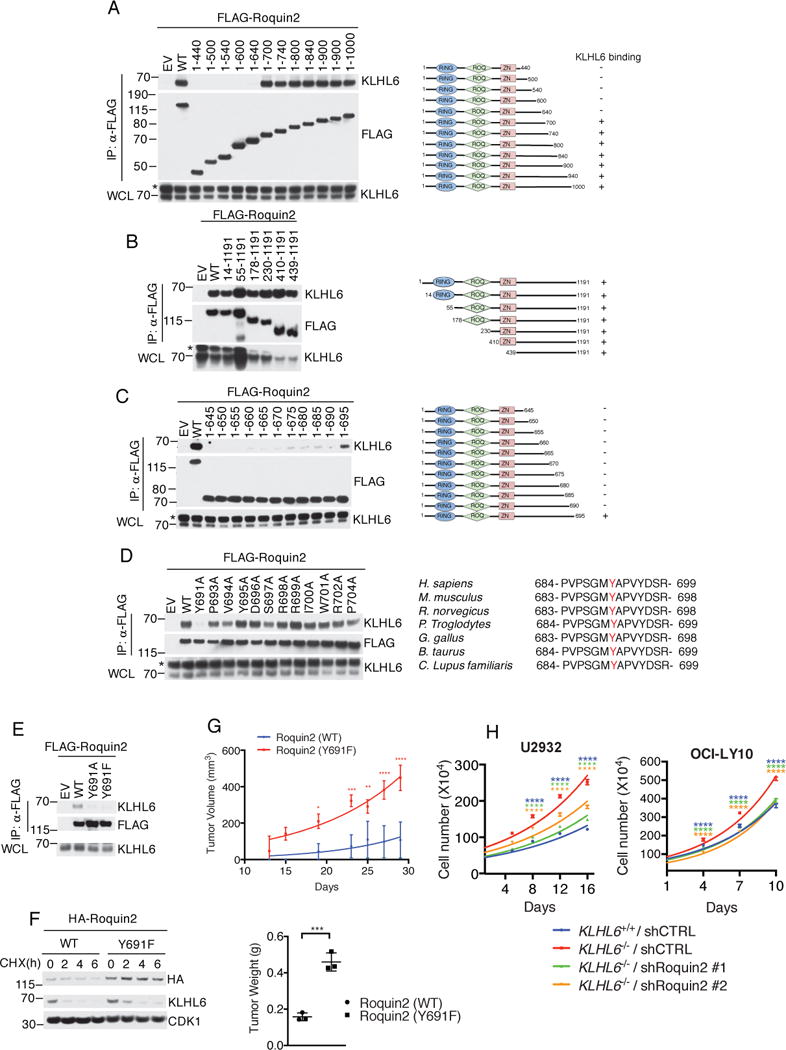
(a) Left panel shows immunoblot analysis of immunoprecipitated FLAG-tagged Roquin2 wild type (WT) or mutants in HEK293T cells stably expressing KLHL6. EV, Empty vector. Right panel shows a schematic representation of Roquin2 mutants. Roquin2 mutants that interact (+) or do not interact (-) with KLHL6 are shown. A representative blot from two independent experiments is shown. Asterisk indicates non-specific bands.
(b–d) Same as in (a).
(e) Immunoblot analysis of immunoprecipitated FLAG-tagged Roquin2 wild type (WT) or mutants, as indicated, in HEK293T cells stably expressing KLHL6. EV, Empty vector.
(f) Immunoblot analysis of whole cell lysates from a DLBCL cell line, BJAB, retrovirally transduced with cDNAs encoding Roquin2(WT) or Roquin2(Y691F) carrying a puromycin cassette. Cells were treated with cycloheximide (CHX) for the indicated times. Quantification and statistical analysis is shown in Fig. S4e.
(g) Top panel shows tumor volume from subcutaneously injected NSG mice with U2932 cells stably expressing retroviruses encoding HA-Roquin2(WT) or HA-Roquin2(Y691F) carrying a puromycin cassette (mean±s.d., n=3 mice per group, two-way ANOVA, *P value≤0.05; **P value≤0.01; *** P value≤0.001; ****P value≤0.0001). Bottom panel shows tumor weight (mean±s.d., n=3 mice per group, two-tailed t-test, *** P value≤0.001).
(h) Cell counts of GFP-sorted U2932 (left panel) or OCI-LY10 (right panel) KLHL6+/+ and KLHL6−/− (clone-derived) cells infected with scramble shRNA(shCTRL) or shRNA targeting Roquin2 (ShRoquin2#1 or #2) carrying a GFP marker. GFP+ cells were grown in media containing 1μg/ml of F(ab′)2-IgM. (mean±s.d., n=3 independent experiments, two-way ANOVA, ****P value≤0.0001).
Unprocessed original scan of immunoblots for (a,b,c,d,e,f) are shown in Supplementary Fig. 8, and statistical source data and exact P values for (g,h) can be found in Supplementary Table 6. Unless otherwise noted, immunoblots are representative of three independent experiments.
We retrovirally-transduced BJAB with Roquin2(WT) or Roquin2(Y691F) to investigate whether tyrosine 691 controls Roquin2 stability in DLBCL cells. Roquin2(Y691F) displayed increased protein levels at steady state as well as an extended half-life when compared to that of Roquin2(WT) (Fig. 4f and S4e). Furthermore, KLHL6(WT) effectively down-regulated protein levels of Roquin2(WT), but not those of Roquin2(Y691F) (Fig. S4f). KLHL6 BTB-mutant(L65P) had no effect on protein levels of Roquin2(WT) and Roquin2(Y691F) (Fig. S4f).
Next, we analyzed the effect of Roquin2 stabilization on DLBCL growth. Expression of the non-degradable Roquin2(Y691F) mutant increased tumor burden as monitored by tumor volume and weight at the experimental endpoint (Fig. 4g). This effect was not an artifact of overexpression because the levels of Roquin2(Y691F) were comparable to those of endogenous Roquin2 in KLHL6−/− cells (Fig. S4g). Moreover, knockdown of Roquin2 impaired the cell growth advantage of KLHL6−/− U2932 and OCI-LY10 cells (Fig. 4h, and S4h and S4i). Importantly, loss of Roquin2 increased toxicity of KLHL6−/− cells preferentially (Fig. S4h), suggesting that loss of KLHL6 promotes cell proliferation in a Roquin2-dependent manner.
KLHL6 is a BCR/NF-κB target gene that links Roquin2 degradation to BCR signaling
KLHL6 is a member of the B-cell Receptor (BCR)-signalosome31 and is induced upon antigen stimulation in the germinal center3. Thus, we investigated whether mRNA and protein levels of KLHL6 and Roquin2 were affected by BCR stimulation. First, we found ABC-DLBCL cells predominantly expressed an IgM-BCR as opposed to GCB-DLBCL cells, which are positive for IgG-BCR (Fig. S5a)32. Then, we analyzed levels of KLHL6 and Roquin2 in IgM-positive ABC-DLBCL cell lines (U2932, OCI-LY10 and HBL1) (Fig 5a). BCR stimulation using the fragment affinity-purified antibody F(ab′)2-IgM induced up-regulation of KLHL6 and a corresponding down-regulation of Roquin2 protein levels in OCI-LY10 and U2932, but not in HBL1 [cell line with low KLHL6 for both mRNA and protein levels (Fig. 5a)].
To demonstrate that BCR-dependent down-regulation of Roquin2 protein depends on CRLs, we pre-treated U2932 cells with MLN4924, a NEDD8-activating enzyme (NAE) inhibitor that blocks Cullin neddylation33. MLN4924 treatment rescued Roquin2 down-regulation induced by BCR-crosslinking, suggesting that a functional CRL-complex is required to promote Roquin2 degradation (Fig. 5b). Notably, BCR stimulation induced KLHL6 up-regulation both at transcriptional and protein levels (Fig. 5a, 5b and 5c).
Since BCR signaling converges on NF-κB activation10, we investigated whether KLHL6 is an NF-κB target gene. Analysis of CHIP-Seq datasets of NF-κB factors34 revealed enrichment of p50, p52, RelA and RelB at the KLHL6 gene locus (Fig. 5d). Correspondingly, treatment of cells with an IKK or a BTK inhibitor (ibrutinib)13, resulted in a down-regulation of KLHL6 both at mRNA and protein levels (Fig. 5e). Interestingly, a reduced sensitivity of KLHL6−/− cells to ibrutinib was observed (Fig. S5b).
BCR-induced Roquin2 degradation was impaired in KLHL6−/− (Fig. 5f) and KLHL6-knockdown cells (Fig. S5c and S5d). Correspondingly, while exogenous Roquin2(WT) was degraded in a dose-dependent manner upon BCR stimulation, Roquin2(Y691F) mutant was not affected (Fig 5g), supporting BCR signaling promotes Roquin2 degradation in a KLHL6-dependent manner.
