

Abstract
BACKGROUND. Myeloid-derived suppressor cells (MDSCs) are a population of immature immune cells with several protumorigenic functions. CD38 is a transmembrane receptor–ectoenzyme expressed by MDSCs in murine models of esophageal cancer. We hypothesized that CD38 could be expressed on MDSCs in human colorectal cancer (CRC), which might allow for a new perspective on therapeutic targeting of human MDSCs with anti-CD38 monoclonal antibodies in this cancer.
METHODS. Blood samples were collected from 41 CRC patients and 8 healthy donors, followed by peripheral blood mononuclear cell (PBMC) separation. Polymorphonuclear (PMN-) and monocytic (M-) MDSCs and CD38 expression levels were quantified by flow cytometry. The immunosuppressive capacity of M-MDSCs from 10 CRC patients was validated in a mixed lymphocyte reaction (MLR) assay.
RESULTS. A significant expansion of CD38+ M-MDSCs and a trend of expansion of CD38+ PMN-MDSCs (accompanied by a trend of increased CD38 expression on both M- and PMN-MDSCs) were observed in PBMCs of CRC patients when compared with healthy donors. The CD38+ M-MDSCs from CRC patients were found to be immunosuppressive when compared with mature monocytes. CD38+ M- and PMN-MDSC frequencies were significantly higher in CRC patients who previously received treatment when compared with treatment-naive patients.
CONCLUSIONS. This study provides a rationale for an attempt to target M-MDSCs with an anti-CD38 monoclonal antibody in metastatic CRC patients.
FUNDING. NCI P01-CA14305603, the American Cancer Society, Scott and Suzi Lustgarten Family Colon Cancer Research Fund, Hansen Foundation, and Janssen Research and Development.
Keywords: Gastroenterology, Oncology
Keywords: Cancer, Cancer immunotherapy, Colorectal cancer
Monocytic myeloid-derived suppressor cells that express transmembrane receptor CD38 are increased in colorectal cancer patients.
Introduction
Myeloid-derived suppressor cells (MDSCs) are known to support the progression of multiple types of cancer through immunosuppression, angiogenesis, tumor cell survival, and metastasis (1) and by activating fibroblasts (2) in the tumor microenvironment (TME). MDSCs are separated into 2 groups based upon their histological characteristics: polymorphonuclear MDSCs (PMN-MDSCs) are similar to neutrophils, while monocytic MDSCs (M-MDSCs) share phenotypic characteristics of monocytes. Both PMN- and M-MDSCs have been implicated in the pathogenesis of human cancer (3), where they function not only within the primary tumor, but also in peripheral lymphoid organs (4).
We have previously reported high expression levels of the ectoenzyme CD38 on the surface of MDSCs (particularly M-MDSCs) in a murine model of esophageal cancer (5), and expression of CD38 on MDSCs has been reported in multiple myeloma (6). It should be noted that CD38 is also expressed on T cells, B cells, and NK cells (7). Since there are several monoclonal antibodies targeting human CD38 available (8–10), with daratumumab already being approved by the FDA for treatment of multiple myeloma (11), we set out to characterize CD38+ MDSCs in colorectal cancer (CRC).
CRC is the third most common cancer in the United States (12) and may be associated with chronic inflammation (13). There is nascent evidence (14, 15) supporting expansion of MDSC-like cells in patients with advanced-stage CRC, yet these reports do not provide enough information regarding the nature of the cells, nor do they indicate any options for targeted therapy. Currently, adjuvant therapy remains to be established in stage II CRC; adjuvant therapy is approved for stage III CRC, and therapy for stage IV CRC is complex, involving chemotherapy or targeted agents or immunotherapy (16).
The objective of this study was to evaluate MDSC frequencies and CD38 expression patterns in peripheral blood mononuclear cells (PBMCs) from CRC patients at stages I–IV, with the goal of proposing selective targeting of CD38+ cells to ablate MDSCs in CRC patients. Herein, we demonstrate the expansion of functional CD38+ M-MDSCs in peripheral blood of CRC patients. Furthermore, the expansion of CD38+ M-MDSCs was more pronounced in patients with previous therapy.
Results
The M-MDSC population is expanded in the peripheral blood mononuclear cells of CRC patients.
We assessed the myeloid fraction within PBMCs from 41 CRC patients (Tables 1 and 2), and Supplemental Table 1; supplemental material available online with this article; https://doi.org/10.1172/jci.insight.97022DS1) and 8 healthy volunteers using the previously published classification (Figure 1, A and B) (17).
Table 1. Characteristics of study population.

