

Abstract
Reactive oxygen species (ROS) are well known for their role in mediating both physiological and pathophysiological signal transduction. Enzymes and subcellular compartments that typically produce ROS are associated with metabolic regulation, and diseases associated with metabolic dysfunction may be influenced by changes in redox balance. In this review, we summarize the current literature surrounding ROS and their role in metabolic and inflammatory regulation, focusing on ROS signal transduction and its relationship to disease progression. In particular, we examine ROS production in compartments such as the cytoplasm, mitochondria, peroxisome and endoplasmic reticulum, and discuss how ROS influence metabolic processes such as proteasome function, autophagy and general inflammatory signaling. We also summarize and highlight the role of ROS in the regulation metabolic/inflammatory diseases including atherosclerosis, diabetes and stroke. To successfully develop improved therapies that target oxidative signaling, it is vital to understand the balance ROS signaling plays in both physiology and pathophysiology, and how manipulation of this balance and the identity of the ROS may influence cellular and tissue homeostasis. An increased understanding of specific sources of ROS production and an appreciation for how ROS influence cellular metabolism may help guide us in this effort to treat cardiovascular diseases.
Keywords: ROS, Metabolism, Inflammation, Cardiovascular
Introduction
Reactive oxygen species (ROS) regulate cellular homeostasis and act as prime modulators of cellular dysfunction contributing to disease pathophysiology. ROS are byproducts of numerous enzymatic reactions in various cell compartments, including the cytoplasm, cell membrane, endoplasmic reticulum (ER), mitochondria and peroxisome, as part of basal metabolic function. They are also generated specifically by enzymes such as NADPH oxidases and serve a signaling function in the cell. Depending upon the source of ROS, cell type and tissue environment, ROS signaling may participate in normal physiological processes or contribute to a maladaptive response that leads to metabolic dysfunction and inflammatory signaling. Diseases associated with elevated inflammatory signaling and metabolic dysfunction such as atherosclerosis, diabetes and stroke are associated with an altered redox balance.1–3 Understanding the role of ROS signaling in the regulation of metabolic activity, inflammatory activation and diseases associated with metabolic dysfunction is important in our pursuit of novel therapies to treat these diseases. This review highlights the role of ROS signaling in basic metabolic processes and inflammatory signaling, and focuses on how this regulation contributes to disease development.
1. Sources of ROS and their role in metabolic function
I. Cytoplasmic
Cytoplasmic ROS (cytoROS) production is a cornerstone of cellular signaling and disease pathophysiology. One of the most well-known sources of cytoROS is the NADPH oxidase (NOX) family of enzymes. NOX2 (or gp91phox) is well-characterized for its role in phagocytic function;4 however, it and three homologs, NOX1, NOX4, and NOX5, are expressed throughout the cardiovascular system.5 NOX proteins produce O2− through NADPH electron exchange, and NOX-dependent ROS production influence many metabolic processes and disease states.6 Briefly, endothelial cell (EC) NOX-dependent ROS production drives hypoxia inducible factor 1α (HIF1α)-mediated glucose transporter 1 (GLUT1) expression, hexokinase activity and resultant glycolysis in response to low oxygen tension as part of the angiogenic response.7 In response to inflammatory stimuli, neutrophil phosphofructokinase 2 colocalizes with NOX2, inducing its activation. NADP+ produced as a byproduct of NOX2 O2− generation is used to facilitate an elevated glycolytic rate. A locally increasing NADPH concentration as a by-product of increased glycolysis is hypothesized to further enhance NOX2 activity; however, this has yet to be proven.8
In addition to NOX-dependent ROS production, the nitric oxide synthases (endothelial, neuronal and inducible) are sources of cytoplasmic ROS. Endothelial nitric oxide synthase (eNOS) produces O2− through its oxygenase domain in a Ca2+/calmodulin-dependent reaction in the absence of tetrahydrobiopterin (BH4).9 Dysregulated or uncoupled eNOS is a hallmark of cardiovascular and metabolic diseases.10 The oxygenase domain is vital for O2− production from neuronal and inducible NOS (nNOS and iNOS, respectively), and O2− generation is dependent upon reduced L-arginine.11, 12 NOS uncoupling in various disease states results in a dysregulated NO response whereby synthesized NO combines with O2− to produce peroxynitrite. Peroxynitrite enhances pentose phosphate pathway (PPP) activity through stimulation of glucose-6-phosphate dehydrogenase (G6PD), leading to elevated NADPH.13 A similar mechanism has been noted in response to H2O2, a downstream metabolite of O2−.14 Thus, in the transient setting, peroxynitrite, and even H2O2, may promote a protective response through PPP-dependent increases in reducing equivalents. However, chronic stimulation of the PPP by ROS may instigate a toxic feedback loop whereby increased NADPH production results in excessive O2− through NOX.15 In support of both hypotheses, G6PD deficiency and overexpression are protective against oxidative stress16,17 However, further investigation into how ROS and metabolic diseases affect NADPH shuttling is needed.
CytoROS also induce AMP-activated protein kinase (AMPK) activity.18 AMPK is a central regulator of cellular metabolism implicated in multiple metabolic functions including glycolysis, lipid metabolism, mitochondrial function, cell growth and autophagy (discussed below).19 Hypoxia can regulate AMPK activity through an ROS-dependent ER Ca2+/Stromal interaction molecule 1/Ca2+/calmodulin-dependent protein kinase kinase beta pathway in addition to indirect regulation via a change in the AMP/ATP ratio.20 H2O2 can directly modulate AMPK activity and downstream metabolic function through oxidation and S-glutathionylation of the α- and β-subunits of AMPK by targeting cysteines 299 and 304.21 ROS-mediated impairment of the mitochondrial respiratory chain can also increase the AMP/ATP ratio which can activate AMPK,22 and peroxynitrite induces AMPK activation in bovine aortic ECs through a c-SRC(Tyr416)/phosphoinositide 3-kinase (PI-3K)/phosphoinositide-dependent kinase-1 (Ser241) pathway. Peroxynitrite-dependent AMPK phosphorylation at Thr172 leads to phosphorylation of acetyl CoA carboxylase at Ser79, resulting in inactivation and increased fatty acid oxidation.23
Low oxygen tension in ECs increases glycolysis as part of the angiogenic response through NOX-mediated, ROS-induced HIF1α signaling.7 However, increased ROS signaling in some disease states inactivates the glycolysis rate-limiting enzyme pyruvate kinase M2, and diverts glycolytic substrates into the PPP pathway to generate reducing equivalents needed for ROS detoxification, thereby acting as a protective mechanism.24 In total, cytoROS signaling plays a critical role in the PPP and glycolytic pathways (Figure 1). Below we will outline the contributions of various cytoROS-producing entities in the development of metabolic disorders.
Figure 1. Cytosolic ROS production and regulation of cytosolic metabolic pathways.
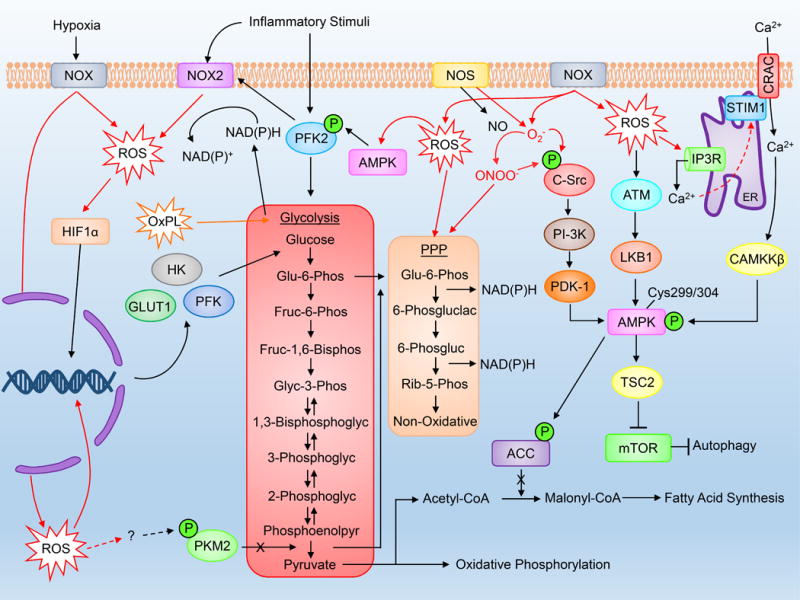
Cytosolic ROS are formed most notably through NOX activity and influence metabolic processes including glycolysis and downstream oxidative phosphorylation, pentose phosphate pathway activity and autophagy. Please refer to the abbreviation table for full names of listed proteins.
II. Mitochondria
Mitochondria are central regulators of aerobic energy production. Proper respiratory chain function requires a delicate balance between pro-oxidant and anti-oxidant systems. Importantly, mitochondrial respiration relies on electron transfer and a proton gradient to drive ATP production. ROS are a natural byproduct of this process; however, inflammatory and metabolic diseases are associated with perturbed mitoROS production.25, 26 MitoROS are generated by numerous mechanisms including Complexes I-III (Figure 2). Complex I serves as an entry point for electrons from NADH into the respiratory chain. O2− is produced from the interaction of O2 with reduced FMN when the matrix NADH/NAD+ ratio is high, resulting in O2− release into the matrix.27 In addition, mitoROS is produced through complex I via reverse electron transfer (RET). RET occurs in a 2-step process involving (1) reduced coenzyme Q (CoQ), and (2) a change in proton motive force that drives electrons back into complex I. Complex III is also a source of mitoROS. Under normal conditions, electrons flow from the CoQ pool to cytochrome C. Although complex III produces very low levels of O2−, in the presence of Antimycin A, the Qi site is inhibited, which promotes O2− production from the Qo site due to the interaction between O2 and a ubisemiquinone bound to the Qo site.28 O2− generated from complex III is mainly released into the intermembrane space, but upon dismutation, H2O2 may diffuse into the matrix.29
Figure 2. Mitochondrial ROS production.
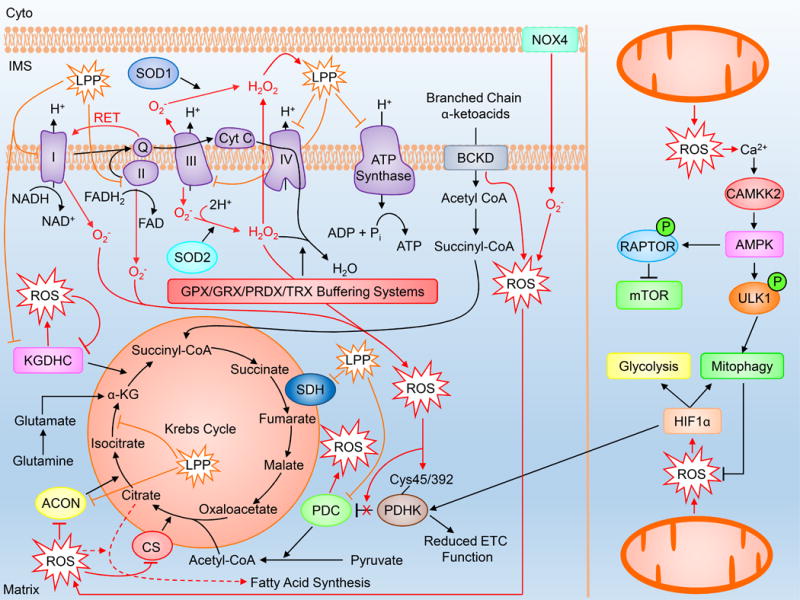
MitoROS are produced as a normal byproduct of mitochondrial respiration and metabolic enzymatic activity. Under settings of increased ROS generation as a result of dysregulated enzymatic activity and cellular stress, mitoROS can influence metabolic pathways including the Krebs cycle, fatty acid synthesis, ATP generation, glycolysis and mitophagy. Cyto=cytoplasm, IMS=intermembrane space, LPP=lipid peroxidation product. Please refer to the abbreviation table for full names of listed proteins.
While complex I and III are well known for their contribution to mitoROS production, complex II may also serve as an ROS producing complex.30 Complex II oxidizes succinate to fumarate as part of the Krebs cycle and acts as the site of ubiquinone reduction in the electron transport chain (ETC). ROS are produced from complex II when both complex III and complex I are inhibited. Complex II ROS production is believed to proceed through a forward mechanism involving electrons from succinate, or in a reverse mechanism where electrons are provided through a reduced ubiquinone pool. Both mechanisms result in ROS production from the complex II flavin site.30
Other than complex-derived mitoROS, enzymes involved in metabolic reactions produce mitoROS (reviewed in 31, 32). Of importance, the α-ketoglutarate dehydrogenase (KGDHC) and pyruvate dehydrogenase (PDC) complexes produce ROS as a result of both forward electron transfer and RET.33, 34 KGDHC, PDC and branched-chain α-keto acid dehydrogenase complex, a dehydrogenase complex that catalyzes the oxidative decarboxylation of α-ketoacids, are thought to produce significantly more ROS than complex I.35 Recent evidence indicates PDC and KGDHC-derived ROS may be regulated through protein-S-glutathionylation. Interestingly, S-glutathionylation during forward electron transfer may attenuate ROS production, whereas S-glutathionylation during RET may increase ROS from PDC and KGDHC36 In addition to generating ROS, KGDHC is also an early target of oxidative stress. Similarly, aconitase, an enzyme responsible for the isomerization of citrate to isocitrate, is responsive to ROS.37, 38 In conditions of low oxidative stress, when aconitase activity and Krebs cycle substrate production is diminished, α-ketoglutarate levels and NADH production can be maintained through glutamate via glutaminolysis; however, in the presence of high oxidative stress, KGDHC is inhibited, reducing NADH production and mitochondrial respiratory capacity.38 RET-induced mitoROS can inactivate aconitase and pyruvate dehydrogenase kinase 2 (negative regulator of PDC) through reversible oxidation of cysteine 45 and 392 when the NADH/NAD+ ratio is elevated. This regulation is hypothesized to promote acetyl-CoA production via PDC from carbohydrates while simultaneously inhibiting β-oxidation, resulting in cytoplasmic export of citrate and stimulation of fatty acid synthesis.39 Reduced aconitase activity is associated with aging and diseases associated with metabolic dysfunction.40
Recent evidence indicates that a subset of NOX4 is localized to the mitochondria and is a regulator of mitoROS generation. NOX4 expression is increased in kidney cortex from diabetic rats and facilitates glucose-induced mitoROS production. Likewise, NOX4 induces cysteine oxidation, and resultant decreased activity, of aconitase and citrate synthase in cardiac myocytes,41 and influences mitochondrial morphology and complex I activity.42 Additionally, ATP production from normal mitochondrial respiration negatively regulates mitochondrial NOX4-induced ROS production through direct interaction between ATP and the NOX4 Walker A binding motif,43 which suggests mitochondrial NOX4 may play a dynamic role in metabolic function.
Apart from ROS producing pathways, impairment of mitoROS scavenging pathways results in ROS accumulation leading to organelle and cell dysfunction. Within the mitochondria, the predominant ROS buffering systems include the glutaredoxin (Grx), glutathione and thioredoxin (Trx) systems.44 Dismutation of O2− into H2O2 occurs through the superoxide dismutase (SOD) family of proteins.45 In the matrix, dismutation primarily proceeds through SOD2 (MnSOD), whereas in the intermembrane space, dismutation is carried out by SOD1 (Cu, Zn-SOD). H2O2 decomposition into O2 and H2O then occurs via the GSH redox system, which includes glutathione reductases, peroxidases (GPX) and peroxiredoxins (Prdx).46
The Trx and Grx systems also play a prominent role in mitochondrial ROS buffering. The mitochondrial Trx system involves thioredoxin-2 (Trx2), thioredoxin reductase-2 (TrxR2) and members of the Prdx family of proteins. Importantly, the antioxidant effect of Trx2 activity results from reduction of other oxidized proteins, mainly the Prdxs. Oxidization of Trx2 is reversible and is remedied through TrxR2 using NADPH as an electron donor. 47 Homozygous knockout (KO) of Trx2 is embryonically lethal, and heterozygous mice, while viable, show decreased mitochondrial respiratory function and increased mitoROS production,.48 Likewise, cardiac-specific TrxR2 KO mice exhibit cardiac structural changes, dysregulation of autophagy, decreased oxygen consumption and a change in their metabolic profile.49
Mitochondrial Grx family members include Grx2 and Grx5. Grx2 regulates O2− production from complex 1 by catalyzing glutathionylation of two thiol groups, and Grx5 regulates iron/sulfur enzymes.50–52 However, information regarding how the GRX and Trx systems regulate cardiovascular function is limited. Cumulatively, mitochondria maintain a delicate balance between oxidant and anti-oxidant systems, and dysregulation can result in organelle dysfunction resulting in metabolic stress.
MitoROS can also influence HIF1α stabilization and cell proliferation.53, 54 Hypoxia-induced HIF1α promotes a metabolic shift favoring anaerobic glycolysis and reduced mitochondrial respiration by upregulating glucose transporters and glycolytic enzymes while also inhibiting PDC through activation of pyruvate dehydrogenase kinase 1.55, 56 Reduced PDC activity causes decreased Krebs cycle and ETC flux, which may in turn attenuate mitoROS production in hypoxic conditions. Likewise, HIF1α induces mitophagy resulting in decreased mitochondrial mass, O2 consumption and resultant ROS generation. Thus, in hypoxic conditions, mitoROS favor the stabilization of HIF1α, which induces a metabolic shift towards glycolysis, leading to reduced mitochondrial activity and mitoROS production.57, 58 In normoxic conditions, inhibition of ETC function using rotenone (complex I inhibitor) or TTFA (complex II inhibitor) induces cell death through induction of mitoROS-mediated autophagy.59 AMPK, a regulator of uncoordinated 51-like kinase 1 (ULK1) activity and downstream autophagy, can be regulated by mitoROS, and mitoROS contribute to autophagy through AMPK/ULK1-dependent signaling (discussed below).60, 61 Similarly, AMPK is responsible for the mitoROS/HIF1α-dependent extension of life span observed in Caenorhabditis elegans (C. elegans).62 Slightly elevated levels of mitoROS in mice are also associated with extension of lifespan.63 Interestingly, mice with heterozygote deletion of mouse clock-1, a protein involved in CoQ biosynthesis, exhibit increased longevity, increased inflammatory cytokine production and macrophage activation that is dependent upon elevated mitoROS. These mice also show enhanced resistance to infection, which may be due to the observed increase in macrophage phagocytic activity.64
Mitochondrial fission and fusion are closely associated with mitochondrial function and can influence, and be influenced by, mitoROS production. (For a comprehensive review of mitochondrial fission and fusion, please refer to references.65, 66 With regard to mitochondrial fission, the small cytoplasmic GTPase dynamin-related protein-1 (Drp1) is implicated in ROS signaling. Drp1 activity regulates mitoROS production and downstream mitochondrial functional changes in a variety of environments,67–69 and oxidative stress influences Drp1 mitochondrial translocation and resultant fission.70 Mitochondrial fission also precedes mitophagy, which can be regulated by oxidative stress and is thought to be a negative regulator of mitoROS signaling through selective degradation of dysfunctional mitochondria.71, 72 Drp1 undergoes numerous post-translational modifications including S-nitrosylation and phosphorylation, and data suggest that ROS signaling contributes to serine 616 phosphorylation and activation of Drp1 GTPase activity.73–75 However, the contribution of ROS signaling to the regulation of other fission proteins is currently unknown and is an area ripe for future investigation.
Similar to Drp1 and mitochondrial fission, ROS signaling may regulate mitochondrial fusion. The inner mitochondrial membrane GTPase optic atrophy protein-1 (OPA1) is regulated by reactive oxygen species modulator 1 (ROMO1). In response to ROS, ROMO1 is inactivated leading to OPA1 cleavage, cristae remodeling and mitochondrial fission.76 Likewise, deletion of OPA1 induces morphological irregularities, respiratory defects and ROS generation.77 In addition to OPA1, mitofusin 1 and 2 activity can regulate and be regulated by ROS.78–80 The precise mechanism and contribution of fission/fusion-regulated ROS signaling to metabolic function remains to be explored; however, the contribution of ROS and mitochondrial morphology regulation in the setting of metabolic diseases will be explored below.
In summary, the mitochondria are dynamic players in metabolic regulation and signaling. MitoROS are produced as part of normal mitochondrial function, but various cellular stresses augment ROS levels either through increased oxidant production or decreased antioxidant activity. As discussed, mitoROS can regulate cellular metabolic function (illustrated in Figure 2), and in section 2, we will discuss the contribution of mitoROS to metabolic and cardiovascular diseases.
III. Peroxisome
Peroxisomes, like mitochondria, are vital organelles in aerobic metabolism that regulate key processes such as α- and β-oxidation, glyoxylate metabolism, amino acid catabolism, the pentose phosphate pathway, ketogenesis, polyamine oxidation and isoprenoid and cholesterol metabolism.81 Peroxisomes are also a significant source of ROS. In particular, peroxisomes produce H2O2 due to an abundance of O2-consuming oxidases, which include acyl-CoA oxidases (ACOX), D-amino acid oxidase, D-aspartate oxidase, polyamine oxidase, xanthine oxidase (also produces O2−), L-α-hydroxyacid oxidase and L-pipecolic oxidase.82, 83 Unlike the mitochondria, peroxisomal electron transfer does not lead to ATP generation. Instead, free electrons are transferred to H2O to form H2O2.83 In addition, peroxisomes can produce nitric oxide through NOS.84 However, similar to mitochondria, peroxisomes contain numerous oxidant scavenging enzymes including GPX, catalase, Prdx1 and 5, peroxisomal membrane associated protein 20, SOD1 and SOD2.82 For an in-depth review of peroxisomal enzymes, please refer to85.
Recently, a new role for peroxROS in mTOR complex (mTORC) 1 activity was defined. In response to elevated peroxisomal β-oxidation, peroxROS activate tuberous sclerosis proteins 1 and 2 (TSC1 and TSC2), which are bound to the peroxisomal assembly proteins peroxin 19 and 5 (Pex19 and Pex5), respectively. ROS induce TSC2-mediated GTPase activity of Rheb, leading to mTORC1 inhibition and autophagy induction.86 Pex5 binds ataxia-telangiectasia mutated (ATM) and localizes it to the peroxisomal membrane. In response to peroxROS, ATM activation (Ser1981) induces AMPK and TSC2 activity and downstream mTORC1 inhibition, ULK1 activation and pexophagy. As part of this mechanism, ATM activation also induces the phosphorylation of Pex5 (Ser141), triggering ubiquination at lysine 209 and subsequent binding of p62, which is required for peroxisomal targeting to autophagosomes (Figure 3).87 In addition to pexophagy regulation, peroxROS can also disrupt the mitochondrial redox balance and promote mitochondrial fission/fragmentation88 and mitochondrial-mediated cell death.89
Figure 3. Peroxisomal ROS and metabolism.
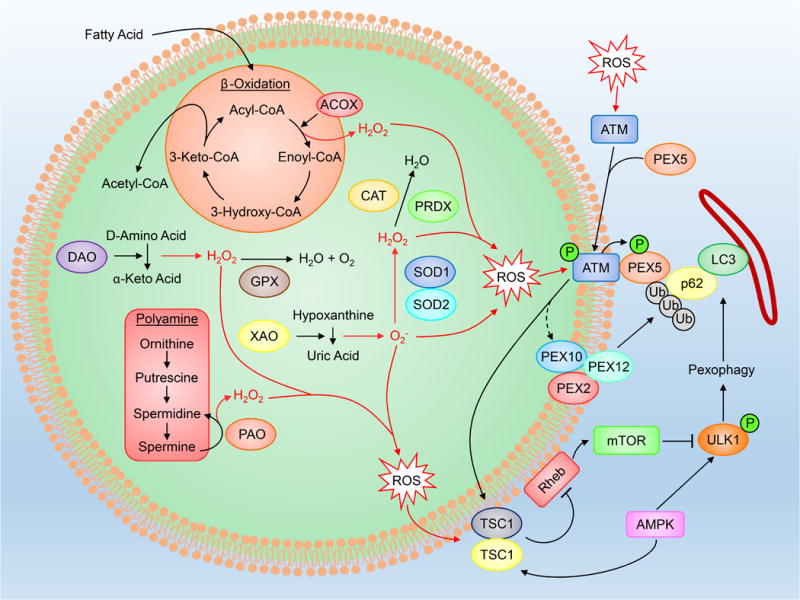
PeroxROS are produced as byproducts of enzymatic reactions within β-oxidation, polyamine synthesis, D-amino acid deamination and hypoxanthine oxidation, and have been found to be key regulators of pexophagy. Full names of abbreviations are listed in the accompanying table.
IV. ROS and the Endoplasmic Reticulum
The ER has a well-established role in metabolic and cardiovascular diseases90, 91 due to its roles in Ca2+ handling, protein synthesis/folding and regulation of the secretory pathway. Protein-folding is highly sensitive to ER redox status and dysregulation of disulfide bond formation in response to ER stress increases luminal oxidative stress leading to a decline in ER function.92 One of the most well understood routes for disfulfide bond introduction into folded proteins is the protein disulfide-isomerase (PDI) and ER oxidoreductin 1 (ERO1) pathway. PDI introduces disulfide bonds through thiol oxidation in folding substrates leaving PDI in a reduced state. Reduced PDI is re-oxidized through ERO1, which transfers acquired electrons through a flavin adenine dinucleotide cofactor to molecular oxygen, forming H2O2. ER H2O2 can further be used by Prdx4 to re-oxidize PDI, thereby increasing the efficiency of ERO1-mediated disulfide bond transfer.93, 94 Overexpression of a human hyperactive mutant of ERO1 induces severe oxidative stress and induction of the unfolded protein response (UPR), an ER stress response involved in the pathogenesis of metabolic and cardiovascular diseases, highlighting the sensitivity of the ER to changes in redox balance.95 Furthermore, UPR activation can induce ERO1 activation leading to increased oxidative stress and sustained UPR signaling, and administration of antioxidants can attenuate the UPR and improve downstream protein secretion.96 In addition to the PDI/ERO1 pathway, ROS is produced through the membrane associated monooxygenase system via cytochrome p450, cytochrome b5 reductase and through ER-localized NOX4 (Figure 4).97–99
Figure 4. Endoplasmic reticulum and ROS.