Stabilization of Roquin2 down-regulate BCR responsive genes
To investigate whether the pro-proliferative effect of the non-degradable Roquin2(Y691F) mutant depends on its RNA binding ability, we generated a double mutant Roquin2(Y691FΔROQ) lacking the ROQ domain. Notably, deletion of the ROQ domain abolished the growth advantage induced by Roquin2(Y691F) expression (Fig. 6a).
Figure 6. Stabilization of Roquin2 down-regulates BCR responsive genes.
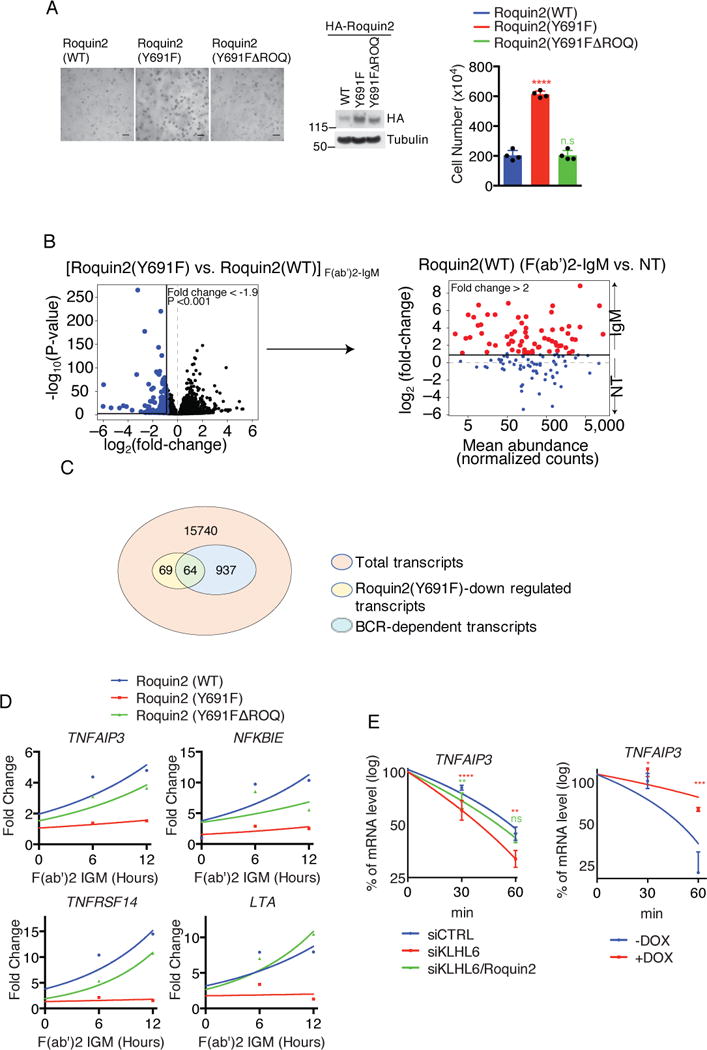
(a) Representative image of U2932 cells expressing HA-Roquin2(WT), HA-Roquin2(Y691F) or HA-Roquin2(Y691FΔROQ) with puromycin cassette plated into a matrigel (Left panel). Middle panel shows immunoblot analysis of whole cell lysates and right panel shows cell counts from the matrigel (mean±s.d., n=4 independent experiments, one-way ANOVA, ****P value≤0.0001, n.s, not significant). Scale bar 150μm.
(b) Volcano plot (left panel) showing down-regulated mRNAs (blue) in U2932 cells expressing Roquin2(Y691F) vs Roquin2(WT) upon 12 hours treatment with 10 μg/ml of F(ab′)2-IgM [log2(fold-change)<–0.9]. Down-regulated mRNAs were further plotted in an MA (log ratio, mean average)-plot (right panel). The mRNAs up-regulated upon treatment with F(ab′)2-IgM in cells expressing Roquin2(WT) [log2(fold-change)>1] are shown in red. (n=3 independent experiments, DEseq2, P value<0.001). NT, non treated.
(c) Venn diagram showing the overlap between genes down-regulated by expression of Roquin2(Y691F) and BCR responsive genes.
(d) qPCR analysis of the indicated mRNAs in U2932 cells expressing HA-Roquin2(WT, Y691F or Y691FΔROQ) treated with 10 μg/ml of F(ab′)2-IgM for indicated times. Value for PCR product present at time 0 hour was set as 1 for each condition. A representative graph from two independent experiments is shown.
(e) qPCR analysis of TNFAIP3 mRNA in U2932 cells electroporated with indicated siRNAs (left panel) and treated with actinomycinD for the indicated times. The value for PCR product present at time 0 hour was arbitrarily set as 100% (mean±s.d., n=3 independent experiments, two-way ANOVA, **P value ≤0.01, ****P value≤0.0001, n.s, not significant). Same analysis was performed in VAL cells expressing KLHL6 under a DOX-inducible promoter (Right panel). Cells were pre-treated with DOX for 12 hours and actinomycinD for the indicated times (mean±s.d., n=3 independent experiments, two-way ANOVA, *P value ≤0.05; *** P value≤0.001).
Unprocessed original scan of immunoblots for (a) are shown in Supplementary Fig. 8, and source data for (d) and statistical source data and exact P values for (a,e) and (b) can be found in Supplementary Table 6 and 3, respectively. Unless otherwise noted, immunoblots are representative of three independent experiments.
Next, we investigated whether misregulation of Roquin2 degradation would result in a deregulation of the BCR transcriptional program. We measured differential gene expression in U2932 cells expressing Roquin2(WT) or Roquin2(Y691F) upon BCR ligation via RNA-seq (Fig. 6b and Supplementary Table 3). Pairwise comparison revealed that 133 mRNAs were significantly down-regulated in Roquin2(Y691F) expressing cells as compared to Roquin2(WT). 64 of these 133 mRNAs overlapped with BCR-responsive genes defined as those with at least a two-fold up-regulation in expression upon BCR stimulation in cells expressing Roquin2(WT) (Fig. 6b and 6c). These 64 genes represented BCR responsive genes that failed to be up-regulated upon BCR stimulation in presence of a non-degradable Roquin2 mutant.
Gene ontology (GO) enrichment analysis (Fig. S6a and Supplementary Table 4) revealed Roquin2 regulated genes are involved in immune and inflammatory responses and implicated as regulators of the NF-κB pathway and lymphoid tumor suppressors (e.g., TNF, NFKBIE, TNFAIP3, LTA, TNFRSF14)14, 35–38. To identify targets relevant to DLBCL biology, we ranked these genes by the percentage of genetic alterations in human DLBCL (TCGA, http://cancergenome.nih.gov/) and the base mean expression in our RNA-Seq analysis (Fig. S6b and Supplementary Table 3). We validated the top 11 candidates in a secondary screen and found 7 of them were dependent on a functional ROQ domain (Fig. S6b and c).
Amongst these targets, we focused on tumor necrosis factor-α-inducible gene 3 (TNFAIP3) because it is frequently inactivated in ABC-DLBCLs12–15 and is a direct target of the Roquin proteins19. We found that TNFAIP3 was up-regulated upon BCR stimulation consistent with its function as a negative regulator of the NF-κB program (Fig. 6d)39. This response was abolished in cells expressing Roquin2(Y691F), suggesting that stabilization of Roquin2 contributes to a reduction of TNFAIP3 mRNA levels during BCR signaling. This effect was rescued partially in cells expressing Roquin2(Y691FΔROQ) as a build-up of TNFAIP3 mRNA levels was observed. Notably, other NF-κB target genes (NFKBIE and LTA) and the tumor suppressor gene TNFRSF14 displayed a similar pattern.
Lastly, we investigated whether the KLHL6-Roquin2 axis directly controls TNFAIP3 mRNA stability. Ablation of KLHL6 shortened the half-life of TNFAIP3, which was partially rescued by concomitant ablation of Roquin2 (Fig. 6e). Re-expression of KLHL6(WT) in VAL, which carries endogenous BTB-mutations, also increased the half-life of TNFAIP3 (Fig. 6e).
KLHL6-Roquin2 axis controls NF-κB activation
Since KLHL6 regulates TNFAIP3 mRNA levels, we hypothesized that loss of KLHL6 would lead to increased NF-κB activation in ABC-DLBCL. First, we investigated whether the TNFAIP3 transcriptional changes would reflect similar changes in protein levels. BCR dependent degradation of Roquin2(WT) inversely correlated with the up-regulation of TNFAIP3 levels, which was reduced in cells stably expressing Roquin2(Y691F) (Fig. 7a). Correspondingly, TNFAIP3 protein levels were down-regulated in KLHL6−/− cells, both at steady state and in response to BCR stimulation (Fig. 7b). Knockdown of Roquin2 in U2932 KLHL6−/− cells rescued TNFAIP3 levels similar to those of KLHL6+/+ cells (Fig. S7a). Likewise, depletion of Roquin2 more robustly upregulated of TNFAIP3 protein levels in HBL1 cells (Fig. 7c). Correspondingly, re-expression of KLHL6(WT) in U2932 KLHL6−/− cells increased TNFAIP3 levels (Fig. S7b).
