

ABSTRACT
Releasing the patient's immune system against their own malignancy by the use of checkpoint inhibitors is delivering promising results. However, only a subset of patients currently benefit from them. One major limitation of these therapies relates to the inability of T cells to detect or penetrate into the tumor resulting in unresponsiveness to checkpoint inhibition. Virotherapy is an attractive tool for enabling checkpoint inhibitors as viruses are naturally recognized by innate defense elements which draws the attention of the immune system. Besides their intrinsic immune stimulating properties, the adenoviruses used here are armed to express tumor necrosis factor alpha (TNFa) and interleukin-2 (IL-2). These cytokines result in immunological danger signaling and multiple appealing T-cell effects, including trafficking, activation and propagation. When these viruses were injected into B16.OVA melanoma tumors in animals concomitantly receiving programmed cell-death protein 1 (PD-1) blocking antibodies both tumor growth control (p < 0.0001) and overall survival (p < 0.01) were improved. In this set-up, the addition of adoptive cell therapy with OT-I lymphocytes did not increase efficacy further. When virus injections were initiated before antibody treatment in a prime-boost approach, 100% of tumors regressed completely and all mice survived. Viral expression of IL2 and TNFa altered the cytokine balance in the tumor microenvironment towards Th1 and increased the intratumoral proportion of CD8+ and conventional CD4+ T cells. These preclinical studies provide the rationale and schedule for a clinical trial where oncolytic adenovirus coding for TNFa and IL-2 (TILT-123) is used in melanoma patients receiving an anti-PD-1 antibody.
KEYWORDS: Immunotherapy, Virotherapy, Immunovirotherapy, Checkpoint blockade, Adoptive cell therapy, Adenovirus, Melanoma, Solid tumors, anti-PD1, T cell therapy, Adoptive T cell therapies, Models of immunostimulation, Therapeutic antibodies
Introduction
The immunosuppressive microenvironment of solid tumors is one of the key features that protects them from effective antitumor immune responses.1-3 In such an environment, both endogenously induced or adoptively transferred antitumor T cells switch into an exhausted state where their function is impaired.4,5 Checkpoint inhibitors are monoclonal antibodies designed to block specific suppressive pathways upregulated in many tumors.6 These antibodies have revolutionized the field of immunotherapy resulting in regulatory approval for the treatment of different types of cancers including melanoma, lung, renal, urothelial and other cancers, with more to follow. Despite impressive proof-of-concept long term efficacy in some patients, in fact only a relatively small subpopulation (10–50% of patients depending on tumor type) gain measurable benefit from checkpoint inhibition as a single-agent modality.6,7 Amongst the major limitations of checkpoint blockade are situations where the tumor is invisible to the immune system (“cold” and “excluded” tumors),8 or situations where immune suppression is exerted through several pathways concurrently.9,10 It is increasingly appreciated that checkpoint inhibition tends to work in “hot” tumors characterized by CD8+ lymphocyte infiltration, neoantigens and PD-L1 expression, while little efficacy is seen in “cold” or “immune excluded” tumors.10
Given the increased understanding of the limitations of checkpoint blockade therapy, and the emerging mechanism-of-action data relating to oncolytic immunotherapy, the use of viral platforms is an appealing approach to overcome these obstacles. Due to mechanisms conserved during evolution, viruses are recognized by innate defense mechanisms resulting in two major consequences which are not subject to resistance mediated by tumor immunosuppression. Recognition of conserved viral patterns recruits immune cells to the tumor and converts the microenvironment towards a proimmunogenic one.11,12 Immunogenic viruses such as adenovirus, are able to affect signaling in the tumor through several mechanisms, promoting the creation of new adaptive responses including those against tumor specific antigens.13-15 Of note, anti-viral responses seem to contribute to anti-tumor responses through epitope spreading and danger signaling.16 Translational benefits of these viral platforms for conversion of tumor-associated immunosuppression is embodied in the use of oncolytic viruses. They are an attractive tool for tumor immunotherapy as they are self-amplifying and their replication is restricted to the tumor, thus achieving high specificity for the target with limited adverse events.17,18 Systemic effects are achieved through the immune system and in the case of some viruses such as 5/3 chimeric adenovirus also through vascular dissemination.19
There is currently a number of checkpoint inhibitors approved by regulatory agencies7 and the first oncolytic virus was approved by the FDA and EMA in 2015 for the treatment of unresectable melanoma.20 There are, also, ongoing clinical trials where the effect after the combination of these two therapies is being studied.21 Initial clinical data supports the notion that oncolytic virus can increase response rates without increasing toxicity,22 in stark contrast to combinations of checkpoint inhibitors which increase both or in some cases just toxicities.6 Since there is a plethora of checkpoints and their inhibitors, it was hypothesized that their combinations might improve efficacy. Given the frequency of patients that do not respond to checkpoint inhibition (50–80% depending on tumor type6), technologies are urgently needed to increase response rates.