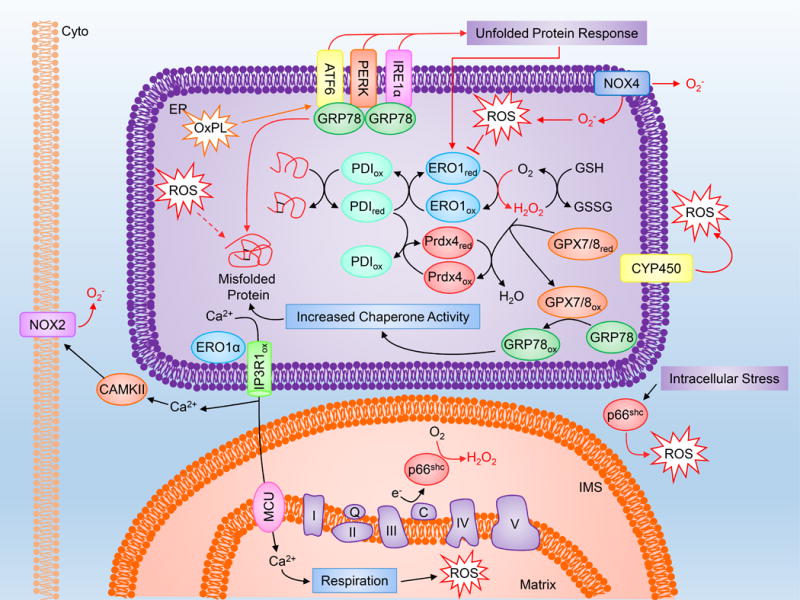
The ER is highly sensitive to redox status, and altered ROS signaling can influence protein folding, Ca2+ release and mitochondrial respiration. Please refer to the abbreviation table for full names of depicted proteins.
As part of this redox balance, the ER contains an antioxidant system that is vital to proper ER function: peroxiredoxins (Prdx4, mentioned above) and glutathione peroxidases (GPX7 and GPX8). GPX7/8 contain a KDEL ER localization sequence, making them ER-specific ROS scavengers. GPX7/8 both interact with ERO1α, one of two isoforms of ERO1, at cysteine208/cysteine41. Reduced GPX7/8 scavenges ERO1-derived H2O2, leaving GPX7/8 in an oxidized form.100 Oxidized GPX7, through its cysteine 86 residue, can bind to the cysteine41/420 residue of glucose-regulated protein, 78 kDa (GRP78). This binding promotes GRP78 oxidation and enhanced protein refolding chaperone activity. Silencing of GPX7 induces oxidative stress, accumulation of misfolded proteins and induction of the UPR.101 PDI oxidation is regulated by GPX7/8 as well.100
The ER and mitochondria sit in close proximity to one another, and changes in the ER redox balance influence mitochondrial function. In response to ER stress, activating transcription factor 4 and C/EBP homologous protein (CHOP) induce ERO1α. In mitochondrial-associated ER membranes (MAMs), ERO1α oxidizes the type 1 inositol 1,4,5-trisphosphate receptor (IP3R) inducing mitochondrial Ca2+ uptake and mitoROS production through mitochondrial respiration.102, 103 ERO1α-mediated cytoplasmic Ca2+ efflux through the ER IP3R is also hypothesized to contribute to NOX2-dependent ROS stimulation via CAMKII.100 SHC-transforming protein 1 isoform p66 (p66shc) is a regulator of cellular oxidative stress104 that translocates to MAMs in response to cellular stress and produces mitoROS through cytochrome C oxidation,105 and is capable of inducing ER stress106 and inhibiting mTOR-dependent anabolic metabolism (Figure 4).107
In summary, maintenance of the ER redox balance is critical to proper ER function, and alterations in ER ROS producing and scavenging pathways provoke ER stress and contribute to metabolic dysfunction.
V. ROS and Inflammation
Inflammation and metabolism are intricately intertwined, considering that numerous metabolic and cardiovascular disorders exhibit chronic low-grade inflammation.108 Canonical NF-κB signaling is associated with insulin resistance, obesity and atherosclerosis,108–110 and circulating dietary factors such as fatty acids and glucose can trigger inflammatory signaling. It has also been suggested that NF-κB may regulate metabolic reprogramming favoring aerobic glycolysis.111 The influence of ROS on NF-κB signaling may depend upon the cellular location of oxidation (cytoplasmic vs nuclear).112 In general, ROS are known to activate NF-κB in response to inflammatory agonists.113 NF-κB nuclear translocation occurs in response to H2O2114 through a mechanism involving IκBα tyrosine phosphorylation (Tyr42), phosphorylation of the serine/threonine PEST domain with subsequent degradation via calpain, and p65 phosphorylation (ser529).115, 116 ROS-induced NF-κB is inhibited by SOD2 overexpression,117 and the NOX family of proteins also influence,118 and are influenced by, NF-κB activity.119, 120
Specific inflammatory agonists utilize ROS as part of their signaling cascades. IL-1β induces active endosomal IL-1R complex assembly that involves MyD88 and NOX2 ROS-induced tumor necrosis factor (TNF) receptor associated factor (TRAF) 6 endosomal recruitment.121 NOX4 activity is required for lipopolysaccharide (LPS)-induced NF-κB activation,122 and TNFα-induced NF-κB activation increases antioxidant expression leading to decreased TNFα-induced apoptotic signaling through a ROS/JNK pathway.123
Flow-induced activation of NF-κB is regulated by ROS signaling as well. Flow-mediated EC dysfunction and monocyte adhesion is dependent upon NOX-derived O2− regulation of NF-κB inducing kinase (NIK) and IKK signaling leading to NF-κB activation.124–126 As part of this response, bone morphogenic protein 4 and p21-activated kinase act as upstream regulators of flow-induced ROS generation and downstream NF-κB signaling, contributing to oscillatory shear stress-induced vascular dysfunction and atherosclerotic lesion development.126, 127
An abundance of evidence points to a role for mitoROS in regulating inflammatory signaling. In response to inflammatory stimuli, proinflammatory cytokines are synthesized and released from cells. Importantly, mitoROS contribute to LPS-induced cytokine release25, thrombin induced NF-κB activation via IP3R Ca2+ signaling,128 and lysophosphatidylcholine-induced AP-1 activity and downstream endothelial activation.129 Likewise, mitochondrial H2O2 production contributes to endothelial NF-κB activation in aged rat arteries,130 and inhibition of mitoROS through ETC inhibition abrogates hypoxia-induced endothelial NF-κB activation and IL-6 secretion.131 Evidence also indicates that mitoROS may be a downstream result of NF-κB activation as well.132
RET-induced mitoROS are involved in metabolic changes associated with macrophage activation during inflammation. Macrophage proinflammatory signaling is supported in part by metabolic repurposing that favors glycolysis-derived ATP production and RET-induced mitoROS generation through mitochondrial hyperpolarization and succinate oxidation.133 RET-derived mitoROS promote HIF1α stabilization,134 leading to regulation of glycolytic capacity and IL-1β mRNA and protein expression.135,133 In response to LPS, immune cells secrete IL-1β through the inflammasome. MitoROS signaling is a primary regulator of NLR Family Pyrin Domain Containing 3 (NLRP3) inflammasome activation. NLRP3 inflammasome activation consists of two phases: (1) a priming phase where agonists such as LPS stimulate NF-κB-mediated NLRP3, IL-1β and interleukin-18 (IL-18) transcription, and (2) an activation phase where a multi-protein complex consisting of NLRP3, Apoptosis-associated speck-like protein containing a CARD, and caspase 1 is assembled.136 The fully assembled NLRP3 inflammasome, along with activation via potassium efflux,137 lysosomal destabilization and mitoROS,138, 139 regulates the maturation of IL-1β and IL-18.140 Thus, in LPS-stimulated macrophages, RET-induced mitoROS production, as a result of a metabolic shift towards glycolysis, regulates 1L-1β transcription (inflammasome priming), but may also regulate the maturation and secretion of IL-1β (inflammasome activation). However, alternative pathways of IL-1β processing independent of inflammasome activity also exist.141
Thioredoxin-interacting protein (TxNIP), a negative regulator of Trx, is an ROS-regulated proapoptotic factor that mediates mitoROS and NOX4 activity and influences glucose-induced inflammasome activation in two ways.142, 143 First, TxNIP-dependent inhibition of Trx induces ROS which further exacerbate inflammasome activity and inflammatory cytokine signaling. Second, during inflammasome activation, TxNIP dissociates from Trx and interacts with NLRP3, which is required for proper inflammasome activity in response to glucose stimulation.142 In addition, IRE1α, an ER stress protein, increases inflammasome activity by regulating TxNIP mRNA stability.144, 145
Apart from ROS-dependent inflammasome activation, ROS are critical to macrophage phagocytic activity. Importantly, metabolic and cardiovascular diseases associated with chronic inflammation display impairments in macrophage phagocytic/efferocytic activity that are correlated with changes in ROS signaling. NOX2-dependent oxidative signaling is required for sufficient phagocytic function and pathogen/apoptotic cell degradation,146 as shown by increased lung inflammation and abdominal aortic aneurysm progression in myeloid NOX2 KO mice.147, 148
Macrophage NOX-derived ROS induce microtubule-associated protein 1A/1B-light chain 3 (LC3) translocation to the phagosome, which is required for lysosomal fusion and phagosomal clearance in LC3-associated phagocytosis (LAP).149 Toll-like receptor signaling promotes mitochondrial recruitment to phagosomes where augmented mitoROS kill phagocytosed bacteria and enhance NOX-dependent ROS production.150 As part of LAP, Drp1-mediated mitochondrial fission increases cytosolic Ca2+ through inhibition of the mitochondrial Ca2+ uniporter and mitoROS production, which are required for efficient phagosomal sealing and LAP-mediated apoptotic cell degradation.151 Drp1-dependent mitochondrial fission and resultant ROS production also contribute to NF-κB activation in T cells,152 and changes in mitochondrial dynamics regulate T cell metabolic reprogramming.153
Mer tyrosine kinase (MerTK) is an essential membrane protein in macrophages that participates in efferocytosis. In response to inflammatory stimuli, proteolytic cleavage and inhibition of MerTK activity impair efferocytosis and downstream resolution of inflammation.154 In response to LPS, TLR4-TIR-domain-containing adapter-inducing interferon-β signaling induces MerTK shedding through a NOX2/PKCα/ p38 MAPK and a Disintegrin and metalloproteinase domain-containing protein 17 (ADAM17) pathway, and macrophage-specific ADAM17 deletion protects against MerTK shedding in vivo.155 MitoROS and AMPK also play a significant role in macrophage efferocytosis. Apoptotic cell-released lysophosphatidylcholine diminishes mitochondrial membrane potential and ATP production coupled with concomitant mitoROS generation and downstream AMPK activation. AMPK facilitates both metabolic reprograming towards glycolysis and tubulin synthesis that are needed for macrophage chemokinesis and efficient efferocytosis.156 Macrophages experience defective efferocytosis in response to TNFα as well, potentially through phospholipase A2/arachidonic acid-dependent ROS and Rho activity.157
Overall, inflammation is an underlying component to many diseases including those that exhibit metabolic distress. ROS act as central regulators of inflammatory signaling, particularly with respect to NF-κB activation and inflammasome signaling (Figure 5). The following sections will highlight the contribution of ROS to specific cellular functions and their role in regulating metabolic dysfunction in various diseases.
Figure 5. Inflammation and ROS.
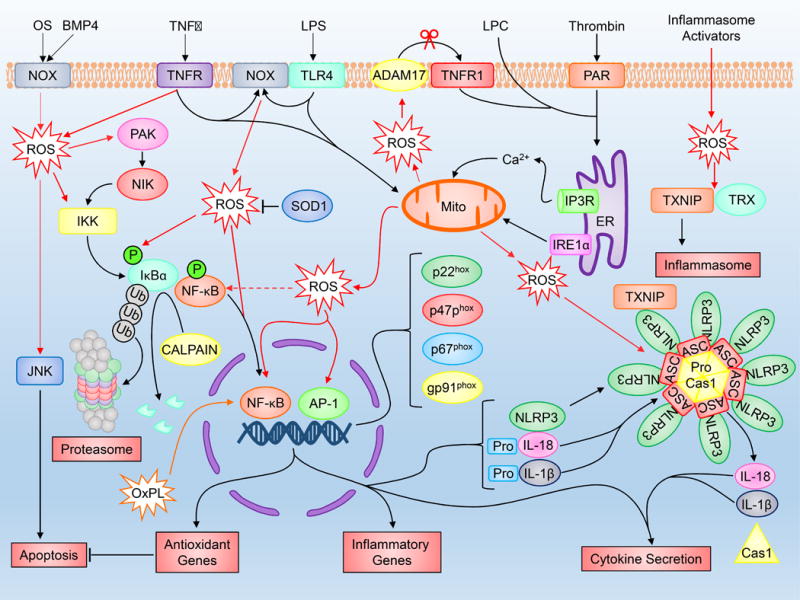
Various inflammation-inducing stimuli including TNFα, LPS, thrombin and oscillatory shear stress influence ROS production through sources including NOX and the mitochondria. Elevated ROS production as a result of inflammatory signaling can mediate canonical NF-κB activation and downstream inflammatory gene induction, proteasome activity, antioxidant gene transcription, inflammasome activation and cytokine secretion. Full names for abbreviations are listed in the accompanying table.
VI. Secondary Products of ROS
Secondary products of ROS also mediate inflammatory signaling in a variety of cellular environments. For example, lipid peroxidation products, such as 4-hydroxynonenal, and oxidized phospholipids (OxPLs) regulate NF-κB activation and inflammatory signaling through numerous pathways,158–160 and can propagate ROS signaling; however, whether secondary ROS product signaling is protective or instigates a pathophysiological response may depend on ROS concentration, cell-type and cellular stress.159 In regards to pathophysiology, lipid peroxidation and OxPLs have been implicated in atherogenesis.161,162 Mechanistically, both products can regulate NF-κB activity and chemokine release resulting in arterial wall inflammation and immune cell recruitment.163–165 However, incubation of LPS-treated mice or macrophages with OxPLs suppresses NF-κB activation and TNFα secretion, suggesting that OxPLs may be protective in some conditions.166 It should also be noted that in a single agonist condition, OxPLs may influence macrophage polarization and contribute to the macrophage pro-inflammatory response.167
Secondary products of ROS can also regulate metabolic function in various tissues and cellular environments. In response to OxPLs, EC glycolytic and proliferative capacity are increased and are dependent upon NRF2 signaling.168 Similarly, oxidation of cholesterol may promote a protective response in macrophages by increasing autophagy and promoting effective efferocytosis.169 Lipid peroxidation can increase autophagy in rat VSMCs through an ER stress/JNK-dependent mechanism.170 In the mitochondria, lipid peroxidation affects membrane fluidity and is capable of modulating ETC complex activity, Krebs cycle enzymes, proteostasis and mitochondrial membrane potential (Figure 2).171 Overall, while secondary products are known to influence various enzymes and contribute to diseased states such as atherosclerosis and cardiac diseases, specific signal transduction pathways have yet to be fully elucidated. Nevertheless, secondary products of ROS signaling have a clear role in metabolic and inflammatory signaling.
VII. ROS and Autophagy
Autophagy is a highly conserved catabolic process in which cytoplasmic macromolecules and organelles are delivered to lysosomes for degradation.172 Constitutive basal levels of autophagy support metabolic homeostasis by promoting a balance between protein synthesis and degradation, as well as organelle biogenesis and degradation; however, dysregulated autophagic flux contributes to metabolic and inflammatory diseases.172,173. For an extensive review on the regulation of autophagic machinery, please refer to174.
Oxidative stress regulates autophagic flux through its influence on autophagic gene transcription, protein activity and organellular degradation.175 ROS induce macroautophagy (commonly referred to as autophagy), and selective degradation of oxidized proteins through chaperone-mediated autophagy (CMA) is important for cellular viability during periods of oxidative stress.176. Oxidative modification of CMA substrates increases their susceptibility to degradation, and CMA is enhanced through LAMP2a upregulation in cells challenged with ROS.176 Little is known about the effect of ROS on microautophagy and further work is needed to distinguish the roles of macroautophagy, microautophagy, and CMA in response to oxidative stress.
In addition to the role of ROS in autophagic flux, ROS are required for autophagy induced by starvation,177 dopamine,178 sodium selenite,179 mitochondrial electron transport chain inhibitors TTFA and rotenone,180 TNFα181 and LPS.182 The source and identity of ROS that mediate these effects remain unclear. Sources of ROS located at or near the plasma membrane, such as NOXs, are prime candidates to transduce signals from external stimuli into the cell. However, this hypothesis has only been verified in macrophages through LAP. 149
As mentioned, ROS can influence AMPK activation. In response to ROS, cytoplasmic ATM promotes liver kinase B1 (LKB1)-dependent AMPK activation and downstream autophagy through the regulation of TSC2 activity and mTOR inhibition.183,184 Furthermore, overexpression of catalase blocks H2O2-induced autophagy,177,182 and starvation-induced H2O2 production regulates cysteine oxidation of Atg4, a negative regulator of autophagy that delipidates LC3-II to LC3-1 and inhibits autophagosome formation.177 Starvation methods including the removal of glucose, L-glutamine, pyruvate and selenium or amino acids can also increase O2− levels.185 Interestingly, amino acid starvation alone increases intracellular H2O2 levels, and overexpression of SOD2 in serum-starved, amino acid-starved and H2O2-treated cells attenuates ROS production and inhibits autophagy.185 Mitochondrial antioxidants, such as SS-31 and SOD2, can block autophagy induced by stressors including mitochondrial inhibitors,180 sodium selenite,179 and immobilization.186 Given the critical role of ROS in the regulation of autophagy, further work is needed to clarify the specific mechanisms by which ROS influence autophagic machinery.
ROS are also implicated in regulating the selective degradation of mitochondria through mitophagy. MitoROS can trigger mitochondrial permeability transition pore opening and a burst of mitoROS, which has been described as ROS-induced ROS release.187, 188 Thus, ROS produced by damaged mitochondria may act as a self-removal signal,175 and while this hypothesis needs further verification, the benefits of selectively recycling mitochondria with oxidized proteins and damaged DNA as a secondary defense against oxidative stress are clear and have been demonstrated in yeast.72,189 This is supported by recent work suggesting that ROS are required for Phosphatase and tensin homolog-induced kinase 1/Parkin-dependent mitophagy.190
VIII. ROS and the Proteasome
The proteasome is a highly organized multimeric complex responsible for the selective hydrolysis of cytoplasmic, nuclear and ER proteins191 which supports metabolic homeostasis by maintaining a stable pool of free amino acids for protein synthesis.192 The proteasome also promotes cellular viability in response to oxidative stress.175, 193 Amino acids are continuously subjected to oxidative modification as a consequence of aerobic respiration, and most of these modifications are irreversible and irreparable. Studies using antisense oligonucleotides against critical proteasome subunits 194 or proteasome inhibitors 195 have implicated the proteasome as the primary pathway for degrading oxidized proteins. Thus, the proteasome serves as an important secondary defense against oxidants.
The central catalytic component of the proteasome is the barrel-shaped 20S core particle (CP) composed of four heptameric rings consisting of two outer “gate-keeping” α rings and two identical inner β rings that contain catalytically active/proteolytic β subunits.191,196 The 20S CP may be flanked by regulatory subunits through α subunit interaction193 which modulate 20S CP activity and substrate specificity.197 The most common of these subunits is the 19S regulatory particle (RP),197 which along with the 20S CP, comprises the 26S proteasome. The 26S proteasome is a key component of the ubiquitin proteasome pathway (UPP) and regulates the degradation of ubiquitinated proteins.197,198 The 19S RP recognizes polyubiquitinated substrates and hydrolyzes ATP to unfold and translocate target proteins into the 20S CP.197 The 20S CP may also associate with other regulatory proteins, such as PA28αβ, PA28γ, and PA200, for ubiquitin/ATP-independent protein degradation.198,199
Mildly oxidized proteins selectively and rapidly undergo proteolysis, and strong evidence suggests the 20S proteasome is largely responsible for this degradation.193,195,200 H2O2 impairs ubiquitin conjugation and reduces ubiquitin/ATP-dependent proteolysis;201 however, disruption of the ubiquitin system does not affect oxidized protein degradation,195 suggesting that the 20S ubiquitin/ATP-independent proteasome is responsible for protein degradation during oxidative stress. Concordant with these findings, in vitro studies using purified 26S show that the 26S proteasome, even in the presence of ATP and a functional ubiquitination system, does not degrade oxidized proteins.193 However, both the 26S and 20S proteasome are sensitive to oxidative stress.202 In fact, in the presence of the activator PA28αβ, exogenous ROS enhances 20S protease activity202 while H2O2 disrupts 26S proteasome complex integrity and reversibly dislocates the 20S CP from the 19S RP.200. This dissociation is accompanied by a loss of 26S proteasome activity and enhances cell survival following H2O2-induced oxidative stress.200 Interestingly, dissociation of the 26S proteasome increases the fraction of 20S proteasome, which may account for an increase in cell survival.200 Consistent with this notion is the observation that yeast deficient in 26S assembly are more resistant to oxidative stress than their wild type counterparts.203
Some evidence, however, supports a role for the 26S proteasome in degrading oxidized proteins. Ubiquitin carboxyl-terminal hydrolase 14 (USP14), a 26S proteasome-associated deubiquitinating enzyme, decreases ubiquitin-protein conjugate degradation by disassembling polyubiquitin chains.204 Inhibition of USP14 enhances cell survival and reduces the accumulation of oxidized proteins in cells challenged by menadione,204 suggesting that ubiquitination and the 26S proteasome are important for degrading oxidized proteins. Moreover, the expression of Ubiquitin conjugating enzyme (UBC) 4, an E2 enzyme, promotes the degradation of glutathionylated proteins in lens fiber cells, and this degradation is blocked by a dominant negative form of ubiquitin and proteasome inhibitors.205 Several studies also suggest that the 26S proteasome is critical for cellular viability during recovery from oxidative stress. Cells treated with H2O2 exhibit a transient increase in proteolytic activity and in ubiquitin conjugation post-treatment.201, 206 While oxidative stress negatively regulates 26S activity, 26S proteasome activity is almost completely restored 24 hours post-treatment.202 Finally, it has been observed that certain oxidized proteins are preferentially ubiquitinated.207 Thus, there is evidence to support a role for both the 20S and 26S proteasome in degrading oxidized proteins. However, further work may help to distinguish the different roles of the 20S and 26S proteasome in dealing with specific oxidized substrates.
The proteasome plays an important role in mitigating the effects of ROS, and oxidative modification of proteasome subunits modulates proteasome activity. Carboxylation of Regulatory Particle Triphosphate (RPT) 3, an ATPase subunit in the 19S RP, impairs ATPase activity and decreases 26S proteasome activity.208. Additionally, both carbonylation and 4-hydroxy-2-nonenal (HNE) modification of two α-subunits in the 20S proteasome impair ubiquitin/ATP-independent proteolysis.209 Interestingly, S-glutathionylation directly modulates specific proteolytic activity of the 20S proteasome.210 Low doses of GSH or GSSG enhance the chymotrypsin-like activity of purified proteasome, while higher doses inhibit this activity.210 The effect of S-glutathionylation seems specific to the chymotrypsin-like activity, as GSH and GSSG do not affect the trypsin-like activity of purified proteasomes.210
Other post-translational modifications also modulate proteasome activity in response to oxidative stress. Poly-ADP ribosylation of the 20S proteasome by the redox-sensitive enzyme poly-ADP ribose polymerase enhances the chymotrypsin-like activity of the 20S proteasome in K562 leukemia cells.211 In addition, apoptosis signal-regulating kinase 1 (Ask1) is activated by oxidative stress,212 and Ask1 phosphorylation of RPT5, a 19S subunit, negatively regulates 26S proteasome activity.213 Significantly, Ask1 is required for ROS-induced inhibition of 26S proteasome activity.214 Thus, Ask1-dependent proteasome phosphorylation may act as a critical regulatory mechanism for proteasome activity during oxidative stress.
Following oxidative stress, E1 activating enzyme expression and ubiquitin conjugation are increased in bovine lens epithelial cells.206 Increased expression appears to be due to increased translation of E1 mRNA.206 Consistent with the 20S proteasome playing a critical role in degrading oxidized proteins, H2O2 treatment increases α3, α4, β1, and β2 20S proteasome subunit expression without affecting the 19S RPS subunit S4.215 Interestingly, upregulation of immunoproteasome subunits has also been reported.215, 216 Low-molecular-mass protein (LMP) 2, LMP7, LMP10 are β subunits that may replace constitutive β subunits in the 20S proteasome upon interferon-γ stimulation,217 and increased expression of these subunits has been reported in neural cells in response to exogenous ROS.216 The transcription factors that regulate immunoproteasome and standard proteasome subunit expression following oxidative stress require further study.
