

Abstract
Systems medicine is a holistic approach to deciphering the complexity of human physiology in health and disease. In essence, a living body is constituted of networks of dynamically interacting units (molecules, cells, organs, etc.) that underlie its collective functions. Declining resilience due to aging and other chronic environmental exposures drives the system to transition from a health state to a disease state; these transitions, triggered by acute perturbations or chronic disturbance, manifest as qualitative shifts in the interactions and dynamics of the disease-perturbed networks. Understanding health-to-disease transitions poses a high-dimensional nonlinear reconstruction problem that requires deep understanding of biology as well as innovation in study design, technology, and data analysis. With a focus on the principles of systems medicine, this Review discusses approaches for deciphering this biological complexity from a novel perspective, namely understanding how disease-perturbed networks function; their study provides insights into fundamental disease mechanisms. The immediate goals for systems medicine are to identify early transitions to cardiovascular (and other chronic) diseases, and to accelerate the translation of new preventive, diagnostic or therapeutic targets into clinical practice, a critical step in the development of personalized, predictive, preventive and participatory (P4) medicine.
Keywords: Cardiovascular disease, Systems Biology, Systems Medicine, Translational Studies
Subject Terms: Biomarkers, Functional Genomics, Genetics
Introduction
Excessive spending and poor outcomes in health care and drug development necessitate the search for new research paradigms to translate cutting-edge, scientific discoveries of systems medicine into the clinic more quickly and efficiently.1,2 Despite the advent of technological breakthroughs in collecting and analyzing big data for human physiology, there are serious concerns about whether the research enterprise is ready to deliver on its promise for transforming health and health care.3 Meeting this challenge is particularly pressing for cardiovascular diseases (CVDs) that account for ~17% of the US national health spending and impact the life of three out of five adults over the age of 60.4 Surprisingly, <15% of clinical guidelines are based on high-quality research-based evidence.5,6 Moreover, guideline-driven care, although effective on a population scale, fails to account for the individual’s unique susceptibility to disease and their response to therapeutic interventions. Therefore, healthcare stakeholders seek research solutions that offer personalization yet apply to the population scale and translate easily and safely into clinical practice. Herein, we review new approaches rooted in the principles of systems medicine that can provide the foundational framework for a novel translational research paradigm. At the heart of this new paradigm is the individual-specific interpretation of a high-dimensional data space for personalized and precise health management.
Systems Medicine: A new paradigm in medicine based on systems theory
A fundamental concept of systems medicine is the human body as a “network of networks” (Figure 1). Each level of biological complexity, from the genome to molecules, to cells, to organs and finally to individuals, can be modeled as networks with specific components (nodes) and interactions between them (edges). The architecture of highly interconnected biochemical, molecular and cellular networks of living systems contradicts the reductionist philosophy of deterministic linear relationships between causal agents and physiological responses that underlies current clinical practice and pharmaceutical research. The nonlinear physiological responses to internal and external stimuli do not enable an intuitive interpretation. Hence, there is a need to establish a new conceptual framework that takes into consideration the high-dimensional, multi-scale, dynamical nature of our bodies, and to study human physiology with respect to the fundamental characteristics of systems, such as resilience and state transitions (Table 1). These concepts are not foreign to medicine. For instance, the physiological underpinning of resilience is homeostasis: the adaptation of physiological interactions of regulatory networks following a perturbation to maintain consistency of the tissue and organ composition and function.7 This optimal, tightly regulated physiological steady state is self-stabilizing (a systems set-point of “attractor state”) yet adaptive, thereby establishing a health continuum that depends on both the genetic makeup of each individual and the environmental exposures over their lifetimes, as we discuss below. Therefore, disease can be conceptualized as a failure to maintain the health state for an individual system. Eventually, this results in a transition towards a suboptimal and unfortunately self-stabilizing state that we recognize as a disease state and is maintained by distinct disease-perturbed network architectures.8,9
Figure 1.
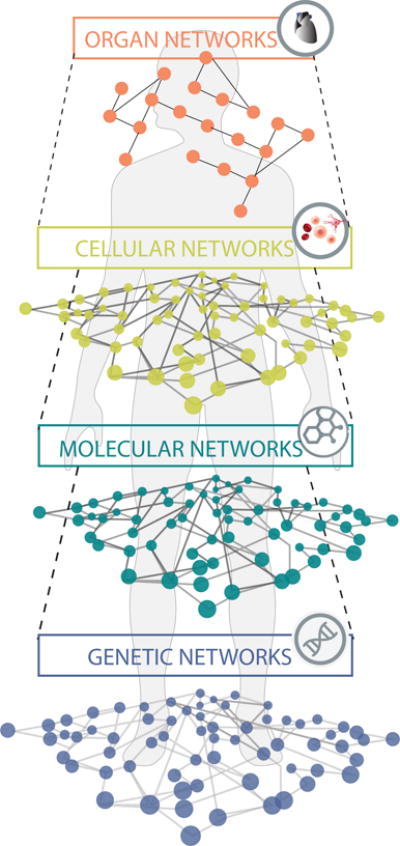
The Network-of-networks. Our bodies are made up of many networks that are integrated at and communicating on multiple scales.
Table 1.
Basic principles of Systems Biology that drive the proposed working framework for Systems Medicine.
| Principle | Definition | Open Challenges |
|---|---|---|
| Equilibrium | Systems move towards a stable steady state balancing internal or external forces that are strongly opposed to one another | Define steady states (health, disease): How and why do they destabilize? |
| Critical State Transitions | A sudden system shift from one stable steady state to another when certain parameters reach a threshold (see Table 2) |
Identify health-to-disease transitions: When has the transition become irreversible (“tipping point” hypothesis)? How do perturbations drive the transition? |
| Robustness (or Resilience) |
The system’s ability to maintain functions and behaviors in the face of changing internal and external environments (perturbations) | Define the limits of a stable steady state: How to design clinical studies that test homeostatic mechanisms and include the concept of resilience? |
| Equifinality | The existence of multiple trajectories that converge to a common end state (e.g., disease) | Identify possible trajectories towards a disease state: What does this mean for biomarker discovery? How to stratify individuals and their trajectories to disease? |
| Mutuality | Since system’s components are reciprocally interdependent, it is impossible to know what “causes” something | Understand the limits of prediction: How to develop multiple interventions protocols and tailor them for specific individuals? |
Resilience, Reversibility and Health-to-Disease Transitions
Within the formal framework of homeostasis as a manifestation of systems dynamics, we can conceptually visualize health and disease as the basins of attraction (Table 2) of two alternative stable states of the system (Figure 2).10 The “width” and the “steepness” of the health state attractor basin determine the system’s resilience – its capacity to absorb a perturbation without shifting to an alternative state but instead to relax back to the healthy state. In contrast to acute perturbations that may or may not kick the system out of the basin of attraction, chronic exposure to environmental risk factors can alter the architecture of system’s regulatory networks that mathematically “shape the potential landscape”. Thus, in formal terms, such disturbance of the network architecture can result in the flattening of the basin of attraction, which translates into system’s destabilization, and therefore eventually facilitate a transition to a disease state – or at least reduce the barrier to such a transition. This process represents a critical state transition (Table 2) that can be observed as an abrupt qualitative (discontinuous) shift of a system’s state. Such behavior, long known in ecological systems, was recently experimentally validated at the biochemical and cellular levels of human physiology—and represents a powerful new conceptual tool for dealing with biological complexity of organism at many levels.11–13
Table 2.
Glossary for Critical State Transitions and Attractors in Systems Medicine.
| Term | Definition | Example |
|---|---|---|
| Alternative stable states | Different steady states of the very same system can be realized under the same external conditions, depending on history | Healthy State (fasting glucose <115 mg/dL) vs. Diabetic State (fasting glucose >180 mg/dL) Since the pathological state is also a stable state (a new equilibrium) reversal is difficult, as best illustrated in the inherent challenge of weight loss by diet |
| Bifurcation (Critical State Transitions) |
The current stable state of a system disappears due to (slow) change of system characteristics and the system is forced to move to an alternative stable state | During the process of gradual health deterioration (e.g., fasting glucose >115 mg/dL – prediabetic state), poor diet results in a sudden catastrophic shift to a disease state that self-stabilizes in the new equilibrium (Figure 2) |
| Attractor | An equilibrium state to which a system converges after some time; a stable steady state. | After an oral glucose tolerance test, blood glucose increases to an unstable value (>200 mg/dL) that will finally decrease to the health steady state value (<115 mg/dL) |
| Basin of attraction | The entire set of initial conditions from which the system automatically moves to an attractor | Temporary deviations from an equilibrium state following acute perturbations after which the systems resettle in the steady state manifest the basin of attraction – e.g., normoglycemia following a large meal that caused a peak in blood glucose |
| Potential U(x) | A mathematical quantity that captures the “driving force” in a dynamical system, and can be graphed as the elevation over each state space position × (a state variable) to obtain a landscape picture (potential well) | Can be approximated as the inverse of the t probability P(x) to find individual in a population at that state space position x, where × is a state variable, e.g., x = fasting glucose (mg/dL) |
| Threshold |
A point where the system is very sensitive to changing conditions, e.g., at the “cusp” between two basins of attraction. In a critical transition, the threshold becomes a tipping point |
In a prediabetic individual, when the basin of attraction for glucose homeostasis is flat and blood glucose reaches a borderline value, e.g., >115 mg/dL, the individual is more sensitive to glucose challenge. |
| Tipping point | The point in a critical transition at which the system flips to another attractor state | A specific value of a parameter × (characterizing disease progression) at which the system undergoes a critical state transition and moves to a new attractor. |
Figure 2.