Figure 7. KLHL6-Roquin2 axis controls NF-κB activation.
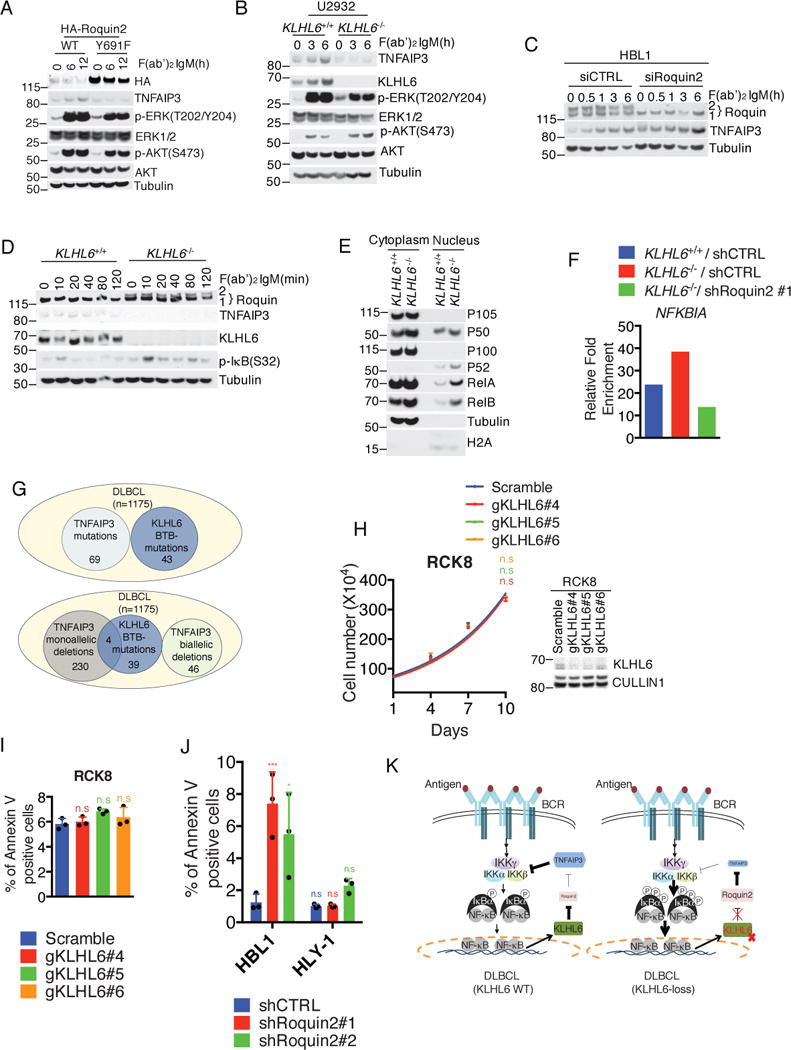
(a) Immunoblot analysis of whole cell lysates from U2932 cells expressing HA-Roquin2(WT) or HA-Roquin2(Y691F) treated with 10μg/ml of F(ab′)2-IgM for the indicated times.
(b) Immunoblot analysis of whole cell lysates from U2932 KLHL6+/+ or KLHL6−/− cells (clone-derived) treated with 10μg/ml of F(ab′)2-IgM for the indicated times.
(c) Immunoblot analysis of whole cell lysates from HBL1 cells electroporated with indicated siRNAs and treated as in (a) for the indicated times.
(d) Immunoblot analysis of whole cell lysates from U2932 KLHL6+/+ or KLHL6−/− cells (clone-derived) treated with 10μg/ml of F(ab′)2-IgM for the indicated times.
(e) Immunoblot analysis of fractionated U2932 KLHL6+/+ or KLHL6−/− (clone-derived) cells.
(f) RelA ChIP-qPCR for the NFBKIA promoter in U2932 KLHL6+/+, KLHL6−/− or KLHL6−/− cells infected with indicated shRNAs. Data are displayed as fold enrichment relative to IgG control. A representative graph from two independent experiments is shown.
(g) Overlap of BTB-associated mutations of KLHL6 withTNFAIP3 alterations in DLBCLs. Top panel shows tumors sequenced at UNMC6 and DCI7 (n=1175) with deleterious mutations of KLHL6 in the BTB-domain and TNFAIP3 mutations. In the bottom panel, the TNFAIP3 subset is shown as biallelic and monoallelic deletions.
(h) Cell counts of GFP-sorted RCK8 cells expressing Cas9, the indicated gRNAs and a GFP marker. Cells were grown in media containing 1μg/ml of F(ab′)2-IgM (mean±s.d., n=3 independent experiments, two-way ANOVA, n.s, not significant). Right panel shows immunoblot analysis of whole cell lysates.
(i) Apoptosis analysis of cells from (h) is shown (mean±s.d., n=3 independent experiments, one-way ANOVA, n.s, not significant).
(j) The percentage of GFP+ and AnnexinV+ HBL1 and HLY-1 cells expressing the indicated shRNAs is shown (mean±s.d., n=3 independent experiments, one-way ANOVA, *P value≤0.05; *** P value≤0.001, n.s, not significant).
(k) Model of KLHL6-Roquin2 axis in ABC-DLBCL.
Unprocessed original scan of immunoblots for (a,b,c,d,e,h) are shown in Supplementary Fig. 8, and source data for (f) and statistical source data for (h,i,j) and exact P values for (j) can be found in Supplementary Table 6. Unless otherwise noted, immunoblots are representative of three independent experiments.
Next, we investigated whether loss of KLHL6 resulted in increased IKK activation. The amount of IκBα phosphorylation upon BCR stimulation was higher in KLHL6−/− cells, suggesting increased IKK activity (Fig. 7d). Importantly, the increase in phosphorylation was reduced by re-expression of KLHL6(WT) (Fig. S7c). We also detected an increase in the nuclear translocation of the NF-κB transcriptional factors in KLHL6−/− cells (Fig. 7e), which was partially mitigated by the concomitant knockdown of Roquin2 (Fig. S7d). Moreover, ablation of KLHL6 increased RelA DNA-binding at the NFKBIA promoter, an effect reversed by simultaneous knockdown of Roquin2 (Fig. 7f).
We analyzed the mutual exclusivity of BTB-associated KLHL6 mutations with TNAFAIP3 alterations in DLBCL patients. Deleterious KLHL6 BTB-mutations showed no overlap with TNFAIP3 biallelic deletion or mutation (Fig. 7g and S7e). Using a weighted test40, we found exclusivity with a trend toward significance (P-value 0.085, Supplementary Table 5). Instead, 4 BTB-mutations had a coincident monoallelic deletion in TNFAIP3, suggesting that these mutations might confer additional NF-κB activation when only one TNFAIP3 allele is lost. Interestingly, tumour cells from patients with KLHL6 mutations revealed a higher NF-κB activity, although this signature was not only confined to KLHL6 mutated cases (Fig. S7f).
To investigate whether the tumor suppressor role of KLHL6 would be diminished in TNFAIP3-null ABC-DLBCLs, we ablated KLHL6 in RCK8 cells14 and observed no significant effects on cellular proliferation (Fig. 7h) and apoptosis (Fig. 7i). Additionally, utilizing shRNAs targeting Roquin2 in both HBL1(TNFAIP3 WT) and HLY1 cells (TNFAIP3-null41), we observed elevated apoptosis in HBL1 only (Fig. 7j), indicating the relevance of Roquin2 in cells harboring a functional TNFAIP3 gene.
Discussion
KLHL6 is a BTB-Kelch domain protein mutated in human DLBCL. Somatic mutations localize to the BTB domain with relevant hotspots at amino acids 65 and 904–7. Most KLHL6 alterations in DLBCL include monoallelic missense mutations and infrequent copy loss4–7. Mutations of KLHL6 are likely the consequence of aberrant hypersomatic mutation42, similarly to those of BCL6 and MYC43. Importantly, the hotspots and other deleterious BTB-domain mutations result in loss of both CULLIN3 interaction and E3 ligase activity. Data from VAL cells suggest that mutations might be accompanied by a transcriptional down-regulation, resulting in a loss of function. Indeed, about 6% of DLBCL tumors display down-regulation of KLHL6 transcript. In ABC-DLBCL cell lines, ablation of KLHL6 promotes cell growth both in vitro and in vivo, supporting a tumor suppressor role. These findings are consistent with low KLHL6 expression levels correlating with poorer survival in ABC-DLBCL patients29, 30.