2. Cardiovascular and Metabolic Diseases
I. Atherosclerosis
NOX
Atherosclerosis is mediated in large part through subendothelial lipoprotein retention, endothelial dysfunction, vascular remodeling and a heightened inflammatory response, and is an underlying cause of heart disease and stroke.218,219 Of importance, metabolic disease risk factors typically observed in patients with obesity and diabetes (changes in cholesterol, elevated triglycerides, insulin resistance) increase the risk for atherosclerosis.220,221 Although the link between oxidized LDL and atherosclerosis was discovered nearly 30 years ago,222 the NOX enzyme family was only defined in the mid-1990s. Some of the first studies performed exploring NOX-induced ROS concluded that NOX-derived ROS have little influence on lesion development in ApoE−/− mice on normal chow diets. Neither p47phox−/− (NOX1 and NOX2 activator) nor NOX2 (gp91phox−/y) mice crossed with ApoE−/− mice show changes in aortic sinus lesion area, although NOX2−/yApoE−/− mice exhibit reduced plasma triglyceride and cholesterol levels.223,224 However, these findings appear to be aortic sinus-specific, as further analysis of p47phox−/− ApoE−/− and NOX2−/y ApoE−/− mice (on chow and high-fat/western-type diets) revealed a reduction in descending aortic lesion development.225–227
NOX1−/y ApoE−/− mice are protected from both atherogenic diet-induced and diabetes-induced (atherogenic diet + streptozatocin, STZ) atherosclerosis,228–230 associated with decreased ROS production, inflammatory signaling and aortic macrophage infiltration. NOX1 likely exerts its effects in part by reducing VSMC proliferation and migration,231 and its phosphorylation on threonine 429 (Th4429) may be a contributing factor.232 However, the role of NOX1 in the initiation and progression of atherosclerosis remains controversial because recent findings in NOX1-deficient ApoE−/− mice indicate NOX1 may be protective against hyperlipidemia and plaque instability in response to a western-type diet.233
Similar to NOX1, the role of NOX2 in lesion etiology remains controversial. As mentioned, NOX2−/y ApoE−/− mice fed an atherogenic diet show reductions in the development and progression of atherosclerosis.226 Likewise, treatment of high fat diet fed ApoE−/− mice with a NOX2-specific inhibitor reduces aortic lesion area,234 and while there are compelling data regarding a role for global NOX2-induced ROS in plaque development, the tissue-specific role of NOX2 remains ill-defined. In ApoE−/− mice, EC-specific overexpression of NOX2 increases inflammatory signaling and macrophage infiltration in early lesions, but does not influence diet-induced lesion progression.235 Bone marrow transplantation between p47phox −/− ApoE−/− mice and p47+/+ ApoE−/− mice shows that suppression of either bone marrow cell or vascular wall O2− production attenuates western-type diet induced atherosclerosis.236 Moreover, suppression of NOX2 in bone marrow cells is associated with reduced plasma oxLDL, suggesting NOX2-derived ROS may regulate oxLDL production. oxLDL is scavenged and internalized by the lectin-like oxidized low density lipoprotein receptor 1 (LOX-1) and contributes to atherogenesis through ROS-dependent mechanisms involving NF-κB activation,237 inhibition of AKT/eNOS signaling,238 macrophage proinflammatory cytokine production and cell death,239,240 and VSMC apoptosis.241 The importance of NOX2-derived ROS within macrophages is also seen in its role in pathogen degradation after phagocytosis through a process called macropinocytosis. 242, 243
Given the diverse roles of both NOX1 and NOX2 in vascular disease, it is not surprising that NOX4 can also be both a harbinger of lesion development and a protector. As a protector, NOX4-derived ROS appear to be critical in maintaining vessel homeostasis in mouse models of atherosclerosis, because NOX4−/− Ldlr−/− and NOX4−/− ApoE−/− mice experience endothelial dysfunction and increased plaque burden as well as increased plaque formation in partial ligation plus high fat diet models.244,245 However, in diabetes-induced atherosclerosis, global NOX4 deletion may be protective or augment diabetes-induced lesion development, depending on the time frame.227, 229 NOX4 deletion in a 10-week STZ model of diabetes reduces plaque burden associated with T cell activation and infiltration,227 but in a 20-week model of STZ-induced diabetes, NOX4 deletion augments VSMC collagen deposition and proliferation, which may exacerbate lesion progression.246 The contribution of NOX4 in diabetic lesion development may also differ between early and advanced lesions, as NOX4 deletion has a minimal effect on early diabetes-induced plaque progression in the aortic arch,229 but is critical to suppressing the inflammatory response in advanced lesions.230
In tissue-specific NOX4 KO models, a more specific role for NOX4 signaling in atherosclerosis emerges. Expression of an EC-specific NOX4 mutation (human P437H dominant negative mutation) in ApoE−/− mice exacerbates lesion progression in response to STZ;247 whereas EC overexpression of wild-type NOX4 attenuates high-fat diet-induced lesion development.248 As mentioned, NOX4 is upregulated in advanced lesions, as opposed to the upregulation of NOX1 and NOX2 in the early phases of plaque development.225,249,250 While these studies indicate that this upregulation may be protective, other studies have shown NOX4 upregulation may promote lesion progression. Importantly, upregulated NOX4 in advanced plaques correlates with VSMC dysfunction and plaque instability,249 and smooth muscle-specific NOX4 deletion protects against western-type diet-induced atherosclerosis.251 Aged ApoE−/− mice fed a western-type diet and aged human carotid VSMCs exhibit increased mitochondrial NOX4 induction with parallel increases in mitoROS production, and inhibition of NOX4 increases aged VSMC mitochondrial complex I and II activity, decreases mitoROS and attenuates VCAM-1 induction.250 ER-derived NOX4 signaling is also implicated in VSMC apoptosis in response to 7-ketocholesterol, a major cholesterol oxidation product found in human plaques. 7-ketocholesterol upregulates IRE1 activity leading to JNK/AP-1-dependent NOX4 gene induction. Increased ROS production through NOX4 induces ER stress proteins GRP78 and CHOP and the apoptosis regulator Bcl-2-associated X protein (Bax).252 A role for NOX2 in CHOP induction has also been observed,253 and in vivo silencing of CHOP attenuates lesion area and plaque necrosis in hyperlipidemic mice.254,255
The PPP pathway can serve as both a biosynthetic pathway for nucleotides and a factory for NADPH production resulting in increased reducing capacity or NOX activity and O2− production. G6PD catalyzes the first step of the PPP and generates NADPH as a by-product. With regard to atherosclerosis, deficiency in G6PD abrogates O2− production and resultant inflammatory signaling and lesion growth in ApoE−/− mice fed a western-type diet.256 However, G6PD overexpression in ECs reduces TNFα-induced ROS production and increases eNOS activity, suggesting a protective role for this protein.257 Future studies should aim to clarify the circumstances by which G6PD contributes to pro- and anti-oxidant signaling, as well as the role of other PPP enzymes, in NOX activity and atherogenesis.257
As noted above, a plethora of evidence indicates a role for NOX-induced ROS in atherogenesis, although these molecules play both a positive and negative role in lesion progression. It is becoming increasingly clear that no model of atherosclerosis is the same. Changes in diet (composition and timing), genetic background, and age all influence NOX signaling. Tissue-specific knockouts have helped to create a clearer picture of the role of NOX in various tissues as it pertains to atherosclerosis, and continued investigation will determine the suitability of NOX enzymes as potential therapeutic targets.
Mitochondrial ROS
MitoROS signaling plays a major role in atherosclerosis and associated vascular complications, and induction of mitoROS is correlated with human plaque development.53 In general, scavenging of mitoROS attenuates atherosclerosis development and complications associated with atherosclerosis.250,258 Genetic inhibition of mitoROS may also have beneficial effects on lesion development. Suppression of mitoROS signaling in macrophages through the overexpression of mito-targeted catalase reduces lesion area, inflammatory signaling and immune cell infiltration into the aortic root of Ldlr−/− mice fed a western-type diet. In cultured mitoCatalase-overexpressing macrophages, LPS and oxLDL-induced mitoROS promote IKKβ phosphorylation (serine 177) and downstream p65 phosphorylation (serine 536), and inhibition of this pathway blocks macrophage MCP-1 induction.259 Furthermore, bone marrow transfer from mitoCatalase-expressing mice to aged WT Ldlr−/− mice diminishes neutrophil extracellular traps and lesion area, but is unable to perturb increased plasma cholesterol levels.260
As discussed previously, a dynamic interplay exists between mitoROS production and mitochondrial morphology. With regard to atherosclerosis and metabolic dysfunction, high glucose can induce mitoROS through Drp1-dependent mitochondrial fission in human ECs, and inhibition of fission with the pharmacological inhibitor mitochondrial division inhibitor-1 (Mdivi-1) attenuates diabetes-induced aortic oxidative stress, cell adhesion molecule expression and aortic root lesion development.261 However, there appears to be a cell type-specific role for mitochondrial fission in disease progression, as myeloid Drp1 silencing in Ldlr−/− mice fed a western-type augments plaque necrosis due to impaired efferocytosis.151
Uncoupling protein 2 (UCP2) is a mitochondrial inner membrane protein and is reported to regulate ROS generation through feedback inhibition involving ROS-induced uncoupling and proton leak.262 UCP2 expression is increased in C57BL6 mice in response to an atherogenic diet, and deletion of UCP2 increases oxidative stress, endothelial dysfunction, VCAM-1 expression, macrophage infiltration and atherogenesis.263,264 Likewise, xanthine oxidase is found in various cellular compartments including the mitochondria, and pharmacological inhibition via febuxostat or tungsten reduces chemokine expression, endothelial dysfunction and lesion development.265, 266
Altogether, mitoROS play a significant role is lesion etiology. Given their unique position in regulating cellular energy metabolism, it is no surprise that alteration in mitoROS production influences mitochondrial function and cellular homeostasis and is a possible cause of lesion progression. There are numerous ROS producing complexes within the mitochondria, and teasing out their role in atherosclerosis is of paramount importance.
ROS Scavenging
Similar to inhibition of ROS producing enzymes, increasing ROS scavenging appears to have a beneficial effect on lesion development. Mice overexpressing catalase and catalase + SOD1,267 Prdx4,268 and Trx2 (EC-specific)269 all exhibit decreased lesion development. In contrast, deletion of ROS scavenging systems exacerbates lesion progression, as seen in mice with deletion of NF-E2 related factor 2,270 GPX1,271,272, SOD2,273 and Prdx1 and 2.274,275
The paraoxanase (PON) family of proteins may also serve an anti-oxidant role in atherosclerosis through their ability to hydrolyze lipid peroxides.276 Overexpression of the PON transgenic cluster (PONs 1,2 and 3 together) promotes plaque stability via increased collagen synthesis, decreased necrotic core area and reduced oxLDL and inflammatory markers. PON1 overexpression may also reduce monocyte-to-macrophage differentiation and promote macrophage resistance against oxLDL-induced foam cell formation277 and induction of the LPS + IFNγ inflammatory phenotype.278,279 In contrast, deletion of PON1 increases vascular oxidative stress and leukocyte adhesion,280 and deletion of PON2 exacerbates lesion development.281,282 Recent evidence suggests that PON2 may localize to the mitochondrial inner membrane where it is bound to CoQ and contributes to ROS scavenging and maintenance of complex I and III activity.281
Cumulatively, ROS signaling has been shown to be an underlying causative factor that contributes to the complex etiology of atherosclerosis in animal models (Supplementary Table I). The studies presented highlight a unique role for ROS in the regulation of cellular responses to various atherogenic stimuli, and tissue-specific studies have begun to elucidate the role ROS signaling might play in various cell types and their contribution to lesion progression. However, even in the presence of an abundance of data indicating a causative role for ROS in atherosclerosis, mechanistic details regarding metabolic regulation and its role in ROS signaling are lacking.
II. Diabetes/Obesity
Obesity creates an increased risk for the development of cardiovascular and metabolic diseases including atherosclerosis, diabetes mellitus and hypertension.283 Obesity is associated with insulin resistance and hyperglycemia, which contribute to the development of type II diabetes mellitus, and clinical investigation has yielded novel insight into how diabetes-related complications contribute to systemic vascular and metabolic dysfunction. Clinical and animal research consistently point to oxidative stress as an underlying factor that mediates obesity- and diabetes-related health ailments.284,285 Importantly, glucose metabolism can produce ROS via sorbitol metabolism, hexosamine metabolism, α-ketoaldehyde production, PKC activation, glycation and oxidative phosphorylation.286
Vascular System
Hyperglycemia is a consequence of diabetes and insulin resistance and plays a significant role in the accompanying vascular complications (atherogenesis, endothelial dysfunction, inflammation, altered vascular tone). Early studies highlighted the ability of glucose and free fatty acids to stimulate ROS production in vascular cells including VSMCs, ECs and human leukocytes, and showed that high glucose can impair PPP activity and generation of reducing equivalents.287–290 Since then, investigation has focused on unique pathways by which ROS may regulate vascular complications in diabetes and obesity.
ROS can regulate vascular tone in response to hyperglycemia via regulation of PKCβ and inhibition of eNOS activity.291 Hyperglycemia-induced ROS production activates the 26S proteasome leading to ubiquitination and degradation of the BH4 synthesis rate limiting enzyme guanosine 5′-triphosphate cyclohydrolase I (GTPCH). BH4 deficiency dysregulates eNOS activity which impairs endothelial-dependent relaxation, and treatment of STZ-injected mice with TEMPOL restores GTPCH, BH4 and attenuates endothelial dysfunction.292 Furthermore, PKCβII induces endothelial dysfunction through p66shc-mediated mitoROS production.291,293 Hyperglycemia increases p66shc acetylation (Lys81), which precedes PKCβII-induced phosphorylation (Ser36) of p66shc, and inhibition of p66shc reduces mitoROS, aortic lipid peroxidation and restores eNOS activity and endothelial-dependent relaxation in diabetic mice.291, 293 PKCβ also regulates vascular tone through impairment in the large conductance Ca2+-activated K+ channel. In response to high glucose, the β1 subunit of the Ca2+-activated K+ channel is degraded through PKCβ-stimulated NOX1 and NOX4 activation, inhibition of AKT and activation of the forkhead box protein O3a/F-box only degradation pathway.294
As with atherosclerotic lesion development, NOX-derived ROS play an important role in vascular homeostasis in diabetes and obesity. Inhibition of NOX activity attenuates diabetes-induced impairment of endothelial-dependent relaxation,295 and aged NOX2−/y mice fed a high fat diet are protected from obesity, dyslipidemia, insulin resistance and endothelial dysfunction.296 Similar results have been observed in arteries from diabetic NOX1−/y mice,297 and mice treated with siRNA targeting p22phox,298 as well as NOX2−/y mice with EC-specific overexpression of a human dominant negative (DN) variant of the insulin receptor that causes EC-specific insulin resistance and reduced vascular relaxation.299 Proinflammatory stimuli such as TNFα can induce NOX activity in diabetic mice resulting in impaired vasorelaxation,300 and NOX-derived ROS downregulate cGMP-dependent protein kinase I, which normally promotes vascular relaxation.301 Furthermore, high glucose induces NOX4 upregulation via a PKCζ and NF-κB-dependent pathway.302 Insulin like growth factor 1 (IGF-1) enhances high glucose-induced NOX4/p22phox complex formation and activation of VSMCs, and induces NOX4 Tyr491 phosphorylation leading to NOX4 binding to the SH2 domain of growth factor receptor-bound protein 2 (Grb2). NOX4/Grb2 association is needed for NOX4 localization to the SHPS-1 plasma membrane scaffold in VSMCs and STZ-injected mouse vasculature. Disruption of the NOX4/Grb2 association inhibits Src oxidation in vivo as well as VSMC proliferation.303
ROS produced by NOX also reduce vascular endothelial growth factor (VEGF) expression and post-ischemic neovascularization in diabetic mice. NOX2−/y mice show increased bone marrow mononuclear cell (BM-MNC) to EC differentiation and injection of BM-MNCs from NOX2−/y mice into diabetic mice enhances neovascularization.304 Furthermore, endothelial progenitor cells from diabetic patients show a reduced reendothelialization capacity that can be normalized through inhibition of p47phox.305 Together, these data suggest a major role for ROS-mediated signaling in both facilitating dysregulated vascular tone and impairment of angiogenesis/neovascularization in diabetic ischemic tissue.
One consequence of hyperglycemia is the generation of advanced glycation end products (AGEs), covalent adducts formed between glucose and plasma proteins. AGEs contribute to diabetic-related maladies including neuropathy, retinopathy and cardiomyopathy.306 Given its direct contact with blood, the endothelium is a prime target for AGE-mediated signaling. Importantly, aortic ECs express the receptor for AGEs (RAGE), which is increased in response to high glucose via mitoROS production,307 and incubation of ECs with diabetic red blood cells expressing AGEs increases VCAM-1 induction and tissue factor production through RAGE-induced NOX activation.308 AGE stimulation may also dysregulate eNOS activity via increased NOX and mitoROS within the coronary endothelium of diabetic patients.309 Silencing of NOX2 in macrophages prevents AGE-induced tissue factor expression as well.308 NOX silencing is also beneficial in reducing AGE-induced apoptosis and NF-κB signaling in VSMCs.308, 310 Further, high glucose-induced mitoROS production increases PKC activation, hexosamine pathway activation and AGE formation through DNA strand break-mediated poly(ADP-ribose) polymerase activation and GAPDH poly(ADP-ribosyl)ation.311
As mentioned, mitoROS play a significant role in modulating vascular function in diabetes and obesity through both AGE signaling and modulating vascular tone, and potentially through changes in mitochondrial morphology. Isolated coronary ECs from diabetic mice exhibit increased mitochondrial fission that can be attenuated with in vivo TEMPOL delivery.312 High glucose-induced mitochondrial fission induces mitoROS production,312,313 which may be a result of increased pyruvate uptake following increased fission,314 as well as an instigating step in apoptosis.313 Silencing of Drp1 or Fis1 inhibits high glucose-induced mitoROS and restores eNOS activity and cGMP production, suggesting a role for mitochondrial fission-induced ROS in regulating vascular tone.315 Likewise, high glucose-induced VSMC proliferation can be inhibited by attenuating mitochondrial fission-dependent NOX activity.316 Low blood glucose also commonly occurs in diabetic patients who lack tight glycemic control, and low glucose enhances mitoROS production through Drp1-dependent mitochondrial fission. Suppression of mitochondrial fission in response to low glucose inhibits mitoROS production, increases NO bioavailability and restores endothelial-dependent vascular relaxation.317 Moreover, NOX-induced Drp1-driven mitochondrial fission contributes to inflammasome activation by palmitate and elevated free fatty acids, which are often observed in diabetes.318
Much evidence supports an ROS contribution to the regulation of vascular tone and inflammatory signaling in diabetes and obesity (Supplementary Table II). The interaction of ROS and the metabolic changes that occur in obesity and diabetes is less clear, and is a subject for further investigation.
III. Stroke
Ischemic stroke is a leading cause of death and long term disability in the United States,319 and patients who exhibit metabolic risk factors including diabetes mellitus, obesity and dyslipidemia are at a greater risk of experiencing stroke-related events.320 Within minutes of hypoxia and glucose deprivation, a complex cascade of molecular events ensues, involving depolarization of neurons, increased Ca2+ influx, ATP depletion and release of the excitatory neurotransmitter glutamate.321 Activation of glutamate receptors leads to a further increase in intracellular Ca2+, activation of NOS and NOX signaling, mitochondrial dysfunction and neuronal death. Although hypoxia and glucose deprivation play a major role in the neurodegeneration induced by stroke, a role for ROS is clear. Indeed, several clinical studies have shown a correlation between elevated oxidative stress and brain ischemia,322,323 and decreasing oxidative stress may be protective against stroke-induced complications.324,325.
MitoROS are involved in the pathophysiology of cerebral ischemia as well as reperfusion injury.326 Importantly, depletion of SOD2 increases mortality, stroke volume, brain edema and cytochrome C-mediated neuronal apoptosis following transient and permanent middle cerebral artery occlusion (MCAO).327,328 In response to stroke, mitoROS arise from various sources including, complex I and IV, monoamine oxidase, p66shc, mitochondrial BKCa and mKATP channels, cytochrome b5 reductase and dihydroorotate dehydrogenase.329–333 In patients who experience acute ischemic stroke, p66shc gene expression is increased in peripheral blood monocytes, and post-ischemic knockdown of p66shc in mice undergoing transient MCAO improves survival and functional outcomes and reduces stroke lesion volume.334 p66shc knockdown attenuates transient MCAO (tMCAO)-induced blood-brain barrier (BBB) permeability, which is associated with a reduction in NOX ROS-induced claudin-5 degradation.335 NOX signaling represents an important source of ROS in the pathophysiology of stroke. Several studies have correlated NOX2 and NOX4 depletion with decreased infarct volume, oxidative stress, BBB permeability and neutrophil infiltration after MCAO.324,336–339 Similar results have been reported with non-specific Nox2 inhibitors, apocynin and diphenylene iodonium,337,340 and injection of glutathione into rats subjected to tMCAO reduces infarct volume and increases cell survival signaling.341 Further, NOX2 deletion prevents tMCAO-induced IL-1β, TNFα, and CC-chemokine ligand 2/3 upregulation,342 as well as glutamate toxicity through suppression of complexin II/soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) interaction.343 However, a recent study hypothesizes that deletion of NOX2 only delays infarct progression while simultaneously increasing angiogenesis, but is unable to prevent neuronal loss.344 Similarly, NOX2 deletion may have a protective effect only in models of transient occlusion, as NOX2 deletion has no effect on permanent MCAO-induced neurological dysfunction.345
A role for tissue-specific NOX4 signaling in cerebral ischemia has also recently been elucidated. Overexpression of NOX4 in pericytes increases BBB permeability following permanent MCAO in mice, which coincides with increased MMP-9 activity and phosphorylation of NFκB.346 Similarly, in response to tMCAO, EC NOX4−/− mice have increased BBB stability associated with less autophagy-related stress, but show no reduction in neuronal cell death. In contrast, neuronal NOX4−/− mice display reduced neuronal cell death but no change in BBB stability.339 Separate from NOX2 and NOX4, there have been conflicting reports regarding the role of NOX1 in stroke pathophysiology. NOX1 depletion decreases infarct volume, improves neurological outcome and reduces cerebral edema in mice subjected to tMCAO; however, these effects may be independent of NOX1-ROS signaling.347 Adenoviral-mediated knockdown of NOX1 in rats undergoing tMCAO promotes increased functional recovery, which is associated with reduced infarct size and neuronal cell death.348 However, two different groups have shown that NOX1 depletion does not affect infarct volume, brain edema or neurological score following tMCAO.324,349
As mentioned, diabetes is a risk factor for stroke,350 and elevated blood glucose at stroke onset predisposes individuals to more severe functional outcomes and increased mortality.351 Mechanistically, glucose injection into mice at the time of reperfusion after tMCAO increases NOX-dependent ROS production and neuronal cell death compared to normoglycemic controls, and p47phox−/− mice are protected from this response.352 Furthermore, systemic glucose is regulated by the ventromedial nucleus of the hypothalamus (VMH), and glucose sensing in the VMH is partly regulated by Drp1-dependent mitoROS signaling.353 Glucose load induces Drp1 activity through UCP2 in VMH, and UCP2-mediated mitochondrial fission reduces ROS, a mechanism that seemingly argues against the hypothesis that mitochondrial fission induces ROS, although increased Drp1-dependent mitophagy in response to permanent brain ischemia may play a role in mitigating ROS production.354 Depletion of UCP2 increases infarct volume in mice after tMCAO355 and alters whole body glucose utilization and insulin sensitivity. Re-expression of UCP2 in the VMH rectifies changes in glucose utilization and insulin sensitivity,356 and UCP2 overexpression in mice subjected to tMCAO reduces brain damage and neurological dysfunction.357
Oxygen sensing and glucose metabolism are central to regulating brain homeostasis and contribute to ischemic pathophysiology. Prolyl hydroxylase domain proteins (PHDs) regulate hypoxic signaling, most notably through regulation of HIF1α activity. PHD1−/− mice are protected against the deleterious consequences of permanent brain ischemia.358 PHD1 deficiency reduces neuronal glycolysis and glucose consumption while concomitantly increasing glutamine oxidation, thereby maintaining mitochondrial respiration and energy homeostasis. This is vital for protection against ischemic-related injury as glucose is diverted towards the PPP pathway, thus increasing oxidative PPP flux and reducing equivalents needed for ROS scavenging in response to an ischemic insult.358 A role for oxidant scavenging has also been noted in ischemic pathophysiology. Prdx2 overexpression reduces infarct size and neurological deficits in mice through attenuation of DNA damage and PARP1/p53 pro-death signaling.359 GPX3 deficiency promotes vascular dysfunction and platelet-dependent arterial thrombosis and increases cerebral infarct size in mice with permanent MCAO.360 Conversely, overexpression of GPX1 attenuates tMCAO-induced edema, microglial activation, neutrophil infiltration and neuronal cell death compared to WT mice.361 Similar results have been observed in SOD1 mice.362
Cumulatively, although evidence supports a major role for ROS signaling in stroke pathophysiology, few studies have delved into cell-type specific roles of ROS. Given the dynamic interplay between ECs, pericytes, VSMCs, astrocytes and immune cells in the development and progression of stroke, additional experiments are warranted. In particular, with the unique metabolic alteration that occurs during stroke, more emphasis should be placed on how ROS signaling affects nutrient shuttling and metabolism in the ischemic tissue.
3. Antioxidant Trials
Animal studies have unequivocally suggested that ROS are a viable target for therapeutic intervention in the treatment of cardiovascular disease. However, clinical trials have yielded less than favorable results,363,364 and although these results are negative, they have helped to identify why current approaches fail and how to target future interventions. Most antioxidant trials were conducted with various vitamins (A, C and E), and although vitamins have antioxidant properties, their effects may be too broad and concentrations may never reach the levels needed to attenuate ROS production, nor be as effective in ROS scavenging by tissue antioxidant defenses. Concerns have arisen regarding efficacy of synthetic compared to natural antioxidant formulations, as well as mode of delivery (e.g., pill vs diet). Furthermore, disease progression takes time, especially in the case of atherosclerosis and heart failure. To date, most studies have only looked at a small window of time (~5 years) during more advanced stages of disease, which may not be long enough to uncover positive effects. It has also become increasingly clear that ROS are vital to maintaining physiological function, and thus general scavenging of ROS may compromise cellular homeostasis and augment disease progression in some cases. This is especially important as we consider the location and source of ROS, the specific roles of various types of ROS and how changes in concentration may affect cellular signaling. For instance, H2O2 has been found to induce both vasoconstriction and vasorelaxation in blood vessels treated with KCL and phenylephrine, respectively.365 Likewise, suppression of endothelial H2O2 in mice results in hypotension and prevents the upregulation of eNOS in response to exercise.366 On a similar note, O2−, while largely known for its role in arterial vasoconstriction and mediating EC and VSMC dysfunction, has also been found to regulate both vasoconstriction and vasodilation in a concentration-dependent manner in the cerebral vasculature,367 and is critical to platelet activation and immune cell phagocytic function/recruitment.368,369 ROS signaling in the setting of hypoxia has also been speculated to both instigate and inhibit angiogenesis, which may be dependent upon the local tissue environment and disease setting.305,369 Finally, in many instances, antioxidants scavenge free radicals but do not affect H2O2, which as described throughout this review, is responsible for many of the pathophysiological effects on the vasculature.