Previous data-driven work determined which clinically feasible cytokines provide the best synergy with tumor-recognizing T cells.23 Tumor necrosis factor alpha (TNFa) and interleukin-2 (IL-2) were identified as the best arming devices in the context of tumor infiltrating lymphocyte (TIL) therapy and adoptive therapy with genetically modified anti-tumor T cells.24,25 Since checkpoint blockade exerts its effects through T cells,6 it became logical to study whether TNFa and IL-2 armed adenoviruses could be useful in this setting, with or without additional adoptive T cell transfer.
Materials and methods
Cell line and viruses. B16.OVA, a murine melanoma cell line, was kindly provided by Professor Richard Vile (Mayo Clinic, Rochester, MN, USA) and was cultured in RPMI 1640, supplemented with FBS (10%), L-Glutamine (2 mM), penicillin (100 U/mL), streptomycin (100 μg/mL) and G-418 (5 mg/mL) under recommended conditions. The construction of Ad5-CMV-mIL2 and Ad5-CMV-mTNFα adenoviruses is described elsewhere.24
In vivo studies. For each of the three animal experiments, 4–6 week old C57 BL/6JOlaHsd mice were obtained (Envigo, Indianapolis, IN, USA) and housed in Biosafety level 2 facilities. After one week of quarantine the animals were subcutaneously engrafted with 2.5 × 105 B16.OVA cells in 100 μl of plain RPMI 1640 in the left flank. When the tumors had a volume over 3 mm (around day 11) the animals were randomly divided into groups and treatments started. Tumors were measured at least every 3 days after starting the treatments during the first 15 days and then once per week with an electronic caliper. Tumor volume was calculated as 0.5 × longest diameter × (shortest diameter).2 Mice were observed daily and euthanized when tumor diameter exceeded 18 mm, or at selected time-points for collection of biological samples. Animals for biological sample analysis were selected randomly and subsequently checked that they were representative of the whole group (no significant differences, after running t-test, between the original group and the selected for collection or the remaining animals).
Treatments. Viral treatments were given intratumorally with 30G insulin needles in 50 μL of phosphate buffered saline (PBS) with doses of 0.05-1 × 108 vp (with equal amounts of Ad5-CMV-mIL2 and Ad5-CMV-mTNFα viruses) or PBS only in control groups. Anti-murine PD-1 (Clone RMPI-14, BioXCell) and the adoptive cell transfer therapy (CD8+ enriched population obtained from OT-I transgenic mice23) were injected intraperitoneally in 100 μL of RPMI 1640. The frequency of administration of the treatments in each experiment is provided in the respective figures. All injections were performed under isoflurane anesthesia.
Flow cytometric analyses. After collection, tumors and spleens were passed through 70 μm cell strainers in order to get a single cell suspension and then centrifuged and resuspended in freezing media (90% FBS, 10% DMSO) for storage at -80°C until flow cytometric analysis. Antibody staining for CD3 (PE-Cy5 conjugated, clone 145-2C11, Biolegend), CD8 (FITC conjugated, clone 53–6.7, Biolegend), CD4 (FITC conjugated, clone GK1.5, Biolegend), PD-1 (PE-Cy7 conjugated, clone 29 F.1A12), CTLA-4 (PE-dazzle conjugated, clone UC10-4B9, Biolegend), TIM-3 (PerCP-Cy5.5 conjugated, clone RMT3-23, Biolegend), CD25 (PE-Cy7 conjugated, clone 3C7, Biolegend) and FoxP3 (PE conjugated, clone MF-14, Biolegend) were performed following manufacturer instructions. Recombinant MHC pentamers for the analysis of antigen-specific T cells (H-2Kb/SIINFEKL, H-2Db/KVPRNQDWL, and H-2 Kb/SVYDFFVWL all of them PE conjugated from Proimmune) were used according to manufacturer instructions. At least 100,000 events were analyzed with the Sony SH800Z cytometer (Sony, Tokyo, Japan) under recommended use instructions.
