

ABSTRACT
Therapeutic role of NK cells in solid tumors was challenged previously even though their role in hematological malignancies has clearly been established. Furthermore, functions and numbers of NK cells are greatly suppressed in oral cancer patients necessitating effective future NK immunotherapeutic strategies to aid in the control of disease. The humanized-BLT (hu-BLT) mice were used to implant stem-like/undifferentiated oral tumors to study the role of super-charged NK cells with and without feeding with AJ2 probiotic bacteria. Implanted CSC/undifferentiated tumors resected from NK-injected mice exhibited differentiated phenotype, grew slowly, and did not cause weight loss, whereas those from tumor-bearing mice without NK-injection remained relatively more stem-like/poorly-differentiated, grew faster, and caused significant weight loss. Moreover, in vitro NK-differentiated tumors were sensitive to chemotherapeutic drugs, and when implanted in the oral-cavity grew no or very small tumors in mice. When NK-mediated differentiation of tumors was blocked by IFN-γ and TNF-α antibodies before implantation, tumors grew rapidly, remained stem-like/poorly-differentiated and became resistant to chemotherapeutic drugs. Loss of NK cytotoxicity and decreased IFN-γ secretion in tumor-bearing mice in PBMCs, splenocytes, bone marrow derived immune cells and enriched NK cells was restored by the injection of super-charged NK cells with or without feeding with AJ2. Much greater infiltration of CD45+ and T cells were observed in tumors resected from the mice, along with the restored secretion of IFN-γ from purified T cells from splenocytes in NK-injected tumor-bearing mice fed with AJ2 probiotic bacteria. Thus, super-charged NK cells prevent tumor growth by restoring effector function resulting in differentiation of CSCs/undifferentiated-tumors in hu-BLT mice.
KEYWORDS: Differentiation, BLT-NSG, IFN-γ, NK, OSCSCs, OSCCs
Introduction
Significant challenges remain in finding effective therapeutic modalities to eliminate oral cancer.1,2 It has been shown that cancer stem cells (CSCs)/undifferentiated tumors are the cause of relapse, metastasis and resistance of tumors to chemo- and radiotherapy.3-5 CSCs/undifferentiated tumors evade host immunity through absence or downregulation of MHC class I (MHC-I).6 Lower/no expression of MHC-I on head and neck on cancer cells correlates positively with disease progression and therapeutic unresponsiveness7,8; therefore, growth and expansion of CSCs/undifferentiated tumors with no or low expression of MHC-I could be the reason for the limited success of T cell-based immunotherapies, however, such tumors remain targets of NK cells.7,9,10 It has been shown that freshly isolated tumor infiltrating NK cells or NK cells from peripheral blood of cancer patients are defective in both number and function.11-13 The density of NK cells is lower in oral tumors graded as poor prognostic group compared to those in good prognostic group.11
We have shown previously that Oral Squamous Carcinoma Stem Cells (OSCSCs) express CD44high EpCAMhigh CD26high CD166low CD338+ oral stem cell markers14,15 and are targets of NK cell-mediated cytotoxicity, and trigger IFN-γ secretion, whereas their differentiated counterpart, Oral Squamous Cell Carcinoma (OSCCs), are more resistant and trigger no or lower IFN-γ secretion by the NK cells.16 We have also shown that NK cells play a significant role in tumor differentiation by providing critical signals via secreted cytokines and direct cell-cell contact.15 We have recently demonstrated that NK-differentiated CSCs/undifferentiated tumors are resistant to NK cell-mediated cytotoxicity and trigger lower cytokine secretion from the NK cells.17 Such effects mediated by NK cells are shown to aid in decreasing growth, invasion and metastasis of undifferentiated tumors. Recently, several humanized mouse models have been developed, with the humanized BLT (hu-BLT) mouse model being one of the best preclinical models that exhibits a full repertoire of interactions that occur between the tumor, its environment and the immune system.18,19
In this paper, we demonstrate that CSCs/undifferentiated tumor cells isolated from oral tumor patient can give rise to solid tumors in hu-BLT mice, in the presence of a reconstituted, competent T and B cells. We also demonstrate that injection of super-charged NK cells in vivo decreases tumor growth by selection and differentiation of CSCs/undifferentiated tumors. Furthermore, NK-differentiated tumors become susceptible to chemotherapeutic drugs. Accordingly, we propose that combining autologous or allogeneic super-charged NK cell immunotherapy with chemotherapy may represent an effective strategy for treating patients with oral tumors.
Results
Single injection of super-charged NK cells inhibited OSCSCs tumor growth, and significantly improved health of the mice
Hu-BLT mice were generated and human OSCSCs were implanted in the floor of the mouth of NSG and hu-BLT mice (Fig. 1A and 1B) and weight loss was monitored on a weekly basis (Fig. 1C). Single injection of super-charged NK cells resulted in lower weight loss of mice implanted with OSCSCs (Fig. 1C). Mice implanted with OSCSCs and injected with NK cells did not exhibit morbidity, and were able to intake food; whereas mice with oral tumors in the absence of NK injection became morbid, had complications in ingesting food due to growing tumors (data not shown) and exhibited rapid weight loss (Fig. 1C). Interestingly, tumor-bearing hu-BLT mice without NK injection had less weight loss when compared to tumor-bearing NSG mice, indicating that reconstituted human immune cells were able to limit tumor growth slightly but not efficiently (Fig. 1C). Therapeutic effect of NK injection in hu-BLT mice was also seen when the tumor sizes were compared after tumor resection. Tumors from tumor-bearing hu-BLT mice without NK injection were much larger than those of NK-injected tumor-bearing hu-BLT mice (Fig. 1D and 1E). Tumor weights remained substantially less in NK or NK-injected/AJ2 fed mice (Fig. 1F), in comparison to the large tumors, which were formed in tumor-bearing mice that did not receive NK treatment (Fig. 1D–1F). In addition, in agreement with the weight loss data tumor-bearing hu-BLT mice without NK injection had slightly smaller tumors when compared to tumor-bearing NSG mice, indicating that reconstituted human immune cells were able to limit tumor growth slightly but not efficiently (data not shown).
Figure 1.
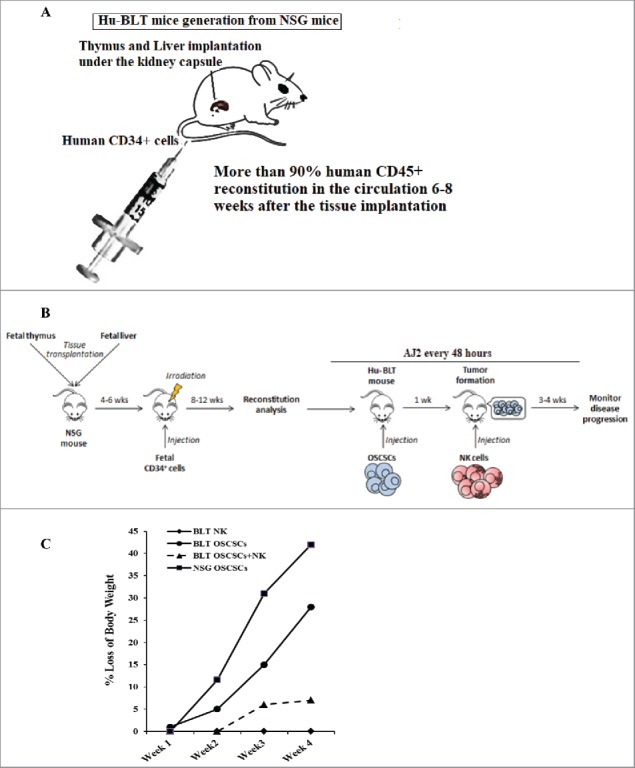
Single injection of super-charged NK cells with/without feeding AJ2 inhibited tumor growth in hu-BLT mice. Hu-BLT mice were generated as described in Materials and Methods, and shown in figure (A). Hu-BLT and NSG mice were implanted orthotopically with 1 × 106 human OSCSCs into the floor of the mouth, and after 7–10 days a group of hu-BLT mice were injected with 1.5 × 106 super-charged NK cells through tail vein, and mice were monitored for disease progression. Another group of hu-BLT mice were fed with AJ2 probiotic bacteria 5 billion/day every 48 hours 2 weeks prior to the implantation of OSCSCs and after implantation of the tumors in the presence and absence of NK injection until the experiments were terminated (B). Weight loss was monitored by weighing the mice on a weekly basis. One of 3 representative experiments is shown in this figure (C). Upon termination of the experiment, mice were sacrificed, and the pictures of tumors were taken after resection (D), and weighed (n = 4) (E). Mice were implanted with human OSCSCs and injected with NK cells and fed with AJ2, as shown in Fig. 1B, and the tumors were resected and weighed post mortem (n = 4) (F). PBMCs were isolated from hu-BLT mice and humans and surface expression of human CD3 (n = 5) (G), CD4 (n = 5) (H), CD8 (n = 5) (I), CD19 (n = 3) (J) and CD16 (n = 5) (K) were determined within CD45+ immune cells using antibody staining followed by flow cytometric analysis.