While numerous studies highlight the physiological and pathophysiological differences in ROS signal transduction and their effect on cellular function, the takeaway remains the same. Instead of broad-spectrum ROS scavenging, it may be more effective to consider therapies that target specific sources of ROS, or limiting the production of specific ROS so as not to disrupt basic physiological function. We also need to consider how specific ROS influence different diseases. For example, in some cases, inhibition of O2− may not be as effective as scavenging H2O2 and vice-versa. However, inhibiting a specific source of ROS such as NOX may be beneficial. To this end, GKT137831, a promising NOX1/4 inhibitor, is currently in phase II clinical trials for diabetic kidney disease.370 Human studies will be needed to test if new therapies targeted to specific sources of ROS, such as mito-tempol, will be beneficial in the treatment of cardiovascular disease.
4. Future Investigation/Concluding Remarks
One of the challenges in dissecting the role of ROS in cardiovascular pathology is that ROS are produced not only as natural by-products of metabolic reactions in various cellular compartments, but they also serve as signaling molecules that regulate specific biochemical pathways in normal cell function and survival. Dysregulation of ROS signaling, or excess non-specific production of ROS, can influence disease pathophysiology. As highlighted in this review, ROS are particularly important in cellular metabolism and inflammatory signaling. Thus, it is not surprising that ROS play a significant role in diseases associated with metabolic dysregulation and inflammation.
There are myriad areas where further investigation is warranted. Importantly, we have seen much emphasis on cytoplasmic and mitochondrial ROS signaling; however, there is a dearth of information regarding the contribution of peroxisomal and endoplasmic reticular ROS to cellular homeostasis. New organelle-targeted probes and antioxidants will help to tease out the contribution of organellular/compartmental-specific ROS and their influence on critical cellular processes including aerobic/anaerobic respiration, β-oxidation, fatty acid synthesis, protein translation and post-translational modifications. Such tools will also provide insight into how ROS-induced inflammatory signaling may affect and be affected by this regulation, given the relationship between inflammatory and metabolic signaling. In addition, with the advent of cre-lox technology and the development of tissue-specific and inducible knockout mouse models, research should continue to delineate the tissue-specific role of ROS signaling in metabolic and cardiovascular diseases. Cardiovascular and metabolic disease etiology is complex, and understanding tissue-specific redox signaling will be important in our effort to develop new and novel therapies to treat disease. Metabolic dysregulation is a primary driver of cellular dysfunction and disease progression, and understanding the contribution and influence of ROS on metabolic processes poses an exciting area for scientific discovery.
Supplementary Material
Acknowledgments
Sources of Funding
SJF is supported by NIH 5T32HL007745-24. MSH is supported by AHA 17SDG33410777. This work was also supported by NIH P01 HL095070.
Abbreviation
- ACOX
Acyl-CoA oxidase
- ADAM17
A disintegrin and metalloproteinase domain-containing protein 17
- AGE
Advanced glycation end product
- AMPK
AMP-activated protein kinase
- AP-1
Activator protein 1
- ASC
Apoptosis-associated speck-like protein containing a CARD
- ASK1
Apoptosis signal-regulating kinase 1
- ATM
ataxia-telangiectasia mutated
- BBB
Blood-brain barrier
- BH4
tetrahydrobiopterin
- BM-MNC
Bone marrow mononuclear cell
- BMP4
Bone morphogenic protein 4
- CaMKKβ
Ca2+/calmodulin-dependent protein kinase kinase Beta
- Cat
Catalase
- CCCP
Carbonyl cyanide 3-chlorophenylhydrazone
- CHOP
C/EBP homologous protein
- CMA
Chaperone-mediated autophagy
- CoQ
Coenzyme Q
- CP
Core particle
- CytoROS
Cytoplasmic ROS
- DN
Dominant negative
- Drp1
Dynamin-related protein 1
- EC
Endothelial cell
- eNOS
Endothelial nitric oxide synthase
- ER
Endoplasmic Reticulum
- ERO1
ER oxidoreductin 1
- ETC
Electron transport chain
- G6PD
Glucose-6-phosphate dehydrogenase
- GlUT1
Glucose transporter 1
- GPX
Glutathione peroxidase
- Grb2
Growth factor receptor-bound protein 2
- GRP78
Glucose-regulate protien 78
- Grx
glutaredoxin
- GTPCH
Guanosine 5′-triphosphate cyclohydrolase I
- HIF1α
Hypoxia inducible factor 1α
- IGF-1
Insulin like growth factor 1
- IKK
IκB kinase
- IL-18
Interleukin 18
- IL-1β
Interleukin 1 Beta
- iNOS
Inducible NOS
- IP3R
Inositol 1,4,5-trisphosphate receptor
- IκBα
Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, alpha
- JNK
c-Jun N-terminal kinase
- KGDHC
α-ketoglutarate dehydrogenase
- KO
Knockout
- LAP
LC3-associated phagocytosis
- LC3
Microtubule-associated protein 1A/1B-light chain 3
- LKB1
Liver kinase B1
- LMP
Low-molecular-mass protein
- LOX-1
Lectin-type oxidized LDL receptor 1
- LPP
Lipid peroxidation product
- LPS
Lipopolysaccharide
- MCU
Mitochondrial calcium uniporter
- MerTK
Mer Tyrosine Kinase
- MICU1
mitochondrial calcium uptake 1
- Mitochondrial-associated ER membrane
MAM
- MitoROS
Mitochondrial ROS
- mTORC
Mammalian target of rapamycin complex
- NAC
N-acetyl cysteine (CC
- NF-κB
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NIK
NF-κB inducing kinase
- NLRP3
NLR family pyrin domain containing 3
- nNOS
Neuronal NOS
- NOX
NADPH oxidase
- ONOO
Peroxynitrite
- OPA1
Optic atrophy protein 1
- OxPL
Oxidized Phospholipid
- P66SHC
SHC-transforming protein 1 isoform p66
- PDC
pyruvate dehydrogenase
- PDHK
Pyruvate dehydrogenase kinase
- PDI
Protein disulfide-isomerase
- Pex
Peroxin
- PHD
Prolyl hydroxylase domain protein
- PINK1
(PTEN)-induced kinase 1
- PON
Paraoxanase
- PPP
Pentose phosphate pathway
- Prdx
peroxiredoxin
- RAGE
Receptor for AGE
- RET
Reverse electron transfer
- ROMO1
ROS modulator 1
- ROS
Reactive oxygen species
- RP
Regulatory particle
- RPT
Regulatory particle triphosphate
- SNARE
Soluble N-ethylmaleimide-sensitive factor attachment protein
- SOD
Superoxide dismutase
- STZ
Streptozatocin
- tMCAO
Transient middle cerebral artery occlusion
- TNFα
Tumor necrosis factor alpha
- TRAF
TNF Receptor associated factor
- Trx
Thioredoxin
- TrxR
Thioredoxin reductase
- TSC
Tuberous sclerosis protein
- TxNIP
Thioredoxin-interacting protein
- UBC
Ubiquitin conjugating enzyme
- UCP2
Uncoupling protein 2
- ULK1
uncoordinated 51-like kinase 1
- UPP
Ubiquitin proteasome pathway
- UPR
Unfolded protein response
- USP14
Ubiquitin carboxyl-terminal hydrolase 14
- VCAM-1
Vascular cell adhesion molecule 1
- VDAC1
Voltage dependent anion channel 1
- VEGF
Vascular endothelial growth factor
- VMH
Ventromedial nucleus of the hypothalamus
- VSMC
Vascular smooth muscle cell
Footnotes
Disclosures
None
References
- 1.Nikolay VG, Pavel VA, Alexander DN, Irina LZ, Richard OJ. Reactive oxygen species in pathogenesis of atherosclerosis. Current Pharmaceutical Design. 2015;21:1134–1146. doi: 10.2174/1381612820666141014142557. [DOI] [PubMed] [Google Scholar]
- 2.Kaneto H, Katakami N, Matsuhisa M, Matsuoka T-a. Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediators of Inflammation. 2010;2010 doi: 10.1155/2010/453892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Allen CL, Bayraktutan U. Oxidative stress and its role in the pathogenesis of ischaemic stroke. International Journal of Stroke. 2009;4:461–470. doi: 10.1111/j.1747-4949.2009.00387.x. [DOI] [PubMed] [Google Scholar]
- 4.Bedard K, Krause K-H. The nox family of ros-generating nadph oxidases: Physiology and pathophysiology. Physiological Reviews. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 5.Panday A, Sahoo MK, Osorio D, Batra S. Nadph oxidases: An overview from structure to innate immunity-associated pathologies. Cell Mol Immunol. 2015;12:5–23. doi: 10.1038/cmi.2014.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cave AC, Brewer AC, Narayanapanicker A, Ray R, Grieve DJ, Walker S, Shah AM. Nadph oxidases in cardiovascular health and disease. Antioxidants & Redox Signaling. 2006;8:691–728. doi: 10.1089/ars.2006.8.691. [DOI] [PubMed] [Google Scholar]
- 7.Paik J-Y, Jung K-H, Lee J-H, Park J-W, Lee K-H. Reactive oxygen species-driven hif1α triggers accelerated glycolysis in endothelial cells exposed to low oxygen tension. Nuclear Medicine and Biology. 2017;45:8–14. doi: 10.1016/j.nucmedbio.2016.10.006. [DOI] [PubMed] [Google Scholar]
- 8.Baillet A, Hograindleur M-A, El Benna J, Grichine A, Berthier S, Morel F, Paclet M-H. S. The FASEB Journal. 2017;31:663–673. doi: 10.1096/fj.201600720R. [DOI] [PubMed] [Google Scholar]
- 9.Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BS, Karoui H, Tordo P, Pritchard KA., Jr Superoxide generation by endothelial nitric oxide synthase: The influence of cofactors. Proc Natl Acad Sci U S A. 1998;95:9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang PL. Enos, metabolic syndrome and cardiovascular disease. Trends in endocrinology and metabolism: TEM. 2009;20:295–302. doi: 10.1016/j.tem.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xia Y, Roman LJ, Masters BSS, Zweier JL. Inducible nitric-oxide synthase generates superoxide from the reductase domain. Journal of Biological Chemistry. 1998;273:22635–22639. doi: 10.1074/jbc.273.35.22635. [DOI] [PubMed] [Google Scholar]
- 12.Pou S, Keaton L, Surichamorn W, Rosen GM. Mechanism of superoxide generation by neuronal nitric-oxide synthase. Journal of Biological Chemistry. 1999;274:9573–9580. doi: 10.1074/jbc.274.14.9573. [DOI] [PubMed] [Google Scholar]
- 13.García-Nogales P, Almeida A, Bolaños JP. Peroxynitrite protects neurons against nitric oxide-mediated apoptosis: A key role for glucose-6-phosphate dehydrogenase activity in neuroprotection. Journal of Biological Chemistry. 2003;278:864–874. doi: 10.1074/jbc.M206835200. [DOI] [PubMed] [Google Scholar]
- 14.Kussmaul L, Hamprecht B, Dringen R. The detoxification of cumene hydroperoxide by the glutathione system of cultured astroglial cells hinges on hexose availability for the regeneration of nadph. Journal of Neurochemistry. 1999;73:1246–1253. doi: 10.1046/j.1471-4159.1999.0731246.x. [DOI] [PubMed] [Google Scholar]
- 15.Ham M, Lee J-W, Choi AH, Jang H, Choi G, Park J, Kozuka C, Sears DD, Masuzaki H, Kim JB. Macrophage glucose-6-phosphate dehydrogenase stimulates proinflammatory responses with oxidative stress. Molecular and Cellular Biology. 2013;33:2425–2435. doi: 10.1128/MCB.01260-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matsui R, Xu S, Maitland KA, Hayes A, Leopold JA, Handy DE, Loscalzo J, Cohen RA. Glucose-6 phosphate dehydrogenase deficiency decreases the vascular response to angiotensin ii. Circulation. 2005;112:257–263. doi: 10.1161/CIRCULATIONAHA.104.499095. [DOI] [PubMed] [Google Scholar]
- 17.Zhang Z, Yang Z, Zhu B, Hu J, Liew CW, Zhang Y, Leopold JA, Handy DE, Loscalzo J, Stanton RC. Increasing glucose 6-phosphate dehydrogenase activity restores redox balance in vascular endothelial cells exposed to high glucose. PLOS ONE. 2012;7:e49128. doi: 10.1371/journal.pone.0049128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang S, Song P, Zou M-H. Amp-activated protein kinase, stress responses and cardiovascular diseases. Clinical Science (London, England : 1979) 2012;122:555–573. doi: 10.1042/CS20110625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mihaylova MM, Shaw RJ. The amp-activated protein kinase (ampk) signaling pathway coordinates cell growth, autophagy, & metabolism. Nature cell biology. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mungai PT, Waypa GB, Jairaman A, Prakriya M, Dokic D, Ball MK, Schumacker PT. Hypoxia triggers ampk activation through reactive oxygen species-mediated activation of calcium release-activated calcium channels. Molecular and Cellular Biology. 2011;31:3531–3545. doi: 10.1128/MCB.05124-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zmijewski JW, Banerjee S, Bae H, Friggeri A, Lazarowski ER, Abraham E. Exposure to hydrogen peroxide induces oxidation and activation of amp-activated protein kinase. The Journal of Biological Chemistry. 2010;285:33154–33164. doi: 10.1074/jbc.M110.143685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cardaci S, Filomeni G, Ciriolo MR. Redox implications of ampk-mediated signal transduction beyond energetic clues. Journal of Cell Science. 2012;125:2115–2125. doi: 10.1242/jcs.095216. [DOI] [PubMed] [Google Scholar]
- 23.Zou M-H, Hou X-Y, Shi C-M, Kirkpatick S, Liu F, Goldman MH, Cohen RA. Activation of 5′-amp-activated kinase is mediated through c-src and phosphoinositide 3-kinase activity during hypoxia-reoxygenation of bovine aortic endothelial cells: Role of peroxynitrite. Journal of Biological Chemistry. 2003;278:34003–34010. doi: 10.1074/jbc.M300215200. [DOI] [PubMed] [Google Scholar]
- 24.Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang J-k, Shen M, Bellinger G, Sasaki AT, Locasale JW, Auld DS, Thomas CJ, Vander Heiden MG, Cantley LC. Inhibition of pyruvate kinase m2 by reactive oxygen species contributes to cellular antioxidant responses. Science (New York, NY) 2011;334:1278–1283. doi: 10.1126/science.1211485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bulua AC, Simon A, Maddipati R, Pelletier M, Park H, Kim K-Y, Sack MN, Kastner DL, Siegel RM. Mitochondrial reactive oxygen species promote production of proinflammatory cytokines and are elevated in tnfr1-associated periodic syndrome (traps) The Journal of Experimental Medicine. 2011;208:519–533. doi: 10.1084/jem.20102049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nishikawa T, Araki E. Impact of mitochondrial ros production in the pathogenesis of diabetes mellitus and its complications. Antioxid Redox Signal. 2007;9:343–353. doi: 10.1089/ars.2006.1458. [DOI] [PubMed] [Google Scholar]
- 27.Kussmaul L, Hirst J. The mechanism of superoxide production by nadh:Ubiquinone oxidoreductase (complex i) from bovine heart mitochondria. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:7607–7612. doi: 10.1073/pnas.0510977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Murphy Michael P. How mitochondria produce reactive oxygen species. Biochemical Journal. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bleier L, Dröse S. Superoxide generation by complex iii: From mechanistic rationales to functional consequences. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2013;1827:1320–1331. doi: 10.1016/j.bbabio.2012.12.002. [DOI] [PubMed] [Google Scholar]
- 30.Quinlan CL, Orr AL, Perevoshchikova IV, Treberg JR, Ackrell BA, Brand MD. Mitochondrial complex ii can generate reactive oxygen species at high rates in both the forward and reverse reactions. The Journal of Biological Chemistry. 2012;287:27255–27264. doi: 10.1074/jbc.M112.374629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rimessi A, Previati M, Nigro F, Wieckowski MR, Pinton P. Mitochondrial reactive oxygen species and inflammation: Molecular mechanisms, diseases and promising therapies. Int J Biochem Cell Biol. 2016;81:281–293. doi: 10.1016/j.biocel.2016.06.015. [DOI] [PubMed] [Google Scholar]
- 32.Finkel T. Signal transduction by reactive oxygen species. The Journal of Cell Biology. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Starkov AA, Fiskum G, Chinopoulos C, Lorenzo BJ, Browne SE, Patel MS, Beal MF. Mitochondrial α-ketoglutarate dehydrogenase complex generates reactive oxygen species. The Journal of Neuroscience. 2004;24:7779–7788. doi: 10.1523/JNEUROSCI.1899-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mailloux RJ, Gardiner D, O’Brien M. 2-oxoglutarate dehydrogenase is a more significant source of o 2·−/h 2 o 2 than pyruvate dehydrogenase in cardiac and liver tissue. Free Radical Biology and Medicine. 2016;97:501–512. doi: 10.1016/j.freeradbiomed.2016.06.014. [DOI] [PubMed] [Google Scholar]
- 35.Quinlan CL, Goncalves RLS, Hey-Mogensen M, Yadava N, Bunik VI, Brand MD. The 2-oxoacid dehydrogenase complexes in mitochondria can produce superoxide/hydrogen peroxide at much higher rates than complex i. The Journal of Biological Chemistry. 2014;289:8312–8325. doi: 10.1074/jbc.M113.545301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Brien M, Chalker J, Slade L, Gardiner D, Mailloux RJ. Protein s-glutathionylation alters superoxide/hydrogen peroxide emission from pyruvate dehydrogenase complex. Free Radical Biology and Medicine. 2017;106:302–314. doi: 10.1016/j.freeradbiomed.2017.02.046. [DOI] [PubMed] [Google Scholar]
- 37.Tretter L, Adam-Vizi V. Alpha-ketoglutarate dehydrogenase: A target and generator of oxidative stress. Philosophical Transactions of the Royal Society B: Biological Sciences. 2005;360:2335–2345. doi: 10.1098/rstb.2005.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tretter L, Adam-Vizi V. Inhibition of krebs cycle enzymes by hydrogen peroxide: A key role of [alpha]-ketoglutarate dehydrogenase in limiting nadh production under oxidative stress. J Neurosci. 2000;20:8972–8979. doi: 10.1523/JNEUROSCI.20-24-08972.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hurd TR, Collins Y, Abakumova I, Chouchani ET, Baranowski B, Fearnley IM, Prime TA, Murphy MP, James AM. Inactivation of pyruvate dehydrogenase kinase 2 by mitochondrial reactive oxygen species. Journal of Biological Chemistry. 2012;287:35153–35160. doi: 10.1074/jbc.M112.400002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin G, Brownsey RW, MacLeod KM. Regulation of mitochondrial aconitase by phosphorylation in diabetic rat heart. Cellular and Molecular Life Sciences. 2009;66:919–932. doi: 10.1007/s00018-009-8696-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ago T, Kuroda J, Pain J, Fu C, Li H, Sadoshima J. Upregulation of nox4 by hypertrophic stimuli promotes apoptosis and mitochondrial dysfunction in cardiac myocytes. Circulation Research. 2010;106:1253–1264. doi: 10.1161/CIRCRESAHA.109.213116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bernard K, Logsdon NJ, Miguel V, Benavides GA, Zhang J, Carter AB, Darley-Usmar VM, Thannickal VJ. Nadph oxidase 4 (nox4) suppresses mitochondrial biogenesis and bioenergetics in lung fibroblasts via a nuclear factor erythroid-derived 2-like 2 (nrf2)-dependent pathway. Journal of Biological Chemistry. 2017 doi: 10.1074/jbc.M116.752261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shanmugasundaram K, Nayak BK, Friedrichs WE, Kaushik D, Rodriguez R, Block K. Nox4 functions as a mitochondrial energetic sensor coupling cancer metabolic reprogramming to drug resistance. Nature Communications. 2017;8:997. doi: 10.1038/s41467-017-01106-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marí M, Morales A, Colell A, García-Ruiz C, Fernández-Checa JC. Mitochondrial glutathione, a key survival antioxidant. Antioxidants & Redox Signaling. 2009;11:2685–2700. doi: 10.1089/ars.2009.2695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fukai T, Ushio-Fukai M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxidants & Redox Signaling. 2011;15:1583–1606. doi: 10.1089/ars.2011.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ribas V, García-Ruiz C, Fernández-Checa JC. Glutathione and mitochondria. Frontiers in Pharmacology. 2014;5:151. doi: 10.3389/fphar.2014.00151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang H, Go Y-M, Jones DP. Mitochondrial thioredoxin-2/peroxiredoxin-3 system functions in parallel with mitochondrial gsh system in protection against oxidative stress. Archives of Biochemistry and Biophysics. 2007;465:119–126. doi: 10.1016/j.abb.2007.05.001. [DOI] [PubMed] [Google Scholar]
- 48.Pérez VI, Lew CM, Cortez LA, Webb CR, Rodriguez M, Liu Y, Qi W, Li Y, Chaudhuri A, Van Remmen H, Richardson A, Ikeno Y. Thioredoxin 2 haploinsufficiency in mice results in impaired mitochondrial function and increased oxidative stress. Free Radical Biology and Medicine. 2008;44:882–892. doi: 10.1016/j.freeradbiomed.2007.11.018. [DOI] [PubMed] [Google Scholar]
- 49.Kiermayer C, Northrup E, Schrewe A, Walch A, de Angelis MH, Schoensiegel F, Zischka H, Prehn C, Adamski J, Bekeredjian R, Ivandic B, Kupatt C, Brielmeier M. Heart-specific knockout of the mitochondrial thioredoxin reductase (txnrd2) induces metabolic and contractile dysfunction in the aging myocardium. Journal of the American Heart Association: Cardiovascular and Cerebrovascular Disease. 2015;4:e002153. doi: 10.1161/JAHA.115.002153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rodríguez-Manzaneque MT, Tamarit J, Bellí G, Ros J, Herrero E. Grx5 is a mitochondrial glutaredoxin required for the activity of iron/sulfur enzymes. Molecular Biology of the Cell. 2002;13:1109–1121. doi: 10.1091/mbc.01-10-0517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Berndt C, Lillig CH, Holmgren A. Thiol-based mechanisms of the thioredoxin and glutaredoxin systems: Implications for diseases in the cardiovascular system. American Journal of Physiology - Heart and Circulatory Physiology. 2007;292:H1227–H1236. doi: 10.1152/ajpheart.01162.2006. [DOI] [PubMed] [Google Scholar]
- 52.Beer SM, Taylor ER, Brown SE, Dahm CC, Costa NJ, Runswick MJ, Murphy MP. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: Implications for mitochondrial redox regulation and antioxidant defense. Journal of Biological Chemistry. 2004;279:47939–47951. doi: 10.1074/jbc.M408011200. [DOI] [PubMed] [Google Scholar]
- 53.Martínez-Reyes I, Diebold LP, Kong H, Schieber M, Huang H, Hensley CT, Mehta MM, Wang T, Santos JH, Woychik R, Dufour E, Spelbrink JN, Weinberg SE, Zhao Y, DeBerardinis RJ, Chandel NS. Tca cycle and mitochondrial membrane potential are necessary for diverse biological functions. Molecular cell. 2016;61:199–209. doi: 10.1016/j.molcel.2015.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Solaini G, Baracca A, Lenaz G, Sgarbi G. Hypoxia and mitochondrial oxidative metabolism. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2010;1797:1171–1177. doi: 10.1016/j.bbabio.2010.02.011. [DOI] [PubMed] [Google Scholar]
- 55.Kim J-w, Tchernyshyov I, Semenza GL, Dang CV. Hif-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metabolism. 2006;3:177–185. doi: 10.1016/j.cmet.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 56.Lum JJ, Bui T, Gruber M, Gordan JD, DeBerardinis RJ, Covello KL, Simon MC, Thompson CB. The transcription factor hif-1α plays a critical role in the growth factor-dependent regulation of both aerobic and anaerobic glycolysis. Genes & Development. 2007;21:1037–1049. doi: 10.1101/gad.1529107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Semenza GL. Hypoxia-inducible factor 1: Regulator of mitochondrial metabolism and mediator of ischemic preconditioning. Biochimica et biophysica acta. 2011;1813:1263–1268. doi: 10.1016/j.bbamcr.2010.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, Gonzalez FJ, Semenza GL. Mitochondrial autophagy is an hif-1-dependent adaptive metabolic response to hypoxia. The Journal of Biological Chemistry. 2008;283:10892–10903. doi: 10.1074/jbc.M800102200. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 59.Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Mitochondrial electron-transport-chain inhibitors of complexes i and ii induce autophagic cell death mediated by reactive oxygen species. Journal of Cell Science. 2007;120:4155–4166. doi: 10.1242/jcs.011163. [DOI] [PubMed] [Google Scholar]