Cytometric Bead Array analysis. Fragments of collected tumors were snap-frozen on dry ice and stored at -80°C until analysis. Protein fraction of the samples was obtained as described previously.23 Samples were stained with Cytometric Bead Array Mouse Th1/Th2/Th17 Cytokine kit (560485, BD) and analyzed on BD Accuri C6 Cytometer (BD, Franklin Lakes, NJ, USA) with FCAP Array Software (BD, Franklin Lakes, NJ, USA) under manufacturer instructions. Cytokine expression was normalized to total protein present in the sample, using Warburg-Christian method after the values were obtained by spectrophotometry with Biophotometer (Eppendorf, Wesbury, NY, USA).
Statistical analyses. SPSS (IBM, New York, NY, USA) version 24 was used to analyze the tumor volume evolution by linear mixed-model analysis of multiple time-correlated log-transformed normalized tumor volumes. GraphPad Prism 7 (GraphPad Software, San Diego, CA, USA) was used to analyze overall survival (Kaplan-Meier survival estimates), and to evaluate differences in the biological samples by unpaired t test with Welch's correction. Synergy assessed by Webb method as described before.26
Ethical statement. All animal experiments in this study were performed in accordance with the recommendations in the Act on the Protection of Animals Used for Scientific or Educational Purpose (497/2013) and Government Decree on the Protection of Animals Used for Scientific or Educational Purposes (564/2013) as well as the European Directive 2010/63/EU. The protocols describing the work and procedures were approved by ethical committee from the National Animal Experiment Board of the Regional State Administrative Agency of Southern Finland.
Results
Virotherapy enables anti-PD-1 blockade and adoptive cell therapy: Single-dose set-up
The first study was designed as a proof of concept experiment in which both the viral treatment and the adoptive cell therapy (ACT) were given only once and the PD-1 blocking monoclonal antibody was given 5 times (Fig. 1A). In this set-up, all the therapies tested displayed superior (p < 0.05) tumor growth control (Fig. 1B), and improved overall survival (Fig. 1C) over the saline treated control. Importantly, also checkpoint blockade alone exhibited a significant improvement in antitumor efficacy as compared with saline treated control, validating the selected administration frequency and dosing of the antibody. The best groups in this experiment were virotherapy combined with OT-I ACT or checkpoint blockade, or the three therapies together. When comparing double treatments, those including virus (i.e. Virus + anti-PD-1 or Virus + ACT) showed better tumor growth control (p < 0.01) and overall survival (p < 0.05) than the double group consisting of anti-PD-1 and ACT, indicating that virotherapy was the most relevant companion for both of the T-cell related therapy approaches tested.
Figure 1.

Proof of concept antitumor efficacy and overall survival after the combination of virotherapy with checkpoint blockade and adoptive cell therapy. (A) 7–8 animals per group received subcutaneous B16.OVA tumors that were grown for 11 days. Then, 1 × 108 viral particles of non-replicating adenoviruses (coding for mIL2 and mTNFa) or PBS were injected intratumorally. The same day, depending on the group they belonged, 2 × 106 CD8+ OT-1 T cells were adoptively transferred and/or 0.1 mg of anti-PD-1 were injected. Anti-PD-1 treatment was repeated 5 more times every 3 days. (B) Normalized mean tumor volume and SEM at day 15. (C) Overall survival.