Table 2. Characteristics of CRC patients.

Figure 1. Study design.

(A) Patients diagnosed with stage I–IV CRC were recruited to the study after providing written informed consent. Blood samples were collected from the patients, and the PBMC fraction was separated for immunophenotyping or cryopreserved for future use in the immunosuppression assay and immunophenotyping. (B) Markers used to define myeloid cell subpopulations.
M-MDSCs (defined as CD14+HLA-DR–/lo) were on average 5 times more frequent among PBMCs from CRC patients, compared with healthy donors (Figure 2 and Table 3). PMN-MDSCs (defined as low density, CD33+CD11b+CD14–CD15+SSChi) were expanded 2-fold in PBMCs from CRC patients; however, this trend failed to reach statistical significance (Figure 2 and Table 3). Monocytes (defined as CD14+HLA-DRhi) did not expand significantly in CRC patients (Supplemental Figure 1). We did not observe any significant differences in the expression of surface marker CD38 when comparing monocytes and M-MDSCs, neutrophils and PMN-MDSCs (Supplemental Figure 2), or monocytes from CRC patients and healthy donors (Supplemental Figure 3). However, a trend toward elevated CD38 expression was observed in both M- and PMN-MDSCs from CRC patients (Figure 3 and Table 4).
Figure 2. The M-MDSC population is expanded in the peripheral blood of CRC patients.

See also Supplemental Figure 1. (A) Peripheral blood mononuclear cells (PBMCs) from colorectal cancer (CRC) patients and healthy donors (HD) were subjected to immunophenotyping by flow cytometry. Analysis of 1 representative sample is shown. (B) The frequency of M-MDSCs (CD14+HLA-DRlo) and PMN-MDSCs (low-density CD14–CD11b+CD15+) in the live subset of PBMCs. Gray dots represent individual values; black squares, mean values. Statistical data are presented in Table 3.
Table 3. Frequency of M-MDSCs and PMN-MDSCs.

Figure 3. CD38+ M- and PMN-MDSCs expand in the peripheral blood of CRC patients when compared with healthy donors.

See also Supplemental Figures 2 and 3. (A and B) Representative plots showing expression levels of CD38 on M-MDSC (A) and PMN-MDSC (B) populations (CD14+HLA-DRlo and low-density CD14–CD11b+CD15+, respectively) from PBMCs of an HD and a CRC patient. CD38 expression levels (MFI) on M- and PMN-MDSCs from HDs and CRC patients; mean values and SDs are plotted as horizontal lines. Statistical data are presented in Table 4.
Table 4. Expression levels of CD38 on the surface of M-MDSCs or PMN-MDSCs from PBMCs of CRC patients or healthy donors, expressed as mean fluorescence intensity.

CD38+ M-MDSCs from CRC patients are immunosuppressive.
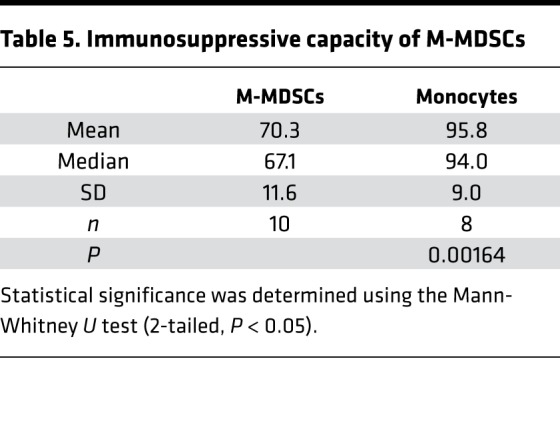
Since the M-MDSC population was significantly expanded in CRC patients, we proceeded to evaluate the immunosuppressive capacity of these cells in a T cell suppression assay using a 3-way allogenic mixed leukocyte reaction (MLR). Indeed, CD38+ M-MDSCs from CRC patients inhibited proliferation of T cells in response to stimulation by dendritic cells in a ratio-dependent manner (Figure 4), and on average, CD38+ M-MDSCs were significantly more immunosuppressive than the control population (monocytes, defined as CD14+HLA-DRhi) (Figure 4A and Table 5). Ideally, CD38– M-MDSCs should have been used as a negative control in the MLR; however, since the majority of M-MDSCs were CD38+ (Figure 3A), such a comparison was not feasible.
Figure 4. CD38+ M-MDSCs from the peripheral blood of CRC patients are immunosuppressive.

