

ABSTRACT
RhoA regulates actin cytoskeleton but recent evidence suggest a role for this conserved Rho GTPase also in other cellular processes, including transcriptional control of cell proliferation and survival. Interestingy, loss of RhoA is synthetic lethal with oncogenic Myc, a master transcription factor that turns on anabolic metabolism to promote cell growth in many cancers. We show evidence indicating that the synthetic lethal interaction between RhoA loss and Myc arises from deficiency in glutamine utilization, resulting from impaired co-regulation of glutaminase expression and anaplerosis by Myc and RhoA – serum response factor (SRF) pathway. The results suggest metabolic coordination between Myc and RhoA/SRF in sustaining cancer cell viability and indicate RhoA/SRF as a potential vulnerability in cancer cells for therapeutic targeting.
KEYWORDS: apoptosis, breast cancer, glutamine metabolism, Myc, RhoA, SRF
Introduction
Tumor cells undergo metabolic transformation to feed the energy and material needs of non-stop proliferation. For example, cells preparing for cell division must increase their mass and replicate DNA, which consumes vast amount of nucleic acids, proteins, lipids and other macromolecules. Therefore, anabolic metabolic processes predominate in cancer cells and there has been substantial recent progress in understanding how cancer critical oncogenes including Myc, H-ras, Src, and Akt engage metabolic processes to stimulate anabolic metabolism in transformed cells.1
Myc is a master regulator of transcription in proliferating cells and its oncogenic deregulation is among the most frequent abberrations observed in human cancers.2 Myc actively coordinates metabolic transformation by stimulating the expression of genes involved in glucose metabolism and ribosome biogenesis. However, one of the most essential metabolic functions of the oncogenic Myc is upregulation of glutamine metabolism, as evidenced by strict glutamine-dependence of the cells with high Myc expression for viability.3,4 Glutamine, once transported to cytosol, is imported into the mitochondrion, where it is converted to glutamate by glutaminase (GLS1), and subsequently metabolized to α-ketoglutarate (α-KG). α-KG is oxidized in the the citric acid (TCA) cycle to generate ATP or provide the carbon skeletons for macromolecular synthesis. Myc stimulates glutamine consumption and metabolism by different mechanisms, including activation of genes involved in glutamine metabolism at the transcriptional level or via small regulating microRNAs.5 Normally glutamine is one of the non-essential amino acids, but Myc can reprogram metabolism so that glutamine is used as the main oxidizable substrate to maintain the TCA cycle activity and cell viability.6 Glutamine is also an indispensable donor of nitrogen in nucleic acid biosynthesis and a primary source of nitrogen for synthesis of many amino acids.7 While the oncogene orchestrated addiction of tumor cells to glutamine exposes a clear cancer cell vulnerability, few concepts have emerged so far as to suggest how to pharmacologically exploit glutamine-dependency of tumor cells in a therapeutic setting.8
Rho GTPases are rarely mutated in cancers, but their expression and activity is often altered, suggesting that their regulation is flawed.9 The altered activity of Rho GTPases influences many key processes in tumorigenesis including cell cycle, proliferation, adhesion, polarity, cell survival and migration.10 These findings have prompted several Rho and Rho pathway targeting strategies, anticipating clinical benefits from inhibited tumor cell proliferation and reduced invasive or metastatic potential of tumor cells.11 Intriguingly, recent evidence has also suggested a role for Rho proteins in generating cancer metabolism type dependencies in the transformed cells. An enticing connection between Rho GTPases and glutamine metabolism was discovered through screening of small molecules that could inhibit the Rho GTPase-dependent transformation and breast cancer cell proliferation. The most efficient inhibitor targeted a specific isoform and splice variant of the mitochondrial glutaminase (GLS1), the enzyme involved in the first step of glutaminolysis turning glutamine to glutamate.12,13 Thus, many independent oncogenic pathways could impinge on the regulation of glutamine metabolism, which is one of the most distinctively altered metabolic pathway between normal and cancer cells.
A recent shRNA gene silencing screen aiming to identify determinants of epithelial integrity-dependent cell cycle restriction in 3-dimensional MCF10A mammary epithelial culture, unexpectedly exposed a strong synthetic lethal interaction between an acute activation of Myc and shRNA silencing of RhoA.14 We present here evidence indicating co-operative action of oncogenic Myc and RhoA-serum response factor (SRF) pathway in upregulation of glutamine metabolism, dependency of the cell survival of Myc and RhoA stimulated glutamine anaplerosis and evidence for pharmacological targetability of RhoA's metabolic function.