Furthermore, we identified Roquin2 as the bona fide substrate of KLHL6. KLHL6 specifically binds, ubiquitylates and triggers Roquin2 (but not Roquin1) degradation in a BCR-dependent manner. Expression of a non-degradable Roquin2(Y691F) mutant phenocopies loss of KLHL6 and concomitant ablation of Roquin2 in KLHL6−/− cells results in an inhibition of cell proliferation. This pro-proliferative effect depends on the ability of Roquin2 to bind RNA18–22. Although Roquin1 and Roquin2 are genetically redundant in the T-cell compartment21, no studies have assessed the functional redundancy in B-cell cancers. Transgenic mice overexpressing Roquin2 or the non-degradable mutant in the GC might be helpful to model the oncogenic role of Roquin2 in DLBCL in vivo.
TNFAIP3 is a relevant target of Roquin2-mediated mRNA decay19. In ABC-DLBCL, BCR-signaling triggers transcription of KLHL6 and degradation of Roquin2, thus releasing TNFAIP3 from mRNA decay. We speculate that KLHL6 inhibits TNFAIP3 decay via Roquin2 degradation contributing in establishing a negative feedback loop to terminate the NF-κB signaling (Fig. 7k). Thus, loss of KLHL6 function promotes BCR-dependent activation of NF-κB in ABC-DLBCL. Correspondingly, mutations of KLHL6 correlate with high NFκB signatures and could serve as a marker of resistance to NFκB pathway targeting drugs such as Ibrutinib13, MLN492433 or bortezomib1.
TNFAIP3 genetic mutations and deletions are frequently observed in ABC-DLBCLs12–15. Functional reconstitution of TNFAIP3 in TNFAIP3-null DLBCL causes apoptosis and growth arrest, supporting a tumor suppressor role in DLBCL44, 45. Interestingly, co-occurrence of KLHL6 BTB-mutations and TNFAIP3 biallelic deletion or mutations was rarely observed in human DLBCL, suggesting that these two genes may have similar downstream components (i.e. IKK activation). A partial overlap between KLHL6 BTB-mutations and TNFAIP3 monoallelic deletion further points to a possible synergy towards NF-κB activation. On the other hand, neither Roquin2 amplification nor sequence alteration of TNFAIP3 mRNA at the Roquin2 binding site has been observed in DLBCLs. This suggests that the tumor suppressor mechanism of KLHL6 might extend beyond Roquin2/TNFAIP3 deregulation. It is possible that KLHL6 might be involved in other biological processes such as cell adhesion, migration or immune surveillance in patients.
Much remains in evaluating the functional impact of the KLHL6-Roquin2 axis in GCB-DLBCLs, where unknown KLHL6 substrates or different Roquin2 mRNA targets can contribute to the proliferation and survival of GCB-DLBCL cells. It is also worth noting that KLHL6 mutations are not observable at high frequency in non-B-cell cancers, suggesting a possibly differential role for KLHL6 in a different genetic and cellular context.
METHODS
Cell culture and drug treatment
HEK293T cells were maintained in Dulbecco’s modified Eagle’s medium containing 10% fetal bovine serum (FBS). U2932, BJAB, SUDHL4, SUDHL6, RAMOS, TMD8, HBL1, Pfeiffer, OCI-LY8, Karpas422, ARP1, RCK8, HLY-1, SUDHL10 and VAL were maintained in RPMI 1640 medium containing 10% FBS. OCI-LY1, OCI-LY7, OCI-LY10, and OCI-LY19 were maintained in Iscove’s modified Dulbecco’s Medium containing 10% FBS. For BCR-crosslinking experiment, Goat-F(ab′)2 anti-human IgM (SouthernBiotech, #2022-01) was used at indicated concentrations. For 3D matrigel colony formation assay, 150μl of Corning Matrigel Basement Membrane Matrix was used in mix of 100μl of DMEM/F12 containing 10% Knockout Serum Replacement (SR) and plated in Millicell EZ slide (Millipore). For quantification of cell numbers from colonies grown for 14 days, 300μl of Corning Dispase(#354235) was used according to manufacturers’ protocols. The following drugs were used: proteasome inhibitor MG132 (Peptide Institute Inc.; 10 μM final concentration), Cycloheximide (Sigma Aldrich; 50μg/ml final concentration), MLN4924 (Active Biochem; 5μM final concentration), IKK-16(Selleckchem; 10μM final concentration), Ibrutinib((Selleckchem; 5μM final concentration), ActinomycinD (Sigma Aldrich; 2μg/ml final concentration), Doxycycline hyclate(Sigma Aldrich; 1 μg/ml final concentration). When indicated, cells were selected with puromycin(Sigma Aldrich; 0.5μg/ml-1μg/ml final concentration) and hygromycin(ThermoFisher; 100μg/ml). MTS assays (Promega, G5421), AnnexinV staining (Thermo fisher Annexin V, Alexa Fluor® 680 conjugate; A35109) was performed according to manufacturers’ protocols. For detection of IgM and IgG surface expression, FITC mouse anti-human IgM (BD, #562029) and APC mouse anti-human IgG (BD, #562025) were used according to manufacturing instruction.
Biochemical methods
Extract preparation, immunoprecipitation, and immunoblotting, fractionation of DLBCLs and chromatin immunoprecipitation were carried out according to ref46. Bands quantification was performed using ImageJ software and plotted using nonlinear-fit curve in Prism. All antibodies were used at a dilution of 1:1000 unless specified. The following antibodies were used: anti-FLAG (Sigma, F7425,1:3000), anti-HA (Biolegend, #901513), anti-Cullin1 (Invitrogen, #71-8700), anti-Cullin3 (Bethyl Laboratories, A301-109A), anti-ubiquitin K48 (EMD Millipore, 05-1307), anti-Roquin1/2 (EMD Millipore, MABF288), anti-Roquin2(Santa Cruz, sc-165026) anti-Roquin2(Bethyl Laboratories, A305-150A) anti-KLHL6 (Abcam, ab182163), anti-KLHL6(Novus Biologicals, NBP1-46128) anti-Tubulin(Santa Cruz, sc-8035), anti-GAPDH(EMD Millipore, MAB374,1:5000), anti-CDK1(Santa Cruz, sc-954), anti-CDK2(Santa Cruz, sc-163), anti-p-AKT S473 (Cell Signaling Technology, #4051), anti-p-ERK T202/Y204 (Cell Signaling Technology, #9101), anti-ERK1/2 (Cell Signaling Technology, #9102), anti-AKT (Cell Signaling Technology, #4691), anti-TNFAIP3(Cell Signaling Technology, #5630), anti-p-IκB S32 (Cell Signaling Technology, #2859,1:500), anti-p100/p52(Cell Signaling Technology, #4882), anti-p105/p50(Santa Cruz, sc-7178), anti-RelA(Santa Cruz, sc-372), anti-RelB(Santa Cruz, sc-226), anti-histone H2A(EMD Millipore, 07-146) and anti-histone H3(Abcam, ab1791,1:5000), ECL Rabbit IgG HRP-linked whole antibody(GE healthcare, NA934-1ML,1:5000), ECL Mouse IgG, HRP-linked whole antibody(GE healthcare, NA931-1ML,1:5000), Anti-Rat IgG (H+L) polyclonal antibody(Jackson Immunoresearch, 112-035-003,1:5000), Anti-Goat IgG (H+L) polyclonal antibody(Jackson Immunoresearch, 705-035-003,1:5000). KLHL6, Roquin1/2, TNFAIP3, CULLIN1, CULLIN3, p100/p52, p105/p50 and RelA antibodies were validated in our lab utilizing RNAi as well as overexpression, and were also validated by manufacturer. All other primary antibodies were validated on manufacturer datasheets.
The following agarose beads were used: anti-FLAG-M2 affinity gel (Sigma, A2220) and Strep-Tactin Superflow 50% suspension (Neuromics).
Xenotransplantation experiments
All animal work was performed following the ethical guidelines and protocols approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. NOD/SCID/IL2Rγ−/− (NSG) mice were purchased from the Jackson Laboratory. Six to eight-week-old NSG mice received sub-cutaneous (s.c.) flank injections of 1×107 U2932 KLHL6−/− (clone-derived) cells re-expressed either with empty vector, KLHL6(WT), or KLHL6(S94I)), and 1×107 U2932 cells infected with retroviruses encoding HA-Roquin2(WT) or HA-Roquin2(Y691F) in 100μl sterile PBS. Tumor burden was monitored weekly by palpation and eye inspection. Tumor volume calculated by caliper measurement. Tumor weight was analyzed on the excised tumors at the experimental endpoint using an analytical scale. After about one month, tumor volume and weight were measured.
Purification and analysis of KLHL6 interactors
Approximately 5×108 HEK293T and ARP-1 cells stably expressing FLAG-tagged KLHL6(WT) or FLAG-tagged KLHL6(L65P) cells were harvested and subsequently lysed in lysis buffer (LB: 50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 50 mM NaF, 0.5% NP40, plus protease and phosphatase inhibitors). KLHL6 was immunopurified with an anti-FLAG agarose resin (Sigma) and washed five times with LB (15 minutes each). After washing, proteins were eluted with FLAG peptides (Sigma). The eluates (1% of the IP) were separated by SDS-PAGE, and proteins were stained by Silver Staining (Life Technology). The final eluate was then precipitated with trichloroacetic acid (TCA).