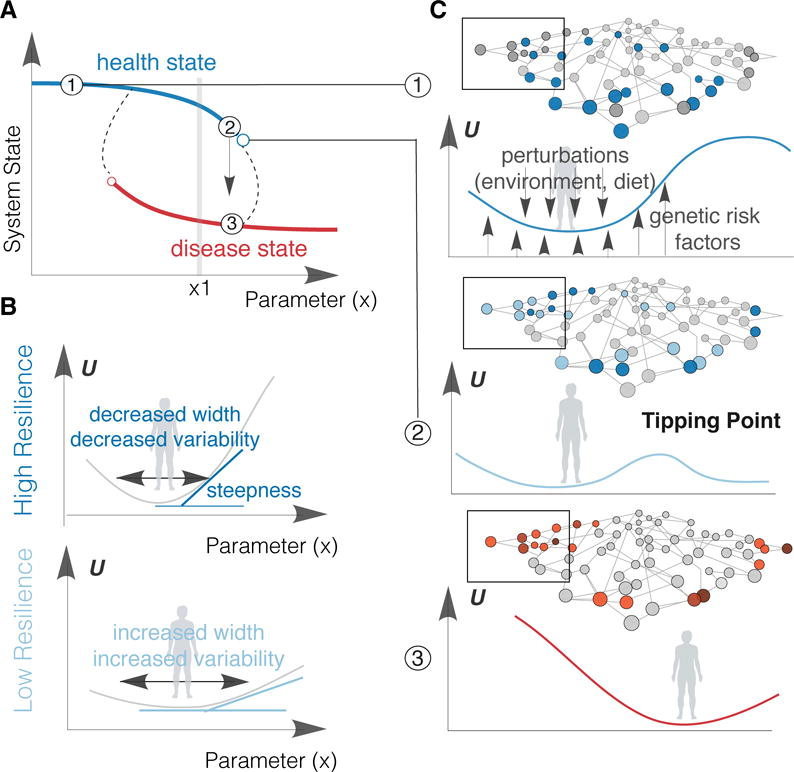
Health-to-Disease as a Critical State Transition. a) Health (blue line) and Disease (red line) are two alternative stable states of the system (reflected in the value of the vertical axis, the system state variable), as a function of a “parameter” × (ho horizontal axis) that characterizes the regime of behavior of a system (“bifurcation diagram). Dashed lines represent a possible path for the abrupt transition from one stable state (health) to another one (disease) as the parameter × increases. The empty circles mark unstable states that forces the system to undergo the switch-like transition to the alternative stable state and represent the critical thresholds (“tipping points”), where the qualitative behavior of the system state changes abruptly. A snapshot based on the parameter value ×1 cannot distinguish between health or disease state without obtaining more information about the actual system state (other variables in omics dimension, represented by y-axis), e.g., 120 mg/dL of fasting glucose can indicate a pre-diabetic individual (blue line) or a diabetic patient under diabetic medication (red line). c) The healthy state (1): The width and steepness of the potential well (around stable lowest “energy” states) are shaped both by genetic and environmental perturbations. The measured variation over time of any active protein, metabolite or other omics measurements (dark blue nodes in the network) informs on individual’s healthy physiological range. At this state, the system is resilient to perturbations. The destabilized healthy (pre-disease) state (2): The potential well has almost flattened allowing access to the disease state. The measured variation over time for certain proteins, metabolites or other omics measurements (light blue nodes) varies significantly and may corresponds to disease state markers (black box). The system has low resilience and is sensitive to perturbations. The disease state (3): The system has shift to a new, resilient steady state. The measured variation over time of any active protein, metabolite or other omics measurements (red nodes in the network) informs on individual’s disease progression. U: landscape potential, x: any systems parameter (e.g., fasting glucose, see Table 2)
Within this framework, a three-stage model of health-to-disease transitions has been proposed.12,13 (A) In the healthy state, the system is resilient to perturbations. (B) In the destabilized but still healthy (pre-disease) state, the system has low resilience and is sensitive to environmental or genetic perturbations. Defined as the limit of a health state that corresponds to what is sometimes considered a pre-disease state by clinicians (e.g., pre-hypertension and pre-diabetes). Very importantly, with appropriate interventions the system can still revert to the health state. (C) In the disease state (a new attractor state), the system cannot spontaneously recover and return to the normal state, which contrasts it to the pre-disease state. In few clear-cut instances, a one-time therapeutic intervention can restore the system back to the health attractor, as best documented in the case of arrhythmia and measures that restore normal rhythm.14 However, in most diseases (other than in surgical treatment) therapy cannot cleanly achieve such reversal to the healthy attractor, but is limited to strengthening compensatory mechanisms that prevent the initial disease state from progressing or causing further damage, as in the case of hypertension and diabetes. Therefore, identifying the “point of no return” at which the pre-disease state inexorably transitions to disease is of utmost importance for clinical practice. This conceptualization could not just revolutionize disease prevention, but also opens a new vista on health and wellness management, and can thus help shape the patient’s expectations. We will discuss this point further below.
The state of the art
Omics biomarkers
Genome-wide association studies (GWAS) have identified single-nucleotide polymorphisms (SNPs) associated with chronic disease-specific clinical phenotypes or their risk factors, for instance blood lipids and C-reactive protein for CVDs.15,16 The general consensus is that while individual genetic variants and risk markers exhibit only modest disease associations, when combined into polygenic or multivariate risk scores they can explain a significant portion of the variation in disease incidence in the general population.15,17,18 Still, in the case of genetic risk factors the heritability explained by the most significant variants is approximately 10%, whereas the estimates of total heritability from family studies are substantially higher, between 30% and 50%.19,20 This “missing heritability” conundrum is typical for complex traits and emphasizes the need for continued development and evaluation of disease risk models that incorporate not only genetic factors or complex genetic phenomena, such as epistasis, that interfere with the effort to pinpoint particular genetic variants as predictors of genetic risk, but also considers non-genetic and dynamical dimensions of human physiology, as well as environmental risk factors.21 By contrast, whole genome sequencing of families with rare cardiovascular disorders, cases where the predominance of a genetic cause is likely, have enabled the rapid identification of new genes related to cardiovascular development and function (BOX 1) and drives the adoption of genome sequencing as a frontline diagnostic tool.22
BOX 1. Rare Family Syndromes and Whole Genome Sequencing.
There are >150 rare heart syndromes or conditions for which there are no basic mechanistic insights beyond the phenotypic manifestation of the disease.91 Rare diseases comprise a heterogeneous set of conditions that affect various organ systems and have wide ranging prognoses. Family genomics is an approach that integrates observed genetic variations in multiple individuals in a family – affected and unaffected – and characterizes these observed variants according to their confidence level, their predicted deleteriousness, the match between their population frequencies and the prevalence of the disease, their mode of transmission through the pedigree, and whether they match the disease status of the individuals.92 This integrative strategy is particularly suited for analysis of rare diseases, where the same set of variants can be assumed to cause the disease in multiple affected individuals in a family, or variants arising de novo in the same gene(s) can explain disease patterns in families with single affected individuals. In the former case, the number of meioses separating the affected individuals determines the power, effectively narrowing the search space within the genome to the regions identical by descent; frequently, a single family may suffice to identify compelling candidate variants and genes. In the latter case, novel mutations are evidenced in individuals relative to their parents.
The family genomics method was successfully applied to study Adams-Oliver Syndrome (AOS), which is defined as aplasia cutis congenita (ACC) with transverse terminal limb defects. AOS has a number of associated developmental anomalies affecting the central nervous system, congenital heart defects (23% of cases) and other vascular anomalies (14% of cases).93 To date, mutations in six genes have been implicated in AOS (ARHGAP31, DOCK6, EOGT, RBPJ, NOTCH1 and DLL4), with both dominant and recessive modes of inheritance and were identified via family genomics analysis.93–98 Additionally, the causal GATA4 variant in a pedigree affected with cardiac septal defects was originally identified by targeted sequencing; the family genomics approach using whole-genome sequencing successfully re-identified this variant with genome-wide significance and objectively confirmed the absence of other candidate causal variants.88,99 Even common diseases like bipolar disorder can be caused by combinations of rare variants.100 In this case, a family-based sequencing strategy can detect the effects of transmitted rare variants with moderate to large effect sizes within a pedigree. Family-based sequencing has certain advantages over sequencing individual, unrelated affected individuals. When variants are observed in more than one family member, there is higher confidence that they are not sequencing errors. Also, under many disease models one expects stronger or more numerous risk variants in familial cases than in sporadic cases.
Despite the potential role of genome sequencing to predict disease risk and stratify patient cohorts into subclasses of diagnosis and to predict the patient-specific response to a therapy, personal genome information is still rarely used, today, for clinical evaluation and management of CVDs.23 Instead, physicians assess a given patient’s physiological state by measuring proteins and other metabolites (e.g., hs-CRP, pro–BNP, and troponin) whose utility have been established only in large cohorts and thus have unknown validity for each individual case. This gap could be soon addressed by adopt additional multi-omics technologies that combines genomics with metabolomics and proteomics in the clinical setting. Overcoming the barrier of CLIA-certified, cost-effective applications for systems medicine technologies (Table 3) and, thus, enabling dense, unbiased, molecular profiling of an individual, it has the potential to (1) support a personalized clinical management of health and disease and (2) accelerate biomarker discovery by an exhaustive characterization of health- and disease-specific network states.24,25 This compendium includes extensive reviews for each of the omics technologies in cardiovascular research.
Table 3.
Major technological advances that have accelerated the adoption of Systems Biology and Systems Medicine.
| Technology | Methods/techniques | Examples |
|---|---|---|
| DNA Microarrays | Transcriptomics, exome capture, epigenetics, small RNA expression, genotyping, metagenomics. Transcriptomics now replaced by “RNA-sequencing” and genotyping increasingly replaced by WGS (see below) |
26 |
| Sequencing (Next-Generation, nanopore, or long-read sequencers; Single-molecule sequencing) |
Whole genome sequencing (WGS), exome analysis, epigenetics, whole transcriptome analysis (RNA-sequencing), small RNA sequencing, GWAS, metagenomics | 15, 27–31 |
| Mass spectrometry | Proteomics, metabolomics, mass spectrometry imaging, lipidomics, glycomics, protein-protein interactions, post-translational modification analysis | 32–39 |
| Microfluidics | Single cell assays including gene expression, whole genome sequencing, protein analysis and epigenome analysis | 13, 40–44 |
| Flow and Mass Cytometry | Single cell protein analysis | 45–48 |
| Genome editing technology | Genome-wide gene deletion assays, screening assays | 31,49,50 |
| iPSC technology | In vitro models for diseases, drug screening | 51–56 |
Herein, we only refer to specific examples that have been powered by the newest technologies and present novel biomarker niches (BOX 2). However, it is important to highlight that despite significant technological advances and the availability of rich data, biomarker discovery has been only marginally successful. The Institute of Medicine report on Omic Diagnostic Technologies enumerated major challenges to biomarker discovery, highlighting the disagreement between the large number of academic research-based, high-profiled publications on biomarkers and the poor translation into clinical practice.57 The aforementioned report specified issues that reduce the signal-to-noise ratio; for instance, unknown genetic and other risk factors that confound the signal to noise increasing the false discovery rate.57 All together, to increase the frequency of successful translational stories, the research enterprise needs to re-design research studies by considering the complexity and variability of human physiology, and by collecting high-dimensional datasets that will allow researchers to identify confounding variables and to stratify populations at early phases of biomarker discovery.58
BOX 2. Cell population structures as a new class of biomarkers.