Results
We originally observed the synthetic lethal interaction between Myc activation and RhoA silencing in 3-dimensional (3D) MCF10A mammary epithelial culture, raising the possibility that the cells were killed by an anoikis type of death mechanism. For example, RhoA loss could have perturbed the arrangement of basement membrane (BM)-integrin contacts.9 However, we found that siRNA mediated silencing of RhoA together with activation of Myc (using tamoxifen-inducible MycER) induced synergistic apoptosis also in traditional 2D monolayer cell culture (Fig. 1A-D), which bears little resemblance to physiological BM environment. In addition, siRNA-mediated silencing of RhoA alone did not induce any visible changes in the cell adhesion to petri dishes or apoptosis. The RhoA loss coupled apoptosis was strictly Myc-dependent. While these observations do not indicate that extracellular matrix would be unimportant for RhoA-dependent survival, they nevertheless motivated us to investigate whether the apoptotic synergy between RhoA-loss and Myc was mechanistically more cell autonomous than originally thought.
Figure 1.
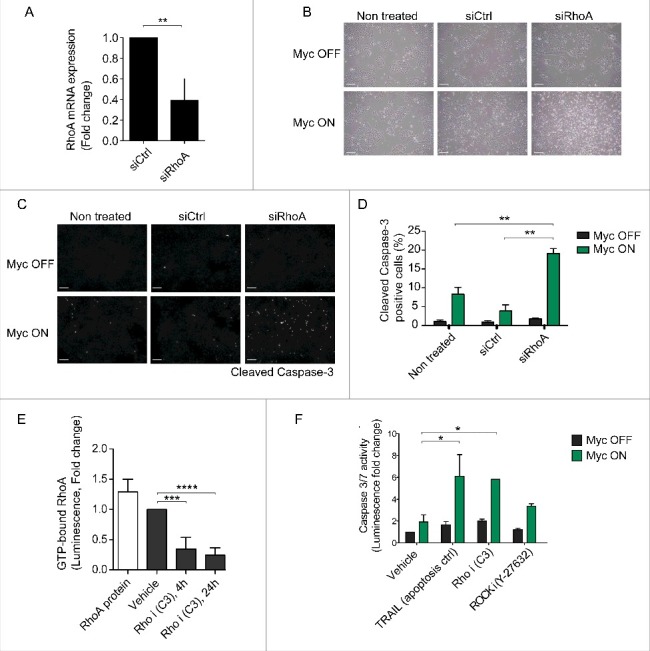
Genetic or pharmacological inhibition of RhoA sensitizes cells to Myc-dependent apoptosis. (A) RhoA mRNA expression in control siRNA (siCtrl) or RhoA siRNA (siRhoA) transfected MCF10A-MycER cells at 72 h posttransfection. The graph presents average and standar deviation (SD) of 3 biological repeats. Student's t-test ** p ≤ 0.01. (B) Phase contrast images demonstrating the level of apoptosis in MCF10A-MycER cell cultures transfected with RhoA targeted siRNA. Cells were cultured for 48 h after siRNA transfection followed by Myc activation with 100 nM 4-OHT. Images were taken 24 h after Myc activation. Scale bar 100 μM. (C) Representative fluorescence microscopy images of the cells treated as above followed by immunostaining with cleaved Caspase-3 antibody. Scale bar 100 μM. (D) Quantification of the apoptotic cells from the images. At least 6 field of views (fov), each containing 500–1000 cells, were analyzed per treatment and the graph represents the average and standard deviation (SD) of 2 biological repeats. Student's t-test ** p ≤ 0.01. (E) Quantification of active GTP-bound RhoA in the cells after treatment with vehicle (MQ water) or with 0.5 μg/ml Rho inhibitor C3 for 4 h or 24 h. RhoA protein denotes an assay control. The graph shows average and SD representing 4 biological replicates. Student's t-test *** p ≤ 0.001, **** p < 0.0001. (F) Quantification of apoptosis in MCF10A-MycER cells treated with 50 ng/ml TRAIL (positive apoptosis control), 0.5 μg/ml C3 or 25 μM Y-27632 (ROCK inhibitor) with or without Myc activation. Cells were seeded to multiwell plates and let adhere for 24 h followed by 24 h Myc activation with 100 nM 4-OHT. Thereafter, the cells were incubated with drugs for another 24 h (or TRAIL 30 min) followed by analysis of caspase-3/7 activity. The graph shows average and SD representing 3 biological replicates. Student's t-test as above, * p ≤ 0.05.