- 60.Sinha RA, Singh BK, Zhou J, Wu Y, Farah BL, Ohba K, Lesmana R, Gooding J, Bay B-H, Yen PM. Thyroid hormone induction of mitochondrial activity is coupled to mitophagy via ros-ampk-ulk1 signaling. Autophagy. 2015;11:1341–1357. doi: 10.1080/15548627.2015.1061849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Scherz-Shouval R, Elazar Z. Ros, mitochondria and the regulation of autophagy. Trends Cell Biol. 2007;17:422–427. doi: 10.1016/j.tcb.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 62.Hwang AB, Ryu E-A, Artan M, Chang H-W, Kabir MH, Nam H-J, Lee D, Yang J-S, Kim S, Mair WB, Lee C, Lee SS, Lee S-J. Feedback regulation via ampk and hif-1 mediates ros-dependent longevity in caenorhabditis elegans. Proceedings of the National Academy of Sciences of the United States of America. 2014;111:E4458–E4467. doi: 10.1073/pnas.1411199111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lapointe J, Hekimi S. Early mitochondrial dysfunction in long-lived mclk1(+/−) mice. The Journal of Biological Chemistry. 2008;283:26217–26227. doi: 10.1074/jbc.M803287200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang D, Malo D, Hekimi S. Elevated mitochondrial reactive oxygen species generation affects the immune response via hypoxia-inducible factor-1α in long-lived mclk1+/− mouse mutants. The Journal of Immunology. 2010;184:582–590. doi: 10.4049/jimmunol.0902352. [DOI] [PubMed] [Google Scholar]
- 65.van der Bliek AM, Shen Q, Kawajiri S. Mechanisms of mitochondrial fission and fusion. Cold Spring Harbor Perspectives in Biology. 2013;5 doi: 10.1101/cshperspect.a011072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wai T, Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab. 2016;27:105–117. doi: 10.1016/j.tem.2015.12.001. [DOI] [PubMed] [Google Scholar]
- 67.Yu T, Robotham JL, Yoon Y. Increased production of reactive oxygen species in hyperglycemic conditions requires dynamic change of mitochondrial morphology. Proc Natl Acad Sci U S A. 2006;103:2653–2658. doi: 10.1073/pnas.0511154103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Watanabe T, Saotome M, Nobuhara M, Sakamoto A, Urushida T, Katoh H, Satoh H, Funaki M, Hayashi H. Roles of mitochondrial fragmentation and reactive oxygen species in mitochondrial dysfunction and myocardial insulin resistance. Exp Cell Res. 2014;323:314–325. doi: 10.1016/j.yexcr.2014.02.027. [DOI] [PubMed] [Google Scholar]
- 69.Hong Z, Kutty S, Toth PT, Marsboom G, Hammel JM, Chamberlain C, Ryan JJ, Zhang HJ, Sharp WW, Morrow E, Trivedi K, Weir EK, Archer SL. Role of dynamin related protein 1 (drp1)-mediated mitochondrial fission in oxygen-sensing and constriction of the ductus arteriosus. Circulation research. 2013;112:802–815. doi: 10.1161/CIRCRESAHA.111.300285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wu S, Zhou F, Zhang Z, Xing D. Mitochondrial oxidative stress causes mitochondrial fragmentation via differential modulation of mitochondrial fission–fusion proteins. FEBS Journal. 2011;278:941–954. doi: 10.1111/j.1742-4658.2011.08010.x. [DOI] [PubMed] [Google Scholar]
- 71.Frank M, Duvezin-Caubet S, Koob S, Occhipinti A, Jagasia R, Petcherski A, Ruonala MO, Priault M, Salin B, Reichert AS. Mitophagy is triggered by mild oxidative stress in a mitochondrial fission dependent manner. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research. 2012;1823:2297–2310. doi: 10.1016/j.bbamcr.2012.08.007. [DOI] [PubMed] [Google Scholar]
- 72.Kurihara Y, Kanki T, Aoki Y, Hirota Y, Saigusa T, Uchiumi T, Kang D. Mitophagy plays an essential role in reducing mitochondrial production of reactive oxygen species and mutation of mitochondrial DNA by maintaining mitochondrial quantity and quality in yeast. Journal of Biological Chemistry. 2012;287:3265–3272. doi: 10.1074/jbc.M111.280156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Giedt RJ, Yang C, Zweier JL, Matzavinos A, Alevriadou BR. Mitochondrial fission in endothelial cells after simulated ischemia/reperfusion: Role of nitric oxide and reactive oxygen species. Free Radic Biol Med. 2012;52:348–356. doi: 10.1016/j.freeradbiomed.2011.10.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lim S, Lee S-Y, Seo H-H, Ham O, Lee C, Park J-H, Lee J, Seung M, Yun I, Han SM, Lee S, Choi E, Hwang K-C. Regulation of mitochondrial morphology by positive feedback interaction between pkcδ and drp1 in vascular smooth muscle cell. Journal of Cellular Biochemistry. 2015;116:648–660. doi: 10.1002/jcb.25016. [DOI] [PubMed] [Google Scholar]
- 75.Kim J-E, Kang T-C. P47phox/cdk5/drp1-mediated mitochondrial fission evokes pv cell degeneration in the rat dentate gyrus following status epilepticus. Frontiers in Cellular Neuroscience. 2017;11:267. doi: 10.3389/fncel.2017.00267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Norton M, Ng AC-H, Baird S, Dumoulin A, Shutt T, Mah N, Andrade-Navarro MA, McBride HM, Screaton RA. Romo1 is an essential redox-dependent regulator of mitochondrial dynamics. Science Signaling. 2014;7:ra10–ra10. doi: 10.1126/scisignal.2004374. [DOI] [PubMed] [Google Scholar]
- 77.Tang S, Le PK, Tse S, Wallace DC, Huang T. Heterozygous mutation of opa1 in drosophila shortens lifespan mediated through increased reactive oxygen species production. PLOS ONE. 2009;4:e4492. doi: 10.1371/journal.pone.0004492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shen T, Zheng M, Cao C, Chen C, Tang J, Zhang W, Cheng H, Chen K-H, Xiao R-P. Mitofusin-2 is a major determinant of oxidative stress-mediated heart muscle cell apoptosis. Journal of Biological Chemistry. 2007;282:23354–23361. doi: 10.1074/jbc.M702657200. [DOI] [PubMed] [Google Scholar]
- 79.Papanicolaou KN, Ngoh GA, Dabkowski ER, O’Connell KA, Ribeiro RF, Stanley WC, Walsh K. Cardiomyocyte deletion of mitofusin-1 leads to mitochondrial fragmentation and improves tolerance to ros-induced mitochondrial dysfunction and cell death. American Journal of Physiology - Heart and Circulatory Physiology. 2012;302:H167–H179. doi: 10.1152/ajpheart.00833.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Zorzano A, Hernández-Alvarez MI, Sebastián D, Muñoz JP. Mitofusin 2 as a driver that controls energy metabolism and insulin signaling. Antioxidants & Redox Signaling. 2015;22:1020–1031. doi: 10.1089/ars.2014.6208. [DOI] [PubMed] [Google Scholar]
- 81.Wanders RJA, Waterham HR. Biochemistry of mammalian peroxisomes revisited. Annual Review of Biochemistry. 2006;75:295–332. doi: 10.1146/annurev.biochem.74.082803.133329. [DOI] [PubMed] [Google Scholar]
- 82.Bonekamp NA, Völkl A, Fahimi HD, Schrader M. Reactive oxygen species and peroxisomes: Struggling for balance. BioFactors. 2009;35:346–355. doi: 10.1002/biof.48. [DOI] [PubMed] [Google Scholar]
- 83.Fransen M, Nordgren M, Wang B, Apanasets O. Role of peroxisomes in ros/rns-metabolism: Implications for human disease. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2012;1822:1363–1373. doi: 10.1016/j.bbadis.2011.12.001. [DOI] [PubMed] [Google Scholar]
- 84.Loughran P, Stolz D, Vodovotz Y, Watkins S, Simmons R, Billiar T. Monomeric inducible nitric oxide synthase localizes to peroxisomes in hepatocytes. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:13837–13842. doi: 10.1073/pnas.0503926102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schrader M, Fahimi HD. Peroxisomes and oxidative stress. Biochim Biophys Acta. 2006;1763:1755–1766. doi: 10.1016/j.bbamcr.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 86.Zhang J, Kim J, Alexander A, Cai S, Tripathi DN, Dere R, Tee AR, Tait-Mulder J, Di Nardo A, Han JM, Kwiatkowski E, Dunlop EA, Dodd KM, Folkerth RD, Faust PL, Kastan MB, Sahin M, Walker CL. A tsc signaling node at the peroxisome regulates mtorc1 and autophagy in response to ros. Nature cell biology. 2013;15:1186–1196. doi: 10.1038/ncb2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang J, Tripathi DN, Jing J, Alexander A, Kim J, Powell RT, Dere R, Tait-Mulder J, Lee J-H, Paull TT, Pandita RK, Charaka VK, Pandita TK, Kastan MB, Walker CL. Atm functions at the peroxisome to induce pexophagy in response to ros. Nat Cell Biol. 2015;17:1259–1269. doi: 10.1038/ncb3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ivashchenko O, Van Veldhoven PP, Brees C, Ho Y-S, Terlecky SR, Fransen M. Intraperoxisomal redox balance in mammalian cells: Oxidative stress and interorganellar cross-talk. Molecular Biology of the Cell. 2011;22:1440–1451. doi: 10.1091/mbc.E10-11-0919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang B, Van Veldhoven PP, Brees C, Rubio N, Nordgren M, Apanasets O, Kunze M, Baes M, Agostinis P, Fransen M. Mitochondria are targets for peroxisome-derived oxidative stress in cultured mammalian cells. Free Radical Biology and Medicine. 2013;65:882–894. doi: 10.1016/j.freeradbiomed.2013.08.173. [DOI] [PubMed] [Google Scholar]
- 90.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Minamino T, Komuro I, Kitakaze M. Endoplasmic reticulum stress as a therapeutic target in cardiovascular disease. Circulation Research. 2010;107:1071–1082. doi: 10.1161/CIRCRESAHA.110.227819. [DOI] [PubMed] [Google Scholar]
- 92.Cao SS, Kaufman RJ. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxidants & Redox Signaling. 2014;21:396–413. doi: 10.1089/ars.2014.5851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Tavender TJ, Springate JJ, Bulleid NJ. Recycling of peroxiredoxin iv provides a novel pathway for disulphide formation in the endoplasmic reticulum. The EMBO Journal. 2010;29:4185–4197. doi: 10.1038/emboj.2010.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Espinosa-Diez C, Miguel V, Mennerich D, Kietzmann T, Sánchez P, Cadenas S, Lamas S. Antioxidant responses and cellular adjustments to oxidative stress. 2015 doi: 10.1016/j.redox.2015.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hansen HG, Schmidt JD, Søltoft CL, Ramming T, Geertz-Hansen HM, Christensen B, Sørensen ES, Juncker AS, Appenzeller-Herzog C, Ellgaard L. Hyperactivity of the ero1α oxidase elicits endoplasmic reticulum stress but no broad antioxidant response. Journal of Biological Chemistry. 2012;287:39513–39523. doi: 10.1074/jbc.M112.405050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Malhotra JD, Miao H, Zhang K, Wolfson A, Pennathur S, Pipe SW, Kaufman RJ. Antioxidants reduce endoplasmic reticulum stress and improve protein secretion. Proc Natl Acad Sci U S A. 2008;105:18525–18530. doi: 10.1073/pnas.0809677105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zangar RC, Davydov DR, Verma S. Mechanisms that regulate production of reactive oxygen species by cytochrome p450. Toxicol Appl Pharmacol. 2004;199:316–331. doi: 10.1016/j.taap.2004.01.018. [DOI] [PubMed] [Google Scholar]
- 98.Samhan-Arias AK, Gutierrez-Merino C. Purified nadh-cytochrome b5 reductase is a novel superoxide anion source inhibited by apocynin: Sensitivity to nitric oxide and peroxynitrite. Free Radic Biol Med. 2014;73:174–189. doi: 10.1016/j.freeradbiomed.2014.04.033. [DOI] [PubMed] [Google Scholar]
- 99.Wu R-F, Ma Z, Liu Z, Terada LS. Nox4-derived h2o2 mediates endoplasmic reticulum signaling through local ras activation. Molecular and Cellular Biology. 2010;30:3553–3568. doi: 10.1128/MCB.01445-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Delaunay-Moisan A, Appenzeller-Herzog C. The antioxidant machinery of the endoplasmic reticulum: Protection and signaling. Free Radic Biol Med. 2015;83:341–351. doi: 10.1016/j.freeradbiomed.2015.02.019. [DOI] [PubMed] [Google Scholar]
- 101.Wei P-C, Hsieh Y-H, Su M-I, Jiang XJ, Hsu P-H, Lo W-T, Jeng Y-M, Wang J-M, Chen P-l, Chang Y-C, Lee K-F, Tsai M-D, Shew J-Y, Lee W-H. Loss of the oxidative stress sensor npgpx compromises grp78 chaperone activity and induces systemic disease. Molecular cell. 2012;48:747–759. doi: 10.1016/j.molcel.2012.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Gorlach A, Bertram K, Hudecova S, Krizanova O. Calcium and ros: A mutual interplay. Redox Biol. 2015;6:260–271. doi: 10.1016/j.redox.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Anelli T, Bergamelli L, Margittai E, Rimessi A, Fagioli C, Malgaroli A, Pinton P, Ripamonti M, Rizzuto R, Sitia R. Ero1α regulates ca2+ fluxes at the endoplasmic reticulum–mitochondria interface (mam) Antioxidants & Redox Signaling. 2011;16:1077–1087. doi: 10.1089/ars.2011.4004. [DOI] [PubMed] [Google Scholar]
- 104.Migliaccio E, Giorgio M, Mele S, Pelicci G, Reboldi P, Pandolfi PP, Lanfrancone L, Pelicci PG. The p66shc adaptor protein controls oxidative stress response and life span in mammals. Nature. 1999;402:309–313. doi: 10.1038/46311. [DOI] [PubMed] [Google Scholar]
- 105.Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, Pinton P, Rizzuto R, Bernardi P, Paolucci F, Pelicci PG. Electron transfer between cytochrome c and p66shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 122:221–233. doi: 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 106.Granatiero V, Gherardi G, Vianello M, Salerno E, Zecchini E, Toniolo L, Pallafacchina G, Murgia M, Blaauw B, Rizzuto R, Mammucari C. Role of p66shc in skeletal muscle function. Scientific Reports. 2017;7:6283. doi: 10.1038/s41598-017-06363-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Soliman MA, Abdel Rahman AM, Lamming DA, Birsoy K, Pawling J, Frigolet ME, Lu H, Fantus IG, Pasculescu A, Zheng Y, Sabatini DM, Dennis JW, Pawson T. The adaptor protein p66shc inhibits mtor-dependent anabolic metabolism. Science signaling. 2014;7:ra17–ra17. doi: 10.1126/scisignal.2004785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Baker RG, Hayden MS, Ghosh S. Nf-κb, inflammation and metabolic disease. Cell metabolism. 2011;13:11–22. doi: 10.1016/j.cmet.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Arkan MC, Hevener AL, Greten FR, Maeda S, Li Z-W, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, Karin M. Ikk-[beta] links inflammation to obesity-induced insulin resistance. Nat Med. 2005;11:191–198. doi: 10.1038/nm1185. [DOI] [PubMed] [Google Scholar]
- 110.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. Journal of Clinical Investigation. 2003;112:1821–1830. doi: 10.1172/JCI19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Mauro C, Leow SC, Anso E, Rocha S, Thotakura AK, Tornatore L, Moretti M, De Smaele E, Beg AA, Tergaonkar V, Chandel NS, Franzoso G. Nf-[kappa]b controls energy homeostasis and metabolic adaptation by upregulating mitochondrial respiration. Nat Cell Biol. 2011;13:1272–1279. doi: 10.1038/ncb2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kabe Y, Ando K, Hirao S, Yoshida M, Handa H. Redox regulation of nf-κb activation: Distinct redox regulation between the cytoplasm and the nucleus. Antioxidants & Redox Signaling. 2005;7:395–403. doi: 10.1089/ars.2005.7.395. [DOI] [PubMed] [Google Scholar]
- 113.Marui N, Offermann MK, Swerlick R, Kunsch C, Rosen CA, Ahmad M, Alexander RW, Medford RM. Vascular cell adhesion molecule-1 (vcam-1) gene transcription and expression are regulated through an antioxidant-sensitive mechanism in human vascular endothelial cells. The Journal of Clinical Investigation. 1993;92:1866–1874. doi: 10.1172/JCI116778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Canty TG, Boyle EM, Farr A, Morgan EN, Verrier ED, Pohlman TH. Oxidative stress induces nf-κb nuclear translocation without degradation of iκbα. Circulation. 1999;100:II-361–Ii-364. doi: 10.1161/01.cir.100.suppl_2.ii-361. [DOI] [PubMed] [Google Scholar]
- 115.Takada Y, Mukhopadhyay A, Kundu GC, Mahabeleshwar GH, Singh S, Aggarwal BB. Hydrogen peroxide activates nf-κb through tyrosine phosphorylation of iκbα and serine phosphorylation of p65: Evidence for the involvement of iκbα kinase and syk protein-tyrosine kinase. Journal of Biological Chemistry. 2003;278:24233–24241. doi: 10.1074/jbc.M212389200. [DOI] [PubMed] [Google Scholar]
- 116.Schoonbroodt S, Ferreira V, Best-Belpomme M, Boelaert JR, Legrand-Poels S, Korner M, Piette J. Crucial role of the amino-terminal tyrosine residue 42 and the carboxyl-terminal pest domain of iκbα in nf-κb activation by an oxidative stress. The Journal of Immunology. 2000;164:4292–4300. doi: 10.4049/jimmunol.164.8.4292. [DOI] [PubMed] [Google Scholar]
- 117.Chen C-J, Fu Y-C, Yu W, Wang W. Sirt3 protects cardiomyocytes from oxidative stress-mediated cell death by activating nf-κb. Biochemical and Biophysical Research Communications. 2013;430:798–803. doi: 10.1016/j.bbrc.2012.11.066. [DOI] [PubMed] [Google Scholar]
- 118.Johar S, Cave AC, Narayanapanicker A, Grieve DJ, Shah AM. Aldosterone mediates angiotensin ii-induced interstitial cardiac fibrosis via a nox2-containing nadph oxidase. The FASEB Journal. 2006;20:1546–1548. doi: 10.1096/fj.05-4642fje. [DOI] [PubMed] [Google Scholar]
- 119.Anrather J, Racchumi G, Iadecola C. Nf-κb regulates phagocytic nadph oxidase by inducing the expression of gp91phox. Journal of Biological Chemistry. 2006;281:5657–5667. doi: 10.1074/jbc.M506172200. [DOI] [PubMed] [Google Scholar]
- 120.Gauss KA, Nelson-Overton LK, Siemsen DW, Gao Y, DeLeo FR, Quinn MT. Role of nf-κb in transcriptional regulation of the phagocyte nadph oxidase by tumor necrosis factor-α. Journal of Leukocyte Biology. 2007;82:729–741. doi: 10.1189/jlb.1206735. [DOI] [PubMed] [Google Scholar]
- 121.Li Q, Harraz MM, Zhou W, Zhang LN, Ding W, Zhang Y, Eggleston T, Yeaman C, Banfi B, Engelhardt JF. Nox2 and rac1 regulate h(2)o(2)-dependent recruitment of traf6 to endosomal interleukin-1 receptor complexes. Molecular and Cellular Biology. 2006;26:140–154. doi: 10.1128/MCB.26.1.140-154.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Park HS, Jung HY, Park EY, Kim J, Lee WJ, Bae YS. Cutting edge: Direct interaction of tlr4 with nad(p)h oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of nf-κb. The Journal of Immunology. 2004;173:3589–3593. doi: 10.4049/jimmunol.173.6.3589. [DOI] [PubMed] [Google Scholar]
- 123.Gloire G, Legrand-Poels S, Piette J. Nf-kappab activation by reactive oxygen species: Fifteen years later. Biochem Pharmacol. 2006;72:1493–1505. doi: 10.1016/j.bcp.2006.04.011. [DOI] [PubMed] [Google Scholar]
- 124.Hwang J, Saha A, Boo YC, Sorescu GP, McNally JS, Holland SM, Dikalov S, Giddens DP, Griendling KK, Harrison DG, Jo H. Oscillatory shear stress stimulates endothelial production of from p47phox-dependent nad(p)h oxidases, leading to monocyte adhesion. Journal of Biological Chemistry. 2003;278:47291–47298. doi: 10.1074/jbc.M305150200. [DOI] [PubMed] [Google Scholar]
- 125.Mohan S, Koyoma K, Thangasamy A, Nakano H, Glickman RD, Mohan N. Low shear stress preferentially enhances ikk activity through selective sources of ros for persistent activation of nf-κb in endothelial cells. American Journal of Physiology - Cell Physiology. 2007;292:C362–C371. doi: 10.1152/ajpcell.00535.2005. [DOI] [PubMed] [Google Scholar]
- 126.Sorescu GP, Song H, Tressel SL, Hwang J, Dikalov S, Smith DA, Boyd NL, Platt MO, Lassègue B, Griendling KK, Jo H. Bone morphogenic protein 4 produced in endothelial cells by oscillatory shear stress induces monocyte adhesion by stimulating reactive oxygen species production from a nox1-based nadph oxidase. Circulation Research. 2004;95:773–779. doi: 10.1161/01.RES.0000145728.22878.45. [DOI] [PubMed] [Google Scholar]
- 127.Orr AW, Hahn C, Blackman BR, Schwartz MA. Pak signaling regulates oxidant-dependent nf-κb activation by flow. Circulation research. 2008;103:671–679. doi: 10.1161/CIRCRESAHA.108.182097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hawkins BJ, Solt LA, Chowdhury I, Kazi AS, Abid MR, Aird WC, May MJ, Foskett JK, Madesh M. G protein-coupled receptor ca(2+)-linked mitochondrial reactive oxygen species are essential for endothelial/leukocyte adherence. Molecular and Cellular Biology. 2007;27:7582–7593. doi: 10.1128/MCB.00493-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Li X, Fang P, Li Y, Kuo Y-M, Andrews AJ, Nanayakkara G, Johnson C, Fu H, Shan H, Du F, Hoffman NE, Yu D, Eguchi S, Madesh M, Koch WJ, Sun J, Jiang X, Wang H, Yang X. Mitochondrial reactive oxygen species mediate lysophosphatidylcholine-induced endothelial cell activation. Arteriosclerosis, thrombosis, and vascular biology. 2016;36:1090–1100. doi: 10.1161/ATVBAHA.115.306964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ungvari Z, Orosz Z, Labinskyy N, Rivera A, Xiangmin Z, Smith K, Csiszar A. Increased mitochondrial h2o2 production promotes endothelial nf-κb activation in aged rat arteries. American Journal of Physiology - Heart and Circulatory Physiology. 2007;293:H37–H47. doi: 10.1152/ajpheart.01346.2006. [DOI] [PubMed] [Google Scholar]
- 131.Pearlstein DP, Ali MH, Mungai PT, Hynes KL, Gewertz BL, Schumacker PT. Role of mitochondrial oxidant generation in endothelial cell responses to hypoxia. Arteriosclerosis, Thrombosis, and Vascular Biology. 2002;22:566–573. doi: 10.1161/01.atv.0000012262.76205.6a. [DOI] [PubMed] [Google Scholar]
- 132.Zhang C, Jiang H, Wang P, Liu H, Sun X. Transcription factor nf-kappa b represses ant1 transcription and leads to mitochondrial dysfunctions. Scientific Reports. 2017;7:44708. doi: 10.1038/srep44708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mills EL, Kelly B, Logan A, Costa AS, Varma M, Bryant CE, Tourlomousis P, Dabritz JH, Gottlieb E, Latorre I, Corr SC, McManus G, Ryan D, Jacobs HT, Szibor M, Xavier RJ, Braun T, Frezza C, Murphy MP, O’Neill LA. Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell. 2016;167:457–470.e413. doi: 10.1016/j.cell.2016.08.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Tannahill GM, Curtis AM, Adamik J, Palsson-McDermott EM, McGettrick AF, Goel G, Frezza C, Bernard NJ, Kelly B, Foley NH, Zheng L, Gardet A, Tong Z, Jany SS, Corr SC, Haneklaus M, Caffrey BE, Pierce K, Walmsley S, Beasley FC, Cummins E, Nizet V, Whyte M, Taylor CT, Lin H, Masters SL, Gottlieb E, Kelly VP, Clish C, Auron PE, Xavier RJ, O/’Neill LAJ. Succinate is an inflammatory signal that induces il-1[bgr] through hif-1[agr] Nature. 2013;496:238–242. doi: 10.1038/nature11986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Cramer T, Yamanishi Y, Clausen BE, Förster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, Firestein GS, Gerber H-P, Ferrara N, Johnson RS. Hif-1α is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.He Y, Hara H, Núñez G. Mechanism and regulation of nlrp3 inflammasome activation. Trends in Biochemical Sciences. 41:1012–1021. doi: 10.1016/j.tibs.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K(+) efflux is the common trigger of nlrp3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38:1142–1153. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Heid ME, Keyel PA, Kamga C, Shiva S, Watkins SC, Salter RD. Mitochondrial ros induces nlrp3-dependent lysosomal damage and inflammasome activation() Journal of immunology (Baltimore, Md: 1950) 2013;191 doi: 10.4049/jimmunol.1301490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in nlrp3 inflammasome activation. Nature. 2011;469:221–225. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 140.Jo E-K, Kim JK, Shin D-M, Sasakawa C. Molecular mechanisms regulating nlrp3 inflammasome activation. Cellular and Molecular Immunology. 2016;13:148–159. doi: 10.1038/cmi.2015.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Netea MG, Simon A, van de Veerdonk F, Kullberg B-J, Van der Meer JWM, Joosten LAB. Il-1β processing in host defense: Beyond the inflammasomes. PLOS Pathogens. 2010;6:e1000661. doi: 10.1371/journal.ppat.1000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhou R, Tardivel A, Thorens B, Choi I, Tschopp J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol. 2010;11:136–140. doi: 10.1038/ni.1831. [DOI] [PubMed] [Google Scholar]