(A) Immunosuppressive capacity of M-MDSCs. M-MDSCs (CD14+HLA-DRlo) and monocytes (CD14+HLA-DRhi) were isolated from PBMCs, and immunosuppressive activity of these cell populations was tested in 3-way allogenic MLR. Gray dots represent individual values (averaged triplicate measures at 1:1 MDSC/monocyte to T cell ratio; black squares, mean values). Statistical data are presented in Table 5. (B) A representative result of MLR with M-MDSCs and monocytes from a single CRC patient. Individual values are plotted as gray squares (Monocyte) or black circles (M-MSDC); horizontal bars represent mean values; error bars indicate SEM; P = 0.02 (1:1), P = 0.01 (1:3), P = 0.009 (1:9).
Table 5. Immunosuppressive capacity of M-MDSCs.
CD38+ M-MDSCs and PMN-MDSCs expand in the peripheral blood of CRC patients who have undergone therapy.
Importantly, we found that CD38+ M-MDSCs were on average twice as abundant in CRC patients who received any form of cancer therapy (surgery, chemotherapy or radiotherapy, targeted therapy, or a combination of these methods; Tables 1 and 2 and Supplemental Table 1), compared with treatment-naive patients (Figure 5A and Table 6). Furthermore, CD38 expression levels were significantly higher on the surface of M-MDSCs from treated CRC patients (Figure 5C and Table 6). Of note, CD38+ PMN-MDSCs and CD38+ monocytes followed the same pattern (Figure 5, B and C; Supplemental Figure 4; and Table 6).
Figure 5. CD38+ M- and PMN-MDSCs expand in peripheral blood of CRC patients who underwent treatment.

See also Supplemental Figure 4. The frequencies of CD38+ M-MDSCs (A) and CD38+ PMN-MDSCs (B) in the live subset of PBMCs from treatment-naive CRC patients or patients who received chemotherapy. Gray dots represent individual values; black squares, mean values. (C) Expression levels of CD38 on the surface of M-MDSCs or PMN-MDSCs from PBMCs of CRC patients, expressed as MFI. Statistical data are presented in Table 6.
Table 6. Frequencies of CD38+ M-MDSCs and PMN-MDSCs, and CD38 expression levels.