We first explored the translational potential of the apoptotic MycON;RhoAOFF switch by testing if the RhoA silencing by siRNA could be substituted with RhoA pathway inhibitors for apoptosis induction. MCF10A cells with or without active Myc were exposed to C3 transferase Rho inhibitor, Rho-associated protein kinase (ROCK) inhibitor Y-27632. TNF-related apoptosis-inducing ligand (TRAIL) with known apoptotic synergy with Myc was used as a control.15 The C3 treatment of MCF10A cells decreased the level of GTP-bound active RhoA but even higher C3 concentrations did not induce significant apoptosis (Fig. 1E and F). In contrast, Rho inhibition by C3 combined with Myc activation induced over 6-fold increase in caspase 3/7 activity, indicating strong apoptotic response. ROCK inhibition also sensitized to Myc apoptosis but less efficiently than RhoA inhibition (Fig. 1F). The results indicate that inhibition of either RhoA expression or its activity is strongly synthetic lethal with high Myc activity and suggest that the survival pathway downstream of RhoA at least partially involves ROCK.
We next asked if the RhoA-deficiency perturbs Myc's ability to stimulate glutamine metabolism, which process is essential for the survival of Myc transformed cells.16,17 Extending previous findings that Myc upregulates GLS1 expression, we found in chromatin immunoprecipitation sequencing (ChIP-seq) assay a strong peak of Myc binding to GLS1 promoter in the proximity of transcription start site (TSS). The binding was further enhanced when inducible Myc (MycER) was activated in the cells (Fig. 2A), suggesting that Myc directly transcriptionally regulates GLS1 expression. Cancer cells and also highly proliferative healthy cells suffer from continual loss of citrate from TCA cycle and become dependent on glutamine as an extra source of TCA intermediates (anaplerosis) for maintaining the mitochondrial integrity and TCA cycle function.18 Glutamine is converted by GLS to glutamate and further metabolized to α-KG, which is the metabolite that enters the TCA cycle to provide energy and macromolecular material sources. We examined whether the MycON;RhoAOFF-induced apoptosis could be rescued by replenishing glutamine carbon to the TCA cycle. MCF10A cells were starved from glutamine for 24 hours followed by Myc activation for another 24 h. As expected, the treatment caused Myc-dependent apoptosis that was completely rescued with exogenous α-KG (Fig. 2B). Strikingly, feeding of the TCA cycle with exogenous α-KG also rescued the RhoA-loss triggered apoptosis of Myc transformed cells (Fig. 2C).
Figure 2.
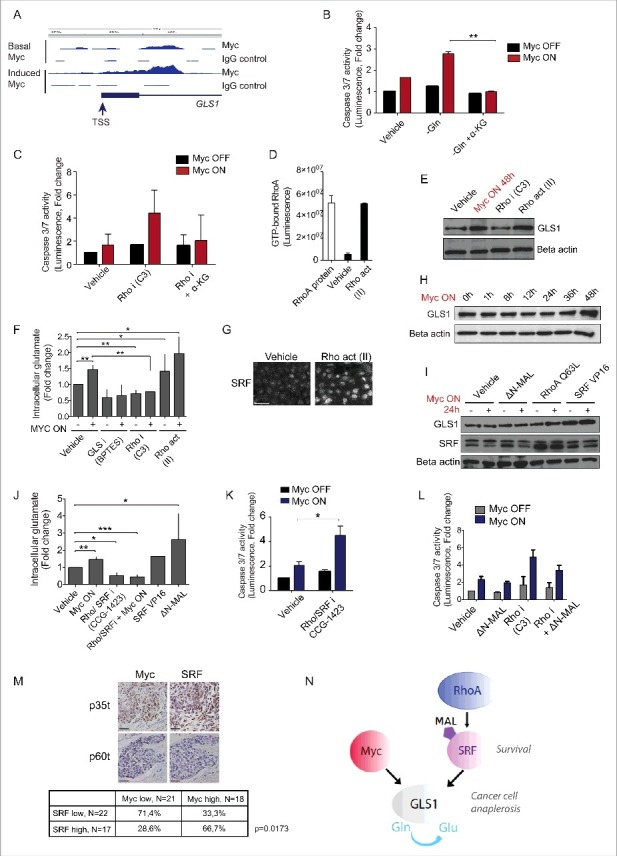
Myc and RhoA/SRF pathway cooperatively support glutamine anaplerosis, which is critical for the survival of transformed cells. (A) ChIP-Seq peaks indicate Myc binding to proximity of GLS1 promoter transcription start site (TSS) in MCF10A-MycER cells. The box denotes 5′UTR region preceding the first intron of GLS1. Note the enhanced binding site occupancy after Myc activation with 100 nM 4-OHT for 24 h. IgG was used as an antibody specificity control. (B) α-KG rescues Myc transformed cells from glutamine-deprivation induced apoptosis. MCF10A-MycER cells were cultured in the precence or absence of glutamine for 24 h followed by 24 h Myc activation with 100 nM 4-OHT. 0.5 mM α-KG was added to the indicated cells at the start of glutamine deprivation. The graph shows average and SD representing 3 biological replicates. (C) α-KG rescues Myc transformed cells from Rho inhibition-induced apoptosis. The experiments were performed as in (B), 0.5 μg/ml C3 was used to inhibit Rho. (D) RhoA activation in MCF10A cells with Rho activator II. The cells were treated for 24 h with 1 μg/ml Rho activator (Rho act; Rho activator II) followed by