Figure 1.

(Continued).
Hu-BLT mice exhibited greater than 96%-99% reconstitution with hCD45+ immune cells in BM, spleen and peripheral blood when considering contaminating mouse CD45+ immune cells (Fig. S1). The profile of CD3+T cells (Fig. 1G), CD3+CD4+T cells (Fig. 1H) and CD3+CD8+T cells (Fig. 1I) in peripheral blood of hu-BLT mice resembled that of human peripheral blood; however, the percentage of B cells was slightly higher (Fig. 1J) while percentage of NK cells was less (Fig. 1K) in hu-BLT mice compared to humans.
Loss of NK cytotoxicity and IFN-γ secretion in tumor-bearing mice within all tissue compartments, and restoration with NK injection and/or feeding AJ2 and anti-PD1 injection
Tumor-bearing mice exhibit lower NK-mediated cytotoxicity in splenocytes (Fig. 2A), BM-derived immune cells (Fig. 2B), PBMCs (Fig. 2C), and CD3+-depleted splenocytes (Fig. 2D). NK-injected tumor-bearing mice, both alone and in combination with feeding AJ2 exhibited elevated NK cytotoxicity in all tissue compartments, with the highest increase observed in mice fed AJ2, injected with NK cells and anti-PD1 antibody (Figs. 2A-2C). When T cells were depleted from the splenocytes by CD3+ positive selection kit, similar profiles of NK cytotoxicity could be seen in CD3+-depleted splenocytes as observed in Fig. 2A (Fig. 2D). Splenocytes (Fig. 2E and Table S1), BM-derived immune cells (Fig. 2F), PBMCs (Fig. 2G), and CD3+ T cells from splenocytes (Fig. 2H) of tumor-bearing hu-BLT mice secreted lower IFN-γ. Injection of anti-PD1 antibody in combination with NK cells and AJ2 either had no or decreased effect on IFN-γ secretion in all tissue compartments when compared to NK and AJ2 groups (Fig. 2E–2G). NK-injected tumor-bearing mice showed elevated IFN-γ secretion within all tissue compartments, with the highest increase observed when mice were also fed AJ2 (Figs. 2E–2H). When T cells were depleted from the splenocytes by CD3+ positive selection kit, the tumor-bearing mice that received NK injection had the highest increase in IFN-γ secretion as compared to tumor-bearing mice in the absence of NK injection, or NK injected non-tumor bearing control mice (Fig. 2I).
Figure 2.
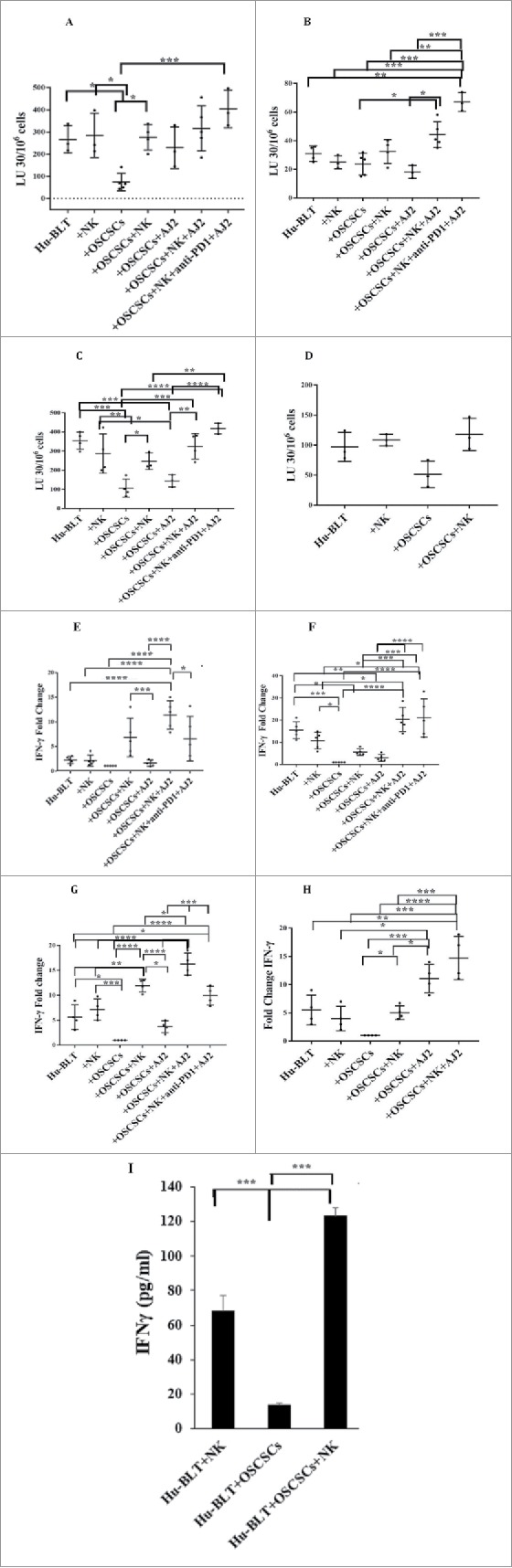
Injection of super-charged NK cells with/without feeding AJ2 restored and increased IFN-γ secretion and cytotoxic function of NK-cells in blood, spleen, BM, enriched-NK cells, and purified CD3+T cells in tumor-bearing hu-BLT mice. Hu-BLT mice were implanted with human OSCSCs and injected with NK cells, and fed AJ2 as described in Fig. 1B, and a week after NK cell injection mice were injected with anti-PD1 (50 µg/mice) via tail-vein injection. Following sacrifice, spleen (n = 5) (A), BM (n = 5) (B) and peripheral blood (n = 5) (C) were collected, single cell suspensions were prepared from each tissue and (1 × 106 cells/ml for spleen and BM and 0.7 × 106 cells/ml for PBMCs) were treated with IL-2 (1000 U/ml) for 7 days. NK enriched cells were isolated from splenocytes, and (1 × 106 cells/ml) were treated with IL-2 (1000 U/ml) for 7 days (n = 3) (D). Cytotoxicity assays were performed using standard 4-hour 51Cr release assay against OSCSCs, and the LU 30/106 cells were determined using inverse number of cells required to lyse 30% of OSCSCs x100. Splenocytes (n = 5) (E), BM cells (n = 5) (F), PBMCs (n = 5) (G) at (1 × 106 cells/ml for spleen and BM and 0.7 × 106 cells/ml for PBMCs) were each treated with IL-2 (1000 U/ml), and positively selected CD3+T cells (n = 4) from the splenocytes at 1 × 106 cells/ml were treated with IL-2 (100 U/ml) (H), T cell depleted splenocytes were cultured at 1×106 cells/ml and treated with IL-2 (1000 U/ml) for 7 days, after which the supernatants were harvested and the levels of IFN-γ were determined using specific ELISAs (I). Fold changes in IFN-γ secretion in each tissue from each group of mice were determined over those obtained from mice injected with OSCSCs alone (E-H).
Increased proportions of CD3+CD8+T cells in various tissue compartments when hu-BLT mice were injected with NK cells and fed AJ2
Increased proportions of CD3+ or CD3+CD8+T cells were seen in BM (Fig. 3A), spleen (Fig. 3B) and blood (data not shown) of mice injected with NK cells alone or in combination with AJ2 feeding (Table S2 and S3). When considering the percentages of T cells at the time of sacrifice, PBMCs, spleen and BM exhibited higher percentages of CD3+ T cells, and BM exhibited an elevation in HLADR+CD11B+ immune subset in tumor-bearing mice injected with NK cells alone or in combination with AJ2 feeding (Table S2). Percentages of CD3+ T cells and CD3+CD8+T cells increased in tumor-bearing mice injected with NK cells alone or in combination with AJ2 feeding when BM and splenocytes were cultured (Table S3). Similarly, tumors dissociated from tumor-bearing mice injected with NK and/or fed AJ2 demonstrated elevated levels of CD3+T cells at the time of sacrifice (Table S4).