Discussion
Targeting MDSCs is an attractive approach in cancer therapy due to the multiple mechanisms that these cells employ in supporting tumor progression. However, because of the heterogeneous nature of this population and lack of a specific marker, this task remains challenging (18). We have previously reported an expansion of CD38+ MDSCs in murine models of esophageal squamous cell carcinoma (5). We also detected expansion of a population of CD38+ MDSC-like (low-density CD15hiCD33lo) cells in the peripheral blood of patients with advanced-stage head and neck cancer and non–small cell lung cancer (5). These data led us to suggest that CD38 could be a target worth pursuing as a novel approach to depletion of these immunosuppressive cells in cancer patients, in particular CRC patients.
In line with this study’s objectives, we analyzed the MDSCs from PBMCs of 41 CRC patients, comparing them to PBMC isolates from 8 healthy donors. Immunophenotyping revealed that the CD38+ M-MDSCs were not only abundant in the PBMCs of CRC patients (Figures 2 and 3), but were functionally immunosuppressive (Figure 4) and could possibly contribute to CRC pathogenesis.
Another striking finding was the association between the patients’ treatment history and the frequency of M-MDSCs, as well as expression of CD38 in this cell population (Figure 5). These observations are consistent with the premise that MDSCs may be mediating resistance to anticancer therapy (19, 20), which implies that targeting MDSCs could prevent or stall the development of treatment resistance.
It is important to note that due to the small size of the patient cohort in this study, we were not able to make conclusions regarding associations between the frequency of CD38+ M-MDSCs and clinical features, such as tumor stage, microsatellite instability (MSI) status, and patient survival. That being said, if expansion of CD38+ M-MDSCs occurs that is independent of MSI status, then one could conjecture that adding anti-CD38 therapy to immune checkpoint inhibitor therapy in metastatic CRC would be beneficial. There were also certain differences between the treated and untreated cohorts (the distribution of tumor staging and differentiation status; Table 2 and Supplemental Table 1) that may have affected our analyses.
This report describes findings that offer a rationale for the use of CD38-targeted therapy in CRC. We have previously demonstrated that administration of an anti-CD38 monoclonal antibody suppressed tumor growth in a murine model of esophageal cancer (5). In addition to MDSC depletion, such an approach can utilize the fact that CD38 can be expressed on the surface of other immunosuppressive cells, such as Tregs and regulatory B cells (Bregs) (21, 22), although the relevance of these cell populations to human CRC progression remains to be determined. In fact, isatuximab, a humanized anti-CD38 antibody currently in clinical trials for multiple myeloma (9), successfully depletes Tregs and reduces Foxp3 and IL-10 expression in these cells and restores the cytotoxic capacity of CD8+ T cells and natural killer cells (23). Daratumumab also depletes MDSCs, Tregs, and Bregs in multiple myeloma patients (6).
Given the extremely favorable safety profile of daratumumab (11), encouraging data from studies with new CD38 antibodies (9), and the diverse mechanisms of actions of anti-CD38 antibodies, our report provides a new perspective and rationale for clinical testing of an anti-CD38 monoclonal antibody in combination with first-line and second-line therapies for metastatic CRC.
Methods
Study design and sample collection.
This study comprised patients from the University of Pennsylvania Health System with stages I–IV CRC who were ≥18 years old, and representative of both sexes and diverse ethnic groups. These patients continued to undergo standard of care. The length of a subject’s participation time in the study was limited to one visit for blood collection. Inclusion in the study was strictly voluntary. Healthy donors (controls) were recruited at the Wistar Institute.
The study proceeded as follows (Figure 1A). After written informed consent was obtained, blood samples were collected, and the following clinical data were obtained: age, sex, diagnosis and treatment, genetic (tumor gene mutations, if available) analysis, and MSI status (if available) (Supplemental Table 1). Blood samples were used fresh for immunophenotyping and/or frozen to be used in functional assays (MLR). Upon study completion, the experimental data were analyzed, and potential correlations with clinical data were evaluated.