In vitro ubiquitylation assay
The ubiquitylation of KLHL6 and Roquin2 was performed in a volume of 10 μl, containing 50 mM Tris pH 7.6, 5 mM MgCl2, 2 mM ATP, 1.5 ng/μl E1 (Boston Biochem), 10 ng/μl UBCH5C, 2.5 μg/μl ubiquitin (Sigma), 1 μM ubiquitin aldehyde, and purified FLAG-KLHL6 or FLAG-KLHL6/HA-Roquin2 complex from HEK293T cells via FLAG immunoprecipitation. The reactions were incubated at 37°C for the indicated times, subjected to SDS-PAGE, then analyzed by immunoblot. When indicated, upon reaction, beads were resuspended in 1% SDS, boiled, then diluted to 0.1% SDS. The eluted proteins were further FLAG-immunoprecipitated, washed and eluted in Laemmli buffer before SDS-PAGE analysis.
In vivo ubiquitylation assay
U2932 KLHL6+/+ and KLHL6−/− (clone-derived) cells were treated with or without MG132 for 6hrs and lysed in 1% SDS. Lysates were then diluted to 0.1% SDS in NP-40 buffer and immunoprecipated (IP) with a polyclonal antibody against Roquin2. The immunocomplexes were subjected to SDS-PAGE and analyzed by immunoblot.
In vitro binding assays
For in vitro binding assays, in vitro-translated FLAG-tagged KLHL6 was added to lysis buffer (50 mM Tris-HCl pH 7.5, 250 mM NaCl 0.1% Triton X-100, 1 mM EGTA) with indicated amounts of Roquin2 peptides. Anti-streptavidin resin was then added to the lysis buffer and incubated at 4°C for 2hrs. Samples were washed three times with the lysis buffer, and complexes were eluted in Laemmli buffer. For the use of HEK293T cells stably expressing KLHL6(WT), cells were lysed with the lysis buffer and anti-streptavidin resin was directly added to the whole cell lysates with the peptides.
Transient transfections and retrovirus-mediated gene transfer
HEK293T cells were transfected using polyethylenimine (PEI). For retrovirus and lenvirius production, GP-293 packaging cells (Clontech) or pCMV-DeltaR8.2 were used respectively. The virus-containing medium was collected after forty-eight hours of transfection and supplemented with 10 μg/ml polybrene (Sigma). Then, cells were spin-infected at 1800rpm for 30 minutes with the viral supernatant for six hours to overnight. For RCK8, cells were electroporated using Neon transfection system according to manufacture’s protocol with LentiCRISPRv2 vector carrying a GFP marker. Transfected cells were sorted by GFP. siRNA oligos transfection was performed with Neon transfection system.
Generating clonal KLHL6−/− cell lines
U2932 or OCI-LY10 Cas9-expressing cells were infected with lentiviruses encoding gRNAs targeting the KLHL6 exon1. Upon infection and puromycin selection, cells were plated in a 96 well plate at a concentration of 0.5 cells/well. Single clones were screened for KLHL6 expression by immunoblotting.
Plasmids
Human Roquin1 and Roquin2 cDNA were kindly provided by Dr. Carola Vinuesa and subcloned into pcDNA3.1-FLAG, pcDNA3.1-HA or pcDNA3.1-FLAG-Streptavidin. Human KLHL6 cDNA was purchased from Dharmacon and subcloned into pcDNA3.1-FLAG, pcDNA3.1-FLAG-Streptavidin, pREV-TRE. C-terminal deletion mutants and point mutants were generated by utilizing the QuikChange Site-directed Mutagenesis kit (Stratagene), and N-terminal deletion mutants were generated by standard PCR methods. For retrovirus production, cDNAs encoding FLAG-tagged or HA-tagged KLHL6 and KLHL6 mutants were subcloned into the retroviral vector MIGR1 and pREV-TRE vector. cDNAs encoding FLAG-HA-tagged or untagged Roquin2 and Roquin2 mutants were subcloned into the retroviral vector pBabe Puro or pMSCV. Lentivirus encoding shRNAs targeting human KLHL6 were subcloned in pSicoR-Puro. Lentivirus encoding shRNAs targeting human Roquin2 were subcloned in pSicoR-GFP.
KLHL6 cDNA was clone into pTRIPZ using AgeI and HpaI.
The target sequences used to knock-down human KLHL6 were:
hKLHL6_shRNA#1:
For: TGCAGCCAGCAACTATTTCATTCAAGAGATGAAATAGTTGCTGGCTGCTTTTTTC
Rev:TCGAGAAAAAAGCAGCCAGCAACTATTTCATCTCTTGAATGAAATAGTTGCTGGCTGCA
hKLHL6_shRNA#2:
For: TGAAGCCTTGAACCCAGAAATTCAAGAGATTTCTGGGTTCAAGGCTTCTTTTTTC
Rev:TCGAGAAAAAAGAAGCCTTGAACCCAGAAATCTCTTGAATTTCTGGGTTCAAGGCTTCA
hKLHL6_shRNA#3:
For: TGCATGATGTTTGGAAATAT TTCAAGAGAATATTTCCAAACATCATGCTTTTTTC
Rev:TCGAGAAAAAA GCATGATGTTTGGAAATAT TCTCTTGAAATATTTCCAAACATCATGCA
hKLHL6_shRNA#4:
For: TGGATTCAGATTGAGTATTT TTCAAGAGAAAATACTCAATCTGAATCCTTTTTTC
Rev:TCGAGAAAAAA GGATTCAGATTGAGTATTT TCTCTTGAAAAATACTCAATCTGAATCCA
The target sequences used to knock-down human Roquin2 were:
hRoquin2 shRNA#1:
For : TGCAGTTGTCTGCCAATCTATTCAAGAGATAGATTGGCAGACAACTGCTTTTTTC
Rev:TCGAGAAAAAAGCAGTTGTCTGCCAATCTATCTCTTGAATAGATTGGCAGACAACTGCA
hRoquin2 shRNA#2:
For: TGGACTCAGATACCCTTTGATTCAAGAGATCAAAGGGTATCTGAGTCCTTTTTTC
Rev:TCGAGAAAAAAGGACTCAGATACCCTTTGATCTCTTGAATCAAAGGGTATCTGAGTCCA
Lentiviruses encoding gRNAs targeting human KLHL6 were subcloned into Lenti-Guide-Puro vector, Lenti-Guide-GFP and LentiCRISPRv2 vector. The target sequences to knockout human KLHL6 were:
| hKLHL6_gRNA#2 | For: CAGAGCGTTTTCCATTCGCA |
| Rev: TGCGAATGGAAAACGCTCTG | |
| hKLHL6_gRNA#3 | For: TCAGAGCGTTTTCCATTCGC |
| Rev: GCGAATGGAAAACGCTCTGA | |
| hKLHL6_gRNA#4 | For: AAAGGTCAAATTTGACGACG |
| Rev: CGTCGTCAAATTTGACCTTT | |
| hKLHL6_gRNA#5 | For: GACTTGGTCGAGATCTTAAA |
| Rev: TTTAAGATCTCGACCAAGTC | |
| hKLHL6_gRNA#6 | For: ACTTGGTCGAGATCTTAAAT |
| Rev: ATTTAAGATCTCGACCAAGT |
Gene silencing by siRNA
For siRNA-mediated silencing, duplexes were purchased from Dharmacon. The target sequence for human KLHL6 siRNA was GCACGAAGGAUGAACGGUU, The target sequence for human Roquin2 siRNA was GCUUGAAAAGUAUCGAUUA. Non-targeting siRNA control sequence was UGGUUUACAUGUCGACUAA.