Figure 3.
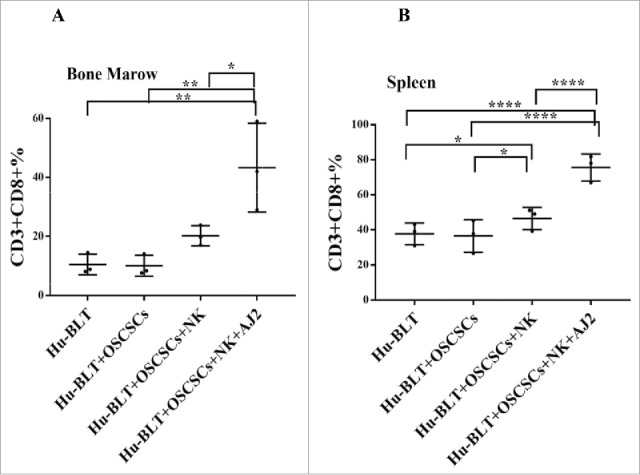
Single injection of super-charged NK cells with/without AJ2 feeding increased numbers of CD8+T cells in hu-BLT mice. Hu-BLT mice were implanted with OSCSCs and injected with NK cells, and fed with AJ2, as described in Fig. 1B, and the percentages of human CD8+ T cells within BM cells (n = 3) (A) and splenocytes (n = 3) (B) were determined using antibody staining followed by flow cytometric analysis.
NK injection and/or feeding AJ2 inhibit growth and progression of stem-like oral tumors, and differentiate CSCs in vivo in hu-BLT mice
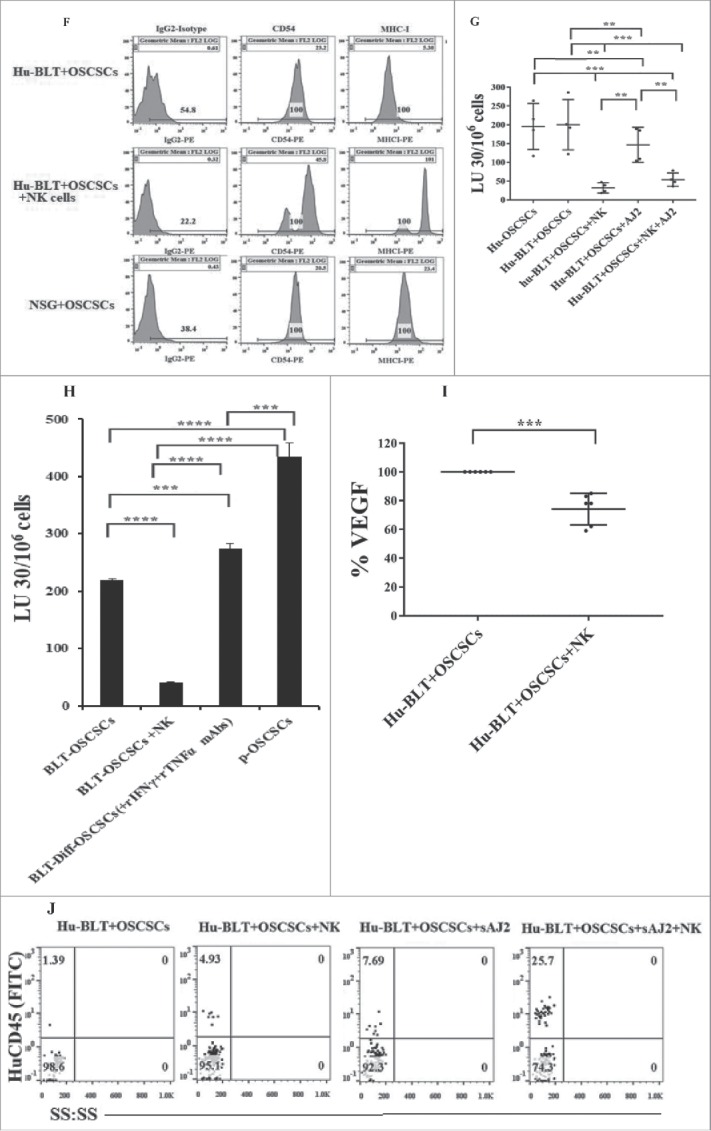
Tumor cells from NSG (Figs. 4A and S2A) and hu-BLT mice without NK injection grew rapidly, whereas those with NK cells did not grow or grew very slowly (Figs. 4A–4C and S2A-S2C). Similarly, tumors from hu-BLT mice implanted with NK supernatant-differentiated tumors did not grow (Fig. S2B and 4C), and blocking tumor differentiation with the combination of anti-IFN-γ and anti-TNF-α antibodies before implantation restored tumor growth in vivo (Figs. S2B, 4B and 4C). Tumor growth was less in NK-injected mice fed AJ2 in comparison to NK alone-injected mice (Figs. S2C and 4C), and both were substantially less than those which only received oral tumor implantation (Figs. S2C and 4C). Nine to 11-fold more hCD45+ immune cells infiltrated in tumors of tumor-bearing mice injected with NK cells when compared to tumor alone mice (Fig. 4D). The percentages of epithelial cells expressing surface EpCAM in tumors were approximately 5-fold higher from tumor-bearing mice without NK injection when compared to NK-injected tumor-bearing mice (Fig. S2D).
Figure 4.
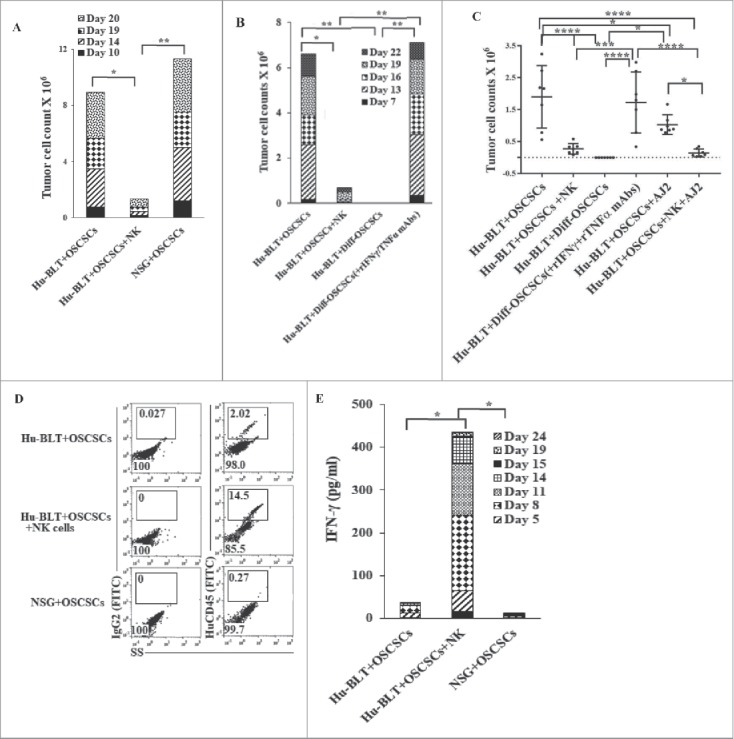
Single injection of super-charged NK cells with/without AJ2 feeding in hu-BLT mice mediated in vivo tumor differentiation, increased IFN-γ secretion and mobilized increased numbers of human immune cells to the tumors, and resulted in decreased ex-vivo tumor growth. Hu-BLT and NSG mice were implanted with OSCSCs and injected with NK cells, as described in Fig. 1B. Following sacrifice, oral tumors were harvested, and single cell suspensions were prepared and the same numbers of cells (total of 3 × 106 cells at 1 × 106 cells/ml) from each group were cultured at day 0. On day 10 supernatants were removed and attached tumor cells were counted, and for subsequent cultures the numbers in each group were adjusted to those obtained from NK injected mice since they expanded the least numbers of tumors. On days 10, 14, 19 and 20 the total numbers of ex-vivo expanding tumor cells were determined in each group. One of several representative experiments is shown in this figure (A). Hu-BLT mice were implanted with OSCSCs or in vitro NK-differentiated-OSCSCs (diff-OSCSCs), or NK-differentiated-OSCSCs treated with antibodies against IFN-γ and TNF-α to block differentiation, followed by NK injection in hu-BLT mice as described in Fig. 1B. Following sacrifice, oral tumors were dissociated, and single cells were prepared and cultured at (total of 3 × 106 cells at 1 × 106 cells/ml), and the numbers of expanding tumor cells were determined as described in Fig. 4A. One of several representative experiments is shown in this figure (B). Hu-BLT mice were implanted with OSCSCs or diff-OSCSCs, or diff-OSCSCs treated with antibodies against IFN-γ and TNF-α, and injected with NK cells and/or fed with AJ2 as described in Fig. 1B. Following sacrifice, oral tumors were harvested and cultured and the numbers of expanding tumor cells were determined as described in Fig. 4A (n = 7) (C). Hu-BLT and NSG mice were implanted with OSCSCs, followed by NK injection in hu-BLT mice as described in Fig. 1A. Oral tumors were harvested, and single cell suspensions were prepared. The percentages of infiltrating hu-CD45+ immune cells within the non-attached cells at day 12 of culture were determined using antibody staining followed by flow cytometric analysis. One of three representative figures is shown in this figure (D). Oral tumors from hu-BLT and NSG mice were cultured as described in Fig. 4A and treated with IL-2 (1000 U/ml), and their supernatants were harvested on days shown in the figure and the levels of IFN-γ were determined using ELISA. One of several representative figures is shown in this figure (E). Expression of human CD54 and MHC-I were assessed on day 10 of oral tumor cultures from hu-BLT and NSG mice using flow cytometric analysis after staining with their respectiv antibodies. One of several representative experiments is shown in this figure (F). Purified NK cells (1 × 106 cells/ml) from the peripheral blood of the healthy human donor were left untreated or treated with IL-2 (1000 U/ml) for 18 hours before they were added to 51Cr labeled OSCSCs cultured from the resected tumors of different experimental groups of hu-BLT mice, and compared it to the cultures of OSCSCs maintained in the lab at various effector to target ratios. NK cell-mediated cytotoxicity was determined using a standard 4-hour 51Cr release assay. LU30/106 cells were determined as described in the Materials and Methods (n = 4) (G and H). Oral tumor cells from hu-BLT mice as described in Fig. 4A were treated with IL-2 (1000 U/ml), and supernatants were harvested after 3 and 7 days and the levels of VEGF secretion were determined using specific ELISAs. The decrease in VEGF secretion by tumors obtained from NK-injected animals (n = 6) were calculated based on the amounts obtained from OSCSCs alone injected mice (I). Infiltrating percentages of hu-CD45+ immune cells within the oral tumors dissociated from different experimental groups of hu-BLT mice as described in Fig. S2C were determined using flow cytometric analysis after staining with antibody. One of several representative experiments is shown in this figure (J).
Figure 4.