Checkpoint blockade delivers better tumor control than adoptive cell therapy when combined with virotherapy
In order to identify the best candidate for the combination with the virotherapy, a second experiment was carried out with modifications in the virus dose and number of anti-PD-1 treatments (Fig. 2A). As the virus was identified as a key component in the proof of concept experiment, a reduced virus dose was used to highlight the role of OT-I cell transfer and anti-PD-1 in the combination set-up, and select the one with stronger impact on the outcome of tumor control. Virus dose reduction resulted in non-significant (p = 0.054) tumor growth control compared to virus monotherapy, but this group was studied later at full dose (see below). Also, the number of administrations of anti-PD-1 was increased to evaluate effects on long-term survival (Fig. 2B and C). When compared with the group treated with virus only, the only dual treatment that showed significant improvement on both tumor growth control (p < 0.0001) and overall survival (p < 0.001) was virus + anti-PD-1 therapy. When the three therapies were combined, better tumor growth control (than virotherapy alone) is achieved (p < 0.0001) but this did not result in a significant improvement in survival (p = 0.072). The groups delivering the best results were virus + anti-PD-1 and the triple combination, with no significant differences between them.
Figure 2.

Antitumor efficacy and overall survival after the combination of low dose virotherapy with checkpoint blockade and adoptive cell therapy. (A) Subcutaneous B16.OVA tumors were grown for 10 days. Then mice received i.t. 5 × 106 viral particles of non-replicating adenoviruses coding for mIL2 and mTNFa or PBS. The same day, depending on the group they belonged, 2 × 106 CD8+ OT-1 T cells were adoptively transferred and/or 0.1 mg of anti-PD-1. Checkpoint blockade treatment was repeated 9 more times every 3 days. 6 random animals from each group were euthanized at day 12 and organs were collected for further analysis, the remaining animals (9-12) were maintained for survival studies. (B) Overall survival and statistical significances. (C) Individual tumor growth lines for the different conditions tested and statistical significance of the differences between groups at day 12.
Tumors treated with anti-PD-1 and virotherapy display the most favorable TIL profile
In the same experiment, 6 animals from each group were euthanized on day 12 to analyze their tumors, particularly the TIL subsets. The groups treated with virotherapy and anti-PD-1 displayed an increase in the frequency of intratumoral cytotoxic CD8+ T cells (Fig. 3A). Considering the set-up of the study, special attention was paid on CD8+ TIL phenotype, especially in terms of expression of different checkpoint markers. Besides PD-1, for its particular involvement with one of the therapies, two other checkpoint pathways CTLA-4 and TIM-3 were studied. The percentage of PD-1+ CD8+ T cells was increased in the virotherapy + anti-PD-1 group (Fig. 3B) and the expression of PD-1 in those cells was also upregulated (Fig. 3C). The virus + anti-PD-1 group and more intensely the group receiving the three therapies, showed an increased amount of CD8+ cells positive for CTLA-4 (Fig. 3D). A trend for increased numbers of TIM-3+ CD8 cells was observed only in the triple treated group (Fig. 3E). Regarding the levels of CTLA-4 and TIM-3 in positive cells, there was no significant difference in expression patterns (mean fluorescence intensity, Supplementary figure 1).
Figure 3.

Phenotypical analysis of tumor infiltrating lymphocytes 13 days after the different treatments started. (A) Percentage CD3+ CD8+ cells of total cells in the tumor. (B) Percentage of PD-1+ lymphocytes of parent population (CD3+ CD8+). (C) Mean fluorescence intensity of the channel used for anti-PD-1. (D) Percentage of CTLA-4+ lymphocytes of parent population (CD3+ CD8+). (E) Percentage of TIM-3+ lymphocytes of parent population (CD3+ CD8+). (F) Percentage of OVA-specific lymphocytes of parent population (CD3+ CD8+). (G) Percentage CD3+ CD4+ cells of total cells in the tumor. (H) Percentage of Regulatory T cells (CD25+ FoxP3+) of parent population (CD3+ CD4+).(I) Percentage of conventional CD4 T cells (CD25+ FoxP3-) of parent population (CD3+ CD4+).