Over the last decade, advances in genome sequencing and microfluidics allow scientist to explore new aspects of human physiology, such as the microbial communities in our gut, skin and other body cavities, as well as the cellular composition of human blood and other tissues. This present the opportunity to consider a new biomarker class that is related to the population structure of the microbial communities, or our own cells. The measured heterogeneity either reflected as different bacteria species or cell types and their states is a new biological observable in human health and disease.
Metagenomics
The recognition that microorganisms may play a critical role in maintaining human health than in generating diseases places metagenomics as one of the most relevant areas of future research.101,102 Several papers over the last decade investigate the impact of qualitative and quantitative changes in gut microbiota on the pathogenesis of cardiometabolic diseases in cohorts of well-phenotyped patients.103–105 The microbial-mammalian symbiosis plays a critical role in metabolic health. Microbial metabolites emerge as key messengers in the complex communication between the gut microbiota and their host.36 Recent studies suggested that choline and phosphatidylcholine from the diet could be metabolized to trimethylamine (TMA) by intestinal microbiota which would be further metabolized to a proatherogenic factor – trimethylamine-N-oxide (TMAO).106 TMAO is associated with increased risks for both prevalent CVD and incident major adverse cardiac events (myocardial infarction, stroke or death).103 Further studies suggest that more plasma and urine metabolites like GlcNAc-6-P and mannitol with increased levels in patients with coronary heart disease might be of microbial origin.32 While association analysis of species and function levels between intestinal microbes and these metabolites can reveal further biological insights highlighting the omics integration.36,107 Finally, there is the hypothesis how oral microbiota may provide a link between periodontal infection and cardiovascular disease, a well-established relationship.108 As impaired dentition accompanies old age, it may be worthwhile to explicitly investigate the oral microbiome as this is likely a contributor to changes in GIT microbiota and consequently health status.101
Immune system populations at single-cell level
The development of single-cell technologies has enabled a new, high-resolution information to model and understand disease progression, as well as drug or other perturbation responses.12,13,40,44 For instance, single-cell analysis has now been used to understand the origin and role of cellular heterogeneity in progression and drug resistance of various types of cancers including colon cancer, glioblastoma, breast cancer and prostate cancer.109 However, cell population heterogeneity becomes an important “biological observable” for other chronic diseases as well. Profiling immune system at single-cell resolution has been demonstrated as a potential biomarker for disease progression or treatment-specific responses.110 For instance, using mass cytometry, which allows single-cell profiling of >30 protein markers, in whole-blood samples from 32 patients undergoing hip replacement, researchers characterized the phenotypic and functional immune response to surgical trauma.45 Similarly, mass cytometry was used to build a functional map of healthy fetal-maternal immunity that will allow identifying adverse maternal and neonatal outcomes.46 As innate and adaptive immune responses have an essential role in the development and progression of many cardiovascular diseases, cellular heterogeneity of immune cells may provide a new set of biomarkers for atherosclerosis, coronary artery disease or cardiac arrhythmia – all associated with chronic inflammation.111
The landscape of translational research studies
The study design of translational and clinical research depends heavily on the project goals and budget (Figure 3). For instance, cohort studies or randomized clinical trials are the cornerstone of quantifying the effect of risk factors or interventions, respectively. Such cohort or “class”-based studies inform on the average health or disease states or the average response to therapeutics for a population (class) presumed to be homogeneous. More importantly, due to their enormous cost, these studies traditionally obtain a “low-dimensional snapshot” of the physiological state for hundreds of participants, limiting our ability to (1) capture information about health-to-disease transitions in the time dimension, (2) perform comparative outcome research across studies that measure different variables and (3) expand our understanding of explanatory end points.60 On the other hand, seminal studies, such as the Framingham Heart Study, the Nurses’ Health Study, and the Women’s Health Initiative, depart from the traditional “class-based” design and epitomize the power of longitudinal data for large population cohorts.61–64 These studies do in fact investigate how healthy individuals transition to a disease state, revealing risk factors and biomarkers that affect such transitions. However, the variables assessed are largely based on traditional clinical established measures or questionnaire – whose design is biased by established knowledge. By contrast, a new study model combines high-dimensional (dense), longitudinal profiling using the scientifically unbiased tools of omics measurements. This approach avoids the self-fulfilling prophecy of circular thinking associated with applying knowledge derived from population-averaging studies to yet another population study and therefore paves the way for new discoveries – such as individual specific biomarkers that may never have been discovered in population-averaging studies. Successful stories for such individualized, dense omics-based longitudinal studies for detecting early signs of disease transition in individuals begin to emerge, as in the case of diabetes and inflammatory bowel disease (IBD).65,66
Figure 3.
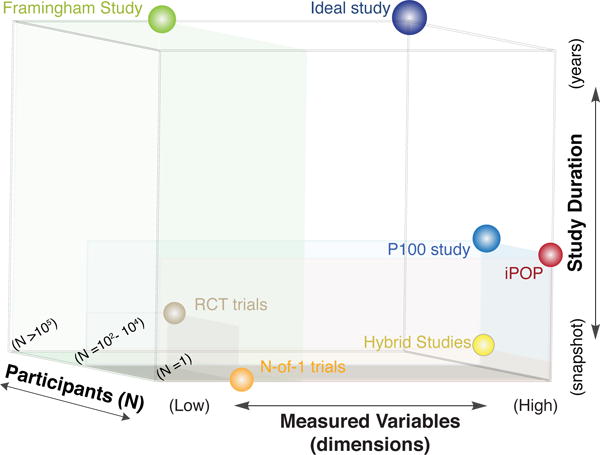
Clinical Research Study Landscape. We can categorize the selected study-designs based on their duration (long-term longitudinal vs. a single measurement/snapshot), the number of participants (single participant vs. thousands/big epidemiological cohorts) and the measures variables -dimensions (a few clinically important variants vs. all-omics-platforms). The P100 study is the closest match to a long-term, high-dimensional longitudinal study for a large population. (iPOP: integrated Personal Omics Profiling, P100 study: Pioneer 100 Wellness study, RCT: Randomized Control Trial)
These two study-design extremes for longitudinal monitoring, the averaging over thousands of individuals, measuring known at relative low dimensionality vs. single individual, dense (omics-wide) unbiased profiling present distinct ways of prior knowledge utilization and thus distinct opportunities for discovery. The second approach brings personalization to a new level because it does not rely on risk indicators that had been derived from averaging large populations but instead rely on the progression of the individual case to establish biomarkers for intervention planning. However, to achieve generalizability of the observed health transitions and outcomes, experience from many such transition in many individuals must be aggregated and explained within a formal framework of molecular pathophysiology.67 The first step is the acquisition of data by longitudinal dense profiling in large populations. Towards such a data-rich Framingham Heart-like study for the digital age, the Pioneer 100 Wellness study (herein, P100 study) prototyped a new research model using personal, dense, dynamical data (pD3) clouds for 108 individuals.17,68 Finally, there are “hybrid” study designs that are perfectly suited to address specific problems that can benefit from low-dimensional continuous data collection at large populations. Between these two extremes is the hybrid model study: It still stratifies healthy individuals into classes and tailors prevention strategies but utilizes their dynamic response to specific perturbations – a novel aspect deeply rooted in the ideas of “N-of-1” and systems medicine.69 Below we illustrate this hybrid model (Strategy 1) and the new multi-omics longitudinal model (Strategy 2).
Strategy 1: The Hybrid Methods
Stress tests, such as the commonly used oral glucose tolerance test and the metabolic exercise stress test, measure the body’s resilience to perturbations. The time it takes to return to the initial state (recovery time, ranging from minutes to hours) can be associated with the width and the steepness of the health state attractor (described above) and has the potential to stratify patients in healthy or pre-disease states (Figure 4). The quantification of recovery time requires monitoring human physiology at higher time resolution than used in many traditional research studies. Also, it raises an interesting question: Is there a proper sampling frequency to investigate health-to-disease transitions? The optimal frequency of study will certainly depend on the nature of the tested trait and disease. External perturbations such as diet and medical interventions can shift blood physiology within minutes or days, while the manifestation of clinical phenotypes may require years.
Figure 4.
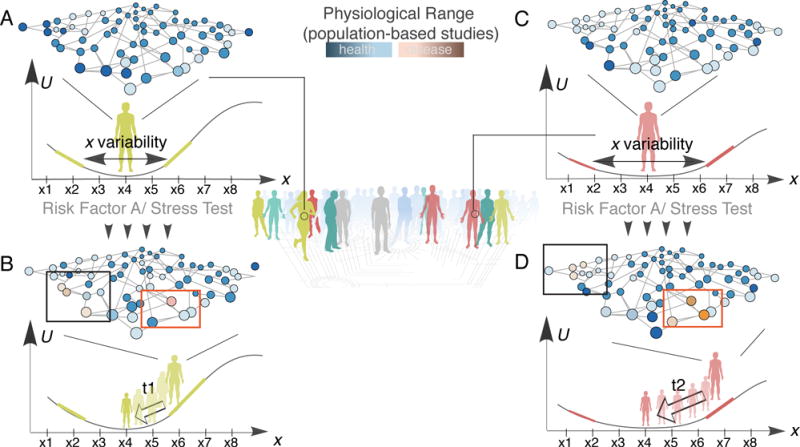
A new strategy to stratify health prevention and identify early disease signs. Recovery time after exposure to a specific risk factor (stress test) can be a new biomarker to stratify patients. A, C) The individuals exhibit a very similar profile before stress test – although their resilience (width and steepness of the potential well) is different. B, D) After the stress test, omics profiling can identify the most variable proteins/metabolites for each individual (red boxes) enabling personalized recommendations. While the recovery time (t1 vs. t2) can identify the high-risk individuals (longer time, higher risk), who can be further profiled for early disease biomarkers (black boxes). U: landscape potential (Table 2).