Forty-seven patients were recruited to the study; however, for technical reasons we could not process blood samples from 6 patients, so these patients had to be excluded from the study (Supplemental Table 1).
PBMC isolation.
PBMCs were isolated from the patients using Ficoll-Paque PLUS (GE Healthcare) and used fresh (immunophenotyping) or frozen in FBS supplemented with 10% DMSO at –80°C. For MLR suppression assays, frozen PBMCs were thawed at 37°C and washed with MACS buffer (DPBS supplemented with 1% FBS and 2 mM EDTA) before use.
Flow cytometry.
Freshly isolated PBMCs were stained, followed by data acquisition on a BD LSR II flow cytometer. Analysis was performed using FlowJo (TreeStar). The following antibodies were used for staining: HLA-DR–FITC (catalog 555811/clone G46-6), CD11b-APC (catalog 550019/ICRF44), CD15-PerCP/Cy5.5 (323020/W6D3), CD14-APC/Cy7 (557831/MφP9), CD33-PE/Cy7 (333946/P67.6), CD38-PE (555460/HIT2), and IgG1 isotype–PE (555749/MOPC-21). HLA-DR and CD11b antibodies were used at 1:10 dilution; CD38 and IgG1 isotype were at 1:20; and the rest were diluted 1:40. Except for CD15 (BioLegend), all antibodies were from BD Pharmingen. Isotype control antibody was used to establish gates for CD38 expression.
Three-way MLR assay.
The PBMCs were incubated with anti–HLA-DR–FITC, anti-CD14–APC/Cy7, and anti-CD38–PE or isotype control PE for 30 minutes at 4°C. CD14+HLA-DRlo/negCD38+ and CD14+HLA-DR+ cells were isolated by cell sorting on a FACSAria cell sorter (BD Biosciences). T lymphocytes from one healthy donor were purified using the Human CD3+ T Cell Enrichment Column Kit (R&D Systems) and used as responder cells. Dendritic cells were generated from purified CD14+ monocytes from another healthy donor in the presence of 25 ng/ml GM-CSF and 25 ng/mL IL-4 (Peprotech) for 6 days and used as stimulator cells. Responder and stimulator cells were then mixed at a 10:1 ratio, followed by the addition of CD14+HLA-DRlo/negCD38+ M-MDSCs or CD14+HLA-DR+ monocytes. T lymphocyte proliferation was assessed after 5 days of culture by 3H-thymidine incorporation.
Statistics.
Unless stated otherwise, all data are presented as means. Nonparametric Mann-Whitney U test (2-tailed, P < 0.05 considered significant) was used to determine significance when comparing unpaired samples. Benjamini-Hochberg adjustment was used on P values to account for multiple measurements, with an FDR of 5%. Student’s 2-tailed t test was used to determine significance when analyzing individual MLR results (P < 0.05 considered significant). All calculations were performed in GraphPad Prism 6.
Study approval.
The studies were approved by the IRBs of the University of Pennsylvania (no. 821415) and Wistar Institute (no. 2908251).
Author contributions
GAD, AH, and TAK contributed to this manuscript equally. GAD, AH, TAK, CC, KS, GLB, DIG, and AKR designed the study. GAD, AH, TAK, and JWL conducted experiments and/or acquired data. GAD, AH, TAK, EWL, and GLB analyzed data. TAK and AKR wrote the manuscript. All authors reviewed and approved the manuscript prior to submission.
Supplementary Material
Acknowledgments
We thank the Molecular Pathology and Imaging Core (J. Katz, A. Bedenbaugh, D. Budo, and C. O’Donnel), the Human Microbial, Analytical and Repository Core (G. Wu and L. Chau), the Genetically Modified Mouse Core, the Cell Culture and iPS Core, the Wistar Flow Cytometry Shared Resource, and Penn Biostatistics Core. We thank members of the Rustgi and Gabrilovich laboratories for discussions.
Version 1. 03/22/2018
Electronic publication
Footnotes
Conflict of interest: AH is an employee of Daiichi Sankyo Co. Ltd.; CC is an employee of Janssen; and KS is an employee of Genmab. DIG reports receiving commercial research grants from Anixa Diagnostics Corp., Bristol-Myers Squibb, Daichi, Janssen, Reata Pharmaceuticals, and Syndax, and is a consultant/advisory board member for Janssen, Merck, Peregrin Pharmaceuticals, and Syndax.
Reference information: JCI Insight. 2018;3(6):e97022. https://doi.org/10.1172/jci.insight.97022.
Contributor Information
Ayumi Hashimoto, Email: hashimoto.ayumi.ac@daiichisankyo.co.jp.
Christopher Chiu, Email: cchiu7@its.jnj.com.
Kate Sasser, Email: ksr@genmab.com.
Dmitry I. Gabrilovich, Email: dgabrilovich@wistar.org.
References