mRNA Analysis
RNA was extracted using the RNeasy Kit (Qiagen) and trizol(Invitrogen). cDNA synthesis was performed using Maxima first strand cDNA synthesis kit (Thermo Fisher) and RNA to cDNA Ecodry Premix kit(Clontech). Quantitative PCR analysis with SYBR Green PCR Master Mix (Applied Biosystems) was performed according to standard procedures. Primer sequences were:
| hGAPDH | FOR: 5′ GGAGCGAGATCCCTCCAAAAT 3′ |
| REV: 5′ GGCTGTTGTCATACTTCTCATGG 3′ | |
| hTNFAIP3 | FOR: 5′ TCCTCAGGCTTTGTATTTGAGC 3′ |
| REV: 5′ TGTGTATCGGTGCATGGTTTTA 3′ | |
| hTNFRSF14 | FOR: 5′ CCACTGGGTATGGTGGTTTC 3′ |
| REV: 5′ TCACCTTCTGCCTCCTGTCT 3′ | |
| hTNF | FOR: 5′ CTGCACTTTGGAGTGATCGGC3′ |
| REV: 5′ CACCAGCTGGTTATCTCTCAGCTCC 3′ | |
| hNFKBIE | FOR: 5′ TCTGGCATTGAGTCTCTGCG 3′ |
| REV: 5′ AGGAGCCATAGGTGGAATCAG 3′ | |
| hLTA | FOR: 5′ GCTGCTGGTTCTGCTGCC 3′ |
| REV: 5′ CAAGGAGAAACCATCCTGGAGGAAG 3′ | |
| hNEDD4L | FOR: 5′ ACTTCCTCCTCCTCCTCTGC 3′ |
| REV: 5′ TCCAAGTCTTCGCTGATGTG 3′ | |
| hABLIM1 | FOR: 5′ ACTGCATCTCTCCCTGGCTA 3′ |
| REV: 5′ TGTTGGTCACCATGAGCATT 3′ | |
| hSYNGAP1 | FOR: 5′ TCTGAGGAAAACTGCGAGGT 3′ |
| REV: 5′ GCAAACACCTCCTTCAGCTC 3′ | |
| hNEIL2 | FOR: 5′ GCCTCCACAAAAAGAAGTGC 3′ |
| REV: 5′ TTGTTGGCTTTCTTGGCTCT 3′ | |
| hLGALS8 | FOR: 5′ CTGGGCATTTATGGCAAAGT 3′ |
| REV: 5′ GACAGTTCTGGGTGCGATTT 3′ | |
| hCD274 | FOR: 5′ TATGGTGGTGCCGACTACAA 3′ |
| REV: 5′ TGCTTGTCCAGATGACTTCG 3′ | |
| hKLHL6 | FOR: 5′ GCAGCCAGCAACTATTTCAGG 3′ |
| REV: 5′ ACGTGTAGTCCAACAGAGTGT 3′ | |
| hNFKBIA | FOR: 5′ TATAAACGCTGGCTGGG 3′ |
| REV: 5′ CCCTAGTGGCTCATCGC 3′ |
Normalization and quantification of protein levels
Protein concentrations of cell extracts were measured using a Bio-Rad DC protein assay (Lowry assay) according to the manufacturer’s protocol. For each experiment, equal amounts of protein (~15μg) were separated by SDS-PAGE and analyzed by immunoblotting. Equal protein levels in each lane were confirmed by Ponceau S staining of the membrane and by immunoblotting a constitutively expressed protein.
Cell Proliferation Assay
2 × 103 cells were plated for MTS assay according to manufacturers’ protocols.
For the long-term cell proliferation assay, 5×104-2×105 cells were plated, counted and re-plated every 3–5 days.
Flow cytometry
Flow cytometry was performed on Attune NxT Flow Cytometer using FITC for GFP-expressing cells, Alexa-680 AnnexinV to detect apoptosis, FITC to detect IgM or APC to detect IgG staining. All stainings were performed according to manufacturers’ protocols. For investigating shRNA or gRNA effects on survival, 5×105 cells were spin-infected in a 24-well plate with 1000 μl lentivirus containing medium in the presence of polybrene(8μg/ml). Media was changed after the spin-infection and the number of infected cells was determined on day 2 when GFP was fully expressed in all infected cells. The number of viable GFP positive cells on day 2 was set to 100% to normalize for transduction efficiency and every consecutive assessment was calculated in relation to day 2. When indicated, AnnexinV positive cells were gated on GFP positive cells (see gating strategy in Supplementary Figures 3 and 5).
MudPIT analysis
TCA-precipitated proteins were urea-denatured, reduced, alkylated and digested with endoproteinase Lys-C (Roche), followed by modified trypsin (Roche), as described47, 48. Peptide mixtures were loaded onto 100-μm fused silica microcapillary columns packed with 5-μm C18 reverse phase (Aqua, Phenomenex), strong cation exchange particles (Luna, Phenomenex), and reverse phase49. Loaded microcapillary columns were placed in-line with a Quaternary Agilent 1100 series HPLC pump and a LTQ linear ion trap mass spectrometer equipped with a nano-LC electrospray ionization source (Thermo Scientific). Fully automated 10-step MudPIT runs were carried out on the electrosprayed peptides, as described47. Tandem mass (MS/MS) spectra were interpreted using SEQUEST50 against a database of 61,318 sequences, consisting of 30,449 non-redundant human proteins (downloaded from NCBI on 2012-08-27, 160 usual contaminants (such as human keratins, IgGs and proteolytic enzymes), and, to estimate false discovery rates, 30,659 randomized amino-acid sequences derived from each non-redundant protein entry. Peptide/spectrum matches were sorted and selected using DTASelect with the following criteria set: spectra/peptide matches were only retained if they had a DeltCn of at least 0.08 and a minimum XCorr of 1.8 for singly-, 2.0 for doubly-, and 3.0 for triply-charged spectra. In addition, peptides had to be fully tryptic and at least seven amino acids long. Combining all runs, proteins had to be detected by at least two such peptides, or one peptide with two independent spectra. Under these criteria the final FDRs at the protein and spectral levels were 2.1%±0.3 and 0.94% ± 0.03, respectively. Peptide hits from multiple runs were compared using CONTRAST51. To estimate relative protein levels, Normalized Spectral Abundance Factors (NSAFs) were calculated for each detected protein, as described52,53,54.
Analysis of KLHL6 expression in DLBCL patients
Raw DNA copy number data from high resolution single nucleotide polymorphism (SNP) microarray analysis of 609 primary DLBCL tumors were utilized form a previously published study26. The data were visualized using the integrative genomics viewer (IGV)55. Cases were sorted according to their KLHL6 copy number status, and those with copy number <1.8 were classified as possessing a deletion, according to previously described criteria56.
Gene expression microarray from 249 tumors with matched DNA copy number data were obtained from a previously published study26. Their cell of origin subtype was determined using the Wright algorithm57, as previously reported56. Row normalized heatmaps for 4 probe sets corresponding to KLHL6 were sorted according to their average expression, and significant reduction in KLHL6 expression defined as being 1 standard deviation below the mean.
Raw cel files for publicly available Affymetrix U133 plus 2.0 gene expression microarray data for diffuse large B-cell lymphoma tumors (GSE10846, GSE34171, GSE31312) were obtained from the gene expression omnibus. Data were RMA normalized using the ExpressionFileCreator module of GenePattern58. Scores to categorize diffuse large B-cell lymphoma tumors by cell of origin subtype were calculated according to the Wright algorithm57, Intensities from the 4 probes for KLHL6 (1555275_a_at, 1560396_at, 1560397_s_at, 228167_at) were averaged for use in the survival analysis. Cases were dichotomized into being above or below the median expression level of KLHL6 expression within each dataset to avoid confounding batch effects. For NF-κB signatures, Affymetrix U133 plus 2 gene expression microarrays were performed on 84 matched DLBCL tumors26,59. Raw cel files were RMA normalized with median scaling using the ExpressionFileCreator module of GenePattern58. Sample-level enrichment of NF-κB target genes was calculated using the single sample gene set enrichment analysis60 and the c3 TFT gene set database of mSigDB61.
RNA-seq
Total RNA was extracted from U2932 cells using RNeasy Mini Kit (QIAGEN, #74104) and polyA+ transcripts isolated with oligo (dT)25-conjugated magnetic Dynabeads (Thermo Fisher). Strand specific RNA-seq libraries were prepared following a published protocol62. Briefly, RNA was chemically fragmented in first strand buffer, converted to cDNA using SuperScript® III reverse transcriptase (Invitrogen), end-repaired, A-tailed and ligated to custom-designed universal adapters using an end-repair mix, klenow fragment, and T4 DNA ligase (all from Enzymatics). After ligation, adapters were removed by SPRI purification using SPRIselect beads (Beckman coulter) and amplified with Q5 Hot Start DNA polymerase (New England Biolabs) while introducing custom dual indexes. Three biological replicates were sequenced on a NextSeq 500 (Illumina) at a depth of at least 2×107 reads each. Reads were mapped and analyzed with a custom bioinformatic pipeline based on STAR63, SAMTOOLS64, and the R packages DEGseq65 and DEseq266. We used human genome version GRCh38 and gene annotations from the ENSEMBL release 83. GO analyses were performed using version 6.8 of the DAVID web server67, 68.
CHIP-seq
The data discussed in this publication were obtained from the NCBI’s Gene Expression Omnibus34 and are accessible through GEO at Series accession numbers GSE55105. FASTQs were downloaded and mapped to hg19 with bowtie2 (v2.1.0). Genome browser tracks were generated using custom scripts. When available, biological replicate were merged by taking the mean of the reads density at each position. The data were visualized using the integrative genomics viewer (IGV)55.