Like CVDs, diabetes is a chronic, complex disease that is strongly associated with individual’s lifestyle, such as poor diet. Evidence-based guidelines have long been used to provide uniform pharmacological and lifestyle management recommendations for the “average” patient. But to begin to expose what individuality is lost in such population-based guidelines, notably with respect to the kinetics of stress-response, Zeevi et al (2015) applied an innovative study design that combines continuous monitoring of blood glucose and dense omics profiling for 800 participants to expose the inter-individual systems differences. Postprandial glycemic response (PPGR) to every meal assessed by standard continuous glucose monitoring allowed researchers to establish a metric of resilience for each individual. The study not only confirmed that high PPGR values are associated with metabolic syndrome but also opened a new vista on individuality of kinetics of homeostatic response because of the additional omics dimensions measured for each proband.70–72 Food diary logs, blood metabolomics and gut metagenomics were integrated for the interpretation of each individuals PPGR/meal. With this additional high-dimensional information of potential response modifiers, researchers were able to predict individual-specific response to diet. The predictive capability comes from a statistical machine learning approach that leverages the large number of individual and their diverse responses.
This study illustrates the power of longitudinal high-dimensional N-of-1 trials for large populations in two distinct ways: (1) The physiological response to diet is highly individualized and general dietary guidelines can be harmful in individual cases, drawing our attention to other critical issues on disease management, such as drug prescription. (2) A relatively small number of individual, high-resolution profiles that exhibits diversity (as opposed the uniformity that is favored in standard statistical analysis) can inform about physiological principles in the response to a perturbation (nutritional and therapeutic interventions).
To design an equivalent study for coronary artery disease (CAD), what would be the single parameter we should and could measure at high temporal resolution? Continuous noninvasive monitoring of complex physiological variables, such as heart rate and heart rate variability (HRV), is an active research field.73–77 Since HRV integrates the responses of the sympathetic and parasympathetic systems, it can serve as a marker for many disease states beyond CVD.78–80 The key point is that the routine measurement of complex phenotypes in combination with a high-dimensional molecular systems characterization using multi-omics (see Strategy 2) can reveal many different aspects of healthy and disease states.
The new type of phenotype-biomarkers such as PPGR or HRV that result from complex non-linear network dynamics, can now be collected through connected digital devices. By leveraging such digital biomarkers, e.g., how patients are responding to a drug or treatment in the real-world, healthcare professionals would be able to adjust and improve tailored care plans. At the same time, new high-throughput microfluidic platforms may soon achieve high-dimensional and dense longitudinal bedside measurement. Recently, Gao et al (2017) published a mass-spectrometry-based lipidomics analysis of human dried blood spots.39 They were able to measure >1,200 lipid molecular species from a single blood sample in postprandial response kinetics experiments, of the kind as Zeevi et al (2015) performed. They identified significant alterations in triacylglyceride species that could be further investigated to reveal nutritional risk factors with atherosclerosis.
Strategy 2: Personal, Dense, Dynamic Data (pD3) Clouds
The Pioneer 100 Wellness study was a feasibility study focused on generating personal, dense, dynamic data clouds for two primary purposes: (1) to show that actionable information could be readily identified from the data that, when acted upon, would result in health benefits to each individual (based on connections that are already known and approved); and (2) to build a dataset that can be mined for novel biological and medical discovery, including early-stage diagnostics and prevention and early-stage disease reversal strategies.17 This project profiled 108 individuals using whole-genome sequencing, proteomics, metabolomics, clinical chemistries and gut microbiomes every three months over the course of nine months. Specifically, these data included 218 clinical laboratory tests, >800 metabolites and proteins in the body fluids, as well as the abundances of >4,000 gut microbial species. There were three primary outcomes of this study. First, it identified over 3,000 statistically significant associations across the diverse data categories. The resulting inter-omic cross-sectional correlation network resulted in 766 nodes connected through 3,470 statistically significant interactions (edges). Second, the whole-genome sequencing data were used to calculate polygenic risk scores for 127 GWAS-assessed traits or conditions, including blood pressure, heart rate, QT interval, and LDL cholesterol. These genomic indicators were then associated with the measurements in the blood (proteins, metabolites, and clinical chemistries) to identify ways in which the genetic risk was manifesting in the body (e.g., the concentration of cystine in the plasma is negatively correlated with the genetic risk for Inflammatory Bowel Disease). These types of observations can provide hypotheses for patient stratification of potential confounding factors, as well for personalized prevention, early-disease reversal or treatment.
Finally, as the size of the pioneer population is increasing and time of observation expanded, we can identify biomarkers of biological as opposed to chronological age. Phenotypes, such as lung function, grip strength or bone mineral density have long been used as indicators of biological aging. However, a large portion of the observed phenotypic variance remains unexplained and a comprehensive understanding of most complex phenotypes is lacking. Erikson et al (2016) pursued genome sequencing of healthy aged individuals (Wellderly) to understand the genetics of disease-free aging without medical intervention.81 The authors concluded that healthy aging is associated with having a lower genetic risk for select diseases, and particularly, cognitive decline. However, they emphasized the difficulty to achieve genome-wide significance due to small sample size and the complexity of a rare phenotype, such as healthy aging, that takes years to accrue. Therefore, monitoring the temporal change of pD3 clouds for each individual will help us to startify the entire population based on individual’s “wellderly” potential (decreased genetic risk for chronic diseases) and follow their aging process to identify how individual’s environmental exposures influence his/her biological aging and chronic disease delay.
The principle of equifinality: early vs. late disease stage biomarkers
The current disease-oriented paradigm of clinical research and practice precludes detailed knowledge about the early presentation of CVDs.82 Biomarkers for clinical decision-making and drug development represent cardiovascular disease endpoints limiting the diagnosis and management of early stages of CVDs, as well as drug discovery and development that reverse or delay the progression of subclinical phenotypes. In general, the clinical efficacy of developed drugs has remained unimpressive, owing in large part to the heterogeneous manifestation and progression of CVDs.83 The latter highlights why longitudinal, omics profiling (pD3 clouds) will provide a window into individual-specific mechanisms that cause or contribute to health-to-disease transition and disease progression, which could ultimately enable the ‘precise’ targeting of these mechanisms.
Investigators have found that neurodegeneration following prion infection in mice, progresses through gradual gene expression shifts that can be presented as disease-perturbed networks.9 When they summarized the results across individuals, they identified specific state transitions (gene network states) as the diseases progresses. However, there was significant heterogeneity in the sequence of the gene expression changes depending on the mouse strain- and infectious agent.9 This is a critical departure from traditional biomarker research, even of multi-marker signatures, because it becomes evident that the network architecture reconfigures, and not just individual nodes favoring a high-dimensional, systems biology approach.
We hypothesize that there are multiple possible trajectories from health to disease that depend on the genetic makeup and environmental exposure of each individual – a view consistent with the systems principle of equifinality (Table 1), which highlights the notion of disease states being stable attractor states as explained earlier. If this is the case (as it seems highly likely), what does this mean for biomarker discovery and how can we analyze the diversity of the possible trajectories, and identity those that pertain go a given patient?
We picture that the array of perturbed regulators across individuals in a pre-disease state, like pre-hypertension, affect collectively a specific regulatory subnetwork (Figure 5). Still, each individual may present changes in distinct nodes of this apparent “disease-perturbed” network, depending on their genetics and environmental exposure. In other words, the “reaction norm” postulated by early geneticists is what is determined by the genome: it shapes the detailed structure of the basin of attraction of the health state and hence, the individual-specific exit trajectory. This concept underscores the notion of not just multi-marker panels for CVD diagnosis and drug discovery but that such panels need to be further stratified by the polygenic risk of individuals. pD3 clouds offer the underlying data needed to define relevant marker panels that represent disease-perturbed networks and their dynamics. Since disease-related physiological parameters and their response to perturbations vary so much between individuals, a new strategy is to define some apparent networks of markers based on pairwise functional relationships between markers. One obvious type of relationship is the correlation between markers, measured as their co-variation across a cohort of individuals, thus precisely exploiting the inter-individual variability that otherwise would appear as noise. In this way, Price et al (2017) built 70 relevant subnetworks that appear as modules of markers (or “community networks”) and essentially represent sets of physiologically related variables.17 For instance, one module contained C-peptide, triglycerides, insulin, homeostatic risk assessment–insulin resistance (HOMA-IR), fasting glucose, high-density lipid (HDL) cholesterol, and small low-density lipid (LDL), which the authors labelled the “cardiometabolic health” network. This network-based data representation is expected to be more sensitive for detecting minute departures from the healthy state in individuals given that each person may follow distinct trajectories when exiting the health state. Without knowledge of data representation, the inter-individual variability in the high-dimensional data space would be considered noise and lost. This method could for instance facilitate the discovery of biomarker that could assist in clinical decision-making and drug development of conditions, such as pre-hypertension or pre-diabetes that epitomize the earliest stage of departure from the health attractor. This is important because, in accordance with the principle of equifinality, the earlier in the disease process, the higher the inter-individual variability. Knowledge of the existence of the “community networks” and their members thus may be key for future multivariate blood diagnostics.
Figure 5.
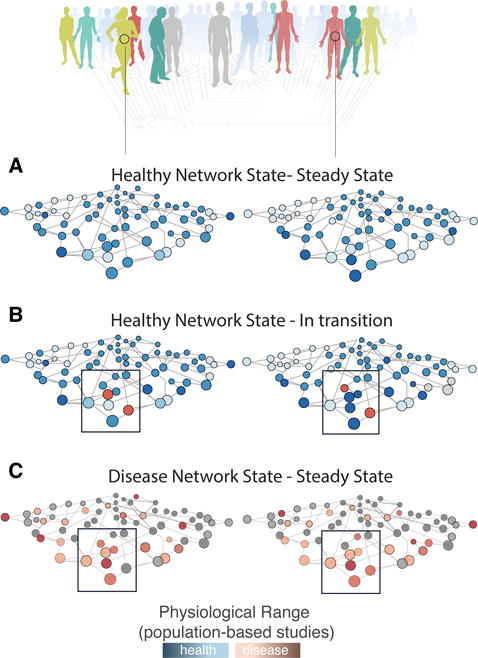
The principle of equifinality. A) The health state variability: Individuals show heterogeneity in the concentration of measured physiological dimensions (e.g., metabolites or proteins in the blood) as indicated by the color shade (dark blue corresponds to higher detected concentrations). B) The pre-disease state variability: Exposure to specific perturbations (e.g., nutrition, infection agent etc.) reveals the pre-disease state profiles. Blue nodes correspond to early molecular signs for a health-to-disease transition signifying the future disease-perturbed network (black box), known as leading network hypothesis. Individuals may show “out-of-range” values for different nodes of the “leading network”. C) The disease state variability: Individuals show heterogeneity in the concentration of measured physiological dimensions as indicated by the color shade (dark red corresponds to higher detected concentrations).