(Continued).
Significantly higher IFN-γ secretion was obtained from tumors of tumor-bearing mice injected with NK cells at different days of cultures when compared to those from tumor-bearing hu-BLT or NSG mice (Fig. 4E). Expression of CD54 and MHC-I were higher on tumors obtained from NK-injected tumor-bearing mice as compared to hu-BLT and NSG mice, which only received tumors (Fig. 4F). Tumors from NK-injected tumor-bearing mice in the absence and presence of feeding with AJ2 were highly resistant to NK cell-mediated cytotoxicity when compared to tumors from tumor-bearing mice without NK injection demonstrating their differentiated phenotype (Fig. S2E and Figs. 4G and 4H). When NK-mediated differentiation of tumor cells was blocked with anti-IFN-γ and anti-TNF-α antibodies before implantation, the susceptibility of tumors to NK cell-mediated cytotoxicity was restored (Fig. 4H). Oral tumors from hu-BLT mice injected with NK cells secreted relatively less VEGF when secretion were set at 100% in tumor-bearing mice and the extent of decrease were assessed in NK injected tumor-bearing mice (Fig. 4I).
Tumors from NK-injected hu-BLT mice had higher levels of hCD45+ infiltrating lymphocytes, and the highest increase was seen in those which were injected with NK cells and fed with AJ2 (Fig. 4J). The majority of infiltrating CD45+ immune cells were CD3+T cells with CD4+T cell subsets having moderately higher proportions than CD8+ T cells, and demonstrating CD3+CD56+CD16+ NKT subsets. When tumors were treated with IL-2, the high intensity CD4, CD8 and CD16/CD56 surface expression, which were down-regulated during interaction with tumors, was restored (Fig. S2F).
Sera from peripheral blood of NK-injected tumor-bearing hu-BLT mice exhibited increased IFN-γ secretion as well as other cytokines/chemokines/growth factors and ligands when compared to those from tumor-bearing hu-BLT mice without NK cells, and, in particular, IFN-γ secretion was further enhanced by feeding mice AJ2 (Fig. 5A-5C and Table S5).
Figure 5.

Single injection of super-charged NK cells with/without AJ2 feeding in tumor bearing mice restored and increased cytokine, chemokine and growth factor secretions within serum obtained from peripheral blood of hu-BLT mice. Serum from peripheral blood was obtained as described in the Materials and Methods section and multiplex arrays were performed to determine secretion of IFN-γ, one of the four representative figure is shown here (A). Fold changes of IFN-γ were determined based on the values obtained from control hu-BLT mice (n = 5) (B). Multiplex arrays were used to determine cytokines, chemokines, and growth factor secretion in sera obtained from the peripheral blood (C).
Cis-diamminedichloridoplatinum(II) (CDDP) or Paclitaxel with and without N-acetylsysteine (NAC) induce significant cell death in OSCSCs differentiated with IL-2+anti-CD16mAb treated NK supernatant
Differentiation of OSCSCs with NK supernatants resulted in significant susceptibility of tumors to CDDP (Fig. 6A). Similarly, Paclitaxel mediated higher cell death of NK-supernatants differentiated OSCSCs, and NAC significantly increased Paclitaxel-mediated cell death (Fig. 6B). Blocking NK-mediated differentiation of OSCSCs with anti-IFN-γ and anti-TNF-α antibodies substantially decreased cell death induced by CDDP or paclitaxel with and without NAC (Fig. 6B). Treatment of OSCCs, patient-derived differentiated oral tumor cells, with CDDP or paclitaxel and NAC exhibited higher cell death (Fig. S3A-S3B).
Figure 6.

CDDP or Paclitaxel with and without NAC induce significant cell death in OSCSCs differentiated with NK-supernatants and not in poorly-differentiated tumors. Highly purified NK cells were treated with the combination of IL-2 (1000 U/ml) and anti-CD16mAb (3μg/ml) for 18 hours, after which the NK supernatants were added to OSCSCs in the presence of anti-TNF-α (1:100) and anti-IFN-γ (1:100) for a period of 5 days. Thereafter, OSCSCs were detached and treated with/without Cisplatin for 18–24 hours. The viability of OSCSCs was then determined using PI staining and flow cytometric analysis. One of 3 representative experiments is shown in this figure (A). OSCSCs were treated with the supernatants from NK cells as described in Fig. 6A. Afterwards, tumors were detached and treated with/without NAC (20 nM) for 24 hours, followed by treatment with Paclitaxel for 18–24 hours. OSCSCs viability was determined by PI staining and flow cytometric analysis. One of 3 representative experiments is shown in this figure (B).
Monocytes and osteoclasts from NK-injected tumor-bearing mice had greater ability to activate NK cells when compared to those from tumor-bearing mice in the absence of NK injection
NK cells from NK injected tumor-bearing mice and NK supernatant-differentiated tumor-bearing mice when cultured with autologous monocytes had significantly augmented cytotoxicity (Fig. 7A) and IFN-γ secretion (Fig. 7B) when compared to those implanted with undifferentiated tumors in the absence of NK cell injection. Blocking NK-mediated differentiation of tumors through the addition of antibodies to TNF-α and IFN-γ before implantation decreased cytotoxicity (Fig. 7A), and IFN-γ secretion (Fig. 7B) of the NK cells. We then generated OCs from BM-derived monocytes of hu-BLT mice and cultured with allogeneic NK cells from healthy human donors to study the extent of NK cells expansion. NK cells expansion (Fig. 7C), and IFN-γ secretion (Fig. 7D and 7E) were augmented in NK-injected tumor-bearing mice or NK supernatant-differentiated tumor-bearing mice when compared to those obtained from tumor-bearing mice without NK injection (Fig. 7C-7E). Thus, monocytes and osteoclasts from NK injected tumor-bearing mice had greater ability to activate NK cells than those from tumor-bearing mice in the absence of NK injection.