Statistics and Reproducibility
All graphs show mean values with error bars signifying standard deviation (s.d.) as indicated in the figure legends. Exact P values for each experiment are provided in Supplementary Table 6. Unless otherwise noted, all immunoblots were successfully repeated at least three times. One-tailed t-test was performed for Fig. 3e (right panel) and two-tailed t-test for Fig 4g. Other analyses performed were either one-way (Fig. 3c, 3e (left panel), 5e, 6a, 7i, 7j, S3b, S3f) or two-way ANOVA (Fig. 1i, 3b, 3d, 4g, 4h, 6e, 7h, S2d, S3d, S4e, S4h, S5b) as indicated in the figure legends. DEseq2 was performed for RNA-seq analysis in Fig. 6b and Supplementary Table 3. Weighted Exclusivity Test (WExT) was performed for Supplementary Table 5. Mantel-Cox was performed for survival analysis in Fig. 3a. Pearson Correlation Coefficient was used in Fig. S2c.
Data availability
RNA–seq data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) under accession code GSE93675. Previously published CHIP-seq data were obtained from GEO under accession code GSE55105. KLHL6 copy number change and expression were calculated through re-analysis of SNP data and microarray from GSE11318, GSE12906, GSE15127, GSE22082, GSE34171 and GSE11318, GSE34171, respectively. Microarray data for DLBCL survival were obtained from GEO under accession code GSE10846, GSE34171, GSE31312. Data for NF-κB signature correlation were retrieved from GSE10846. Raw mass spectrometry data are available in PRIDE (https://www.ebi.ac.uk/pride/archive/) under accession code PXD008963.
Raw data from independent experiments with n<5 can be found in the Statistical source data (Supplementary Table 6). Unprocessed immunoblots are provided in Supplementary Fig. 8. All other data supporting the findings of this study are available from the corresponding author on reasonable request.
Supplementary Material
Acknowledgments
The authors thank Dr. Carola Vinuesa for kindly providing Roquins cDNAs, Dr. Andrei Thomas-Tikhonenko, Dr. Laura Pasqualucci and Dr. Yibin Yang for kindly providing DLBCL cell lines, Dr. Michele Pagano for kindly providing FBPs cDNAs, Dr. Bangjin Kim for helping with 3D matrigel colony formation assay; Rizwan Saffie, Dr. Donita Brady, Dr. Eric Witze and Dr. Roger Greenberg for critically reading the manuscript. This work was supported in part by grant R00-CA166181-04, R01-CA207513-01 from the National Cancer Institute and Gilead Sciences Research Scholars Program in Hematology/Oncology to L.B. R.B. is supported by an NIH Innovator Award (DP2MH107055), the Searle Scholars Program (15-SSP-102), the March of Dimes Foundation (1-FY-15-344), and the W.W. Smith Charitable Trust (C1404).
Footnotes
Author Contributions. L.B. conceived, directed the project and oversaw the results. J.C. designed and performed most experiments. K.L. helped J.C. with experiments in Fig. 1i, 3b, 3d, 4h, 6a, 7h, 7j, S3b, and S4h. K.I. helped J.C. with experiments in Fig. 6b. R.B. helped with the bioinformatics analysis of RNA-seq data. A.S., L.F. and M.P.W. performed the mass spectrometry analysis of the KLHL6 complex purified by L.B.. M.G. and S.T. helped with Fig. 1a, 1b and 3a, 7g, S1a, S1b, S7e, and S7f. L.B. and J.C. wrote the manuscript.
Author Information. The authors declare no competing financial interests.
References
- 1.Yang Y, Staudt LM. Protein ubiquitination in lymphoid malignancies. Immunol Rev. 2015;263:240–256. doi: 10.1111/imr.12247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gupta-Rossi N, et al. Specific over-expression of deltex and a new Kelch-like protein in human germinal center B cells. Mol Immunol. 2003;39:791–799. doi: 10.1016/s0161-5890(03)00002-6. [DOI] [PubMed] [Google Scholar]
- 3.Kroll J, et al. The BTB-kelch protein KLHL6 is involved in B-lymphocyte antigen receptor signaling and germinal center formation. Mol Cell Biol. 2005;25:8531–8540. doi: 10.1128/MCB.25.19.8531-8540.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morin RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011 doi: 10.1038/nature10351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lohr JG, et al. Discovery and prioritization of somatic mutations in diffuse large B-cell lymphoma (DLBCL) by whole-exome sequencing. Proc Natl Acad Sci U S A. 2012;109:3879–3884. doi: 10.1073/pnas.1121343109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Idoia GR, et al. CREBBP Loss Cooperates with BCL2 Over-Expression to Promote Lymphoma in Mice. Blood. 2016;128 [Google Scholar]
- 7.Reddy A, et al. Genetic and Functional Drivers of Diffuse Large B Cell Lymphoma. Cell. 2017;171:481–494 e415. doi: 10.1016/j.cell.2017.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alizadeh AA, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- 9.Rosenwald A, et al. Molecular diagnosis of primary mediastinal B cell lymphoma identifies a clinically favorable subgroup of diffuse large B cell lymphoma related to Hodgkin lymphoma. J Exp Med. 2003;198:851–862. doi: 10.1084/jem.20031074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Staudt LM. Oncogenic activation of NF-kappaB. Cold Spring Harb Perspect Biol. 2010;2:a000109. doi: 10.1101/cshperspect.a000109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Young RM, Shaffer AL, 3rd, Phelan JD, Staudt LM. B-cell receptor signaling in diffuse large B-cell lymphoma. Semin Hematol. 2015;52:77–85. doi: 10.1053/j.seminhematol.2015.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lenz G, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science. 2008;319:1676–1679. doi: 10.1126/science.1153629. [DOI] [PubMed] [Google Scholar]
- 13.Davis RE, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463:88–U97. doi: 10.1038/nature08638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Compagno M, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009;459:717–721. doi: 10.1038/nature07968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kato M, et al. Frequent inactivation of A20 in B-cell lymphomas. Nature. 2009;459:712–716. doi: 10.1038/nature07969. [DOI] [PubMed] [Google Scholar]
- 16.Fu M, Blackshear PJ. RNA-binding proteins in immune regulation: a focus on CCCH zinc finger proteins. Nat Rev Immunol. 2016 doi: 10.1038/nri.2016.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kataoka K, et al. Aberrant PD-L1 expression through 3′-UTR disruption in multiple cancers. Nature. 2016;534:402–406. doi: 10.1038/nature18294. [DOI] [PubMed] [Google Scholar]
- 18.Leppek K, et al. Roquin promotes constitutive mRNA decay via a conserved class of stem-loop recognition motifs. Cell. 2013;153:869–881. doi: 10.1016/j.cell.2013.04.016. [DOI] [PubMed] [Google Scholar]
- 19.Murakawa Y, et al. RC3H1 post-transcriptionally regulates A20 mRNA and modulates the activity of the IKK/NF-kappaB pathway. Nat Commun. 2015;6:7367. doi: 10.1038/ncomms8367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Glasmacher E, et al. Roquin binds inducible costimulator mRNA and effectors of mRNA decay to induce microRNA-independent post-transcriptional repression. Nat Immunol. 2010;11:725–733. doi: 10.1038/ni.1902. [DOI] [PubMed] [Google Scholar]
- 21.Vogel KU, et al. Roquin paralogs 1 and 2 redundantly repress the Icos and Ox40 costimulator mRNAs and control follicular helper T cell differentiation. Immunity. 2013;38:655–668. doi: 10.1016/j.immuni.2012.12.004. [DOI] [PubMed] [Google Scholar]
- 22.Schlundt A, et al. Structural basis for RNA recognition in roquin-mediated post-transcriptional gene regulation. Nat Struct Mol Biol. 2014;21:671–678. doi: 10.1038/nsmb.2855. [DOI] [PubMed] [Google Scholar]
- 23.Yu D, et al. Roquin represses autoimmunity by limiting inducible T-cell co-stimulator messenger RNA. Nature. 2007;450:299–303. doi: 10.1038/nature06253. [DOI] [PubMed] [Google Scholar]
- 24.Puente XS, et al. Non-coding recurrent mutations in chronic lymphocytic leukaemia. Nature. 2015;526:519–524. doi: 10.1038/nature14666. [DOI] [PubMed] [Google Scholar]
- 25.Lohr JG, et al. Widespread genetic heterogeneity in multiple myeloma: implications for targeted therapy. Cancer Cell. 2014;25:91–101. doi: 10.1016/j.ccr.2013.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Green MR, et al. Transient expression of Bcl6 is sufficient for oncogenic function and induction of mature B-cell lymphoma. Nat Commun. 2014;5:3904. doi: 10.1038/ncomms4904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lydeard JR, Schulman BA, Harper JW. Building and remodelling Cullin-RING E3 ubiquitin ligases. EMBO Rep. 2013;14:1050–1061. doi: 10.1038/embor.2013.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lo SC, Li XC, Henzl MT, Beamer LJ, Hannink M. Structure of the Keap1 : Nrf2 interface provides mechanistic insight into Nrf2 signaling. Embo Journal. 2006;25:3605–3617. doi: 10.1038/sj.emboj.7601243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leo Meriranta AP. Alejandra Cervera, Harald Holte Jr., Rainer Lehtonen, Sampsa Hautaniemi and Sirpa Leppä Low Expression and Somatic Mutations of the KLHL6 Gene Predict Poor Survival in Patients with Activated B-Cell like Diffuse Large B-Cell Lymphoma. Blood. 2016;128(22):2926. [Google Scholar]