Data translation to actionable possibilities
As described above, we can design a framework to study disease complexity using principles from systems theory. An important component of the proposed framework is to quantify individual response to nutritional, therapeutic and other interventions (e.g., exercise, inflammation, nutrition, etc.). The participants in the Zeevi et al (2015) study had to follow a specific diet protocol and self-report activity or medication that could affect glucose levels.69 On the other hand, the participants in the P100 study didn’t follow a specific protocol, as this study had a broader goal: to improve participants’ overall health – not a single dimension.17,84 Therefore, a unique aspect of the P100 study was to provide personalized health coaching on actionable information based on the genetics and the longitudinal clinical and ‘omics assessments. Essentially, coaches presented personalized actionable items to participants who could decide on and actively participate in their health amelioration. The key point is that the coaches acted as behavioral specialists and determined before the study the objectives of health care for each individual. The coaches then both explained the actionable possibilities and placed them in the context of each individual’s health objective. Previous reports where individuals actively annotated data with environmental and behavioral information has presented new scientific hypothesis for translational research and improved clinical outcomes.3,85,86 The effect of each “perturbation” was measured in the next two rounds of data collection. This unique design of P100 study demonstrated that it is possible to 1) study human health and disease considering social and psychological interactions, and 2) build a fast-track, data-driven translational research platform. Operating on the nascent field of scientific wellness – the early transitions to disease, a clinically-neglected realm, researchers managed to minimize the distance between “diagnosis” and “intervention” and achieve personalization through patient participation.
Systems medicine strategies to personalized in vitro models
Understanding the underlying molecular and cellular mechanisms of disease manifestation is an important step in the development of efficient and effective therapies for cardiovascular diseases. While the improvements in genome-wide molecular techniques provide a powerful lens to study human disease, there are obvious limitations to what kinds of samples can be collected from humans. For CVD, researchers have access only to what is released from acutely injured tissues (e.g., during myocardial infarction with widespread necrosis, inflammation, and invasion of acute-phase response cells). In other words, the reported networks (Fig 1) represent just snapshots of tissue state that only remotely resemble the normal or diseased tissue under stable conditions. Therefore, designing and developing research strategies to study human health-to-CVD transitions non-invasively at scale will be paramount to implement the new systems medicine in CVD (BOX 3). Pioneering work in Yamanaka’s lab has provided a major breakthrough for stem cell research through the discovery of induced pluripotent stem cells (iPSCs).87 The ability to create individual-specific stem cells and then differentiate them to any cell type provides a unique opportunity to study genetic diseases in vitro. Several studies have now reproduced the disease phenotypes, molecular and electrophysiological abnormalities and drug responses in patient-derived cells for several cardiomyopathies, tachycardias, Barth, Marfan, long QT and LEOPARD syndromes.52,53,87 Recently, Ang et al. generated iPSCs for individuals from the same family with cardiac septal defect, differentiated them to cardiomyocytes, and used a systems approach to uncover how the mutated GATA4 impairs the cardiac gene program.88 The study showed that some of the disease phenotypes such as impaired contractility, calcium handling, and metabolic activity in cardiomyocytes are replicated in the in vitro model; it also unmasked how the mutation in GATA4 disrupts recruitment of TBX5 at the enhancers leading to aberrant chromatin states and a faulty transcriptional program.89
BOX 3. Health-to-disease transitions and their reflections.
Ongoing preventive monitoring of heart physiology requires easily-accessible samples and minimally invasive procedures. Blood is a natural window into human physiology. It contains abundant information concerning the health and disease status of organs in the form of secreted RNAs, proteins, and metabolites, as well as cell population diversity.112 Changes in the concentrations of some of these molecular and cellular components can provide reflections of disease processes. A proof-of-principle study comes from the field of infectious disease.9 Researchers studied the dynamics of prion infection in mice and identified the transition of a health- to a disease- network state through progressive activation of four distinct sub-networks that are related to neurodegeneration.9 Then, they leveraged the organ-specific nature of the perturbed proteins to prove that this transition is reflected in the blood, enabling the early diagnosis and disease stratification based on the stage and infectious agent weeks ahead of the clinical manifestation of the disease.113 Importantly, these distinct network states can point to therapeutic targets or precede clinical symptoms, offering opportunities for intervention in disease progression. A similar ongoing effort for Huntington’s disease in mice aims to prove that this paradigm (reflections of early transitions) holds for complex, non-infectious, chronic diseases.114 Traditional clinical biomarkers, such as Troponin T (TnT) and alanine aminotransferase (ALT) are classified under the paradigm of organ-specific (or organ-enriched) proteins. Their elevated levels in the blood reflect damage in heart or liver, respectively. In fact, TnT is the most significant predictive biomarker for subclinical myocardial injury in 30 days than any other systemic inflammation marker, such as C-reactive protein and cholesterol lipids.115 Still, we lack longitudinal monitoring of TnT of populations at risk.
In the medicine of future, our ability to generate patient-derived cell types will enable predicting their personalized drug response even before administering the drugs. Recently, Matsa et al. demonstrated this in the lab using transcriptome data from iPSC-derived cardiomyocytes from seven patients.56 The study showed that the cells derived from different patients retained patient-specific gene expression profiles and this could be used to predict the drug response.67 Patient-derived cells can serve as a platform for screening effective therapy strategies as well as to study cardiotoxicity using small molecule libraries. One of the main reasons for failure of candidate drugs at late stages of development is cardiotoxicity, which could be avoided by pre-screening such drugs on in vitro cardiomyocyte models or liver toxicity models.90
Future perspectives.
All of the above advocates for new workflows and infrastructure to enable fast and effective translation of research from bench to bedside. We foresee a new model for translational and clinical research, wherein virtually the entire population becomes an ongoing longitudinal study for P4 (predictive, personalized, preventive, and participatory) medicine. Personal, dense, dynamic data (pD3) clouds of an individual before the incidence of disease (baseline) and during subclinical stage of disease are crucial to understand early health-to-disease transitions that mark clinical onset. Expanded electronic medical records are a way to start: genetic data need to be incorporated into the medical record because of their potential to aid clinical decision-making, and to facilitate the adoption of future omics metrics. As efforts for integration of multiple streams of health data (electronic medical data, imaging, omics and biosensors) mature, generating more comprehensive pD3 clouds, it will become possible to measure and integrate new aspects of our physiology and to understand how (chronic) environmental exposures influence it. This can revolutionize the management of wellness and disease when applied to the same health systems and patient populations by designing a personalized, evidence-driven medical practice. The task now is to make actionable the distinct features in the pD3 clouds and to convert this information into effective metrics to be used in clinical settings, leading to improved patient outcomes.
Acknowledgments
Sources of Funding
The authors were supported by National Institute of General Medical Sciences (NIGMS) Grant R01GM109964, and NIGMS National Centers for Systems Biology Grant 2P50GM076547-06A1.
Non-standard Abbreviations and Acronyms
- P4 Medicine
Personalized, Predictive, Preventive and Participatory Medicine
- PD3 Clouds
Personal, Dense, Dynamical Data Clouds
- CLIA
Clinical Laboratory Improvement Amendments
- CVD
Cardiovascular Diseases
- CAD
Coronary Artery Disease
- GWAS
Genome-Wide Association Studies
- SNP
Single-Nucleotide Polymorphism
- hs-CRP
High-Sensitivity C-Reactive Protein
- LDL
Low-Density Lipoprotein
- BNP
Brain Natriuretic Peptide
- PPGR
Postprandial glycemic response
- HRV
Heart Rate Variability
- iPSC
Induced Pluripotent Stem Cell
Footnotes
Disclosures
GG, NDP and LH hold stock options in Arivale, Inc. Arivale, Inc. did not fund the study and was not involved in its design, implementation, or reporting.
References
- 1.Lenfant C. Shattuck lecture–clinical research to clinical practice–lost in translation? N Engl J Med. 2003;349:868–874. doi: 10.1056/NEJMsa035507. [DOI] [PubMed] [Google Scholar]
- 2.Wang JJ, Aboulhosn JA, Hofer IS, Mahajan A, Wang Y, Vondriska TM. Operationalizing Precision Cardiovascular Medicine: Three Innovations. Circ Res. 2016;119:984–987. doi: 10.1161/CIRCRESAHA.116.309776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Califf RM, Robb MA, Bindman AB, et al. Transforming Evidence Generation to Support Health and Health Care Decisions. N Engl J Med. 2016;375:2395–2400. doi: 10.1056/NEJMsb1610128. [DOI] [PubMed] [Google Scholar]
- 4.Benjamin EJ, Blaha MJ, Chiuve SE, et al. Heart Disease and Stroke Statistics-2017 Update: A Report From the American Heart Association. Circulation. 2017;135:e146–e603. doi: 10.1161/CIR.0000000000000485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tricoci P, Allen JM, Kramer JM, Califf RM, Smith SC. Scientific evidence underlying the ACC/AHA clinical practice guidelines. JAMA. 2009;301:831–841. doi: 10.1001/jama.2009.205. [DOI] [PubMed] [Google Scholar]
- 6.Han H, Chao H, Guerra A, Sosa A, Christopoulos G, Christakopoulos GE, Rangan BV, Maragkoudakis S, Jneid H, Banerjee S, Brilakis ES. Evolution of the American College of Cardiology/American Heart Association Clinical Guidelines. J Am Coll Cardiol. 2015;65:2726–2734. doi: 10.1016/j.jacc.2015.04.050. [DOI] [PubMed] [Google Scholar]
- 7.Vodovotz Y, An G, Androulakis IP. A Systems Engineering Perspective on Homeostasis and Disease. Front Bioeng Biotechnol. 2013;1:6. doi: 10.3389/fbioe.2013.00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.del Sol A, Balling R, Hood L, Galas D. Diseases as network perturbations. Curr Opin Biotechnol. 2010;21:566–571. doi: 10.1016/j.copbio.2010.07.010. [DOI] [PubMed] [Google Scholar]
- 9.Hwang D, Lee IY, Yoo H, Gehlenborg N, Cho J-H, Petritis B, Baxter D, Pitstick R, Young R, Spicer D, Price ND, Hohmann JG, Dearmond SJ, Carlson GA, Hood LE. A systems approach to prion disease. Mol Syst Biol. 2009;5:252. doi: 10.1038/msb.2009.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scheffer M. Critical Transitions in Nature and Society. Princeton University Press; [Google Scholar]
- 11.Choi M, Shi J, Jung SH, Chen X, Cho KH. Attractor landscape analysis reveals feedback loops in the p53 network that control the cellular response to DNA damage. Sci Signal. 2012;5:ra83. doi: 10.1126/scisignal.2003363. [DOI] [PubMed] [Google Scholar]
- 12.Mojtahedi M, Skupin A, Zhou J, Castaño IG, Leong-Quong RYY, Chang H, Trachana K, Giuliani A, Huang S. Cell Fate Decision as High-Dimensional Critical State Transition. PLoS Biol. 2016;14:e2000640. doi: 10.1371/journal.pbio.2000640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bargaje R, Trachana K, Shelton MN, McGinnis CS, Zhou JX, Chadick C, Cook S, Cavanaugh C, Huang S, Hood L. Cell population structure prior to bifurcation predicts efficiency of directed differentiation in human induced pluripotent cells. Proc Natl Acad Sci U S A. 2017;114:2271–2276. doi: 10.1073/pnas.1621412114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Acton QA. Arrhythmia: New Insights for the Healthcare Professional: 2011 Edition: ScholarlyBrief. ScholarlyEditions. 2012 Available at https://books.google.com/books?id=6_c1J1U9NtUC.