Figure 7.
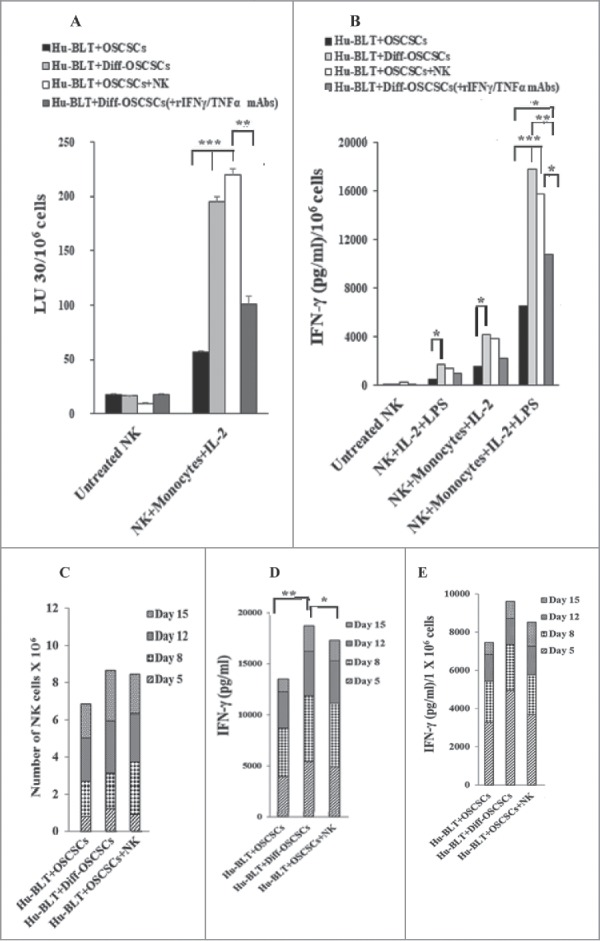
Monocytes or osteoclasts from tumor-bearing mice injected with super-charged NK cells or implanted with only NK-differentiated OSCSC-tumors induced significantly more IFN-γ from autologous or allogeneic NK-tumor co-cultures when compared to those of tumor-alone implanted mice with NK cells. Hu-BLT mice were implanted with OSCSCs and injected with NK cells, and fed AJ2 as described in Fig. 1B. After sacrifice, NK cells from splenocytes and monocytes from BM cells were isolated, as described in Material and Methods section. Autologous NK cells were left untreated or treated with IL-2 (1000 U/ml) in combination with monocytes (NK:monocytes, 2:1) and at day 7 after the co-culture, NK cells were used as effector cells in a standard 4-hour 51Chromium release assay against OSCSCs. The LU 30/106 cells were determined using inverse number of NK cells required to lyse 30% of the target cells X100 (A). Autologous NK cells were left untreated or treated with IL-2 (1000 U/ml) or with the combination of IL-2 (1000 U/ml) and LPS (100 ng/ml) in the absence and presence of monocytes (NK:monocytes, 2:1) for 7 days, after which the supernatants were harvested and IFN-γ secretion were determined using single ELISA (B). OCs were generated from purified hu-BLT monocytes, as described in Material and Methods section. Purified allogeneic NK cells from healthy human donors were pre-treated with IL-2 (1000 U/ml) and anti-CD16mAb (3 µg/ml) for 18 hours and then cultured with hu-BLT-OCs in the presence of sAJ2 (NK:OCs:sAJ2, 2:1:4). After culture, numbers of NK cells in the culture were counted on day 5, 8, 12 and 15 using microscopy (C). The supernatants were harvested from cultures on days 5, 8, 12 and 15 and the IFN-γ secretion was determined using single ELISA (D). The levels of IFN-γ obtained from ELISA were determined in 1 × 106 cells using cell counts from Fig. 7C (E).
Discussion
The therapeutic role of NK cells in hematological malignancies have previously been clearly demonstrated by several groups, however, their effect in solid tumors have been found to be suboptimal in limiting tumor growth by existing strategies.20-25 We have recently established an NK activating strategy with superb tumor-targeting function, which we coined as super-charged NK cells, and used successfully to target several solid tumors in in vitro and in mouse models.26-28 We have shown that NK cells serve as effectors of selection and differentiation of oral tumors,29-31 glioblastomas,32 pancreatic tumors and lung tumors.33 These two functions of NK cells are mediated by two distinct stages of NK maturation, namely cytotoxic (CD16+CD56+) and split-anergized (CD16dim/−CD56+/++) NK cells respectively, resulting in differentiation, increased expression of MHC class I on tumor cells, and subsequent promotion of T cell activation.34-36 The combination of IFN-γ and TNF-α secreted by NK cells is important for lysis of target cells, whereas IFN-γ is primarily responsible for the differentiation of tumors.37 IFN-γ, and to a lesser extent TNF-α, are responsible for limiting tumor growth through differentiation as evidenced by an increased expression of differentiation antigens MHC-I, CD54 and B7H1.37 As shown in this report, tumor differentiation by IFN-γ and TNF-α released from NK cells renders tumors susceptible to chemotherapeutic drugs CDDP and Paclitaxel since the addition of antibodies to IFN-γ and TNF-α, which blocks NK cell-mediated tumor differentiation, restores CSC/undifferentiated tumor resistance to chemotherapy drugs.
To demonstrate in vivo relevance of our in vitro findings, oral CSCs were implanted in the oral mucosa of hu-BLT and NSG mice and tumor generation in the presence and absence of tail-vein injection of super-charged NK cells was studied. Single delivery of potent super-charged NK cells after oral tumor implantation was found to decrease the size of tumors and prevent weight loss of mice when compared to either NSG or hu-BLT tumor-bearing mice without NK injection.
Oral tumors from NK-injected mice remained very small in size and demonstrated approximately 3 to 9-fold more human CD45+ infiltrating immune cells, exhibiting significantly higher amounts of IFN-γ secretion in all time points tested when compared to those obtained from tumor-bearing mice without NK injection. When tumors were dissociated, and equal numbers of tumors were cultured from each group of mice, little to no tumor growth was observed from tumor-bearing mice injected with NK cells, whereas large numbers of tumor growth were obtained from NSG or hu-BLT mice injected with oral tumors without NK injection.
Oral tumors from NK-injected mice exhibited significant resistance to NK cell-mediated cytotoxicity and increased expression of MHC-I, CD54 and B7H1, demonstrating their differentiated phenotype. Whereas those from tumor-bearing hu-BLT and NSG mice without NK injection were highly susceptible to NK cell-mediated cytotoxicity, and had no/low expression of differentiation antigens, demonstrating maintenance of their stem-like phenotype in the absence of NK cells.29,33 These results indicated that IFN-γ secretion by NK cells and/or NK activated CD3+ T cells are crucial for the maintenance of tumor differentiation, for the inhibition of tumor growth.
To determine whether in vitro NK-differentiated tumors grow in vivo, we differentiated tumors with NK cell supernatants and equal numbers of tumors, non-differentiated or NK supernatant-differentiated tumors were implanted in the oral mucosa. Since our in vitro results demonstrated that differentiation is primarily due to the dual functions of IFN-γ and TNF-α, we also differentiated stem-like tumors with NK supernatants in the presence of antibodies to IFN-γ and TNF-α, and implanted the same numbers of tumors as CSCs/undifferentiated tumors to hu-BLT mice. Very few tumors grew from NK supernatant-differentiated tumors, whereas those differentiated in the presence of NK supernatants with antibodies to IFN-γ and TNF-α or implanted with CSCs/undifferentiated tumors grew and proliferated significantly and were susceptible to NK cell-mediated cytotoxicity, demonstrating their stem-like/undifferentiated phenotype. Similarly, few tumors grew from NK-injected tumor-bearing mice, and those, which grew, were highly resistant to NK cell-mediated cytotoxicity exhibiting their differentiated phenotype. Higher numbers of tumor infiltrating NK cells and T cells were found in different tissue compartments including tumors in NK-injected tumor-bearing mice, and those with NK-differentiated tumors. These experiments demonstrated that NK-mediated differentiation limits tumor growth, increases differentiation antigens on tumors and induces tumor resistance to NK cell-mediated cytotoxicity, thereby potentially facilitating in vivo activation of T cells.