analysis of GTP-bound RhoA. (E) Rho upregulates GLS1. Western blot analysis shows GLS1 protein levels in MCF10A-MycER cells after 24 h treatment with control (MQ water), 100 nM 4-OHT to activate Myc, 0.5 μg/ml Rho inhibitor C3 or with 1 μg/ml Rho activator II. Beta actin was used as a loading control. (F) Effect of altered Rho activity on the intracellular glutamate levels in the presence or absence of active Myc. The glutamate levels were measured after the cells were treated for 24 h with 100 nM 4-OHT to activate Myc followed by 24 h treatments with 0.5 μg/ml C3, 1 μg/ml Rho activator II or a positive control 10 μM GLS inhibitor BPTES. The graph shows average and SD representing 3 biological replicates. (G) Immunofluorescence staining of MCF10A-MycER cells shows nuclear accumulation of SRF after 24 h treatment with 1 μg/ml Rho activator II. (H) Time course of GLS1 induction by Myc. Myc was activated with 100 nM 4-OHT. Beta actin serves as a loading control. (I) Rho/SRF pathway cooperates with Myc to induce accumulation of GLS1. The western blot analysis shows the GLS1 and SRF levels in MCF10A cells after 24 h Myc activation, ΔN-MAL induction with 1 μg/ml doxocycline and after the cells were engineered to express RhoA Q63L or SRF VP16. (J) Effect of altered Rho/SRF signaling activity on the intracellular glutamate levels in the presence or absence of active Myc. The intracellular glutamate levels were measured as in (F) using 250 nM GCG-1423. The bars on the right denote MCF10A cells expressing SRF VP16 or activated ΔN-MAL. (K) Inhibition of Rho/SRF signaling sensitizes to Myc-dependent apoptosis. Experiments were performed using 500 nM RhoA/SRF inhibitor. (L) Enforced SRF activity rescues cells from apoptosis triggered by active Myc together with Rho inhibition. In the experiments, 100 nM 4-OHT was used to activate Myc and 1 μg/ml doxocycline to induce the SRF coactivator ΔN-MAL for 24 h followed by 24 h inhibition of Rho with 0.5 μg/ml C3. (M) Representative images from immunohistochemical stainings of breast cancer samples showing examples of Myc and SRF negative and high MYC/SRF positive samples. The table shows quantification of blinded samples. The high expressing group included samples with >50% positive cells and the low expressing/negative group samples with 0–50% positive cells. The scale bar is 50 μm. In the table, the z-score test for 2 population proportions indicates statistically significant difference between the proportions. (N) A model proposing that Myc and RhoA/SRF cooperatively sustain glutamine anaplerosis in cancer cells, which is critical for their survival. Thus, the RhoA/SRF-dependent glutaminolytic pathway may offer new targets for therapeutic interventions aiming to selectively kill metabolically transformed cancer cells.
We next asked whether the Rho activity might affect the glutamine metabolism and validated both Rho inhibitor C3 and a cell permeable Rho activating reagent Rho Activator II (Rho act) for these studies (Fig. 2D). Consistent with the role of Rho pathway in regulation of glutaminolysis, we found that Rho inhibition slightly decreases GLS1 expression. However, more notably, Rho activation increased the GLS1 expression with similar potency as Myc (Fig. 2E). To address the role of Rho pathway in regulation of glutamine metabolism more quantitatively, we examined the levels of intracellular glutamate (metabolite of glutamine converted by GLS1). Interestingly, Rho inhibition decreased the intracellular glutamate levels and rendered Myc unable to elevate glutamate levels. Moreover, Myc and Rho activation additively increased the cellular glutamate levels (Fig. 2F). These findings suggest that Myc requires Rho pathway function for activation of the cell survival critical glutaminolysis.