- 15.Kessler T, Vilne B, Schunkert H. The impact of genome-wide association studies on the pathophysiology and therapy of cardiovascular disease. EMBO Mol Med. 2016;8:688–701. doi: 10.15252/emmm.201506174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sabater-Lleal M, Huang J, Chasman D, et al. Multiethnic meta-analysis of genome-wide association studies in >100 000 subjects identifies 23 fibrinogen-associated Loci but no strong evidence of a causal association between circulating fibrinogen and cardiovascular disease. Circulation. 2013;128:1310–1324. doi: 10.1161/CIRCULATIONAHA.113.002251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Price ND, Magis AT, Earls JC, et al. A wellness study of 108 individuals using personal, dense, dynamic data clouds. Nat Biotechnol. 2017;35:747–756. doi: 10.1038/nbt.3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Whitfield JB. Genetic insights into cardiometabolic risk factors. Clin Biochem Rev. 2014;35:15–36. [PMC free article] [PubMed] [Google Scholar]
- 19.Fischer M, Broeckel U, Holmer S, Baessler A, Hengstenberg C, Mayer B, Erdmann J, Klein G, Riegger G, Jacob HJ, Schunkert H. Distinct heritable patterns of angiographic coronary artery disease in families with myocardial infarction. Circulation. 2005;111:855–862. doi: 10.1161/01.CIR.0000155611.41961.BB. [DOI] [PubMed] [Google Scholar]
- 20.Zdravkovic S, Wienke A, Pedersen NL, Marenberg ME, Yashin AI, De Faire U. Heritability of death from coronary heart disease: a 36-year follow-up of 20 966 Swedish twins. J Intern Med. 2002;252:247–254. doi: 10.1046/j.1365-2796.2002.01029.x. [DOI] [PubMed] [Google Scholar]
- 21.Zuk O, Schaffner SF, Samocha K, Do R, Hechter E, Kathiresan S, Daly MJ, Neale BM, Sunyaev SR, Lander ES. Searching for missing heritability: designing rare variant association studies. Proc Natl Acad Sci U S A. 2014;111:E455–464. doi: 10.1073/pnas.1322563111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stavropoulos DJ, Merico D, Jobling R, et al. Whole Genome Sequencing Expands Diagnostic Utility and Improves Clinical Management in Pediatric Medicine. NPJ Genomic Med. 2016;1 doi: 10.1038/npjgenmed.2015.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Smith JA, Ware EB, Middha P, Beacher L, Kardia SLR. Current Applications of Genetic Risk Scores to Cardiovascular Outcomes and Subclinical Phenotypes. Curr Epidemiol Rep. 2015;2:180–190. doi: 10.1007/s40471-015-0046-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McShane LM, Cavenagh MM, Lively TG, Eberhard DA, Bigbee WL, Williams PM, Mesirov JP, Polley M-YC, Kim KY, Tricoli JV, Taylor JMG, Shuman DJ, Simon RM, Doroshow JH, Conley BA. Criteria for the use of omics-based predictors in clinical trials. Nature. 2013;502:317–320. doi: 10.1038/nature12564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Horak P, Klink B, Heining C, et al. Precision oncology based on omics data: The NCT Heidelberg experience. Int J Cancer. 2017;141:877–886. doi: 10.1002/ijc.30828. [DOI] [PubMed] [Google Scholar]
- 26.Joehanes R, Ying S, Huan T, Johnson AD, Raghavachari N, Wang R, Liu P, Woodhouse KA, Sen SK, Tanriverdi K, Courchesne P, Freedman JE, O’Donnell CJ, Levy D, Munson PJ. Gene expression signatures of coronary heart disease. Arterioscler Thromb Vasc Biol. 2013;33:1418–1426. doi: 10.1161/ATVBAHA.112.301169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akat KM, Moore-McGriff D, Morozov P, Brown M, Gogakos T, Correa Da Rosa J, Mihailovic A, Sauer M, Ji R, Ramarathnam A, Totary-Jain H, Williams Z, Tuschl T, Schulze PC. Comparative RNA-sequencing analysis of myocardial and circulating small RNAs in human heart failure and their utility as biomarkers. Proc Natl Acad Sci. 2014;111:11151–11156. doi: 10.1073/pnas.1401724111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang K-C, Yamada KA, Patel AY, Topkara VK, George I, Cheema FH, Ewald GA, Mann DL, Nerbonne JM. Deep RNA Sequencing Reveals Dynamic Regulation of Myocardial Noncoding RNAs in Failing Human Heart and Remodeling With Mechanical Circulatory Support. Circulation. 2014;129:1009–1021. doi: 10.1161/CIRCULATIONAHA.113.003863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nikpay M, Goel A, Won H-H, et al. A comprehensive 1000 Genomes–based genome-wide association meta-analysis of coronary artery disease. Nat Genet. 2015;47:1121–1130. doi: 10.1038/ng.3396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dehghan A, Bis JC, White CC, et al. Genome-Wide Association Study for Incident Myocardial Infarction and Coronary Heart Disease in Prospective Cohort Studies: The CHARGE Consortium. PLOS ONE. 2016;11:e0144997. doi: 10.1371/journal.pone.0144997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Muka T, Koromani F, Portilla E, O’Connor A, Bramer WM, Troup J, Chowdhury R, Dehghan A, Franco OH. The role of epigenetic modifications in cardiovascular disease: A systematic review. Int J Cardiol. 2016;212:174–183. doi: 10.1016/j.ijcard.2016.03.062. [DOI] [PubMed] [Google Scholar]
- 32.Feng Q, Liu Z, Zhong S, Li R, Xia H, Jie Z, Wen B, Chen X, Yan W, Fan Y, Guo Z, Meng N, Chen J, Yu X, Zhang Z, Kristiansen K, Wang J, Xu X, He K, Li G. Integrated metabolomics and metagenomics analysis of plasma and urine identified microbial metabolites associated with coronary heart disease. Sci Rep. 2016;6 doi: 10.1038/srep22525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.McGregor E, Dunn MJ. Proteomics of heart disease. Hum Mol Genet. 2003;12:R135–R144. doi: 10.1093/hmg/ddg278. [DOI] [PubMed] [Google Scholar]
- 34.Beck HC, Overgaard M, Melholt Rasmussen L. Plasma proteomics to identify biomarkers – application to cardiovascular diseases. Transl Proteomics. 2015;7:40–48. [Google Scholar]
- 35.Arab S, Gramolini AO, Ping P, Kislinger T, Stanley B, van Eyk J, Ouzounian M, MacLennan DH, Emili A, Liu PP. Cardiovascular Proteomics. J Am Coll Cardiol. 2006;48:1733–1741. doi: 10.1016/j.jacc.2006.06.063. [DOI] [PubMed] [Google Scholar]
- 36.Fu J, Bonder MJ, Cenit MC, et al. The Gut Microbiome Contributes to a Substantial Proportion of the Variation in Blood Lipids. Circ Res. 2015;117:817–824. doi: 10.1161/CIRCRESAHA.115.306807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stegemann C, Pechlaner R, Willeit P, Langley SR, Mangino M, Mayr U, Menni C, Moayyeri A, Santer P, Rungger G, Spector TD, Willeit J, Kiechl S, Mayr M. Lipidomics Profiling and Risk of Cardiovascular Disease in the Prospective Population-Based Bruneck Study. Circulation. 2014;129:1821–1831. doi: 10.1161/CIRCULATIONAHA.113.002500. [DOI] [PubMed] [Google Scholar]
- 38.Chugh S, Suen C, Gramolini A. Proteomics and Mass Spectrometry: What Have We Learned About The Heart? Curr Cardiol Rev. 2010;6:124–133. doi: 10.2174/157340310791162631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gao F, McDaniel J, Chen EY, Rockwell HE, Drolet J, Vishnudas VK, Tolstikov V, Sarangarajan R, Narain NR, Kiebish MA. Dynamic and temporal assessment of human dried blood spot MS/MSALL shotgun lipidomics analysis. Nutr Metab. 2017;14 doi: 10.1186/s12986-017-0182-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Heath JR, Ribas A, Mischel PS. Single-cell analysis tools for drug discovery and development. Nat Rev Drug Discov. 2016;15:204–216. doi: 10.1038/nrd.2015.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abbaspourrad A, Zhang H, Tao Y, Cui N, Asahara H, Zhou Y, Yue D, Koehler SA, Ung LW, Heyman J, Ren Y, Ziblat R, Chong S, Weitz DA. Label-free single-cell protein quantification using a drop-based mix-and-read system. Sci Rep. 2015;5 doi: 10.1038/srep12756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Darmanis S, Gallant CJ, Marinescu VD, Niklasson M, Segerman A, Flamourakis G, Fredriksson S, Assarsson E, Lundberg M, Nelander S, Westermark B, Landegren U. Simultaneous Multiplexed Measurement of RNA and Proteins in Single Cells. Cell Rep. 2016;14:380–389. doi: 10.1016/j.celrep.2015.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DeLaughter DM, Bick AG, Wakimoto H, McKean D, Gorham JM, Kathiriya IS, Hinson JT, Homsy J, Gray J, Pu W, Bruneau BG, Seidman JG, Seidman CE. Single-Cell Resolution of Temporal Gene Expression during Heart Development. Dev Cell. 2016;39:480–490. doi: 10.1016/j.devcel.2016.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wills QF, Livak KJ, Tipping AJ, Enver T, Goldson AJ, Sexton DW, Holmes C. Single-cell gene expression analysis reveals genetic associations masked in whole-tissue experiments. Nat Biotechnol. 2013;31:748–752. doi: 10.1038/nbt.2642. [DOI] [PubMed] [Google Scholar]