It is noteworthy that while PD-1 was abundantly expressed in all groups (average values between 40–70%), the values were much lower for CTLA-4 (average values below 2%) and TIM-3 (average values below 1%). When studying the presence of tumor-specific T cells the two groups with best tumor control (virus + anti-PD-1 with or without ACT) showed an increase in OVA-specific TILs (Fig. 3F). These tumor specific cells showed a similar pattern on PD-1, CTLA-4 and TIM-3 as the ungated CD8+ cell population (Supplementary Figure 2). It should be noted that in the triple and virus + ACT groups, some of the OVA specific T cells were probably cells of the OT-1 graft. Regarding the CD4+ compartment (Fig. 3G), a significant increase in total CD3+ CD4+ cells was assessed only in the group treated with virus + anti-PD-1 while T-regulatory CD4+ cells (i.e. “Treg”) were increased only in the group treated with the virus as monotherapy (Fig. 3H), while the conventional CD4+ T helper cell population, characterized as CD3+ CD4+ CD25+ FoxP3- cells, was increased in all groups receiving viral therapy combined with either anti-PD-1 or ACT (Fig. 3I).
Optimized treatment regimen with virotherapy and PD-1 blockade results in complete responses
In the third experiment (Fig. 4A) virus and anti-PD-1 were administered at full dose. There were two groups treated with both the virus and the checkpoint inhibitor. In one of them, 15 rounds of both treatments were given simultaneously (through different administration routes). In the other group, the virus treatment was given 15 times while the anti-PD-1 treatment was started only with the third virus administration, resulting in a total of 13 cycles. Interestingly, optimizing the treatment protocol to mimic multiple rounds of both therapies as typical in patient treatment improved overall survival (Fig. 4B) and led to better tumor growth control (Fig. 4C) when anti-PD-1 and virotherapy were combined.
Figure 4.

Antitumor efficacy and overall survival in a clinically relevant set up after the combination of virotherapy with checkpoint blockade. (A) 12–16 animals per group received subcutaneous B16.OVA tumors that were grown for 10 days. Then mice received i.t. 1 × 108 viral particles of non-replicating adenoviruses coding for mIL2 and mTNFa or PBS. The same day, depending on the group they belonged, they received 0.1 mg of anti-PD-1. Checkpoint blockade treatment was repeated every 3 days for a total of 13–15 times. At day 4, 7 untreated animals engrafted with the same tumors were sacrificed to study the status of the tumor at “baseline” time point. 6–7 random animals from each group were euthanized at day 11 (grey line) and organs were collected for further analysis, remaining animals were maintained for survival studies. (B) Overall survival and statistical significances. (C) Individual tumor growth lines for the different conditions tested and statistical significance of the differences between groups at day 39.
Both double treatments were superior over any monotherapy and treatment synergy was observed (Supplementary Figure 3). Moreover, in double-treated groups, tumors were increasing in size for the first 10 days, up to 15-fold over the original volume, but after that, antitumor effects seemed to appear reducing large tumors to scars (probably lacking viable tumor cells), as seen in human immunotherapy trials.27
Tumor growth was controlled significantly better (p < 0.01) with the prime and boost approach. This approach resulted in 100% of the animals remaining alive without tumors by day 90. Analysis of the survival curves indicated that prime and boost virotherapy with “delayed” anti-PD-1 resulted in the lowest hazard ratio when compared to anti-PD-1 alone (HR = 0.026 [0,005; 0,139]) or virotherapy alone (HR = 0.069 [0,015; 0,327])
Cytokine-armed adenovirus drives the microenvironment of melanoma tumors towards a proinflammatory state
The tumor microenvironment was studied by analyzing cytokine expression levels on day 11 (gray dotted line in Fig. 4 indicates this time point). As expected, intratumoral expression of TNF and IL-2 (Fig. 5A and 5B) were 10 times higher in groups that were treated with cytokine-expressing viruses. This is most easily explained by the expression of these transgenes by the viruses. Interferon gamma on the other hand did not show any relevant changes across the groups (Fig. 5C). The group delivering the best results in tumor control (virus + anti-PD-1 with the prime and boost strategy) also showed an increase in IL-4, IL-6 and IL-10 (Fig. 5D-F). IL-17 A expression (Fig. 5G) was significantly upregulated in groups which received both anti-PD-1 and virotherapy. The expression of IL-17 A is typically linked to “helper Th17” responses, so this data hints on the possible relevance of that T cell subset. When cytokines were grouped into proinflammatory Th1 (TNF, IL-2 and IFNg) or anti-inflammatory Th2 (IL4, IL6 ad IL10), cytokine-coding viruses were found to skew the cytokine balance of the tumors towards Th1 (Fig. 5H), suggesting that alteration of tumor microenvironment contributes to the anti-tumor effect seen.