Recent results have suggested that RhoA/ROCK pathway regulates serum response factor (SRF)-dependent transcriptional responses, including those important for cell survival.19,20 In agreement, Rho activation induced a strong nuclear accumulation of SRF in MCF10A cells (Fig. 2G). We examined whether RhoA pathway could augment Myc's ability to induce GLS1 expression and performed experiments at 24 h timepoint before full Myc induction of GLS1 (Fig. 2H). Activation of RhoA/SRF pathway with a constitutively active form of RhoA (RhoA Q63L) or SRF (SRF VP16) synergized with Myc in induction of GLS1 protein accumulation (Fig. 2I). At the early timepoint, a conditionally active SRF co-activator MAL/MRTF (ΔN-MAL) did not have similar effect on GLS1 accumulation. We also explored the role of Rho/SRF pathway in glutaminolysis with a small molecule inhibitor CCG-1423, which targets Rho/SRF transcriptional signaling.21 Similar to Rho-specific inhibitor (Fig. 2F), also CCG-1423 reduced the intracellular glutamate levels and blocked Myc's capacity to stimulate glutaminolysis (Fig. 2J). Furthermore, consistent with the findings that SRF upregulates GLS1 (Fig. 2I), both the constitutively active SRF VP16 and the induciby activated ΔN-MAL upregulated the cellular glutamate levels (Fig. 2J). The inhibition of Rho/SRF transcriptional signaling was strongly synthetic lethal with Myc (Fig. 2K) and, vice versa, the enforced activation of SRF with ΔN-MAL partially rescued the MycON;RhoAOFF-induced apoptosis (Fig. 2L). The results suggest that RhoA/SRF signaling and Myc cooperate to establish a pattern of glutaminolysis that is critical for the survival of transformed cells.
The results from the cell based assays suggested that Myc expressing cancer cells could strongly benefit from SRF co-expression. We addressed the question by determining the status of Myc and SRF expression in a panel of 39 clinical breast cancer samples. The expression status of Myc and SRF was blindly evaluated in immunohistochemically stained samples and the samples were categorized to high (>50% cells Myc or SRF positive) and low/negative expression groups (0–50% cells positive). Interestingly, 67% of the samples with high Myc levels also harbored high level SRF expression (Fig. 2M). The data suggest mutual requirement of Myc and SRF in context of breast cancer.
Together, we show evidence that RhoA/SRF signaling regulates glutaminolysis in epithelial cells and that this pathway plays a critical role in cancer cell anaplerosis (Fig. 2N). Our findings that oncogenic Myc is dependent on RhoA/SRF signaling to satisfy the glutamine needs of metabolic transformation also exposes a potential cancer cell vulnerability that could be therapeutically exploited with RhoA/SRF pathways inhibitors.
Discussion
The present study clarifies the mechanistic basis for synthetic lethal interaction between loss of RhoA and oncogenic Myc, originally found in a shRNA screen aiming to expose interactions between the epithelial integrity and cell cycle machinery in 3D mammary epithelial culture.14 Synthetic lethal interactions, especially pharmacological treatments found to interact with cancer-specific genetic lesions to selectively induce death of cancer cells, hold significant clinical potential in cancer therapeutics. For example, clinical proof of concept has been achieved by targeting PARP inhibitors against BRCA mutated tumors.22,23 Interestingly, recent evidence also suggests that antiproliferative responses to targeted therapies can be transformed into more therapeutically beneficial apoptotic (synthetic lethal) responses through combining antiproliferative intervention with inhibition of cancer cell specific metabolic processes, such as glutamine utilization.24
We show in the present study that not only siRNA mediated silencing of RhoA, but also a pharmacological inhibition of Rho or Rho-SRF pathway activity is robustly synthetic lethal with oncogenic Myc. The findings suggest that drugs directed to inhibit RhoA pathway possess a translational potential as cancer cell apoptosis inducing modalities. Moreover, the present study shows evidence that the RhoA-dependent synthetic lethal activity may not be due to loss of classical RhoA functions in regulation of cytoskeleton dynamics but instead, the lethal effects are coupled to very recently documented functions of Rho GTPases in regulation of mitochondrial glutamine metabolism.25-27 The present findings suggest that RhoA and Myc together feed glutamine-dependent anaplerosis, which is critical for TCA function in cancer cells to provide energy and macromolecular material sources (Fig. 2N). The increased occupancy of GLS1 transcription regulating sites by activated Myc suggests that Myc contributes to glutaminolysis via GLS1 upregulation. While RhoA-SRF signaling cooperated with Myc in induction of GLS1, we could not obtain evidence for direct SRF regulation of GLS1. Therefore, the exact mechanisms of RhoA/SRF regulation of GLS1 remain to be clarified by future studies.
In summary, our results expose a new cancer cell vulnerability that derives from the specific requirement of Myc transformed cells to sustain RhoA/SRF signaling to maintain glutamine-dependent anaplerosis. Myc is among the commonest oncogenes in all cancers and a key driver of metabolic transformation.28 Therefore, RhoA/SRF signaling could present a new target for therapeutic strategies aiming to exploit the metabolic vulnerability of transfomed cells that is prevalent in cancers with high Myc expression level
Materials and methods
Cell culture
MCF10A cells were obtained from American Type Culture Collection (ATCC) and were previously transduced with pBabe-puro-MycERtm construct. Cells were cultured in MCDB-170 (US Biological) with 70 μg/ml BPE, 5 μg/ml insulin, 0.5 μl/ml hydrocortison, L-glutamine, 5 ng/ml human EGF, 5 μg/ml transferrin and 0.01 μM isopropretenol. ΔN-MAL and SRF-VP16 plasmids were a kind gift from Prof. Martin Eilers Laboratory (University of Würzburg).