- 143.Shah A, Xia L, Goldberg H, Lee KW, Quaggin SE, Fantus IG. Thioredoxin-interacting protein mediates high glucose-induced reactive oxygen species generation by mitochondria and the nadph oxidase, nox4, in mesangial cells. Journal of Biological Chemistry. 2013;288:6835–6848. doi: 10.1074/jbc.M112.419101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Lerner AG, Upton J-P, Praveen PVK, Ghosh R, Nakagawa Y, Igbaria A, Shen S, Nguyen V, Backes BJ, Heiman M, Heintz N, Greengard P, Hui S, Tang Q, Trusina A, Oakes SA, Papa FR. Ire1α induces thioredoxin-interacting protein to activate the nlrp3 inflammasome and promote programmed cell death during endoplasmic reticulum stress. Cell metabolism. 2012;16:250–264. doi: 10.1016/j.cmet.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bronner DN, Abuaita BH, Chen X, Fitzgerald KA, Nuñez G, He Y, Yin X-M, O’Riordan MXD. Endoplasmic reticulum stress activates the inflammasome via nlrp3-caspase-2 driven mitochondrial damage. Immunity. 2015;43:451–462. doi: 10.1016/j.immuni.2015.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Forman HJ, Torres M. Reactive oxygen species and cell signaling: Respiratory burst in macrophage signaling. Am J Respir Crit Care Med. 2002;166:S4–8. doi: 10.1164/rccm.2206007. [DOI] [PubMed] [Google Scholar]
- 147.Kigawa Y, Miyazaki T, Lei XF, Nakamachi T, Oguchi T, Kim-Kaneyama JR, Taniyama M, Tsunawaki S, Shioda S, Miyazaki A. Nadph oxidase deficiency exacerbates angiotensin ii-induced abdominal aortic aneurysms in mice. Arterioscler Thromb Vasc Biol. 2014;34:2413–2420. doi: 10.1161/ATVBAHA.114.303086. [DOI] [PubMed] [Google Scholar]
- 148.Segal BH, Han W, Bushey JJ, Joo M, Bhatti Z, Feminella J, Dennis CG, Vethanayagam RR, Yull FE, Capitano M, Wallace PK, Minderman H, Christman JW, Sporn MB, Chan J, Vinh DC, Holland SM, Romani LR, Gaffen SL, Freeman ML, Blackwell TS. Nadph oxidase limits innate immune responses in the lungs in mice. PLoS One. 2010;5:e9631. doi: 10.1371/journal.pone.0009631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Martinez J, Malireddi RKS, Lu Q, Cunha LD, Pelletier S, Gingras S, Orchard R, Guan J-L, Tan H, Peng J, Kanneganti T-D, Virgin HW, Green DR. Molecular characterization of lc3-associated phagocytosis (lap) reveals distinct roles for rubicon, nox2, and autophagy proteins. Nature cell biology. 2015;17:893–906. doi: 10.1038/ncb3192. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 150.West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, Ghosh S. Tlr signalling augments macrophage bactericidal activity through mitochondrial ros. Nature. 2011;472:476–480. doi: 10.1038/nature09973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Wang Y, Subramanian M, Yurdagul A, Jr, Barbosa-Lorenzi VC, Cai B, de Juan-Sanz J, Ryan TA, Nomura M, Maxfield FR, Tabas I. Mitochondrial fission promotes the continued clearance of apoptotic cells by macrophages. Cell. doi: 10.1016/j.cell.2017.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Röth D, Krammer PH, Gülow K. Dynamin related protein 1-dependent mitochondrial fission regulates oxidative signalling in t cells. FEBS Letters. 2014;588:1749–1754. doi: 10.1016/j.febslet.2014.03.029. [DOI] [PubMed] [Google Scholar]
- 153.Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang C-H, Sanin DE, Qiu J, Kretz O, Braas D, van der Windt GJW, Chen Q, Ching-Cheng Huang S, O’Neill CM, Edelson BT, Pearce EJ, Sesaki H, Huber TB, Rambold AS, Pearce EL. Mitochondrial dynamics controls t cell fate through metabolic programming. Cell. 2016;166:63–76. doi: 10.1016/j.cell.2016.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sather S, Kenyon KD, Lefkowitz JB, Liang X, Varnum BC, Henson PM, Graham DK. A soluble form of the mer receptor tyrosine kinase inhibits macrophage clearance of apoptotic cells and platelet aggregation. Blood. 2007;109:1026–1033. doi: 10.1182/blood-2006-05-021634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Thorp E, Vaisar T, Subramanian M, Mautner L, Blobel C, Tabas I. Shedding of the mer tyrosine kinase receptor is mediated by adam17 protein through a pathway involving reactive oxygen species, protein kinase cδ, and p38 mitogen-activated protein kinase (mapk) The Journal of Biological Chemistry. 2011;286:33335–33344. doi: 10.1074/jbc.M111.263020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Jiang S, Park DW, Stigler WS, Creighton J, Ravi S, Darley-Usmar V, Zmijewski JW. Mitochondria and amp-activated protein kinase-dependent mechanism of efferocytosis. The Journal of Biological Chemistry. 2013;288:26013–26026. doi: 10.1074/jbc.M113.489468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.McPhillips K, Janssen WJ, Ghosh M, Byrne A, Gardai S, Remigio L, Bratton DL, Kang JL, Henson P. Tnf-α inhibits macrophage clearance of apoptotic cells via cytosolic phospholipase a<sub>2</sub> and oxidant-dependent mechanisms. The Journal of Immunology. 2007;178:8117–8126. doi: 10.4049/jimmunol.178.12.8117. [DOI] [PubMed] [Google Scholar]
- 158.Kumagai T, Matsukawa N, Kaneko Y, Kusumi Y, Mitsumata M, Uchida K. A lipid peroxidation-derived inflammatory mediator: Identification of 4-hydroxy-2-nonenal as a potential inducer of cyclooxygenase-2 in macrophages. Journal of Biological Chemistry. 2004;279:48389–48396. doi: 10.1074/jbc.M409935200. [DOI] [PubMed] [Google Scholar]
- 159.Yadav UCS, Ramana KV. Regulation of nf-b-induced inflammatory signaling by lipid peroxidation-derived aldehydes. Oxidative Medicine and Cellular Longevity. 2013;2013:11. doi: 10.1155/2013/690545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Furnkranz A, Leitinger N. Regulation of inflammatory responses by oxidized phospholipids: Structure-function relationships. Current Pharmaceutical Design. 2004;10:915–921. doi: 10.2174/1381612043452929. [DOI] [PubMed] [Google Scholar]
- 161.Zarkovic K, Jakovcevic A, Zarkovic N. Contribution of the hne-immunohistochemistry to modern pathological concepts of major human diseases. Free Radical Biology and Medicine. 2017;111:110–126. doi: 10.1016/j.freeradbiomed.2016.12.009. [DOI] [PubMed] [Google Scholar]
- 162.Berliner JA, Watson AD. A role for oxidized phospholipids in atherosclerosis. New England Journal of Medicine. 2005;353:9–11. doi: 10.1056/NEJMp058118. [DOI] [PubMed] [Google Scholar]
- 163.Parhami F, Fang ZT, Fogelman AM, Andalibi A, Territo MC, Berliner JA. Minimally modified low density lipoprotein-induced inflammatory responses in endothelial cells are mediated by cyclic adenosine monophosphate. Journal of Clinical Investigation. 1993;92:471–478. doi: 10.1172/JCI116590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.van der Valk FM, Bekkering S, Kroon J, Yeang C, Van den Bossche J, van Buul JD, Ravandi A, Nederveen AJ, Verberne HJ, Scipione C, Nieuwdorp M, Joosten LAB, Netea MG, Koschinsky ML, Witztum JL, Tsimikas S, Riksen NP, Stroes ESG. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation. 2016 doi: 10.1161/CIRCULATIONAHA.116.020838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ayala Antonio, Mu#x000F1;oz Mario F, Argüelles Sandro. Lipid peroxidation: Production, metabolism, and signaling mechanisms of malondialdehyde and 4-hydroxy-2-nonenal. Oxidative Medicine and Cellular Longevity. 2014;2014:31. doi: 10.1155/2014/360438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Ma Z, Li J, Yang L, Mu Y, Xie W, Pitt B, Li S. Inhibition of lps- and cpg DNA-induced tnf-α response by oxidized phospholipids. American Journal of Physiology-Lung Cellular and Molecular Physiology. 2004;286:L808–L816. doi: 10.1152/ajplung.00220.2003. [DOI] [PubMed] [Google Scholar]
- 167.Serbulea V, DeWeese D, Leitinger N. The effect of oxidized phospholipids on phenotypic polarization and function of macrophages. Free Radical Biology and Medicine. 2017;111:156–168. doi: 10.1016/j.freeradbiomed.2017.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Kuosmanen SM, Kansanen E, Kaikkonen MU, Sihvola V, Pulkkinen K, Jyrkkänen H-K, Tuoresmäki P, Hartikainen J, Hippeläinen M, Kokki H, Tavi P, Heikkinen S, Levonen AL. Nrf2 regulates endothelial glycolysis and proliferation with mir-93 and mediates the effects of oxidized phospholipids on endothelial activation. Nucleic Acids Research. 2017:gkx1155–gkx1155. doi: 10.1093/nar/gkx1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, Robbins J, Martinez J, Tabas I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metabolism. 2012;15:545–553. doi: 10.1016/j.cmet.2012.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Haberzettl P, Hill BG. Oxidized lipids activate autophagy in a jnk-dependent manner by stimulating the endoplasmic reticulum stress response. Redox Biology. 2013;1:56–64. doi: 10.1016/j.redox.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Anderson EJ, Katunga LA, Willis MS. Mitochondria as a source and target of lipid peroxidation products in healthy and diseased heart. Clinical and experimental pharmacology & physiology. 2012;39 doi: 10.1111/j.1440-1681201105641.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kaur J, Debnath J. Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol. 2015;16:461–472. doi: 10.1038/nrm4024. [DOI] [PubMed] [Google Scholar]
- 173.Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Bento CF, Renna M, Ghislat G, Puri C, Ashkenazi A, Vicinanza M, Menzies FM, Rubinsztein DC. Mammalian autophagy: How does it work? Annual Review of Biochemistry. 2016;85:685–713. doi: 10.1146/annurev-biochem-060815-014556. [DOI] [PubMed] [Google Scholar]
- 175.Scherz-Shouval R, Elazar Z. Regulation of autophagy by ros: Physiology and pathology. Trends Biochem Sci. 2011;36:30–38. doi: 10.1016/j.tibs.2010.07.007. [DOI] [PubMed] [Google Scholar]
- 176.Kiffin R, Christian C, Knecht E, Cuervo AM. Activation of chaperone-mediated autophagy during oxidative stress. Mol Biol Cell. 2004;15:4829–4840. doi: 10.1091/mbc.E04-06-0477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of atg4. Embo j. 2007;26:1749–1760. doi: 10.1038/sj.emboj.7601623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Gomez-Santos C, Ferrer I, Santidrian AF, Barrachina M, Gil J, Ambrosio S. Dopamine induces autophagic cell death and alpha-synuclein increase in human neuroblastoma sh-sy5y cells. J Neurosci Res. 2003;73:341–350. doi: 10.1002/jnr.10663. [DOI] [PubMed] [Google Scholar]
- 179.Kim EH, Sohn S, Kwon HJ, Kim SU, Kim MJ, Lee SJ, Choi KS. Sodium selenite induces superoxide-mediated mitochondrial damage and subsequent autophagic cell death in malignant glioma cells. Cancer Res. 2007;67:6314–6324. doi: 10.1158/0008-5472.CAN-06-4217. [DOI] [PubMed] [Google Scholar]
- 180.Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Mitochondrial electron-transport-chain inhibitors of complexes i and ii induce autophagic cell death mediated by reactive oxygen species. J Cell Sci. 2007;120:4155–4166. doi: 10.1242/jcs.011163. [DOI] [PubMed] [Google Scholar]
- 181.Djavaheri-Mergny M, Amelotti M, Mathieu J, Besançon F, Bauvy C, Souquère S, Pierron G, Codogno P. Nf-κb activation represses tumor necrosis factor-α-induced autophagy. Journal of Biological Chemistry. 2006;281:30373–30382. doi: 10.1074/jbc.M602097200. [DOI] [PubMed] [Google Scholar]
- 182.Yuan H, Perry CN, Huang C, Iwai-Kanai E, Carreira RS, Glembotski CC, Gottlieb RA. Lps-induced autophagy is mediated by oxidative signaling in cardiomyocytes and is associated with cytoprotection. American journal of physiology. Heart and circulatory physiology. 2009;296:H470–479. doi: 10.1152/ajpheart.01051.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Alexander A, Cai S-L, Kim J, Nanez A, Sahin M, MacLean KH, Inoki K, Guan K-L, Shen J, Person MD, Kusewitt D, Mills GB, Kastan MB, Walker CL. Atm signals to tsc2 in the cytoplasm to regulate mtorc1 in response to ros. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:4153–4158. doi: 10.1073/pnas.0913860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Hardie DG, Ross FA, Hawley SA. Ampk: A nutrient and energy sensor that maintains energy homeostasis. Nature Reviews Molecular Cell Biology. 2012;13:251–+. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chen Y, Azad MB, Gibson SB. Superoxide is the major reactive oxygen species regulating autophagy. Cell death and differentiation. 2009;16:1040–1052. doi: 10.1038/cdd.2009.49. [DOI] [PubMed] [Google Scholar]
- 186.Talbert EE, Smuder AJ, Min K, Kwon OS, Szeto HH, Powers SK. Immobilization-induced activation of key proteolytic systems in skeletal muscles is prevented by a mitochondria-targeted antioxidant. Journal of Applied Physiology. 2013;115:529. doi: 10.1152/japplphysiol.00471.2013. [DOI] [PubMed] [Google Scholar]
- 187.Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. Reactive oxygen species (ros)-induced ros release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 2000;192:1001–1014. doi: 10.1084/jem.192.7.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ros) and ros-induced ros release. Physiological Reviews. 2014;94:909–950. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Lemasters JJ. Selective mitochondrial autophagy, or mitophagy, as a targeted defense against oxidative stress, mitochondrial dysfunction, and aging. Rejuvenation research. 2005;8:3–5. doi: 10.1089/rej.2005.8.3. [DOI] [PubMed] [Google Scholar]
- 190.Xiao B, Goh J-Y, Xiao L, Xian H, Lim K-L, Liou YC. Reactive oxygen species trigger parkin/pink1 pathway-dependent mitophagy by inducing mitochondrial recruitment of parkin. Journal of Biological Chemistry. 2017 doi: 10.1074/jbc.M117.787739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Tanaka K. The proteasome: Overview of structure and functions. Proceedings of the Japan Academy. Series B, Physical and Biological Sciences. 2009;85:12–36. doi: 10.2183/pjab.85.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Vabulas RM, Hartl FU. Protein synthesis upon acute nutrient restriction relies on proteasome function. Science. 2005;310:1960. doi: 10.1126/science.1121925. [DOI] [PubMed] [Google Scholar]
- 193.Davies KJ. Degradation of oxidized proteins by the 20s proteasome. Biochimie. 2001;83:301–310. doi: 10.1016/s0300-9084(01)01250-0. [DOI] [PubMed] [Google Scholar]
- 194.Grune T, Reinheckel T, Davies KJ. Degradation of oxidized proteins in k562 human hematopoietic cells by proteasome. J Biol Chem. 1996;271:15504–15509. doi: 10.1074/jbc.271.26.15504. [DOI] [PubMed] [Google Scholar]
- 195.Shringarpure R, Grune T, Mehlhase J, Davies KJA. Ubiquitin conjugation is not required for the degradation of oxidized proteins by proteasome. Journal of Biological Chemistry. 2003;278:311–318. doi: 10.1074/jbc.M206279200. [DOI] [PubMed] [Google Scholar]
- 196.Kopp F, Hendil KB, Dahlmann B, Kristensen P, Sobek A, Uerkvitz W. Subunit arrangement in the human 20s proteasome. Proceedings of the National Academy of Sciences. 1997;94:2939–2944. doi: 10.1073/pnas.94.7.2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Lecker SH, Goldberg AL, Mitch WE. Protein degradation by the ubiquitin–proteasome pathway in normal and disease states. Journal of the American Society of Nephrology. 2006;17:1807–1819. doi: 10.1681/ASN.2006010083. [DOI] [PubMed] [Google Scholar]
- 198.Glickman MH, Ciechanover A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiological Reviews. 2002;82:373–428. doi: 10.1152/physrev.00027.2001. [DOI] [PubMed] [Google Scholar]
- 199.Rechsteiner M, Hill CP. Mobilizing the proteolytic machine: Cell biological roles of proteasome activators and inhibitors. Trends in Cell Biology. 15:27–33. doi: 10.1016/j.tcb.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 200.Wang X, Yen J, Kaiser P, Huang L. Regulation of the 26s proteasome complex during oxidative stress. Science signaling. 2010;3:ra88–ra88. doi: 10.1126/scisignal.2001232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Shang F, Taylor A. Oxidative stress and recovery from oxidative stress are associated with altered ubiquitin conjugating and proteolytic activities in bovine lens epithelial cells. Biochemical Journal. 1995;307:297–303. doi: 10.1042/bj3070297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.REINHECKEL T, SITTE N, ULLRICH O, KUCKELKORN U, DAVIES KJA, GRUNE T. Comparative resistance of the 20s and 26s proteasome to oxidative stress. Biochemical Journal. 1998;335:637–642. doi: 10.1042/bj3350637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Inai Y, Nishikimi M. Increased degradation of oxidized proteins in yeast defective in 26 s proteasome assembly. Arch Biochem Biophys. 2002;404:279–284. doi: 10.1016/s0003-9861(02)00336-3. [DOI] [PubMed] [Google Scholar]
- 204.Lee B-H, Lee MJ, Park S, Oh D-C, Elsasser S, Chen P-C, Gartner C, Dimova N, Hanna J, Gygi SP, Wilson SM, King RW, Finley D. Enhancement of proteasome activity by a small-molecule inhibitor of usp14. Nature. 2010;467:179–184. doi: 10.1038/nature09299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Zetterberg M, Zhang X, Taylor A, Liu B, Liang JJ, Shang F. Glutathiolation enhances the degradation of γc-crystallin in lens and reticulocyte lysates, partially via the ubiquitin–proteasome pathway. Investigative ophthalmology & visual science. 2006;47:3467–3473. doi: 10.1167/iovs.05-1664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Shang F, Gong X, Taylor A. Activity of ubiquitin-dependent pathway in response to oxidative stress: Ubiquitin-activating enzyme is transiently up-regulated. Journal of Biological Chemistry. 1997;272:23086–23093. doi: 10.1074/jbc.272.37.23086. [DOI] [PubMed] [Google Scholar]
- 207.Yamanaka K, Ishikawa H, Megumi Y, Tokunaga F, Kanie M, Rouault TA, Morishima I, Minato N, Ishimori K, Iwai K. Identification of the ubiquitin-protein ligase that recognizes oxidized irp2. Nat Cell Biol. 2003;5:336–340. doi: 10.1038/ncb952. [DOI] [PubMed] [Google Scholar]
- 208.Ishii T, Sakurai T, Usami H, Uchida K. Oxidative modification of proteasome: Identification of an oxidation-sensitive subunit in 26 s proteasome. Biochemistry. 2005;44:13893–13901. doi: 10.1021/bi051336u. [DOI] [PubMed] [Google Scholar]
- 209.Bulteau AL, Lundberg KC, Humphries KM, Sadek HA, Szweda PA, Friguet B, Szweda LI. Oxidative modification and inactivation of the proteasome during coronary occlusion/reperfusion. J Biol Chem. 2001;276:30057–30063. doi: 10.1074/jbc.M100142200. [DOI] [PubMed] [Google Scholar]
- 210.Demasi M, Shringarpure R, Davies KJ. Glutathiolation of the proteasome is enhanced by proteolytic inhibitors. Arch Biochem Biophys. 2001;389:254–263. doi: 10.1006/abbi.2001.2332. [DOI] [PubMed] [Google Scholar]
- 211.Ullrich O, Reinheckel T, Sitte N, Hass R, Grune T, Davies KJ. Poly-adp ribose polymerase activates nuclear proteasome to degrade oxidatively damaged histones. Proc Natl Acad Sci U S A. 1999;96:6223–6228. doi: 10.1073/pnas.96.11.6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Fujino G, Noguchi T, Matsuzawa A, Yamauchi S, Saitoh M, Takeda K, Ichijo H. Thioredoxin and traf family proteins regulate reactive oxygen species-dependent activation of ask1 through reciprocal modulation of the n-terminal homophilic interaction of ask1. Molecular and Cellular Biology. 2007;27:8152–8163. doi: 10.1128/MCB.00227-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Um JW, Im E, Park J, Oh Y, Min B, Lee HJ, Yoon JB, Chung KC. Ask1 negatively regulates the 26 s proteasome. J Biol Chem. 2010;285:36434–36446. doi: 10.1074/jbc.M110.133777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Noguchi T, Takeda K, Matsuzawa A, Saegusa K, Nakano H, Gohda J, Inoue J, Ichijo H. Recruitment of tumor necrosis factor receptor-associated factor family proteins to apoptosis signal-regulating kinase 1 signalosome is essential for oxidative stress-induced cell death. J Biol Chem. 2005;280:37033–37040. doi: 10.1074/jbc.M506771200. [DOI] [PubMed] [Google Scholar]
- 215.Pickering AM, Koop AL, Teoh CY, Ermak G, Grune T, Davies KJA. The immunoproteasome, the 20s proteasome, and the pa28αβ proteasome regulator are oxidative-stress-adaptive proteolytic complexes. The Biochemical journal. 2010;432:585–594. doi: 10.1042/BJ20100878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Ding Q, Reinacker K, Dimayuga E, Nukala V, Drake J, Butterfield DA, Dunn JC, Martin S, Bruce-Keller AJ, Keller JN. Role of the proteasome in protein oxidation and neural viability following low-level oxidative stress. FEBS Letters. 2003;546:228–232. doi: 10.1016/s0014-5793(03)00582-9. [DOI] [PubMed] [Google Scholar]
- 217.Nandi D, Jiang H, Monaco JJ. Identification of mecl-1 (lmp-10) as the third ifn-gamma-inducible proteasome subunit. J Immunol. 1996;156:2361–2364. [PubMed] [Google Scholar]
- 218.Tabas I, García-Cardeña G, Owens GK. Recent insights into the cellular biology of atherosclerosis. The Journal of Cell Biology. 2015;209:13–22. doi: 10.1083/jcb.201412052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Lusis AJ. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Isomaa B, Almgren P, Tuomi T, Forsen B, Lahti K, Nissen M, Taskinen MR, Groop L. Cardiovascular morbidity and mortality associated with the metabolic syndrome. Diabetes Care. 2001;24:683–689. doi: 10.2337/diacare.24.4.683. [DOI] [PubMed] [Google Scholar]
- 221.Steinberger J, Daniels SR. Obesity, insulin resistance, diabetes, and cardiovascular risk in children. An American Heart Association Scientific Statement From the Atherosclerosis, Hypertension, and Obesity in the Young Committee (Council on Cardiovascular Disease in the Young) and the Diabetes Committee (Council on Nutrition, Physical Activity, and Metabolism) 2003;107:1448–1453. doi: 10.1161/01.cir.0000060923.07573.f2. [DOI] [PubMed] [Google Scholar]
- 222.Steinberg D, Parthasarathy S, Carew TE, Khoo JC, Witztum JL. Beyond cholesterol. New England Journal of Medicine. 1989;320:915–924. doi: 10.1056/NEJM198904063201407. [DOI] [PubMed] [Google Scholar]
- 223.Kirk EA, Dinauer MC, Rosen H, Chait A, Heinecke JW, LeBoeuf RC. Impaired superoxide production due to a deficiency in phagocyte nadph oxidase fails to inhibit atherosclerosis in mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2000;20:1529–1535. doi: 10.1161/01.atv.20.6.1529. [DOI] [PubMed] [Google Scholar]
- 224.Hsich E, Segal BH, Pagano PJ, Rey FE, Paigen B, Deleonardis J, Hoyt RF, Holland SM, Finkel T. Vascular effects following homozygous disruption of p47phox. An Essential Component of NADPH Oxidase. 2000;101:1234–1236. doi: 10.1161/01.cir.101.11.1234. [DOI] [PubMed] [Google Scholar]