Hu-BLT mice contain lower frequencies of NK cells with decreased function in their peripheral blood as compared to healthy humans, which could be one reason why stem-like/poorly differentiated oral tumors are easily implantable in these mice. In addition, NK-injected tumors contain higher percentages of CD3+ T cells, indicating the significance of NK cells in recruiting T cells to the tumor microenvironment.26 The reason for decreased human-NK cell frequency in Hu-BLT mice is not known at present but it could be due to targeting of the mouse tissues and/or down-modulation of key NK receptors used to identify NK cells, in addition to previously hypothesized decline in hu-IL-15 secretion in these mice.38 Indeed, when NK cells were stained with the red dye and injected to the BLT-hu mice, even though lower frequencies of CD16/CD56 expressing NK cells were observed, higher frequencies of red cells were obtained.28
OCs or monocytes from NK-injected tumor-bearing mice or from NK-differentiated oral tumor recipients increased allogeneic and autologous NK cell expansion respectively more than OCs or monocytes from tumor-bearing mice without NK injection. Differences between the magnitude of allogeneic and autologous NK suppression when interacting with OCs vs. monocytes respectively, indicate more severe inhibition of NK cells due to the combined defects in both NK and monocytes, whereas less NK suppression is seen when the defect is only in OCs. Thus, the results presented in this paper demonstrate the significance of super-charged NK cells in treatment and prevention of CSCs growth and expansion in a relevant human pre-clinical model. In addition, the mechanism of inhibition of tumor growth was found to be dependent on the selection and differentiation of tumors initiated by the NK cells.
Materials and methods
Cell lines, reagents, and antibodies
RPMI 1640 supplemented with 10% Fetal Bovine Serum (FBS) (Gemini Bio-Products, CA) was used for the cultures of immune cells. OSCSCs and OSCCs were dissociated and grown from the tongue tumors of patients at UCLA,29 and were cultured with RPMI 1640 supplemented with 10% FBS. Recombinant human IL-2 was obtained from NIH-BRB. Flow cytometry antibodies used in this study were obtained from Biolegend (San Diego, CA). Monoclonal anti-TNF-α and monoclonal anti-IFN-γ antibodies were either obtained from commercial sources or prepared in our laboratory and 1:100 dilution was found to be the optimal concentration to use for blocking experiments as described previously.37
Purification of human NK cells and monocytes
Written informed consents approved by UCLA Institutional Review Board (IRB) were obtained and all procedures were approved by UCLA-IRB. PBMCs from healthy human donors were isolated, and NK cells and monocytes were purified using isolation kits obtained from Stem Cell Technologies, as described before.39,40 The purity of NK cells and monocyte populations was found to be 95% or higher, respectively, based on the flow cytometric analysis.
Probiotic bacteria
AJ2 is a combination of 8 different strains of gram-positive probiotic bacteria (Streptococcus thermophiles, Bifidobacterium longum, Bifidobacterium breve, Bifidobacterium infantis, Lactobacillus acidophilus, Lactobacillus plantarum, Lactobacillus casei, and Lactobacillus bulgaricus) used to induce differentiation of stem cells and are selected for their superior ability to induce optimal secretion of both pro-inflammatory and anti-inflammatory cytokines in NK cells. In addition, each strain was grown, and specific colonies were selected after three rounds of sub-cloning based on the ability to withstand environmental pressures such as temperature and acidity.37
Generation of osteoclasts and expansion of super-charged human and hu-BLT-derived NK cells
Purified monocytes were cultured in alpha-MEM medium containing M-CSF (25 ng/ml) and RANKL (25 ng/ml) for 21 days, or otherwise specified. Medium was refreshed every 3 days with fresh alpha-MEM containing M-CSF and RANKL. Human purified and hu-BLT enriched NK cells were activated with IL-2 (1000 U/ml) and anti-CD16mAb (3 ug/ml) for 18–20 hours before they were co-cultured with osteoclasts and sonicated AJ2. The culture media was refreshed with IL-2 every three days as described previously.26 Since ex-vivo expanded NK cells with osteoclasts and sAJ2 had superior cytotoxicity and IFN-γ secretion when compared to other expansion methodologies, and survived for a longer period, they were called super-charged NK cells.26 For sonication, AJ2 was weighed and re-suspended in RPMI 1640 Medium containing 10% FBS at a concentration of 10 mg/ml. The bacteria were thoroughly vortexed, then sonicated on ice for 15 seconds, at 6 to 8 amplitudes. Sonicated samples were then incubated for 30 seconds on ice. After every five pulses, a sample was taken to observe under the microscope until at least 80 percent of cell walls were lysed. It was determined that approximated 20 rounds of sonication/incubation on ice, were conducted to achieve complete sonication. Finally, the sonicated samples (sAJ2) were aliquoted and stored in a -80 degrees Celsius freezer until use.
Analysis of human oral cancer cell growth in immunodeficient and humanized mice
Animal research was performed under the written approval of the UCLA Animal Research Committee (ARC) (2012-101-13A). Humanized-BLT (hu-BLT; human bone marrow/liver/thymus) mice were prepared in our core facility as previously described.18,41
In vivo growth of oral tumors was done by orthotopically implanting tumor cells into 8–10 weeks-old NSG mice or hu-BLT mice in the floor of the mouth. Mice were anesthetized using isoflurane and tumor cells were then transferred by direct injection in the floor of mouth with 10μl HC Matrigel (Corning, NY, USA). 7 to 10 days after the surgery mice received 1.5 × 106 super-charged NK cells via tail vein. Mice were fed AJ2 (5 billion bacteria/dose), began feedings one or two weeks before tumor implantation and were fed every 48 hours, throughout the experiment. Mice were euthanized when signs of morbidity were evident. Oral tumors, BM, spleen and peripheral blood were harvested.
Dissociation and culture of cells from tissues of hu-BLT and NSG mice
To prepare a single cell suspension of mouse tissues for subsequent analyses, animals were sacrificed BM, spleen, peripheral blood and oral tumor were obtained. The oral tumor was immediately cut into 1 mm3 pieces and placed into a digestion buffer containing 1 mg/ml collagenase II (for fat tissue), 10 U/ml DNAse I, and 1% bovine serum albumin in DMEM and incubated for 20 minutes at 37°C oven on a 150 rpm shaker. After digestion, the sample was filtered through a 40 µm cell strainer and centrifuged at 1500 rpm for 10 minutes at 4°C. The pellet was re-suspended in DMEM and cells counted. Single cell suspensions from BM and spleen were obtained by digesting tissues, as described previously.40 PBMCs were obtained using ficoll-Hypaque centrifugation of heparinized blood specimens. The buffy coat containing PBMCs, were harvested, washed and re-suspended in RPMI 1640 medium.
Purification of NK cells, CD3T cells, and monocytes from hu-BLT mice
CD3+ T cells were isolated from hu-BLT splenocytes using positive T cells selection kit (Stem-Cell Technologies), and the cells depleted of T cells were used as NK-enriched cells; NK cells from hu-BLT mice were isolated using the human CD56+ positive selection kit (Stem-Cell Technologies, Canada). Monocytes from hu-BLT mice were isolated from BM cells using human CD14 positive selection kit (eBioscience, San Diego, CA).
ELISA
Single ELISAs were performed, as described previously.39 To analyze and obtain the cytokine and chemokine concentration, a standard curve was generated by either two or three-fold dilution of recombinant cytokines provided by the manufacturer.
Surface Staining and cell death assays
Staining was performed by labeling the cells with antibodies or propidium iodide, as described previously.39,42,43 The cells were washed twice with ice-cold PBS containing 1% BSA. Predetermined optimal concentrations of specific human monoclonal antibodies were added to 1 × 104 cells in 50 µl of cold-BSA and cells were incubated on ice for 30 min. Thereafter cells were washed in cold PBS-BSA and brought to 500 µl with PBS-BSA. For cell death assay 1 × 104 cells in 50 µl of cold-BSA were stained with 8 μg/ml propidium iodide and cells were incubated on ice for 10 min, and brought to 500 µl with PBS-BSA. Flow cytometry analysis was performed using Beckman Coulter Epics XL cytometer (Brea, CA) and results were analyzed in FlowJo vX software (Ashland, OR).