Reagents and antibodies
The following reagents were used: 4-Hydroxytamoxifen (4-OHT, Sigma-Aldrich), Rho inhibitor C3, Rho activator II (CT04, Cytoskeleton), ROCK inhibitor Y-27632, RhoA/SRF inhibitor CCG-1423, Myosin lightchain kinase inhibitor ML-7 (Sigma-Aldrich) and recombinant human TRAIL (R&D systems). The following antibodies were used in immunoblotting and immunofluorescence stainings: Cleaved caspase-3 Asp175 (#9661, Cell Signaling), SRF G-20 (sc-335, Santa Cruz), Beta actin (Santa Cruz) and GLS1 (ab93434, AbCam). In immunohistochemistry we used Myc Y69 (ab32072, AbCam) and SRF (OAAF00701, Aviva Systems Biology).
RNA interference
For 6-well 15 nM of human specific RhoA siRNA (Hs_RhoA_6 FlexiTube siRNA, Qiagen) was diluted to 400 μl medium. 12 μl HiPerFect Transfection reagent was added and the mix was incubated 10 min RT, after which the mix was plated and 250 000 cells were added on top. The plate was incubated 48–72 h and the cells were lysed for analysis or fixed for stainings. RNA was extracted with RNeasy mini kit (Qiagen) according to manufacturer's instructions and siRNA efficacy was validated with real-time qPCR in Biomedicum Functional Genomics Unit (FuGU).
Immunofluorescence stainings and imaging
Cells grown on coverslips were washed with PBS and fixed with 4% PFA for 15 minutes. Then the cells were permeabilized with 0,1% Triton-X-PBS, after which they were blocked for non-specific binding sites using 1% BSA-PBS. The cells were incubated for 60 minutes with primary antibody and washed 3 times with PBS, incubated with secondary antibody (Life Technologies, Alexa fluor conjugated) for 45 minutes, washed and the nuclei were counterstained with Hoechst. The coverslips were mounted using Immu-Mount mounting reagent (Thermo Scientific). Microscopy analyses were performed with Zeiss Axioplan fluorescence microscope with filters for Alexa 488 and Hoechst and imaged with Zeiss AxioCam HRc color camera (Biomedicum Imaging Unit, BIU).
G-LISA RhoA activity assay
RhoA GTPase activity was measured using G-LISA RhoA Activation Assay Kit (Cytoskeleton) according to the manufacturer's instructions. Cells were cultured on 6-well plates (300 000 cells/well) and lysed with 70 μl/well cell lysis buffer included in the kit. Luminescence was measured with VICTORTMX3 (Perking Elmer).
Apoptosis assay
Cells were seeded on 96-well plates (40 000 cells/well), treated, and apoptosis was detected with Caspase-Glo® 3/7 luminescence kit (Promega). Luminescence was measured with VICTORTMX3 (Perking Elmer).
Lysates and immunoblot analysis
The cells were lysed with RIPA buffer supplemented with protease inhibitor cocktail (complete mini-EDTA free, Roche) and harvested with scraper. Cells were incubated on ice for 10 minutes, after which the nuclei were broken using a needle and syringe. Lysates were centrifuged at +4°C and the supernatants collected. The protein concentration was measured using BioRad DCtm Protein assay and VICTORTMX3 96-well plate reader (Perking Elmer). SDS-page and Western blot analysis were performed with standard protocols.
ChIP-sequencing
ChIP-sequencing was performed as described,29 MCF10A-MycER cells were treated with control ethanol or 100 nM 4-OHT to activate Myc for 24 h, after which the cells were crosslinked with 10 min incubation at room temperature with 1% formaldehyde, and the reaction was stopped by adding glycine. The cells were collected with scraping, centrifugated and lysed with RIPA-buffer. The crosslinked chromatin was sonicated into 100–400 bp fragments followed by centrifugation to remove cellular debris. 25 μg of antibodies against Myc (Millipore 06–340) or control rabbit IgG (sc-2027) was added to the lysate and samples were rotated overnight at +4°C. After immunoprecipitations, samples were RNAse A & Proteinase K treated and reverse-crosslinked at +64°C, followed by phenol:chloroform:isoamyl alcohol (25:24:1)-extraction and ethanol precipitation. Fragmented IP-DNA was end-repaired by using Klenow and T4 DNA polymerases and T4 polynucleotide kinase (Thermo), A-nucleotide was added with Klenow fragment exo (Thermo) and Illumina adapters were ligated with T4 DNA ligase (NEB). The amplified 200–300 bp fragments were sequenced using Illumina HiSeq2000 (single 36 bp reads) system. Sequence reads were mapped to the hg18 human reference genome by bwa. Duplicate reads were removed and sequencing depth was normalized by randomly sampling the mapped reads to match the sample with the fewest reads. Peak-calling was performed as in ref. 30 with 200 bp average fragment length.