- 225.Barry-Lane PA, Patterson C, van der Merwe M, Hu Z, Holland SM, Yeh ETH, Runge MS. P47phox is required for atherosclerotic lesion progression in apoe(−/−) mice. Journal of Clinical Investigation. 2001;108:1513–1522. doi: 10.1172/JCI11927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Judkins CP, Diep H, Broughton BRS, Mast AE, Hooker EU, Miller AA, Selemidis S, Dusting GJ, Sobey CG, Drummond GR. Direct evidence of a role for nox2 in superoxide production, reduced nitric oxide bioavailability, and early atherosclerotic plaque formation in apoe−/− mice. American Journal of Physiology - Heart and Circulatory Physiology. 2010;298:H24–H32. doi: 10.1152/ajpheart.00799.2009. [DOI] [PubMed] [Google Scholar]
- 227.Di Marco E, Gray Stephen P, Chew P, Kennedy K, Cooper Mark E, Schmidt Harald HHW, Jandeleit-Dahm Karin AM. Differential effects of nox4 and nox1 on immune cell-mediated inflammation in the aortic sinus of diabetic apoe−/− mice. Clinical Science. 2016;130:1363–1374. doi: 10.1042/CS20160249. [DOI] [PubMed] [Google Scholar]
- 228.Sheehan AL, Carrell S, Johnson B, Stanic B, Banfi B, Miller FJ. Role for nox1 nadph oxidase in atherosclerosis. Atherosclerosis. 2011;216:321–326. doi: 10.1016/j.atherosclerosis.2011.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Gray SP, Di Marco E, Okabe J, Szyndralewiez C, Heitz F, Montezano AC, de Haan JB, Koulis C, El-Osta A, Andrews KL, Chin-Dusting JPF, Touyz RM, Wingler K, Cooper ME, Schmidt HHHW, Jandeleit-Dahm KA. Nadph oxidase 1 plays a key role in diabetes mellitus–accelerated atherosclerosis. Circulation. 2013;127:1888–1902. doi: 10.1161/CIRCULATIONAHA.112.132159. [DOI] [PubMed] [Google Scholar]
- 230.Gray SP, Di Marco E, Kennedy K, Chew P, Okabe J, El-Osta A, Calkin AC, Biessen EAL, Touyz RM, Cooper ME, Schmidt HHHW, Jandeleit-Dahm KAM. Reactive oxygen species can provide atheroprotection via nox4-dependent inhibition of inflammation and vascular remodeling. Arteriosclerosis, Thrombosis, and Vascular Biology. 2016;36:295–307. doi: 10.1161/ATVBAHA.115.307012. [DOI] [PubMed] [Google Scholar]
- 231.Lee MY, Martin AS, Mehta PK, Dikalova AE, Garrido AM, Datla SR, Lyons E, Krause K-H, Banfi B, Lambeth JD, Lassègue B, Griendling KK. Mechanisms of vascular smooth muscle nadph oxidase 1 (nox1) contribution to injury-induced neointimal formation. Arteriosclerosis, Thrombosis, and Vascular Biology. 2009;29:480–487. doi: 10.1161/ATVBAHA.108.181925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Streeter J, Schickling BM, Jiang S, Stanic B, Thiel WH, Gakhar L, Houtman JCD, Miller FJ. Phosphorylation of nox1 regulates association with noxa1 activation domain. Circulation research. 2014;115:911–918. doi: 10.1161/CIRCRESAHA.115.304267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Sobey CG, Judkins CP, Rivera J, Lewis CV, Diep H, Lee HW, Kemp-Harper BK, Broughton BRS, Selemidis S, Gaspari TA, Samuel CS, Drummond GR. Nox1 deficiency in apolipoprotein e-knockout mice is associated with elevated plasma lipids and enhanced atherosclerosis. Free Radical Research. 2015;49:186–198. doi: 10.3109/10715762.2014.992893. [DOI] [PubMed] [Google Scholar]
- 234.Quesada IM, Lucero A, Amaya C, Meijles DN, Cifuentes ME, Pagano PJ, Castro C. Selective inactivation of nadph oxidase 2 causes regression of vascularization and the size and stability of atherosclerotic plaques. Atherosclerosis. 2015;242:469–475. doi: 10.1016/j.atherosclerosis.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Douglas G, Bendall JK, Crabtree MJ, Tatham AL, Carter EE, Hale AB, Channon KM. Endothelial-specific nox2 overexpression increases vascular superoxide and macrophage recruitment in apoe(−/−) mice. Cardiovascular Research. 2012;94:20–29. doi: 10.1093/cvr/cvs026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Vendrov AE, Hakim ZS, Madamanchi NR, Rojas M, Madamanchi C, Runge MS. Atherosclerosis is attenuated by limiting superoxide generation in both macrophages and vessel wall cells. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27:2714–2721. doi: 10.1161/ATVBAHA.107.152629. [DOI] [PubMed] [Google Scholar]
- 237.Cominacini L, Pasini AF, Garbin U, Davoli A, Tosetti ML, Campagnola M, Rigoni A, Pastorino AM, Lo Cascio V, Sawamura T. Oxidized low density lipoprotein (ox-ldl) binding to ox-ldl receptor-1 in endothelial cells induces the activation of nf-κb through an increased production of intracellular reactive oxygen species. Journal of Biological Chemistry. 2000;275:12633–12638. doi: 10.1074/jbc.275.17.12633. [DOI] [PubMed] [Google Scholar]
- 238.Tsai K-L, Chen L-H, Chiou S-H, Chiou G-Y, Chen Y-C, Chou H-Y, Chen L-K, Chen H-Y, Chiu T-H, Tsai C-S, Ou H-C, Kao C-L. Coenzyme q10 suppresses oxldl-induced endothelial oxidative injuries by the modulation of lox-1-mediated ros generation via the ampk/pkc/nadph oxidase signaling pathway. Molecular Nutrition & Food Research. 2011;55:S227–S240. doi: 10.1002/mnfr.201100147. [DOI] [PubMed] [Google Scholar]
- 239.Bae YS, Lee JH, Choi SH, Kim S, Almazan F, Witztum JL, Miller YI. Macrophages generate reactive oxygen species in response to minimally oxidized ldl: Tlr4- and syk-dependent activation of nox2. Circulation research. 2009;104:210–218. doi: 10.1161/CIRCRESAHA.108.181040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Asmis R, Begley JG. Oxidized ldl promotes peroxide-mediated mitochondrial dysfunction and cell death in human macrophages. A Caspase-3–Independent Pathway. 2003;92:e20–e29. doi: 10.1161/01.res.0000051886.43510.90. [DOI] [PubMed] [Google Scholar]
- 241.Hsieh CC, Yen MH, Yen CH, Lau YT. Oxidized low density lipoprotein induces apoptosis via generation of reactive oxygen species in vascular smooth muscle cells. Cardiovasc Res. 2001;49:135–145. doi: 10.1016/s0008-6363(00)00218-2. [DOI] [PubMed] [Google Scholar]
- 242.Kruth HS, Jones NL, Huang W, Zhao B, Ishii I, Chang J, Combs CA, Malide D, Zhang W-Y. Macropinocytosis is the endocytic pathway that mediates macrophage foam cell formation with native low density lipoprotein. Journal of Biological Chemistry. 2005;280:2352–2360. doi: 10.1074/jbc.M407167200. [DOI] [PubMed] [Google Scholar]
- 243.Ghoshal P, Singla B, Lin H, Feck DM, Cantu-Medellin N, Kelley EE, Haigh S, Fulton D, Csányi G. Nox2-mediated pi3k and cofilin activation confers alternate redox control of macrophage pinocytosis. Antioxidants & Redox Signaling. 2016;26:902–916. doi: 10.1089/ars.2016.6639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Langbein H, Brunssen C, Hofmann A, Cimalla P, Brux M, Bornstein SR, Deussen A, Koch E, Morawietz H. Nadph oxidase 4 protects against development of endothelial dysfunction and atherosclerosis in ldl receptor deficient mice. European Heart Journal. 2016;37:1753–1761. doi: 10.1093/eurheartj/ehv564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Schürmann C, Rezende F, Kruse C, Yasar Y, Löwe O, Fork C, van de Sluis B, Bremer R, Weissmann N, Shah AM, Jo H, Brandes RP, Schröder K. The nadph oxidase nox4 has anti-atherosclerotic functions. European Heart Journal. 2015;36:3447–3456. doi: 10.1093/eurheartj/ehv460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Di Marco E, Gray SP, Kennedy K, Szyndralewiez C, Lyle AN, Lassègue B, Griendling KK, Cooper ME, Schmidt HHHW, Jandeleit-Dahm KAM. Nox4-derived reactive oxygen species limit fibrosis and inhibit proliferation of vascular smooth muscle cells in diabetic atherosclerosis. Free radical biology & medicine. 2016;97:556–567. doi: 10.1016/j.freeradbiomed.2016.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Hu P, Wu X, Khandelwal AR, Yu W, Xu Z, Chen L, Yang J, Weisbrod RM, Lee KSS, Seta F, Hammock BD, Cohen RA, Zeng C, Tong X. Endothelial nox4-based nadph oxidase regulates atherosclerosis via soluble epoxide hydrolase. Biochim Biophys Acta. 2017;1863:1382–1391. doi: 10.1016/j.bbadis.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Craige SM, Kant S, Reif M, Chen K, Pei Y, Angoff R, Sugamura K, Fitzgibbons T, Keaney JF. Endothelial nadph oxidase 4 protects apoe−/− mice from atherosclerotic lesions. Free Radical Biology and Medicine. 2015;89:1–7. doi: 10.1016/j.freeradbiomed.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Xu S, Chamseddine AH, Carrell S, Miller FJ. Nox4 nadph oxidase contributes to smooth muscle cell phenotypes associated with unstable atherosclerotic plaques. Redox Biology. 2014;2:642–650. doi: 10.1016/j.redox.2014.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Vendrov AE, Vendrov KC, Smith A, Yuan J, Sumida A, Robidoux J, Runge MS, Madamanchi NR. Nox4 nadph oxidase-dependent mitochondrial oxidative stress in aging-associated cardiovascular disease. Antioxidants & Redox Signaling. 2015;23:1389–1409. doi: 10.1089/ars.2014.6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Tong X, Khandelwal AR, Wu X, Xu Z, Yu W, Chen C, Zhao W, Yang J, Qin Z, Weisbrod RM, Seta F, Ago T, Lee KSS, Hammock BD, Sadoshima J, Cohen RA, Zeng C. Pro-atherogenic role of smooth muscle nox4-based nadph oxidase. Journal of molecular and cellular cardiology. 2016;92:30–40. doi: 10.1016/j.yjmcc.2016.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Pedruzzi E, Guichard C, Ollivier V, Driss F, Fay M, Prunet C, Marie J-C, Pouzet C, Samadi M, Elbim C, O’Dowd Y, Bens M, Vandewalle A, Gougerot-Pocidalo M-A, Lizard G, Ogier-Denis E. Nad(p)h oxidase nox-4 mediates 7-ketocholesterol-induced endoplasmic reticulum stress and apoptosis in human aortic smooth muscle cells. Molecular and Cellular Biology. 2004;24:10703–10717. doi: 10.1128/MCB.24.24.10703-10717.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Li G, Scull C, Ozcan L, Tabas I. Nadph oxidase links endoplasmic reticulum stress, oxidative stress, and pkr activation to induce apoptosis. The Journal of Cell Biology. 2010;191:1113–1125. doi: 10.1083/jcb.201006121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Thorp E, Li G, Seimon TA, Kuriakose G, Ron D, Tabas I. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of apoe−/− and ldlr−/− mice lacking chop. Cell metabolism. 2009;9:474–481. doi: 10.1016/j.cmet.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Zhou A-X, Wang X, Lin CS, Han J, Yong J, Nadolski MJ, Borén J, Kaufman RJ, Tabas I. C/ebp-homologous protein (chop) in vascular smooth muscle cells regulates their proliferation in aortic explants and atherosclerotic lesions. Circulation research. 2015;116:1736–1743. doi: 10.1161/CIRCRESAHA.116.305602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Matsui R, Xu S, Maitland KA, Mastroianni R, Leopold JA, Handy DE, Loscalzo J, Cohen RA. Glucose-6-phosphate dehydrogenase deficiency decreases vascular superoxide and atherosclerotic lesions in apolipoprotein e−/− mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2006;26:910–916. doi: 10.1161/01.ATV.0000205850.49390.3b. [DOI] [PubMed] [Google Scholar]
- 257.Leopold JA, Zhang Y-Y, Scribner AW, Stanton RC, Loscalzo J. Glucose-6-phosphate dehydrogenase overexpression decreases endothelial cell oxidant stress and increases bioavailable nitric oxide. Arteriosclerosis, Thrombosis, and Vascular Biology. 2003;23:411–417. doi: 10.1161/01.ATV.0000056744.26901.BA. [DOI] [PubMed] [Google Scholar]
- 258.Mercer JR, Yu E, Figg N, Cheng KK, Prime TA, Griffin JL, Masoodi M, Vidal-Puig A, Murphy MP, Bennett MR. The mitochondria-targeted antioxidant mitoq decreases features of the metabolic syndrome in atm+/−/apoe−/− mice. Free Radic Biol Med. 2012;52:841–849. doi: 10.1016/j.freeradbiomed.2011.11.026. [DOI] [PubMed] [Google Scholar]
- 259.Wang Y, Wang GZ, Rabinovitch PS, Tabas I. Macrophage mitochondrial oxidative stress promotes atherosclerosis and nf-κb-mediated inflammation in macrophages. Circulation research. 2014;114:421–433. doi: 10.1161/CIRCRESAHA.114.302153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Wang Y, Wang W, Wang N, Tall AR, Tabas I. Mitochondrial oxidative stress promotes atherosclerosis and neutrophil extracellular traps in aged mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2017;37:e99–e107. doi: 10.1161/ATVBAHA.117.309580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Wang Q, Zhang M, Torres G, Wu S, Ouyang C, Xie Z, Zou M-H. Metformin suppresses diabetes-accelerated atherosclerosis via the inhibition of drp1-mediated mitochondrial fission. Diabetes. 2017;66:193–205. doi: 10.2337/db16-0915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Mailloux RJ, Harper M-E. Uncoupling proteins and the control of mitochondrial reactive oxygen species production. Free Radical Biology and Medicine. 2011;51:1106–1115. doi: 10.1016/j.freeradbiomed.2011.06.022. [DOI] [PubMed] [Google Scholar]
- 263.Moukdar F, Robidoux J, Lyght O, Pi J, Daniel KW, Collins S. Reduced antioxidant capacity and diet-induced atherosclerosis in uncoupling protein-2-deficient mice. Journal of Lipid Research. 2009;50:59–70. doi: 10.1194/jlr.M800273-JLR200. [DOI] [PubMed] [Google Scholar]
- 264.Blanc J, Alves-Guerra MC, Esposito B, Rousset S, Gourdy P, Ricquier D, Tedgui A, Miroux B, Mallat Z. Protective role of uncoupling protein 2 in atherosclerosis. Circulation. 2003;107:388–390. doi: 10.1161/01.cir.0000051722.66074.60. [DOI] [PubMed] [Google Scholar]
- 265.Nomura J, Busso N, Ives A, Matsui C, Tsujimoto S, Shirakura T, Tamura M, Kobayashi T, So A, Yamanaka Y. Xanthine oxidase inhibition by febuxostat attenuates experimental atherosclerosis in mice. Scientific Reports. 2014;4:4554. doi: 10.1038/srep04554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Schroder K, Vecchione C, Jung O, Schreiber JG, Shiri-Sverdlov R, van Gorp PJ, Busse R, Brandes RP. Xanthine oxidase inhibitor tungsten prevents the development of atherosclerosis in apoe knockout mice fed a western-type diet. Free Radic Biol Med. 2006;41:1353–1360. doi: 10.1016/j.freeradbiomed.2006.03.026. [DOI] [PubMed] [Google Scholar]
- 267.Yang H, Roberts LJ, Shi MJ, Zhou LC, Ballard BR, Richardson A, Guo ZM. Retardation of atherosclerosis by overexpression of catalase or both cu/zn-superoxide dismutase and catalase in mice lacking apolipoprotein e. Circulation Research. 2004;95:1075–1081. doi: 10.1161/01.RES.0000149564.49410.0d. [DOI] [PubMed] [Google Scholar]
- 268.Guo X, Yamada S, Tanimoto A, Ding Y, Wang K-Y, Shimajiri S, Murata Y, Kimura S, Tasaki T, Nabeshima A, Watanabe T, Kohno K, Sasaguri Y. Overexpression of peroxiredoxin 4 attenuates atherosclerosis in apolipoprotein e knockout mice. Antioxidants & Redox Signaling. 2012;17:1362–1375. doi: 10.1089/ars.2012.4549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Zhang H, Luo Y, Zhang W, He Y, Dai S, Zhang R, Huang Y, Bernatchez P, Giordano FJ, Shadel G, Sessa WC, Min W. Endothelial-specific expression of mitochondrial thioredoxin improves endothelial cell function and reduces atherosclerotic lesions. The American Journal of Pathology. 2007;170:1108–1120. doi: 10.2353/ajpath.2007.060960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Barajas B, Che N, Yin F, Rowshanrad A, Orozco LD, Gong KW, Wang X, Castellani LW, Reue K, Lusis AJ, Araujo JA. Nf-e2–related factor 2 promotes atherosclerosis by effects on plasma lipoproteins and cholesterol transport that overshadow antioxidant protection. Arteriosclerosis, thrombosis, and vascular biology. 2011;31:58–66. doi: 10.1161/ATVBAHA.110.210906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Torzewski M, Ochsenhirt V, Kleschyov AL, Oelze M, Daiber A, Li H, Rossmann H, Tsimikas S, Reifenberg K, Cheng F, Lehr H-A, Blankenberg S, Förstermann U, Münzel T, Lackner KJ. Deficiency of glutathione peroxidase-1 accelerates the progression of atherosclerosis in apolipoprotein e-deficient mice. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27:850–857. doi: 10.1161/01.ATV.0000258809.47285.07. [DOI] [PubMed] [Google Scholar]
- 272.Chew P, Yuen DYC, Stefanovic N, Pete J, Coughlan MT, Jandeleit-Dahm KA, Thomas MC, Rosenfeldt F, Cooper ME, de Haan JB. Antiatherosclerotic and renoprotective effects of ebselen in the diabetic apolipoprotein e/gpx1-double knockout mouse. Diabetes. 2010;59:3198–3207. doi: 10.2337/db10-0195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Ballinger SW, Patterson C, Knight-Lozano CA, Burow DL, Conklin CA, Hu Z, Reuf J, Horaist C, Lebovitz R, Hunter GC, McIntyre K, Runge MS. Mitochondrial integrity and function in atherogenesis. Circulation. 2002;106:544–549. doi: 10.1161/01.cir.0000023921.93743.89. [DOI] [PubMed] [Google Scholar]
- 274.Kisucka J, Chauhan AK, Patten IS, Yesilaltay A, Neumann C, Van Etten RA, Krieger M, Wagner DD. Peroxiredoxin1 prevents excessive endothelial activation and early atherosclerosis. Circulation research. 2008;103:598–605. doi: 10.1161/CIRCRESAHA.108.174870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Park J-G, Yoo J-Y, Jeong S-J, Choi J-H, Lee M-R, Lee M-N, Lee JH, Kim HC, Jo H, Yu D-Y, Kang SW, Rhee SG, Lee M-H, Oh GT. Peroxiredoxin 2 deficiency exacerbates atherosclerosis in apolipoprotein e–deficient mice. Circulation research. 2011;109:739–749. doi: 10.1161/CIRCRESAHA.111.245530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Durrington PN, Mackness B, Mackness MI. Paraoxonase and atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology. 2001;21:473–480. doi: 10.1161/01.atv.21.4.473. [DOI] [PubMed] [Google Scholar]
- 277.Rosenblat M, Volkova N, Ward J, Aviram M. Paraoxonase 1 (pon1) inhibits monocyte-to-macrophage differentiation. Atherosclerosis. 219:49–56. doi: 10.1016/j.atherosclerosis.2011.06.054. [DOI] [PubMed] [Google Scholar]
- 278.She Z-G, Zheng W, Wei Y-S, Chen H-Z, Wang A-B, Li H-L, Liu G, Zhang R, Liu J-J, Stallcup WB, Zhou Z, Liu D-P, Liang C-C. Human paraoxonase gene cluster transgenic overexpression represses atherogenesis and promotes atherosclerotic plaque stability in apoe-null mice. Circulation Research. 2009;104:1160–1168. doi: 10.1161/CIRCRESAHA.108.192229. [DOI] [PubMed] [Google Scholar]
- 279.Aharoni S, Aviram M, Fuhrman B. Paraoxonase 1 (pon1) reduces macrophage inflammatory responses. Atherosclerosis. 2013;228:353–361. doi: 10.1016/j.atherosclerosis.2013.03.005. [DOI] [PubMed] [Google Scholar]
- 280.Ng DS, Chu T, Esposito B, Hui P, Connelly PW, Gross PL. Paraoxonase-1 deficiency in mice predisposes to vascular inflammation, oxidative stress, and thrombogenicity in the absence of hyperlipidemia. Cardiovascular Pathology. 17:226–232. doi: 10.1016/j.carpath.2007.10.001. [DOI] [PubMed] [Google Scholar]
- 281.Devarajan A, Bourquard N, Hama S, Navab M, Grijalva VR, Morvardi S, Clarke CF, Vergnes L, Reue K, Teiber JF, Reddy ST. Paraoxonase 2 deficiency alters mitochondrial function and exacerbates the development of atherosclerosis. Antioxidants & Redox Signaling. 2011;14:341–351. doi: 10.1089/ars.2010.3430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Ng CJ, Bourquard N, Grijalva V, Hama S, Shih DM, Navab M, Fogelman AM, Lusis AJ, Young S, Reddy ST. Paraoxonase-2 deficiency aggravates atherosclerosis in mice despite lower apolipoprotein-b-containing lipoproteins: Anti-atherogenic role for paraoxonase-2. J Biol Chem. 2006;281:29491–29500. doi: 10.1074/jbc.M605379200. [DOI] [PubMed] [Google Scholar]
- 283.Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444:875–880. doi: 10.1038/nature05487. [DOI] [PubMed] [Google Scholar]
- 284.Tabit CE, Chung WB, Hamburg NM, Vita JA. Endothelial dysfunction in diabetes mellitus: Molecular mechanisms and clinical implications. Reviews in Endocrine and Metabolic Disorders. 2010;11:61–74. doi: 10.1007/s11154-010-9134-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. The Journal of Clinical Investigation. 2017;114:1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Robertson RP. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. Journal of Biological Chemistry. 2004;279:42351–42354. doi: 10.1074/jbc.R400019200. [DOI] [PubMed] [Google Scholar]
- 287.Mohanty P, Hamouda W, Garg R, Aljada A, Ghanim H, Dandona P. Glucose challenge stimulates reactive oxygen species (ros) generation by leucocytes. The Journal of Clinical Endocrinology & Metabolism. 2000;85:2970–2973. doi: 10.1210/jcem.85.8.6854. [DOI] [PubMed] [Google Scholar]
- 288.Inoguchi T, Li P, Umeda F, Yu HY, Kakimoto M, Imamura M, Aoki T, Etoh T, Hashimoto T, Naruse M, Sano H, Utsumi H, Nawata H. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase c-dependent activation of nad(p)h oxidase in cultured vascular cells. Diabetes. 2000;49:1939–1945. doi: 10.2337/diabetes.49.11.1939. [DOI] [PubMed] [Google Scholar]
- 289.Cosentino F, Hishikawa K, Katusic ZS, Lüscher TF. High glucose increases nitric oxide synthase expression and superoxide anion generation in human aortic endothelial cells. Circulation. 1997;96:25–28. doi: 10.1161/01.cir.96.1.25. [DOI] [PubMed] [Google Scholar]
- 290.Asahina T, Kashiwagi A, Nishio Y, Ikebuchi M, Harada N, Tanaka Y, Takagi Y, Saeki Y, Kikkawa R, Shigeta Y. Impaired activation of glucose oxidation and nadph supply in human endothelial cells exposed to h2o2 in high-glucose medium. Diabetes. 1995;44:520–526. doi: 10.2337/diab.44.5.520. [DOI] [PubMed] [Google Scholar]
- 291.Paneni F, Mocharla P, Akhmedov A, Costantino S, Osto E, Volpe M, Lüscher TF, Cosentino F. Gene silencing of the mitochondrial adaptor p66<sup>shc</sup> suppresses vascular hyperglycemic memory in diabetes. Circulation Research. 2012;111:278–289. doi: 10.1161/CIRCRESAHA.112.266593. [DOI] [PubMed] [Google Scholar]
- 292.Xu J, Wu Y, Song P, Zhang M, Wang S, Zou M-H. Proteasome-dependent degradation of guanosine 5′-triphosphate cyclohydrolase i causes tetrahydrobiopterin deficiency in diabetes mellitus. Circulation. 2007;116:944–953. doi: 10.1161/CIRCULATIONAHA.106.684795. [DOI] [PubMed] [Google Scholar]
- 293.Kumar S, Kim Y-R, Vikram A, Naqvi A, Li Q, Kassan M, Kumar V, Bachschmid MM, Jacobs JS, Kumar A, Irani K. Sirtuin1-regulated lysine acetylation of p66shc governs diabetes-induced vascular oxidative stress and endothelial dysfunction. Proceedings of the National Academy of Sciences of the United States of America. 2017;114:1714–1719. doi: 10.1073/pnas.1614112114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Lu T, Chai Q, Yu L, d’Uscio LV, Katusic ZS, He T, Lee H-C. Reactive oxygen species signaling facilitates foxo-3a/fbxo-dependent vascular bk channel β(1) subunit degradation in diabetic mice. Diabetes. 2012;61:1860–1868. doi: 10.2337/db11-1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Martín AS, Du P, Dikalova A, Lassègue B, Aleman M, Góngora MC, Brown K, Joseph G, Harrison DG, Taylor WR, Jo H, Griendling KK. Reactive oxygen species-selective regulation of aortic inflammatory gene expression in type 2 diabetes. American Journal of Physiology - Heart and Circulatory Physiology. 2007;292:H2073–H2082. doi: 10.1152/ajpheart.00943.2006. [DOI] [PubMed] [Google Scholar]