51Cr. release cytotoxicity assay
The 51Cr release assay was performed, as described previously.44 Briefly, different numbers of effector cells were incubated with 51Cr–labeled target cells. After a 4-hour incubation period the supernatants were harvested from each sample and counted for released radioactivity using the gamma counter. The percentage specific cytotoxicity was calculated as follows:
LU 30/106 is calculated by using the inverse of the number of effector cells needed to lyse 30% of tumor target cells ×100.
Stem cell differentiation with NK cell supernatants
NK cells were treated with a combination of anti-CD16 monoclonal antibody (3μg/ml) and IL-2 (1,000 U/ml) for 18 hours before supernatants were removed and used for differentiation experiments. The amounts of IFN-γ produced by activated NK cells were measured with IFN-γ ELISA (Biolegend, CA, USA). OSCSCs were differentiated with gradual daily addition of increasing amounts of NK cell supernatants. On average, to induce differentiation, a total of 3,500 pg of IFN-γ containing supernatants were added for 4 days to induce differentiation and resistance of OSCSCSs to NK cell-mediated cytotoxicity. Afterwards, target cells were washed with 1xPBS, detached and used for experiments as described previously.30
Statistical analysis
An unpaired, two-tailed student t-test was performed for the statistical analysis. One-way ANOVA with a Bonferroni post-test was used to compare different groups. (n) denotes the number of mice used for the experiment. For cytotoxicity and cytokine analysis either duplicate or triplicate samples were used for assessment. The following symbols represent the levels of statistical significance within each analysis, ***(p value <0.001), **(p value 0.001-0.01), *(p value 0.01–0.05).
Supplementary Material
Funding Statement
NIH- RO1 DE12880; UCLA Academic senate grant, Seed grants, XO club.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
A.K.K. was supported by Polish Ministry of Science and Higher Education (Mobility Plus Award) and The National Centre for Research and Development (Innomed program).
The authors acknowledge excellent technical help of Jessica Cook and Brett Ploussard.
Authors contribution
KK performed the majority of experiments, data analysis and preparation of the manuscript. PT, NO, JC assisted KK in performing the experiments and edited the manuscript. AKK performed experiments, data analysis, drafted and edited the manuscript. PM, SP, and MK performed some experiments. CF and IN reviewed and edited the manuscript. AJ oversaw the design of the experiments, data analysis, and preparation of the manuscript.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer Statistics, 2012. Ca-a Cancer J Clin. 2012;62(1):p. 10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Gibson MK, Forastiere AA. Reassessment of the role of induction chemotherapy for head and neck cancer. Lancet Oncol. 2006;7(7):p. 565–74. doi: 10.1016/S1470-2045(06)70757-4. [DOI] [PubMed] [Google Scholar]
- 3.Visvader JE, Lindeman GJ. Cancer stem cells: current status and evolving complexities. Cell Stem Cell. 2012;10(6):p. 717–28. doi: 10.1016/j.stem.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 4.Yu CC, Tsai LL, Wang ML, Yu CH, Lo WL, Chang YC, Chiou GY, Chou MY, Chiou SH. miR145 targets the SOX9/ADAM17 axis to inhibit tumor-initiating cells and IL-6-mediated paracrine effects in head and neck cancer. Cancer Res. 2013;73(11):p. 3425–40. doi: 10.1158/0008-5472.CAN-12-3840. [DOI] [PubMed] [Google Scholar]
- 5.Chiou S.H., Yu CC, Huang CY, Lin SC, Liu CJ, Tsai TH, Chou SH, Chien CS, Ku HH, Lo JF. Positive correlations of Oct-4 and Nanog in oral cancer stem-like cells and high-grade oral squamous cell carcinoma. Clin Cancer Res. 2008;14(13):p. 4085–95. doi: 10.1158/1078-0432.CCR-07-4404. [DOI] [PubMed] [Google Scholar]
- 6.Schatton T, Frank MH. Cancer stem cells and human malignant melanoma. Pigment Cell Melanoma Res. 2008;21(1):p. 39–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Grandis JR, Falkner DM, Melhem MF, Gooding WE, Drenning SD, Morel PA. Human leukocyte antigen class I allelic and haplotype loss in squamous cell carcinoma of the head and neck: clinical and immunogenetic consequences. Clin Cancer Res. 2000;6(7):p. 2794–802. [PubMed] [Google Scholar]
- 8.Cabrera CM, Nieto A, Cortes JL, Montes RM, Catalina P, Cobo F, Barroso-Del-Jesus A, Concha A. The low rate of HLA class I molecules on the human embryonic stem cell line HS293 is associated with the APM components' expression level. Cell Biol Int. 2007;31(9):p. 1072–1078. doi: 10.1016/j.cellbi.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 9.Uppaluri R, Dunn GP, Lewis Jr JS. Focus on TILs: prognostic significance of tumor infiltrating lymphocytes in head and neck cancers. Cancer Immun. 2008;8:p. 16. [PMC free article] [PubMed] [Google Scholar]
- 10.Staveley-O'Carroll K, Sotomayor E, Montgomery J, Borrello I, Hwang L, Fein S, Pardoll D, Levitsky H. Induction of antigen-specific T cell anergy: An early event in the course of tumor progression. Proc Natl Acad Sci U S A. 1998;95(3):p. 1178–83. doi: 10.1073/pnas.95.3.1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turkseven MR, Oygur T. Evaluation of natural killer cell defense in oral squamous cell carcinoma. Oral Oncol. 2010;46(5):p. e34–7. doi: 10.1016/j.oraloncology.2010.02.019. [DOI] [PubMed] [Google Scholar]
- 12.Accomando WP, Wiencke JK, Houseman EA, Butler RA, Zheng S, Nelson HH, Kelsey KT. Decreased NK cells in patients with head and neck cancer determined in archival DNA. Clin Cancer Res. 2012;18(22):p. 6147–54. doi: 10.1158/1078-0432.CCR-12-1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mickel RA, Kessler DJ, Taylor JM, Lichtenstein A. Natural killer cell cytotoxicity in the peripheral blood, cervical lymph nodes, and tumor of head and neck cancer patients. Cancer Res. 1988;48(17):p. 5017–22. [PubMed] [Google Scholar]
- 14.Tseng HC, Arasteh A, Paranjpe A, Teruel A, Yang W, Behel A, Alva JA, Walter G, Head C, Jewett A, et al., Increased Lysis of Stem Cells but Not Their Differentiated Cells by Natural Killer Cells; De-Differentiation or Reprogramming Activates NK Cells. Plos One. 2010;5(7):e11590. doi: 10.1371/journal.pone.0011590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tseng H-C, Bui V, Man YG, Cacalano N, Jewett A. Induction of Split Anergy Conditions Natural Killer Cells to Promote Differentiation of Stem Cells through Cell-Cell Contact and Secreted Factors. Front immunol. 2014;5:p. 269–269. doi: 10.3389/fimmu.2014.00269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jewett A, Man Y-G, Tseng H-C, Dual Functions of Natural Killer Cells in Selection and Differentiation of Stem Cells; Role in Regulation of Inflammation and Regeneration of Tissues. J Cancer. 2013;4(1):p. 12–24. doi: 10.7150/jca.5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tseng HC, Cacalano N, Jewett A. Split anergized Natural Killer cells halt inflammation by inducing stem cell differentiation, resistance to NK cell cytotoxicity and prevention of cytokine and chemokine secretion. Oncotarget. 2015;6(11):p. 8947–59. doi: 10.18632/oncotarget.3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shimizu S, Hong P, Arumugam B, Pokomo L, Boyer J, Koizumi N, Kittipongdaja P, Chen A, Bristol G, Galic Z, et al., A highly efficient short hairpin RNA potently down-regulates CCR5 expression in systemic lymphoid organs in the hu-BLT mouse model. Blood. 2010;115(8):p. 1534–44. doi: 10.1182/blood-2009-04-215855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vatakis DN, Bristol GC, Kim SG, Levin B, Liu W, Radu CG, Kitchen SG, Zack JA. Using the BLT humanized mouse as a stem cell based gene therapy tumor model. J Vis Exp. 2012(70):p. e4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brehm C, Huenecke S, Quaiser A, Esser R, Bremm M, Kloess S, Soerensen J, Krevenberg H, Seidl C, Koehl U, et al., IL-2 stimulated but not unstimulated NK cells induce selective disappearance of peripheral blood cells: concomitant results to a phase I/II study. PLoS One. 2011;6(11):p. e27351. doi: 10.1371/journal.pone.0027351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Venton G, Labiad Y, Colle J, Fino A, Afridi S, Torres M, Monteuil S, Loriod B, Fernandez-Nunez N, Farnault L, et al., Natural killer cells in acute myeloid leukemia patients: from phenotype to transcriptomic analysis. Immunol Res. 2016;64(5-6):p. 1225–1236. doi: 10.1007/s12026-016-8848-0. [DOI] [PubMed] [Google Scholar]