Figure 5.

Cytokine profile expression of tumor samples 11 days after the different treatments started. All values are normalized by the cytokine expression of the mock mean value (A) Tumor necrosis factor. (B) Interleukin-2. (C) Interferon gamma. (D) Interleukin-4. (E) Interleukin-6. (F) Interleukin-10. (G) Interleukin-17 A (also studied as Th17 signal representative cytokine). (H) Comparison of pooled Th1 (TNF, IL-2 and IFNg) and Th2 (IL-4, IL-6 and IL-10) cytokines present in the tumor microenvironment.
Long term survivors display increased presence of systemic tumor specific lymphocytes against different epitopes
The presence and distribution of different tumor specific lymphocytes was studied from spleen samples collected on the experiment described in Fig. 4A. First, 6–7 spleens per group were collected on day 11 and at day 90 all mice with complete responses were killed and their spleens were collected following identical procedures. Three different pentamers were used to study lymphocytes against three different tumor specific antigens: OVA, gp100 and Trp2. OVA is a model specific tumor epitope while the other two are “shared” melanoma epitopes. Those animals that responded to the treatments and had their tumors cleared displayed higher numbers (p < 0.05) of tumor specific lymphocytes. When analyzing both time points separately, there were no remarkable differences between groups. Notably, on day 90, there was no significant variation between the groups treated with and without anti-PD-1. This suggests that anti-PD-1 might influence the activity but not frequency of the studied clones of tumor specific lymphocytes.
Perhaps importantly, the proportion of tumor-recognizing T-cell clones was higher on day 90 than on day 11 in all groups. Probably all animals that made it to the end of the experiment had benefited from therapy. This proposes an association between anti-tumor T-cell immunity and survival in the context of melanoma bearing animals treated with viruses coding for TNFa and IL-2 and receiving anti-PD-1.
Discussion
The objective was to study the feasibility of using adenoviruses coding for TNFa and IL-2 to increase the efficacy of PD-1 checkpoint inhibition. Furthermore, the sequence of administration was optimized with relevance for clinical protocols. Previously, different viruses28-31 have been combined with checkpoint inhibitors but, to our knowledge, 100% long term survival has never been previously achieved in this aggressive melanoma model32 used here. Previous results have, however, displayed benefits after the combination of viral platforms with checkpoint inhibitors such as anti-PD-129,30 or PD-L128 among others. One reason for the promising efficacy seen in our approach over other reports could relate to the transgenes utilized. IL-2 and TNFa were selected in a rigorous data-driven process focusing on T-cell effects.23 Previous results also pointed to an increased efficacy of ACT when they were coupled to this virus24
We saw that virus treatment was able to polarize the tumor microenvironment towards an antitumor status as assessed by cytokine profile (Fig 5) but only when the virotherapy is combined with PD-1 blockade the polarization is also seen at T-cell phenotype level (Fig 3). With regard to interaction between virotherapy and checkpoint blockade, the most likely explanation for synergy is that both treatments work towards the enhancement of anti-tumor specific cytotoxic cells, but without direct interaction between them; as two companions working in parallel towards the same purpose, but with different mechanism-of-action. Virotherapy has been proposed responsible for recruiting and activating cytotoxic T lymphocytes (CTLs) and modulating the immune microenvironment by expression of cytokines, and also through inherent alarm signals triggered after viral infection (pathogen associated molecular pattern receptors33). On the other hand, checkpoint blockade could be essential for preventing exhaustion of T cells recruited and stimulated by the virus. Also, blocking PD-1 may be important for prevention of immunosuppressive counter-responses which follow any prolonged immune response occurring in immune competent animals.6 In normal situations the purpose of counter-response is to protect the body against autoimmunity, but this mechanism is hijacked by tumors for avoiding immune-mediated destruction.6
The presence of TILs is considered one of the most important indicators of anti-tumor immunity.34,35 Among TIL populations, those cells able to specifically recognize antigens (especially neoantigens) expressed by the tumor cells are particularly relevant.36 In the B16-OVA model used here, tumor cells are engineered to express a xenoantigen ovalbumin,37 to enable studies of tumor specific T cells. Both subpopulations (CD3+ CD8+ and CD3+ CD8+ OVA+) were significantly increased in groups displaying the best anti-tumor results (Figs. 3A and 3F). This result is compatible with the notion that virus is able to recruit T cells to the tumor.24 Besides the tumor specific TILs we also studied the presence of the relevant lymphocyte clones on the systemic level (Fig 6). In addition to OVA specific lymphocytes, we studied tumor specific lymphocytes against the Trp2 and gp100 antigens. While the ovalbumin is an antigen model-specific for B16.OVA cell line,37 Trp2 and gp100 are widely expressed in a wide variety of melanomas.38,39 This allowed extension of our findings beyond the cell line used to other melanoma models.