Glutamate assay
Intracellular glutamate was measured using Glutamate Colorimetric Assay Kit (#K629–100, BioVision) according to the manufacturer's instructions.
Funding Statement
This work was funded by the Academy of Finland, TEKES and Finnish Cancer Organizations, H.M.H was funded by Integrative Life Sciences (ILS) doctoral program, Emil Aaltonen foundation and Inkeri & Mauri Vänskä foundation.
Abbreviations
- α-KG
α-ketoglutarate
- Gln
Glutamine
- GLS1
Glutaminase
- Glu
Glutamate
- MLCK
Myosin lightchain kinase
- ROCK
Rho-associated kinase
- SRF
Serum response factor
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Biomedicum Functional Genomics Unit (FuGu) and Biomedicum Imaging Unit (BIU) for core services, Tiina Raatikainen and Tarja Välimäki for the technical support and Leevi Viisanen for help with immunohistochemical stainings.
References
- [1].Jones RG, Thompson CB. Tumor suppressors and cell metabolism: a recipe for cancer growth. Genes Dev 2009; 23(5):537-548; PMID:19270154; https://doi.org/ 10.1101/gad.1756509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Dang CV. MYC on the path to cancer. Cell 2012; 149(1):22-35; PMID:22464321; https://doi.org/ 10.1016/j.cell.2012.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Yuneva M, Zamboni N, Oefner P, Sachidanandam R, Lazebnik Y. Deficiency in glutamine but not glucose induces MYC-dependent apoptosis in human cells. J Cell Biol 2007; 178(1):93-105; PMID:17606868; https://doi.org/ 10.1083/jcb.200703099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, et al.. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A 2008; 105(48):18782-18787; PMID:19033189; https://doi.org/ 10.1073/pnas.0810199105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res 2009; 15(21):6479-6483; PMID:19861459; https://doi.org/ 10.1158/1078-0432.CCR-09-0889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Le A, Lane AN, Hamaker M, Bose S, Gouw A, Barbi J, Tsukamoto T, Rojas CJ, Slusher BS, Zhang H, et al.. Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab 2012; 15(1):110-121; PMID:22225880; https://doi.org/ 10.1016/j.cmet.2011.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lacey JM, Wilmore DW. Is glutamine a conditionally essential amino acid? Nutrition reviews. 1990; 48(8):297-309; PMID:2080048 [DOI] [PubMed] [Google Scholar]
- [8].Dang CV. MYC, metabolism, cell growth, and tumorigenesis. Cold Spring Harb Perspect Med 2013; 3(8):a014217; PMID:23906881; https://doi.org/ 10.1101/cshperspect.a014217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Orgaz JL, Herraiz C, Sanz-Moreno V. Rho GTPases modulate malignant transformation of tumor cells. Small GTPases 2014; 5:e29019; PMID:25036871; https://doi.org/ 10.4161/sgtp.29019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Vega FM, Ridley AJ. Rho GTPases in cancer cell biology. FEBS letters. 2008; 582(14):2093-2101; PMID:18460342; https://doi.org/ 10.1016/j.febslet.2008.04.039 [DOI] [PubMed] [Google Scholar]
- [11].Sahai E, Marshall CJ. RHO-GTPases and cancer. Nat Rev Cancer 2002; 2(2):133-142; PMID:12635176; https://doi.org/ 10.1038/nrc725 [DOI] [PubMed] [Google Scholar]
- [12].Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, Wilson KF, Ambrosio AL, Dias SM, Dang CV, et al.. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010; 18(3):207-219; PMID:20832749; https://doi.org/ 10.1016/j.ccr.2010.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wilson KF, Erickson JW, Antonyak MA, Cerione RA. Rho GTPases and their roles in cancer metabolism. Trends Mol Med 2013; 19(2):74-82; PMID:23219172; https://doi.org/ 10.1016/j.molmed.2012.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Marques E, Klefstrom J. Par6 family proteins in cancer. Oncoscience. 