- 22.Miller JS, Soignier Y, Panoskaltsis-Mortari A, McNearney SA, Yun GH, Fautsch SK, McKenna D, Le C, Defor TE, Burns LJ, et al., Successful adoptive transfer and in vivo expansion of human haploidentical NK cells in patients with cancer. Blood. 2005;105(8):p. 3051–7. doi: 10.1182/blood-2004-07-2974. [DOI] [PubMed] [Google Scholar]
- 23.Bachanova V, Burns LJ, McKenna DH, Curtsinger J, Panoskaltsis-Mortari A, Lindgren BR, Cooley S, Weisdorf D, Miller JS. Allogeneic natural killer cells for refractory lymphoma. Cancer Immunol Immunother. 2010;59(11):p. 1739–44. doi: 10.1007/s00262-010-0896-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ilander M, Olsson-Strömberg U, Schlums H, Guilhot J, Brück O, Lähteenmäki H, Kasanen T, Koskenvesa P, Söderlund S, Höglund M, et al., Increased proportion of mature NK cells is associated with successful imatinib discontinuation in chronic myeloid leukemia. Leukemia. 2017;31(5):p. 1108–1116. doi: 10.1038/leu.2016.360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cooley S, Burns LJ, Repka T, Miller JS. Natural killer cell cytotoxicity of breast cancer targets is enhanced by two distinct mechanisms of antibody-dependent cellular cytotoxicity against LFA-3 and HER2/neu. Exp Hematol. 1999;27(10):p. 1533–41. doi: 10.1016/S0301-472X(99)00089-2. [DOI] [PubMed] [Google Scholar]
- 26.Kaur K, Cook J, Park SH, Topchyan P, Kozlowska A, Ohanian N, Fang C, Nishimura I, Jewett A. Novel Strategy to Expand Super-Charged NK Cells with Significant Potential to Lyse and Differentiate Cancer Stem Cells: Differences in NK Expansion and Function between Healthy and Cancer Patients. Front Immunol. 2017;8:p. 297. doi: 10.3389/fimmu.2017.00297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kozlowska AK, Kaur K, Topchyan P, Jewett A. Novel strategies to target cancer stem cells by NK cells; studies in humanized mice. Front Biosci (Landmark Ed). 2017;22:p. 370–384. [DOI] [PubMed] [Google Scholar]
- 28.Kozlowska AK, Kaur K, Topchyan P, Jewett A. Adoptive transfer of osteoclast-expanded natural killer cells for immunotherapy targeting cancer stem-like cells in humanized mice. Cancer Immunol Immunother. 2016;65(7):p. 835–45. doi: 10.1007/s00262-016-1822-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tseng HC, Arasteh A, Paranjpe A, Teruel A, Yang W, Behel A, Alva JA, Walter G, Head C, Jewett A, et al., Increased lysis of stem cells but not their differentiated cells by natural killer cells; de-differentiation or reprogramming activates NK cells. PLoS One. 2010;5(7):p. e11590. doi: 10.1371/journal.pone.0011590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tseng HC, Bui V, Man YG, Cacalano N, Jewett A. Induction of Split Anergy Conditions Natural Killer Cells to Promote Differentiation of Stem Cells through Cell-Cell Contact and Secreted Factors. Front Immunol. 2014;5:p. 269. doi: 10.3389/fimmu.2014.00269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tseng HC, Cacalano N, Jewett A. Split anergized Natural Killer cells halt inflammation by inducing stem cell differentiation, resistance to NK cell cytotoxicity and prevention of cytokine and chemokine secretion. Oncotarget. 2015;6:8947–8959. doi: 10.18632/oncotarget.3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tseng HC, Inagaki A, Bui VT, Cacalano N, Kasahara N, Man YG, Jewett A. Differential Targeting of Stem Cells and Differentiated Glioblastomas by NK Cells. J Cancer. 2015;6(9):p. 866–76. doi: 10.7150/jca.11527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kozlowska AK, Topchyan P, Kaur K, Tseng HC, Teruel A, Hiraga T, Jewett A. Differentiation by NK cells is a prerequisite for effective targeting of cancer stem cells/poorly differentiated tumors by chemopreventive and chemotherapeutic drugs. J Cancer. 2017;8(4):p. 537–554. doi: 10.7150/jca.15989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sawicki MW, Dimasi N, Natarajan K, Wang J, Margulies DH, Mariuzza RA. Structural basis of MHC class I recognition by natural killer cell receptors. Immunol Rev. 2001;181:p. 52–65. doi: 10.1034/j.1600-065X.2001.1810104.x. [DOI] [PubMed] [Google Scholar]
- 35.Cerwenka A, Lanier LL. NKG2D ligands: unconventional MHC class I-like molecules exploited by viruses and cancer. Tissue Antigens. 2003;61(5):p. 335–43. doi: 10.1034/j.1399-0039.2003.00070.x. [DOI] [PubMed] [Google Scholar]
- 36.Stroynowski I, Clark S, Henderson LA, Hood L, McMillan M, Forman J. Interaction of alpha 1 with alpha 2 region in class I MHC proteins contributes determinants recognized by antibodies and cytotoxic T cells. J Immunol. 1985;135(3):p. 2160–6. [PubMed] [Google Scholar]
- 37.Bui VT, Tseng HC, Kozlowska A, Maung PO, Kaur K, Topchyan P, Jewett A. Augmented IFN-gamma and TNF-alpha Induced by Probiotic Bacteria in NK Cells Mediate Differentiation of Stem-Like Tumors Leading to Inhibition of Tumor Growth and Reduction in Inflammatory Cytokine Release; Regulation by IL-10. Front Immunol. 2015;6:p. 576. doi: 10.3389/fimmu.2015.00576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pek EA, Chan T, Reid S, Ashkar AA. Characterization and IL-15 dependence of NK cells in humanized mice. Immunobiology. 2011;216(1-2):p. 218–24. doi: 10.1016/j.imbio.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 39.Jewett A, Bonavida B. Target-induced inactivation and cell death by apoptosis in a subset of human NK cells. J Immunol. 1996;156(3):p. 907–15. [PubMed] [Google Scholar]
- 40.Tseng HC, Kanayama K, Kaur K, Park SH, Park S, Kozlowska A, Sun S, McKenna CE, Nishimura I, Jewett A. Bisphosphonate-induced differential modulation of immune cell function in gingiva and bone marrow in vivo: role in osteoclast-mediated NK cell activation. Oncotarget. 2015;6(24):p. 20002–25. doi: 10.18632/oncotarget.4755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vatakis DN, Koya RC, Nixon CC, Wei L, Kim SG, Avancena P, Bristol G, Baltimore D, Kohn DB, Zack JA, et al., Antitumor activity from antigen-specific CD8T cells generated in vivo from genetically engineered human hematopoietic stem cells. Proc Natl Acad Sci U S A. 2011;108(51):p. E1408–16. doi: 10.1073/pnas.1115050108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jewett A, Cavalcanti M, Bonavida B. Pivotal role of endogenous TNF-alpha in the induction of functional inactivation and apoptosis in NK cells. J Immunol. 1997;159(10):p. 4815–22. [PubMed] [Google Scholar]
- 43.Jewett A, Bonavida B. Interferon-alpha activates cytotoxic function but inhibits interleukin-2-mediated proliferation and tumor necrosis factor-alpha secretion by immature human natural killer cells. J Clin Immunol. 1995;15(1):p. 35–44. doi: 10.1007/BF01489488. [DOI] [PubMed] [Google Scholar]
- 44.Jewett A, Wang MY, Teruel A, Poupak Z, Bostanian Z, Park NH. Cytokine dependent inverse regulation of CD54 (ICAM1) and major histocompatibility complex class I antigens by nuclear factor kappaB in HEp2 tumor cell line: effect on the function of natural killer cells. Hum Immunol. 2003;64(5):p. 505–20. doi: 10.1016/S0198-8859(03)00039-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.