- 296.Du J, Fan LM, Mai A, Li J-M. Crucial roles of nox2-derived oxidative stress in deteriorating the function of insulin receptors and endothelium in dietary obesity of middle-aged mice. British Journal of Pharmacology. 2013;170:1064–1077. doi: 10.1111/bph.12336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Thompson JA, Larion S, Mintz JD, Belin de Chantemèle EJ, Fulton DJ, Stepp DW. Genetic deletion of nadph oxidase 1 rescues microvascular function in mice with metabolic diseasenovelty and significance. Circulation Research. 2017;121:502–511. doi: 10.1161/CIRCRESAHA.116.309965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Kassan M, Choi S-K, Galán M, Lee Y-H, Trebak M, Matrougui K. Enhanced p22(phox) expression impairs vascular function through p38 and erk1/2 map kinase-dependent mechanisms in type 2 diabetic mice. American Journal of Physiology - Heart and Circulatory Physiology. 2014;306:H972–H980. doi: 10.1152/ajpheart.00872.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Sukumar P, Viswambharan H, Imrie H, Cubbon RM, Yuldasheva N, Gage M, Galloway S, Skromna A, Kandavelu P, Santos CX, Gatenby VK, Smith J, Beech DJ, Wheatcroft SB, Channon KM, Shah AM, Kearney MT. Nox2 nadph oxidase has a critical role in insulin resistance–related endothelial cell dysfunction. Diabetes. 2013;62:2130–2134. doi: 10.2337/db12-1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Gao X, Belmadani S, Picchi A, Xu X, Potter BJ, Tewari-Singh N, Capobianco S, Chilian WM, Zhang C. Tumor necrosis factor-α induces endothelial dysfunction in lepr(db) mice. Circulation. 2007;115:245–254. doi: 10.1161/CIRCULATIONAHA.106.650671. [DOI] [PubMed] [Google Scholar]
- 301.Liu S, Ma X, Gong M, Shi L, Lincoln T, Wang S. Glucose down-regulation of cgmp-dependent protein kinase i expression in vascular smooth muscle cells involves nad(p)h oxidase-derived reactive oxygen species. Free Radical Biology and Medicine. 2007;42:852–863. doi: 10.1016/j.freeradbiomed.2006.12.025. [DOI] [PubMed] [Google Scholar]
- 302.Xi G, Shen X, Maile LA, Wai C, Gollahon K, Clemmons DR. Hyperglycemia enhances igf-i–stimulated src activation via increasing nox4-derived reactive oxygen species in a pkcζ-dependent manner in vascular smooth muscle cells. Diabetes. 2012;61:104–113. doi: 10.2337/db11-0990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Xi G, Shen X-C, Wai C, Clemmons DR. Recruitment of nox4 to a plasma membrane scaffold is required for localized reactive oxygen species generation and sustained src activation in response to insulin-like growth factor-i. The Journal of Biological Chemistry. 2013;288:15641–15653. doi: 10.1074/jbc.M113.456046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Ebrahimian TG, Heymes C, You D, Blanc-Brude O, Mees B, Waeckel L, Duriez M, Vilar J, Brandes RP, Levy BI, Shah AM, Silvestre J-S. Nadph oxidase-derived overproduction of reactive oxygen species impairs postischemic neovascularization in mice with type 1 diabetes. The American Journal of Pathology. 2006;169:719–728. doi: 10.2353/ajpath.2006.060042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Sorrentino SA, Bahlmann FH, Besler C, Müller M, Schulz S, Kirchhoff N, Doerries C, Horváth T, Limbourg A, Limbourg F, Fliser D, Haller H, Drexler H, Landmesser U. Oxidant stress impairs in vivo reendothelialization capacity of endothelial progenitor cells from patients with type 2 diabetes mellitus. Restoration by the Peroxisome Proliferator-Activated Receptor-γ Agonist Rosiglitazone. 2007;116:163–173. doi: 10.1161/CIRCULATIONAHA.106.684381. [DOI] [PubMed] [Google Scholar]
- 306.Singh VP, Bali A, Singh N, Jaggi AS. Advanced glycation end products and diabetic complications. The Korean Journal of Physiology & Pharmacology : Official Journal of the Korean Physiological Society and the Korean Society of Pharmacology. 2014;18:1–14. doi: 10.4196/kjpp.2014.18.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Yao D, Brownlee M. Hyperglycemia-induced reactive oxygen species increase expression of the receptor for advanced glycation end products (rage) and rage ligands. Diabetes. 2010;59:249–255. doi: 10.2337/db09-0801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Wautier M-P, Chappey O, Corda S, Stern DM, Schmidt AM, Wautier J-L. Activation of nadph oxidase by age links oxidant stress to altered gene expression via rage. American Journal of Physiology - Endocrinology And Metabolism. 2001;280:E685–E694. doi: 10.1152/ajpendo.2001.280.5.E685. [DOI] [PubMed] [Google Scholar]
- 309.Ren X, Ren L, Wei Q, Shao H, Chen L, Liu N. Advanced glycation end-products decreases expression of endothelial nitric oxide synthase through oxidative stress in human coronary artery endothelial cells. Cardiovascular Diabetology. 2017;16:52. doi: 10.1186/s12933-017-0531-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Koike S, Yano S, Tanaka S, Sheikh AM, Nagai A, Sugimoto T. Advanced glycation end-products induce apoptosis of vascular smooth muscle cells: A mechanism for vascular calcification. International Journal of Molecular Sciences. 2016;17:1567. doi: 10.3390/ijms17091567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Du X, Matsumura T, Edelstein D, Rossetti L, Zsengellér Z, Szabó C, Brownlee M. Inhibition of gapdh activity by poly(adp-ribose) polymerase activates three major pathways of hyperglycemic damage in endothelial cells. Journal of Clinical Investigation. 2003;112:1049–1057. doi: 10.1172/JCI18127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Makino A, Scott BT, Dillmann WH. Mitochondrial fragmentation and superoxide anion production in coronary endothelial cells from a mouse model of type 1 diabetes. Diabetologia. 2010;53:1783–1794. doi: 10.1007/s00125-010-1770-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Yu T, Sheu S-S, Robotham JL, Yoon Y. Mitochondrial fission mediates high glucose-induced cell death through elevated production of reactive oxygen species. Cardiovascular research. 2008;79:341–351. doi: 10.1093/cvr/cvn104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Yu T, Jhun BS, Yoon Y. High-glucose stimulation increases reactive oxygen species production through the calcium and mitogen-activated protein kinase-mediated activation of mitochondrial fission. Antioxidants & Redox Signaling. 2010;14:425–437. doi: 10.1089/ars.2010.3284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Shenouda SM, Widlansky ME, Chen K, Xu G, Holbrook M, Tabit CE, Hamburg NM, Frame AA, Caiano TL, Kluge MA, Duess M-A, Levit A, Kim B, Hartman M-L, Joseph L, Shirihai OS, Vita JA. Altered mitochondrial dynamics contributes to endothelial dysfunction in diabetes mellitus. Circulation. 2011;124:444–453. doi: 10.1161/CIRCULATIONAHA.110.014506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Maimaitijiang A, Zhuang X, Jiang X, Li Y. Dynamin-related protein inhibitor downregulates reactive oxygen species levels to indirectly suppress high glucose-induced hyperproliferation of vascular smooth muscle cells. Biochemical and Biophysical Research Communications. 2016;471:474–478. doi: 10.1016/j.bbrc.2016.02.051. [DOI] [PubMed] [Google Scholar]
- 317.Tanner MJ, Wang J, Ying R, Suboc TB, Malik M, Couillard A, Branum A, Puppala V, Widlansky ME. Dynamin-related protein 1 mediates low glucose-induced endothelial dysfunction in human arterioles. American Journal of Physiology - Heart and Circulatory Physiology. 2017;312:H515–H527. doi: 10.1152/ajpheart.00499.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Li Y, Zhou Z-H, Chen M-H, Yang J, Leng J, Cao G-S, Xin G-Z, Liu L-F, Kou J-P, Liu B-L, Li P, Wen X-D. Inhibition of mitochondrial fission and nox2 expression prevent nlrp3 inflammasome activation in the endothelium: The role of corosolic acid action in the amelioration of endothelial dysfunction. Antioxidants & Redox Signaling. 2016;24:893–908. doi: 10.1089/ars.2015.6479. [DOI] [PubMed] [Google Scholar]
- 319.Writing Group M, Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER, 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB, American Heart Association Statistics C, Stroke Statistics S Heart disease and stroke statistics-2016 update: A report from the american heart association. Circulation. 2016;133:e38–360. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- 320.O’Donnell MJ, Xavier D, Liu L, Zhang H, Chin SL, Rao-Melacini P, Rangarajan S, Islam S, Pais P, McQueen MJ, Mondo C, Damasceno A, Lopez-Jaramillo P, Hankey GJ, Dans AL, Yusoff K, Truelsen T, Diener HC, Sacco RL, Ryglewicz D, Czlonkowska A, Weimar C, Wang X, Yusuf S, investigators I Risk factors for ischaemic and intracerebral haemorrhagic stroke in 22 countries (the interstroke study): A case-control study. Lancet. 2010;376:112–123. doi: 10.1016/S0140-6736(10)60834-3. [DOI] [PubMed] [Google Scholar]
- 321.Deb P, Sharma S, Hassan KM. Pathophysiologic mechanisms of acute ischemic stroke: An overview with emphasis on therapeutic significance beyond thrombolysis. Pathophysiology. 17:197–218. doi: 10.1016/j.pathophys.2009.12.001. [DOI] [PubMed] [Google Scholar]
- 322.Cojocaru IM, Cojocaru M, Sapira V, Ionescu A. Evaluation of oxidative stress in patients with acute ischemic stroke. Rom J Intern Med. 2013;51:97–106. [PubMed] [Google Scholar]
- 323.Domínguez C, Delgado P, Vilches A, Martín-Gallán P, Ribó M, Santamarina E, Molina C, Corbeto N, Rodríguez-Sureda V, Rosell A, Alvarez-Sabín J, Montaner J. Oxidative stress after thrombolysis-induced reperfusion in human stroke. Stroke. 2010;41:653–660. doi: 10.1161/STROKEAHA.109.571935. [DOI] [PubMed] [Google Scholar]
- 324.Kleinschnitz C, Grund H, Wingler K, Armitage ME, Jones E, Mittal M, Barit D, Schwarz T, Geis C, Kraft P, Barthel K, Schuhmann MK, Herrmann AM, Meuth SG, Stoll G, Meurer S, Schrewe A, Becker L, Gailus-Durner V, Fuchs H, Klopstock T, de Angelis MH, Jandeleit-Dahm K, Shah AM, Weissmann N, Schmidt HH. Post-stroke inhibition of induced nadph oxidase type 4 prevents oxidative stress and neurodegeneration. PLoS Biol. 2010;8 doi: 10.1371/journal.pbio.1000479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Yang G, Chan PH, Chen J, Carlson E, Chen SF, Weinstein P, Epstein CJ, Kamii H. Human copper-zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke. 1994;25:165–170. doi: 10.1161/01.str.25.1.165. [DOI] [PubMed] [Google Scholar]
- 326.Sanderson TH, Reynolds CA, Kumar R, Przyklenk K, Huttemann M. Molecular mechanisms of ischemia-reperfusion injury in brain: Pivotal role of the mitochondrial membrane potential in reactive oxygen species generation. Mol Neurobiol. 2013;47:9–23. doi: 10.1007/s12035-012-8344-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Kim GW, Kondo T, Noshita N, Chan PH. Manganese superoxide dismutase deficiency exacerbates cerebral infarction after focal cerebral ischemia/reperfusion in mice: Implications for the production and role of superoxide radicals. Stroke. 2002;33:809–815. doi: 10.1161/hs0302.103745. [DOI] [PubMed] [Google Scholar]
- 328.Fujimura M, Morita-Fujimura Y, Kawase M, Copin J-C, Calagui B, Epstein CJ, Chan PH. Manganese superoxide dismutase mediates the early release of mitochondrial cytochrome c and subsequent DNA fragmentation after permanent focal cerebral ischemia in mice. The Journal of Neuroscience. 1999;19:3414–3422. doi: 10.1523/JNEUROSCI.19-09-03414.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Hernansanz-Agustin P, Ramos E, Navarro E, Parada E, Sanchez-Lopez N, Pelaez-Aguado L, Cabrera-Garcia JD, Tello D, Buendia I, Marina A, Egea J, Lopez MG, Bogdanova A, Martinez-Ruiz A. Mitochondrial complex i deactivation is related to superoxide production in acute hypoxia. Redox Biol. 2017;12:1040–1051. doi: 10.1016/j.redox.2017.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Niatsetskaya ZV, Sosunov SA, Matsiukevich D, Utkina-Sosunova IV, Ratner VI, Starkov AA, Ten VS. The oxygen free radicals originating from mitochondrial complex i contribute to oxidative brain injury following hypoxia-ischemia in neonatal mice. J Neurosci. 2012;32:3235–3244. doi: 10.1523/JNEUROSCI.6303-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Kalogeris T, Bao Y, Korthuis RJ. Mitochondrial reactive oxygen species: A double edged sword in ischemia/reperfusion vs preconditioning. Redox Biol. 2014;2:702–714. doi: 10.1016/j.redox.2014.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Fitzgerald JC, Ugun-Klusek A, Allen G, De Girolamo LA, Hargreaves I, Ufer C, Abramov AY, Billett EE. Monoamine oxidase-a knockdown in human neuroblastoma cells reveals protection against mitochondrial toxins. FASEB J. 2014;28:218–229. doi: 10.1096/fj.13-235481. [DOI] [PubMed] [Google Scholar]
- 333.Giorgio M, Migliaccio E, Orsini F, Paolucci D, Moroni M, Contursi C, Pelliccia G, Luzi L, Minucci S, Marcaccio M, Pinton P, Rizzuto R, Bernardi P, Paolucci F, Pelicci PG. Electron transfer between cytochrome c and p66shc generates reactive oxygen species that trigger mitochondrial apoptosis. Cell. 2005;122:221–233. doi: 10.1016/j.cell.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 334.Spescha RD, Shi Y, Wegener S, Keller S, Weber B, Wyss MM, Lauinger N, Tabatabai G, Paneni F, Cosentino F, Hock C, Weller M, Nitsch RM, Lüscher TF, Camici GG. Deletion of the ageing gene p66shc reduces early stroke size following ischaemia/reperfusion brain injury. European Heart Journal. 2013;34:96–103. doi: 10.1093/eurheartj/ehs331. [DOI] [PubMed] [Google Scholar]
- 335.Spescha RD, Klohs J, Semerano A, Giacalone G, Derungs RS, Reiner MF, Rodriguez Gutierrez D, Mendez-Carmona N, Glanzmann M, Savarese G, Kränkel N, Akhmedov A, Keller S, Mocharla P, Kaufmann MR, Wenger RH, Vogel J, Kulic L, Nitsch RM, Beer JH, Peruzzotti-Jametti L, Sessa M, Lüscher TF, Camici GG. Post-ischaemic silencing of p66shc reduces ischaemia/reperfusion brain injury and its expression correlates to clinical outcome in stroke. European Heart Journal. 2015;36:1590–1600. doi: 10.1093/eurheartj/ehv140. [DOI] [PubMed] [Google Scholar]
- 336.Walder CE, Green SP, Darbonne WC, Mathias J, Rae J, Dinauer MC, Curnutte JT, Thomas GR. Ischemic stroke injury is reduced in mice lacking a functional nadph oxidase. Stroke. 1997;28:2252–2258. doi: 10.1161/01.str.28.11.2252. [DOI] [PubMed] [Google Scholar]
- 337.Chen H, Song YS, Chan PH. Inhibition of nadph oxidase is neuroprotective after ischemia-reperfusion. J Cereb Blood Flow Metab. 2009;29:1262–1272. doi: 10.1038/jcbfm.2009.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Kahles T, Luedike P, Endres M, Galla HJ, Steinmetz H, Busse R, Neumann-Haefelin T, Brandes RP. Nadph oxidase plays a central role in blood-brain barrier damage in experimental stroke. Stroke. 2007;38:3000–3006. doi: 10.1161/STROKEAHA.107.489765. [DOI] [PubMed] [Google Scholar]
- 339.Casas AI, Geuss E, Kleikers PWM, Mencl S, Herrmann AM, Buendia I, Egea J, Meuth SG, Lopez MG, Kleinschnitz C, Schmidt HHHW. Nox4-dependent neuronal autotoxicity and bbb breakdown explain the superior sensitivity of the brain to ischemic damage. Proceedings of the National Academy of Sciences. 2017 doi: 10.1073/pnas.1705034114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Genovese T, Mazzon E, Paterniti I, Esposito E, Bramanti P, Cuzzocrea S. Modulation of nadph oxidase activation in cerebral ischemia/reperfusion injury in rats. Brain Res. 2011;1372:92–102. doi: 10.1016/j.brainres.2010.11.088. [DOI] [PubMed] [Google Scholar]
- 341.Song J, Park J, Oh Y, Lee JE. Glutathione suppresses cerebral infarct volume and cell death after ischemic injury: Involvement of foxo3 inactivation and bcl2 expression. Oxidative Medicine and Cellular Longevity. 2015;2015:426069. doi: 10.1155/2015/426069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 342.Chen H, Kim GS, Okami N, Narasimhan P, Chan PH. Nadph oxidase is involved in post-ischemic brain inflammation. Neurobiol Dis. 2011;42:341–348. doi: 10.1016/j.nbd.2011.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Wang Z, Wei X, Liu K, Zhang X, Yang F, Zhang H, He Y, Zhu T, Li F, Shi W, Zhang Y, Xu H, Liu J, Yi F. Nox2 deficiency ameliorates cerebral injury through reduction of complexin ii-mediated glutamate excitotoxicity in experimental stroke. Free Radical Biology and Medicine. 2013;65:942–951. doi: 10.1016/j.freeradbiomed.2013.08.166. [DOI] [PubMed] [Google Scholar]
- 344.McCann SK, Dusting GJ, Roulston CL. Nox2 knockout delays infarct progression and increases vascular recovery through angiogenesis in mice following ischaemic stroke with reperfusion. PLoS ONE. 2014;9:e110602. doi: 10.1371/journal.pone.0110602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Kim HA, Brait VH, Lee S, De Silva TM, Diep H, Eisenhardt A, Drummond GR, Sobey CG. Brain infarct volume after permanent focal ischemia is not dependent on nox2 expression. Brain Research. 2012;1483:105–111. doi: 10.1016/j.brainres.2012.09.023. [DOI] [PubMed] [Google Scholar]
- 346.Nishimura A, Ago T, Kuroda J, Arimura K, Tachibana M, Nakamura K, Wakisaka Y, Sadoshima J, Iihara K, Kitazono T. Detrimental role of pericyte nox4 in the acute phase of brain ischemia. J Cereb Blood Flow Metab. 2016;36:1143–1154. doi: 10.1177/0271678X15606456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Kahles T, Kohnen A, Heumueller S, Rappert A, Bechmann I, Liebner S, Wittko IM, Neumann-Haefelin T, Steinmetz H, Schroeder K, Brandes RP. Nadph oxidase nox1 contributes to ischemic injury in experimental stroke in mice. Neurobiology of Disease. 2010;40:185–192. doi: 10.1016/j.nbd.2010.05.023. [DOI] [PubMed] [Google Scholar]
- 348.Choi DH, Kim JH, Lee KH, Kim HY, Kim YS, Choi WS, Lee J. Role of neuronal nadph oxidase 1 in the peri-infarct regions after stroke. PLoS One. 2015;10:e0116814. doi: 10.1371/journal.pone.0116814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Jackman KA, Miller AA, Drummond GR, Sobey CG. Importance of nox1 for angiotensin ii-induced cerebrovascular superoxide production and cortical infarct volume following ischemic stroke. Brain Res. 2009;1286:215–220. doi: 10.1016/j.brainres.2009.06.056. [DOI] [PubMed] [Google Scholar]
- 350.Air EL, Kissela BM. Diabetes, the metabolic syndrome, and ischemic stroke. Epidemiology and possible mechanisms. 2007;30:3131–3140. doi: 10.2337/dc06-1537. [DOI] [PubMed] [Google Scholar]
- 351.Hafez S, Coucha M, Bruno A, Fagan SC, Ergul A. Hyperglycemia, acute ischemic stroke and thrombolytic therapy. Translational stroke research. 2014;5:442–453. doi: 10.1007/s12975-014-0336-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Suh SW, Shin BS, Ma H, Van Hoecke M, Brennan AM, Yenari MA, Swanson RA. Glucose and nadph oxidase drive neuronal superoxide formation in stroke. Annals of neurology. 2008;64:654–663. doi: 10.1002/ana.21511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 353.Carneiro L, Allard C, Guissard C, Fioramonti X, Tourrel-Cuzin C, Bailbé D, Barreau C, Offer G, Nédelec E, Salin B, Rigoulet M, Belenguer P, Pénicaud L, Leloup C. Importance of mitochondrial dynamin-related protein 1 in hypothalamic glucose sensitivity in rats. Antioxidants & Redox Signaling. 2012;17:433–444. doi: 10.1089/ars.2011.4254. [DOI] [PubMed] [Google Scholar]
- 354.Zuo W, Zhang S, Xia C-Y, Guo X-F, He W-B, Chen N-H. Mitochondria autophagy is induced after hypoxic/ischemic stress in a drp1 dependent manner: The role of inhibition of drp1 in ischemic brain damage. Neuropharmacology. 2014;86:103–115. doi: 10.1016/j.neuropharm.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 355.Haines BA, Mehta SL, Pratt SM, Warden CH, Li PA. Deletion of mitochondrial uncoupling protein-2 increases ischemic brain damage after transient focal ischemia by altering gene expression patterns and enhancing inflammatory cytokines. Journal of Cerebral Blood Flow & Metabolism. 2010;30:1825–1833. doi: 10.1038/jcbfm.2010.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Toda C, Kim JD, Impellizzeri D, Cuzzocrea S, Liu Z-W, Diano S. Ucp2 regulates mitochondrial fission and ventromedial nucleus control of glucose responsiveness. Cell. 2016;164:872–883. doi: 10.1016/j.cell.2016.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Mattiasson G, Shamloo M, Gido G, Mathi K, Tomasevic G, Yi S, Warden CH, Castilho RF, Melcher T, Gonzalez-Zulueta M, Nikolich K, Wieloch T. Uncoupling protein-2 prevents neuronal death and diminishes brain dysfunction after stroke and brain trauma. 2003;9:1062. doi: 10.1038/nm903. [DOI] [PubMed] [Google Scholar]
- 358.Quaegebeur A, Segura I, Schmieder R, Verdegem D, Decimo I, Bifari F, Dresselaers T, Eelen G, Ghosh D, Schoors S, Janaki Raman SR, Cruys B, Govaerts K, De Legher C, Bouché A, Schoonjans L, Ramer MS, Hung G, Bossaert G, Cleveland DW, Himmelreich U, Voets T, Lemmens R, Bennett CF, Robberecht W, De Bock K, Dewerchin M, Fendt S-M, Ghesquière B, Carmeliet P. Deletion or inhibition of the oxygen sensor phd1 protects against ischemic stroke via reprogramming of neuronal metabolism. Cell metabolism. 2016;23:280–291. doi: 10.1016/j.cmet.2015.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Leak RK, Zhang L, Luo Y, Li P, Zhao H, Liu X, Ling F, Jia J, Chen J, Ji X. Peroxiredoxin 2 battles parp1- and p53-dependent pro-death pathways following ischemic injury. Stroke; a journal of cerebral circulation. 2013;44:1124–1134. doi: 10.1161/STROKEAHA.111.680157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Jin RC, Mahoney CE, Anderson L, Ottaviano F, Croce K, Leopold JA, Zhang Y, Tang S-S, Handy DE, Loscalzo J. Glutathione peroxidase-3 deficiency promotes platelet-dependent thrombosis in vivo. Circulation. 2011;123:1963–1973. doi: 10.1161/CIRCULATIONAHA.110.000034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Ishibashi N, Prokopenko O, Weisbrot-Lefkowitz M, Reuhl KR, Mirochnitchenko O. Glutathione peroxidase inhibits cell death and glial activation following experimental stroke. Molecular Brain Research. 2002;109:34–44. doi: 10.1016/s0169-328x(02)00459-x. [DOI] [PubMed] [Google Scholar]
- 362.Huang C-Y, Fujimura M, Chang Y-Y, Chan PH. Overexpression of copper-zinc superoxide dismutase attenuates acute activation of activator protein-1 after transient focal cerebral ischemia in mice. Stroke. 2001;32:741–747. doi: 10.1161/01.str.32.3.741. [DOI] [PubMed] [Google Scholar]
- 363.Steinhubl SR. Why have antioxidants failed in clinical trials? The American Journal of Cardiology. 2008;101:S14–S19. doi: 10.1016/j.amjcard.2008.02.003. [DOI] [PubMed] [Google Scholar]
- 364.Bjelakovic G, Nikolova D, Gluud L, Simonetti RG, Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: Systematic review and meta-analysis. JAMA. 2007;297:842–857. doi: 10.1001/jama.297.8.842. [DOI] [PubMed] [Google Scholar]
- 365.Lucchesi P, Belmadani S, Matrougui K. Hydrogen peroxide acts as both vasodilator and vasoconstrictor in the control of perfused mouse mesenteric resistance arteries. 2005 doi: 10.1097/01.hjh.0000160214.40855.79. [DOI] [PubMed] [Google Scholar]
- 366.Suvorava T, Kojda G. Reactive oxygen species as cardiovascular mediators: Lessons from endothelial-specific protein overexpression mouse models. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 2009;1787:802–810. doi: 10.1016/j.bbabio.2009.04.005. [DOI] [PubMed] [Google Scholar]
- 367.Faraci FM. Reactive oxygen species: Influence on cerebral vascular tone. Journal of Applied Physiology. 2006;100:739–743. doi: 10.1152/japplphysiol.01044.2005. [DOI] [PubMed] [Google Scholar]
- 368.Nathan C, Cunningham-Bussel A. Beyond oxidative stress: An immunologist’s guide to reactive oxygen species. Nature reviews. Immunology. 2013;13:349–361. doi: 10.1038/nri3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 369.Vara D, Pula G. Reactive oxygen species: Physiological roles in the regulation of vascular cells. 2014 doi: 10.2174/1566524014666140603114010. [DOI] [PubMed] [Google Scholar]
- 370.Teixeira G, Szyndralewiez C, Molango S, Carnesecchi S, Heitz F, Wiesel P, Wood JM. Therapeutic potential of nadph oxidase 1/4 inhibitors. Br J Pharmacol. 2017;174:1647–1669. doi: 10.1111/bph.13532. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.