2015; 2(11):894-895; PMID:26697513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Nieminen AI, Partanen JI, Hau A, Klefstrom J. c-Myc primed mitochondria determine cellular sensitivity to TRAIL-induced apoptosis. EMBO J. 2007; 26(4):1055-1067; PMID:17268552; https://doi.org/ 10.1038/sj.emboj.7601551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Nieminen AI, Eskelinen VM, Haikala HM, Tervonen TA, Yan Y, Partanen JI, Klefström J. Myc-induced AMPK-phospho p53 pathway activates Bak to sensitize mitochondrial apoptosis. Proc Natl Acad Sci U S A 2013; 110(20):E1839-1848; PMID:23589839; https://doi.org/ 10.1073/pnas.1208530110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Dang CV, Le A, Gao P. MYC-induced cancer cell energy metabolism and therapeutic opportunities. Clin Cancer Res 2009; 15(21):6479-6483; PMID:19861459; https://doi.org/ 10.1158/1078-0432.CCR-09-0889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Chen L and Cui H. Targeting glutamine induces apoptosis: A cancer therapy approach. Int J Mol Sci 2015; 16(9):22830-22855; PMID:26402672; https://doi.org/ 10.3390/ijms160922830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wiese KE, Haikala HM, von Eyss B, Wolf E, Esnault C, Rosenwald A, Treisman R, Klefström J, Eilers M. Repression of SRF target genes is critical for Myc-dependent apoptosis of epithelial cells. EMBO J 2015; 34(11):1554-1571; PMID:25896507; https://doi.org/ 10.15252/embj.201490467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Sotiropoulos A, Gineitis D, Copeland J, Treisman R. Signal-regulated activation of serum response factor is mediated by changes in actin dynamics. Cell 1999; 98(2):159-169; PMID:10428028; https://doi.org/ 10.1016/S0092-8674(00)81011-9 [DOI] [PubMed] [Google Scholar]
- [21].Evelyn CR, Wade SM, Wang Q, Wu M, Iñiguez-Lluhí JA, Merajver SD, Neubig RR. CCG-1423: a small-molecule inhibitor of RhoA transcriptional signaling. Mol Cancer Ther 2007; 6(8):2249-2260; PMID:17699722; https://doi.org/ 10.1158/1535-7163.MCT-06-0782 [DOI] [PubMed] [Google Scholar]
- [22].Helleday T, Bryant HE, Schultz N. Poly(ADP-ribose) polymerase (PARP-1) in homologous recombination and as a target for cancer therapy. Cell Cycle 2005; 4(9):1176-1178; PMID:16123586; https://doi.org/ 10.4161/cc.4.9.2031 [DOI] [PubMed] [Google Scholar]
- [23].Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al.. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434(7035):917-921; PMID:15829967; https://doi.org/ 10.1038/nature03445 [DOI] [PubMed] [Google Scholar]
- [24].Li J, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al.. Synthetic lethality of combined glutaminase and Hsp90 inhibition in mTORC1-driven tumor cells. Proc Natl Acad Sci U S A 2015; 112(1):E21-29; PMID:25524627; https://doi.org/ 10.1073/pnas.1417015112 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [25].Wang JB, Erickson JW, Fuji R, Ramachandran S, Gao P, Dinavahi R, Wilson KF, Ambrosio AL, Dias SM, Dang CV, et al.. Targeting mitochondrial glutaminase activity inhibits oncogenic transformation. Cancer Cell 2010; 18(3):207-219; PMID:20832749; https://doi.org/ 10.1016/j.ccr.2010.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Youngblood VM, Kim LC, Edwards DN, Hwang Y, Santapuram PR, Stirdivant SM, Lu P, Ye F, Brantley-Sieders DM, Chen J. The Ephrin-A1/EPHA2 signaling axis regulates glutamine metabolism in HER2-positive breast cancer. Cancer Res 2016; 76:1825-36; PMID:26833123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Lukey MJ, Greene KS, Erickson JW, Wilson KF, Cerione RA. The oncogenic transcription factor c-Jun regulates glutaminase expression and sensitizes cells to glutaminase-targeted therapy. Nat Commun 2016; 7:11321; PMID:27089238; https://doi.org/ 10.1038/ncomms11321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Miller DM, Thomas SD, Islam A, Muench D, Sedoris K. c-Myc and cancer metabolism. Clin Cancer Res 2012; 18(20):5546-5553; PMID:23071356; https://doi.org/ 10.1158/1078-0432.CCR-12-0977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Tuupanen S, Turunen M, Lehtonen R, Hallikas O, Vanharanta S, Kivioja T, Björklund M, Wei G, Yan J, Niittymäki I, et al.. The common colorectal cancer predisposition SNP rs6983267 at chromosome 8q24 confers potential to enhanced Wnt signaling. Nature Gen 2009; 41(8):885-890; PMID:19561604; https://doi.org/ 10.1038/ng.406 [DOI] [PubMed] [Google Scholar]
- [30].Wei GH, Badis G, Berger MF, Kivioja T, Palin K, Enge M, Bonke M, Jolma A, Varjosalo M, Gehrke AR, et al.. Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J 2010; 29(13):2147-2160; PMID:20517297; https://doi.org/ 10.1038/emboj.2010.106 [DOI] [PMC free article] [PubMed] [Google Scholar]