- 1.Gabrilovich DI, Ostrand-Rosenberg S, Bronte V. Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol. 2012;12(4):253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Stairs DB, et al. Deletion of p120-catenin results in a tumor microenvironment with inflammation and cancer that establishes it as a tumor suppressor gene. Cancer Cell. 2011;19(4):470–483. doi: 10.1016/j.ccr.2011.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Greten TF, Manns MP, Korangy F. Myeloid derived suppressor cells in human diseases. Int Immunopharmacol. 2011;11(7):802–807. doi: 10.1016/j.intimp.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumar V, Patel S, Tcyganov E, Gabrilovich DI. The nature of myeloid-derived suppressor cells in the tumor microenvironment. Trends Immunol. 2016;37(3):208–220. doi: 10.1016/j.it.2016.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Karakasheva TA, et al. CD38-expressing myeloid-derived suppressor cells promote tumor growth in a murine model of esophageal cancer. Cancer Res. 2015;75(19):4074–4085. doi: 10.1158/0008-5472.CAN-14-3639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Krejcik J, et al. Daratumumab depletes CD38+ immune regulatory cells, promotes T-cell expansion, and skews T-cell repertoire in multiple myeloma. Blood. 2016;128(3):384–394. doi: 10.1182/blood-2015-12-687749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Malavasi F, et al. Evolution and function of the ADP ribosyl cyclase/CD38 gene family in physiology and pathology. Physiol Rev. 2008;88(3):841–886. doi: 10.1152/physrev.00035.2007. [DOI] [PubMed] [Google Scholar]
- 8.Chillemi A, et al. Anti-CD38 antibody therapy: windows of opportunity yielded by the functional characteristics of the target molecule. Mol Med. 2013;19:99–108. doi: 10.2119/molmed.2013.00009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van de Donk NW, et al. Clinical efficacy and management of monoclonal antibodies targeting CD38 and SLAMF7 in multiple myeloma. Blood. 2016;127(6):681–695. doi: 10.1182/blood-2015-10-646810. [DOI] [PubMed] [Google Scholar]
- 10.de Weers M, et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J Immunol. 2011;186(3):1840–1848. doi: 10.4049/jimmunol.1003032. [DOI] [PubMed] [Google Scholar]
- 11.Laubach JP, Tai YT, Richardson PG, Anderson KC. Daratumumab granted breakthrough drug status. Expert Opin Investig Drugs. 2014;23(4):445–452. doi: 10.1517/13543784.2014.889681. [DOI] [PubMed] [Google Scholar]
- 12.Beaugerie L, Itzkowitz SH. Cancers complicating inflammatory bowel disease. N Engl J Med. 2015;372(15):1441–1452. doi: 10.1056/NEJMra1403718. [DOI] [PubMed] [Google Scholar]
- 13.Long AG, Lundsmith ET, Hamilton KE. Inflammation and colorectal cancer. Curr Colorectal Cancer Rep. 2017;13(4):341–351. doi: 10.1007/s11888-017-0373-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun HL, et al. Increased frequency and clinical significance of myeloid-derived suppressor cells in human colorectal carcinoma. World J Gastroenterol. 2012;18(25):3303–3309. doi: 10.3748/wjg.v18.i25.3303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang B, et al. Circulating and tumor-infiltrating myeloid-derived suppressor cells in patients with colorectal carcinoma. PLoS One. 2013;8(2):e57114. doi: 10.1371/journal.pone.0057114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Edwards MS, Chadda SD, Zhao Z, Barber BL, Sykes DP. A systematic review of treatment guidelines for metastatic colorectal cancer. Colorectal Dis. 2012;14(2):e31–e47. doi: 10.1111/j.1463-1318.2011.02765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bronte V, et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat Commun. 2016;7:12150. doi: 10.1038/ncomms12150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Waldron TJ, Quatromoni JG, Karakasheva TA, Singhal S, Rustgi AK. Myeloid derived suppressor cells: targets for therapy. Oncoimmunology. 2013;2(4):e24117. doi: 10.4161/onci.24117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pancione M, et al. Immune escape mechanisms in colorectal cancer pathogenesis and liver metastasis. J Immunol Res. 2014;2014:686879. doi: 10.1155/2014/686879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sermeus A, Leonard W, Engels B, De Ridder M. Advances in radiotherapy and targeted therapies for rectal cancer. World J Gastroenterol. 2014;20(1):1–5. doi: 10.3748/wjg.v20.i1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Patton DT, Wilson MD, Rowan WC, Soond DR, Okkenhaug K. The PI3K p110δ regulates expression of CD38 on regulatory T cells. PLoS One. 2011;6(3):e17359. doi: 10.1371/journal.pone.0017359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Blair PA, et al. CD19(+)CD24(hi)CD38(hi) B cells exhibit regulatory capacity in healthy individuals but are functionally impaired in systemic lupus erythematosus patients. Immunity. 2010;32(1):129–140. doi: 10.1016/j.immuni.2009.11.009. [DOI] [PubMed] [Google Scholar]
- 23.Feng X, et al. Targeting CD38 Suppresses induction and function of T regulatory cells to mitigate immunosuppression in multiple myeloma. Clin Cancer Res. 2017;23(15):4290–4300. doi: 10.1158/1078-0432.CCR-16-3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.