- 45.Gaudillière B, Fragiadakis GK, Bruggner RV, et al. Clinical recovery from surgery correlates with single-cell immune signatures. Sci Transl Med. 2014;6:255ra131. doi: 10.1126/scitranslmed.3009701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fragiadakis GK, Baca QJ, Gherardini PF, et al. Mapping the Fetomaternal Peripheral Immune System at Term Pregnancy. J Immunol Baltim Md 1950. 2016;197:4482–4492. doi: 10.4049/jimmunol.1601195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Spitzer MH, Nolan GP. Mass Cytometry: Single Cells, Many Features. Cell. 2016;165:780–791. doi: 10.1016/j.cell.2016.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Thomas GD, Hamers AAJ, Nakao C, Marcovecchio P, Taylor AM, McSkimming C, Nguyen AT, McNamara CA, Hedrick CC. Human Blood Monocyte SubsetsHighlights: A New Gating Strategy Defined Using Cell Surface Markers Identified by Mass Cytometry. Arterioscler Thromb Vasc Biol. 2017;37:1548–1558. doi: 10.1161/ATVBAHA.117.309145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Seeger T, Porteus M, Wu JC. Genome Editing in Cardiovascular Biology. Circ Res. 2017;120:778–780. doi: 10.1161/CIRCRESAHA.116.310197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Strong A, Musunuru K. Genome editing in cardiovascular diseases. Nat Rev Cardiol. 2016;14:11–20. doi: 10.1038/nrcardio.2016.139. [DOI] [PubMed] [Google Scholar]
- 51.Shi Y, Inoue H, Wu JC, Yamanaka S. Induced pluripotent stem cell technology: a decade of progress. Nat Rev Drug Discov. 2016;16:115–130. doi: 10.1038/nrd.2016.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee YK, Ran X, Lai KWH, Lau VYM, Siu DCW, Tse HF. Generation and Characterization of Patient-Specific iPSC Model for Cardiovascular Disease. Methods Mol Biol Clifton NJ. 2016;1353:191–213. doi: 10.1007/7651_2015_273. [DOI] [PubMed] [Google Scholar]
- 53.Bayzigitov DR, Medvedev SP, Dementyeva EV, Bayramova SA, Pokushalov EA, Karaskov AM, Zakian SM. Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes Afford New Opportunities in Inherited Cardiovascular Disease Modeling. Cardiol Res Pract. 2016;2016:1–17. doi: 10.1155/2016/3582380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Avior Y, Sagi I, Benvenisty N. Pluripotent stem cells in disease modelling and drug discovery. Nat Rev Mol Cell Biol. 2016;17:170–182. doi: 10.1038/nrm.2015.27. [DOI] [PubMed] [Google Scholar]
- 55.Martins AM, Vunjak-Novakovic G, Reis RL. The Current Status of iPS Cells in Cardiac Research and Their Potential for Tissue Engineering and Regenerative Medicine. Stem Cell Rev Rep. 2014;10:177–190. doi: 10.1007/s12015-013-9487-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Matsa E, Burridge PW, Yu K-H, et al. Transcriptome Profiling of Patient-Specific Human iPSC-Cardiomyocytes Predicts Individual Drug Safety and Efficacy Responses In Vitro. Cell Stem Cell. 2016;19:311–325. doi: 10.1016/j.stem.2016.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Committee on the Review of Omics-Based Tests for Predicting Patient Outcomes in Clinical Trials, Board on Health Care Services, Board on Health Sciences Policy, Institute of Medicine. Evolution of Translational Omics: Lessons Learned and the Path Forward. Washington, D.C.: National Academies Press; 2012. [DOI] [PubMed] [Google Scholar]
- 58.Johnson KW, Shameer K, Glicksberg BS, Readhead B, Sengupta PP, Björkegren JLM, Kovacic JC, Dudley JT. Enabling Precision Cardiology Through Multiscale Biology and Systems Medicine. JACC Basic Transl Sci. 2017;2:311–327. doi: 10.1016/j.jacbts.2016.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Committee on Diagnostic Error in Health Care, Board on Health Care Services, Institute of Medicine, The National Academies of Sciences, Engineering, and Medicine. Improving Diagnosis in Health Care. Washington (DC): National Academies Press (US); 2015. Available at https://www.ncbi.nlm.nih.gov/books/NBK338600/ [Google Scholar]
- 60.James S, Rao SV, Granger CB. Registry-based randomized clinical trials—a new clinical trial paradigm. Nat Rev Cardiol. 2015;12:312–316. doi: 10.1038/nrcardio.2015.33. [DOI] [PubMed] [Google Scholar]
- 61.Dawber TR, Meadors GF, Moore FE. Epidemiological approaches to heart disease: the Framingham Study. Am J Public Health Nations Health. 1951;41:279–281. doi: 10.2105/ajph.41.3.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mahmood SS, Levy D, Vasan RS, Wang TJ. The Framingham Heart Study and the epidemiology of cardiovascular disease: a historical perspective. Lancet Lond Engl. 2014;383:999–1008. doi: 10.1016/S0140-6736(13)61752-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Eaker E, Hahn RA. Women’s Health Initiative. N Engl J Med. 1994;330:70–71. doi: 10.1056/NEJM199401063300121. [DOI] [PubMed] [Google Scholar]
- 64.Belanger CF, Hennekens CH, Rosner B, Speizer FE. The nurses’ health study. Am J Nurs. 1978;78:1039–1040. [PubMed] [Google Scholar]
- 65.Chen R, Mias GI, Li-Pook-Than J, et al. Personal omics profiling reveals dynamic molecular and medical phenotypes. Cell. 2012;148:1293–1307. doi: 10.1016/j.cell.2012.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Smarr L. Quantifying your body: a how-to guide from a systems biology perspective. Biotechnol J. 2012;7:980–991. doi: 10.1002/biot.201100495. [DOI] [PubMed] [Google Scholar]
- 67.Lillie EO, Patay B, Diamant J, Issell B, Topol EJ, Schork NJ. The n-of-1 clinical trial: the ultimate strategy for individualizing medicine? Pers Med. 2011;8:161–173. doi: 10.2217/pme.11.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hood L, Price ND. Demystifying disease, democratizing health care. Sci Transl Med. 2014;6:225ed5. doi: 10.1126/scitranslmed.3008665. [DOI] [PubMed] [Google Scholar]
- 69.Zeevi D, Korem T, Zmora N, et al. Personalized Nutrition by Prediction of Glycemic Responses. Cell. 2015;163:1079–1094. doi: 10.1016/j.cell.2015.11.001. [DOI] [PubMed] [Google Scholar]
- 70.American Diabetes Association. 5. Prevention or Delay of Type 2 Diabetes. Diabetes Care. 2015;38:S31–S32. doi: 10.2337/dc15-S008. [DOI] [PubMed] [Google Scholar]
- 71.Gallwitz B. Implications of postprandial glucose and weight control in people with type 2 diabetes: understanding and implementing the International Diabetes Federation guidelines. Diabetes Care. 2009;32(Suppl 2):S322–325. doi: 10.2337/dc09-S331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Nishida T, Tsuji S, Tsujii M, Arimitsu S, Haruna Y, Imano E, Suzuki M, Kanda T, Kawano S, Hiramatsu N, Hayashi N, Hori M. Oral Glucose Tolerance Test Predicts Prognosis of Patients with Liver Cirrhosis. Am J Gastroenterol. 2006;101:70–75. doi: 10.1111/j.1572-0241.2005.00307.x. [DOI] [PubMed] [Google Scholar]
- 73.Sonawane A, Manickam P, Bhansali S. Stability of Enzymatic Biosensors for Wearable Applications. IEEE Rev Biomed Eng. 2017 doi: 10.1109/RBME.2017.2706661. [DOI] [PubMed] [Google Scholar]
- 74.Huang M, Tamura T, Yoshimura T, Tsuchikawa T, Kanaya S. Wearable deep body thermometers and their uses in continuous monitoring for daily healthcare. Conf Proc Annu Int Conf IEEE Eng Med Biol Soc IEEE Eng Med Biol Soc Annu Conf. 2016;2016:177–180. doi: 10.1109/EMBC.2016.7590669. [DOI] [PubMed] [Google Scholar]
- 75.Gambi E, Agostinelli A, Belli A, Burattini L, Cippitelli E, Fioretti S, Pierleoni P, Ricciuti M, Sbrollini A, Spinsante S. Heart Rate Detection Using Microsoft Kinect: Validation and Comparison to Wearable Devices. Sensors. 2017;17 doi: 10.3390/s17081776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cai G, Huang Y, Luo S, Lin Z, Dai H, Ye Q. Continuous quantitative monitoring of physical activity in Parkinson’s disease patients by using wearable devices: a case-control study. Neurol Sci Off J Ital Neurol Soc Ital Soc Clin Neurophysiol. 2017 doi: 10.1007/s10072-017-3050-2. [DOI] [PubMed] [Google Scholar]
- 77.Scheffler M, Hirt E. Wearable devices for emerging healthcare applications. Conf Proc Annu Int Conf IEEE Eng Med Biol Soc IEEE Eng Med Biol Soc Annu Conf. 2004;5:3301–3304. doi: 10.1109/IEMBS.2004.1403928. [DOI] [PubMed] [Google Scholar]
- 78.Arroyo-Carmona RE, López-Serrano AL, Albarado-Ibañez A, Mendoza-Lucero FMF, Medel-Cajica D, López-Mayorga RM, Torres-Jácome J. Heart Rate Variability as Early Biomarker for the Evaluation of Diabetes Mellitus Progress. J Diabetes Res. 2016;2016:8483537. doi: 10.1155/2016/8483537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Borrione L, Brunoni AR, Sampaio-Junior B, Aparicio LM, Kemp AH, Benseñor I, Lotufo PA, Fraguas R. Associations between symptoms of depression and heart rate variability: An exploratory study. Psychiatry Res. 2017 doi: 10.1016/j.psychres.2017.09.028. [DOI] [PubMed] [Google Scholar]