- 30.Kunder CA, et al. KLHL6 Is Preferentially Expressed in Germinal Center-Derived B-Cell Lymphomas. Am J Clin Pathol. 2017;148:465–476. doi: 10.1093/ajcp/aqx099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Satpathy S, et al. Systems-wide analysis of BCR signalosomes and downstream phosphorylation and ubiquitylation. Mol Syst Biol. 2015;11:810. doi: 10.15252/msb.20145880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lenz G, et al. Aberrant immunoglobulin class switch recombination and switch translocations in activated B cell-like diffuse large B cell lymphoma. Journal of Experimental Medicine. 2007;204:633–643. doi: 10.1084/jem.20062041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Milhollen MA, et al. MLN4924, a NEDD8-activating enzyme inhibitor, is active in diffuse large B-cell lymphoma models: rationale for treatment of NF-kappa B-dependent lymphoma. Blood. 2010;116:1515–1523. doi: 10.1182/blood-2010-03-272567. [DOI] [PubMed] [Google Scholar]
- 34.Zhao B, et al. The NF-kappa B Genomic Landscape in Lymphoblastoid B Cells. Cell Reports. 2014;8:1595–1606. doi: 10.1016/j.celrep.2014.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhou A, Scoggin S, Gaynor RB, Williams NS. Identification of NF-kappa B-regulated genes induced by TNFalpha utilizing expression profiling and RNA interference. Oncogene. 2003;22:2054–2064. doi: 10.1038/sj.onc.1206262. [DOI] [PubMed] [Google Scholar]
- 36.Tian B, Nowak DE, Jamaluddin M, Wang S, Brasier AR. Identification of direct genomic targets downstream of the nuclear factor-kappaB transcription factor mediating tumor necrosis factor signaling. J Biol Chem. 2005;280:17435–17448. doi: 10.1074/jbc.M500437200. [DOI] [PubMed] [Google Scholar]
- 37.Mansouri L, et al. Frequent NFKBIE deletions are associated with poor outcome in primary mediastinal B-cell lymphoma. Blood. 2016;128:2666–2670. doi: 10.1182/blood-2016-03-704528. [DOI] [PubMed] [Google Scholar]
- 38.Boice M, et al. Loss of the HVEM Tumor Suppressor in Lymphoma and Restoration by Modified CAR-T Cells. Cell. 2016;167:405–418 e413. doi: 10.1016/j.cell.2016.08.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chu YY, et al. B cells lacking the tumor suppressor TNFAIP3/A20 display impaired differentiation and hyperactivation and cause inflammation and autoimmunity in aged mice. Blood. 2011;117:2227–2236. doi: 10.1182/blood-2010-09-306019. [DOI] [PubMed] [Google Scholar]
- 40.Leiserson MDM, Reyna MA, Raphael BJ. A weighted exact test for mutually exclusive mutations in cancer. Bioinformatics. 2016;32:736–745. doi: 10.1093/bioinformatics/btw462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fontan L, et al. MALT1 Small Molecule Inhibitors Specifically Suppress ABC-DLBCL In Vitro and In Vivo. Cancer Cell. 2012;22:812–824. doi: 10.1016/j.ccr.2012.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Puente XS, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475:101–105. doi: 10.1038/nature10113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pasqualucci L, et al. Hypermutation of multiple proto-oncogenes in B-cell diffuse large-cell lymphomas. Nature. 2001;412:341–346. doi: 10.1038/35085588. [DOI] [PubMed] [Google Scholar]
- 44.Honma K, et al. TNFAIP3/A20 functions as a novel tumor suppressor gene in several subtypes of non-Hodgkin lymphomas. Blood. 2009;114:2467–2475. doi: 10.1182/blood-2008-12-194852. [DOI] [PubMed] [Google Scholar]
- 45.Schmitz R, et al. TNFAIP3 (A20) is a tumor suppressor gene in Hodgkin lymphoma and primary mediastinal B cell lymphoma. J Exp Med. 2009;206:981–989. doi: 10.1084/jem.20090528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Busino L, et al. Fbxw7alpha- and GSK3-mediated degradation of p100 is a pro-survival mechanism in multiple myeloma. Nat Cell Biol. 2012;14:375–385. doi: 10.1038/ncb2463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Florens L, Washburn MP. Proteomic analysis by multidimensional protein identification technology. Methods Mol Biol. 2006;328:159–175. doi: 10.1385/1-59745-026-X:159. [DOI] [PubMed] [Google Scholar]
- 48.Washburn MP, Wolters D, Yates JR., 3rd Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat Biotechnol. 2001;19:242–247. doi: 10.1038/85686. [DOI] [PubMed] [Google Scholar]
- 49.McDonald WH, Ohi R, Miyamoto DT, Mitchison TJ, Yates JR. Comparison of three directly coupled HPLC MS/MS strategies for identification of proteins from complex mixtures: single-dimension LC-MS/MS, 2-phase MudPIT, and 3-phase MudPIT. International Journal of Mass Spectrometry. 2002;219:245–251. [Google Scholar]
- 50.Eng JK, McCormack AL, Yates JR. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 51.Tabb DL, McDonald WH, Yates JR., 3rd DTASelect and Contrast: tools for assembling and comparing protein identifications from shotgun proteomics. J Proteome Res. 2002;1:21–26. doi: 10.1021/pr015504q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Florens L, et al. Analyzing chromatin remodeling complexes using shotgun proteomics and normalized spectral abundance factors. Methods. 2006;40:303–311. doi: 10.1016/j.ymeth.2006.07.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Paoletti AC, et al. Quantitative proteomic analysis of distinct mammalian Mediator complexes using normalized spectral abundance factors. Proc Natl Acad Sci U S A. 2006;103:18928–18933. doi: 10.1073/pnas.0606379103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zybailov B, et al. Statistical analysis of membrane proteome expression changes in Saccharomyces cerevisiae. J Proteome Res. 2006;5:2339–2347. doi: 10.1021/pr060161n. [DOI] [PubMed] [Google Scholar]
- 55.Robinson JT, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Monti S, et al. Integrative analysis reveals an outcome-associated and targetable pattern of p53 and cell cycle deregulation in diffuse large B cell lymphoma. Cancer Cell. 2012;22:359–372. doi: 10.1016/j.ccr.2012.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wright G, et al. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci U S A. 2003;100:9991–9996. doi: 10.1073/pnas.1732008100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Reich M, et al. GenePattern 2.0. Nat Genet. 2006;38:500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
- 59.Lenz G, et al. Stromal gene signatures in large-B-cell lymphomas. N Engl J Med. 2008;359:2313–2323. doi: 10.1056/NEJMoa0802885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Barbie DA, et al. Systematic RNA interference reveals that oncogenic KRAS-driven cancers require TBK1. Nature. 2009;462:108–U122. doi: 10.1038/nature08460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Liberzon A, et al. Molecular signatures database (MSigDB) 3.0. Bioinformatics. 2011;27:1739–1740. doi: 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parkhomchuk D, et al. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 2009;37:e123. doi: 10.1093/nar/gkp596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li H, et al. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang L, Feng Z, Wang X, Wang X, Zhang X. DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics. 2010;26:136–138. doi: 10.1093/bioinformatics/btp612. [DOI] [PubMed] [Google Scholar]
- 66.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature Protocols. 2009;4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 68.Huang DW, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Research. 2009;37:1–13. doi: 10.1093/nar/gkn923. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA–seq data that support the findings of this study have been deposited in the Gene Expression Omnibus (GEO) under accession code GSE93675. Previously published CHIP-seq data were obtained from GEO under accession code GSE55105. KLHL6 copy number change and expression were calculated through re-analysis of SNP data and microarray from GSE11318, GSE12906, GSE15127, GSE22082, GSE34171 and GSE11318, GSE34171, respectively. Microarray data for DLBCL survival were obtained from GEO under accession code GSE10846, GSE34171, GSE31312. Data for NF-κB signature correlation were retrieved from GSE10846. Raw mass spectrometry data are available in PRIDE (https://www.ebi.ac.uk/pride/archive/) under accession code PXD008963.
Raw data from independent experiments with n<5 can be found in the Statistical source data (Supplementary Table 6). Unprocessed immunoblots are provided in Supplementary Fig. 8. All other data supporting the findings of this study are available from the corresponding author on reasonable request.