- 80.Quintana DS, Elstad M, Kaufmann T, Brandt CL, Haatveit B, Haram M, Nerhus M, Westlye LT, Andreassen OA. Resting-state high-frequency heart rate variability is related to respiratory frequency in individuals with severe mental illness but not healthy controls. Sci Rep. 2016;6:37212. doi: 10.1038/srep37212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Erikson GA, Bodian DL, Rueda M, Molparia B, Scott ER, Scott-Van Zeeland AA, Topol SE, Wineinger NE, Niederhuber JE, Topol EJ, Torkamani A. Whole-Genome Sequencing of a Healthy Aging Cohort. Cell. 2016;165:1002–1011. doi: 10.1016/j.cell.2016.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.George J, Rapsomaniki E, Pujades-Rodriguez M, Shah AD, Denaxas S, Herrett E, Smeeth L, Timmis A, Hemingway H. How Does Cardiovascular Disease First Present in Women and Men? Incidence of 12 Cardiovascular Diseases in a Contemporary Cohort of 1,937,360 People. Circulation. 2015;132:1320–1328. doi: 10.1161/CIRCULATIONAHA.114.013797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fordyce CB, Roe MT, Ahmad T, et al. Cardiovascular drug development: is it dead or just hibernating? J Am Coll Cardiol. 2015;65:1567–1582. doi: 10.1016/j.jacc.2015.03.016. [DOI] [PubMed] [Google Scholar]
- 84.Hood L, Lovejoy JC, Price ND. Integrating big data and actionable health coaching to optimize wellness. BMC Med. 2015;13:4. doi: 10.1186/s12916-014-0238-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lv N, Xiao L, Simmons ML, Rosas LG, Chan A, Entwistle M. Personalized Hypertension Management Using Patient-Generated Health Data Integrated With Electronic Health Records (EMPOWER-H): Six-Month Pre-Post Study. J Med Internet Res. 2017;19:e311. doi: 10.2196/jmir.7831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Petersen C. Patient-generated health data: a pathway to enhanced long-term cancer survivorship. J Am Med Inform Assoc JAMIA. 2016;23:456–461. doi: 10.1093/jamia/ocv184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yang C, Al-Aama J, Stojkovic M, Keavney B, Trafford A, Lako M, Armstrong L. Concise Review: Cardiac Disease Modeling Using Induced Pluripotent Stem Cells: Cardiac Disease iPSC. STEM CELLS. 2015;33:2643–2651. doi: 10.1002/stem.2070. [DOI] [PubMed] [Google Scholar]
- 88.Ang Y-S, Rivas RN, Ribeiro AJS, et al. Disease Model of GATA4 Mutation Reveals Transcription Factor Cooperativity in Human Cardiogenesis. Cell. 2016;167:1734–1749.e22. doi: 10.1016/j.cell.2016.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Garg V, Kathiriya IS, Barnes R, Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS, Hirayama-Yamada K, Joo K, Matsuoka R, Cohen JC, Srivastava D. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424:443–447. doi: 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- 90.Rodriguez B, Burrage K, Gavaghan D, Grau V, Kohl P, Noble D. The systems biology approach to drug development: application to toxicity assessment of cardiac drugs. Clin Pharmacol Ther. 2010;88:130–134. doi: 10.1038/clpt.2010.95. [DOI] [PubMed] [Google Scholar]
- 91.Jin SC, Homsy J, Zaidi S, et al. Contribution of rare inherited and de novo variants in 2,871 congenital heart disease probands. Nat Genet. 2017 doi: 10.1038/ng.3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Roach JC, Glusman G, Smit AFA, Huff CD, Hubley R, Shannon PT, Rowen L, Pant KP, Goodman N, Bamshad M, Shendure J, Drmanac R, Jorde LB, Hood L, Galas DJ. Analysis of genetic inheritance in a family quartet by whole-genome sequencing. Science. 2010;328:636–639. doi: 10.1126/science.1186802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hassed S, Li S, Mulvihill J, Aston C, Palmer S. Adams-Oliver syndrome review of the literature: Refining the diagnostic phenotype. Am J Med Genet A. 2017;173:790–800. doi: 10.1002/ajmg.a.37889. [DOI] [PubMed] [Google Scholar]
- 94.Lehman A, Stittrich A-B, Glusman G, Zong Z, Li H, Eydoux P, Senger C, Lyons C, Roach JC, Patel M. Diffuse angiopathy in Adams-Oliver syndrome associated with truncating DOCK6 mutations. Am J Med Genet A. 2014;164A:2656–2662. doi: 10.1002/ajmg.a.36685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Meester JAN, Southgate L, Stittrich A-B, et al. Heterozygous Loss-of-Function Mutations in DLL4 Cause Adams-Oliver Syndrome. Am J Hum Genet. 2015;97:475–482. doi: 10.1016/j.ajhg.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Stittrich A-B, Lehman A, Bodian DL, et al. Mutations in NOTCH1 cause Adams-Oliver syndrome. Am J Hum Genet. 2014;95:275–284. doi: 10.1016/j.ajhg.2014.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shaheen R, Aglan M, Keppler-Noreuil K, Faqeih E, Ansari S, Horton K, Ashour A, Zaki MS, Al-Zahrani F, Cueto-González AM, Abdel-Salam G, Temtamy S, Alkuraya FS. Mutations in EOGT confirm the genetic heterogeneity of autosomal-recessive Adams-Oliver syndrome. Am J Hum Genet. 2013;92:598–604. doi: 10.1016/j.ajhg.2013.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Southgate L, Machado RD, Snape KM, et al. Gain-of-function mutations of ARHGAP31, a Cdc42/Rac1 GTPase regulator, cause syndromic cutis aplasia and limb anomalies. Am J Hum Genet. 2011;88:574–585. doi: 10.1016/j.ajhg.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hu H, Roach JC, Coon H, et al. A unified test of linkage analysis and rare-variant association for analysis of pedigree sequence data. Nat Biotechnol. 2014;32:663–669. doi: 10.1038/nbt.2895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ament SA, Szelinger S, Glusman G, et al. Rare variants in neuronal excitability genes influence risk for bipolar disorder. Proc Natl Acad Sci U S A. 2015;112:3576–3581. doi: 10.1073/pnas.1424958112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Greenhalgh K, Meyer KM, Aagaard KM, Wilmes P. The human gut microbiome in health: establishment and resilience of microbiota over a lifetime: The human gut microbiome in health. Environ Microbiol. 2016;18:2103–2116. doi: 10.1111/1462-2920.13318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Al Khodor S, Reichert B, Shatat IF. The Microbiome and Blood Pressure: Can Microbes Regulate Our Blood Pressure? Front Pediatr. 2017;5:138. doi: 10.3389/fped.2017.00138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Koeth RA, Wang Z, Levison BS, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med. 2013;19:576–585. doi: 10.1038/nm.3145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kelly TN, Bazzano LA, Ajami NJ, He H, Zhao J, Petrosino JF, Correa A, He J. Gut Microbiome Associates With Lifetime Cardiovascular Disease Risk Profile Among Bogalusa Heart Study Participants. Circ Res. 2016;119:956–964. doi: 10.1161/CIRCRESAHA.116.309219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Yan Q, Gu Y, Li X, et al. Alterations of the Gut Microbiome in Hypertension. Front Cell Infect Microbiol. 2017;7:381. doi: 10.3389/fcimb.2017.00381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wang Z, Klipfell E, Bennett BJ, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature. 2011;472:57–63. doi: 10.1038/nature09922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang Y-J, Li S, Gan R-Y, Zhou T, Xu D-P, Li H-B. Impacts of Gut Bacteria on Human Health and Diseases. Int J Mol Sci. 2015;16:7493–7519. doi: 10.3390/ijms16047493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ordovas JM, Mooser V. Metagenomics: the role of the microbiome in cardiovascular diseases. Curr Opin Lipidol. 2006;17:157–161. doi: 10.1097/01.mol.0000217897.75068.ba. [DOI] [PubMed] [Google Scholar]
- 109.Baslan T, Hicks J. Unravelling biology and shifting paradigms in cancer with single-cell sequencing. Nat Rev Cancer. 2017;17:557–569. doi: 10.1038/nrc.2017.58. [DOI] [PubMed] [Google Scholar]
- 110.Papalexi E, Satija R. Single-cell RNA sequencing to explore immune cell heterogeneity. Nat Rev Immunol. 2017 doi: 10.1038/nri.2017.76. [DOI] [PubMed] [Google Scholar]
- 111.Fernández-Ruiz I. Immune system and cardiovascular disease. Nat Rev Cardiol. 2016;13:503. doi: 10.1038/nrcardio.2016.127. [DOI] [PubMed] [Google Scholar]
- 112.Burtis CA, Burtis CA, Bruns DE, Sawyer BG, Tietz NW. Tietz fundamentals of clinical chemistry and molecular diagnostics. St. Louis: Elsevier/Saunders; 2015. [Google Scholar]
- 113.Lausted C, Lee I, Zhou Y, Qin S, Sung J, Price ND, Hood L, Wang K. Systems approach to neurodegenerative disease biomarker discovery. Annu Rev Pharmacol Toxicol. 2014;54:457–481. doi: 10.1146/annurev-pharmtox-011613-135928. [DOI] [PubMed] [Google Scholar]
- 114.Ament SA, Pearl JR, Grindeland A, et al. High resolution time-course mapping of early transcriptomic, molecular and cellular phenotypes in Huntington’s disease CAG knock-in mice across multiple genetic backgrounds. Hum Mol Genet. 2017;26:913–922. doi: 10.1093/hmg/ddx006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Osman R, L’Allier PL, Elgharib N, Tardif J-C. Critical appraisal of C-reactive protein throughout the spectrum of cardiovascular disease. Vasc Health Risk Manag. 2006;2:221–237. doi: 10.2147/vhrm.2006.2.3.221. [DOI] [PMC free article] [PubMed] [Google Scholar]