Figure 6.
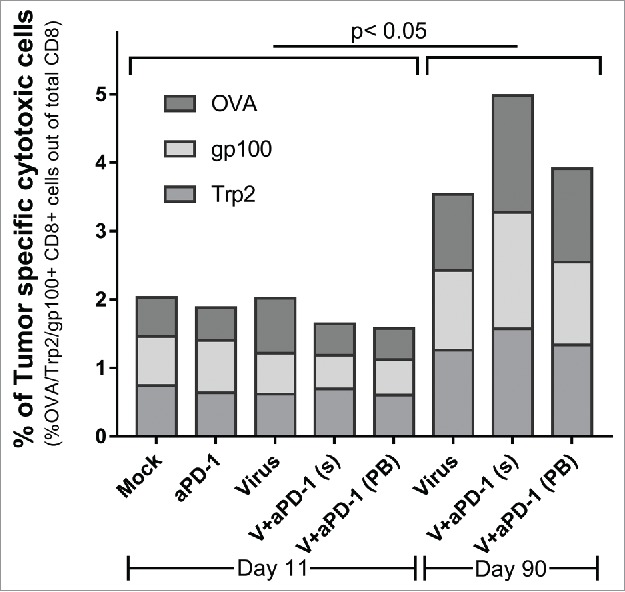
Presence of tumor specific CD8 cells in spleens at two different time points. Stacked percentages of OVA/gp100/Trp2 specific CD8+ CD3+ splenocytes. Samples from day 11 (n = 6-7) and from survivors at day 90 (n = 2 from virus alone group, n = 6 from virus + anti-PD-1[simultaneous] and n = 9 from virus + anti-PD-1 [prime and boost]).
A different view on the presence of tumor specific responses can be obtained by comparing spleens on day 90 to spleens on day 11. The former were obtained from animals that benefited from therapy since they were alive. The latter represent initial stages of induction of the immune response. This comparison is particularly relevant when taken together with the data in Fig. 3F. It seems that when tumors are present the groups treated with virus and anti-PD-1 show an increase in tumor specific lymphocytes at the tumor but not in the spleen. A hypothesis explaining this finding is that the treatments increase not only antitumor responses but also improve the trafficking to and proliferation at the tumor.
Some of the cell types in the CD3+ CD4+ compartment also exert a beneficial role in tumor control,35 while some other subsets like T-regulatory cells may act in an opposite manner.40 Further subsets such as helper CD4+ cells contribute to antitumor responses.41 In line with recent observations on the importance of CD4+ subpopulations, we found that the groups with best tumor control displayed high proportions of helper-like cells versus Tregs (Figs. 3G and 3H). Moreover, IL-17 expression was upregulated in virus-treated tumors, suggesting an additional role for Th17-like CD4+ cells. The fact that Th17 responses have been described to play a bipolar effect42,43 on antitumor immunity might be influenced in this case by antiviral mechanisms enhancing the inflammatory Th17 effects as studied in other immune-related diseases.44,45
Another relevant issue is the selection of the checkpoint inhibitor. There is a relatively wide variety of options (at least preclinically) and there is currently a lack of deep understanding on their respective optimal use. Based on the expression profiles of inhibitory pathways in the melanoma model used here, the most widely expressed inhibitory signal seems to come from the PD-1 pathway rather than CTLA-4 or TIM-3 (Fig 3), albeit every pathway has different mechanisms of action making head-to-head comparisons of absolute inhibitory effect on lymphocytes difficult.6,46 Therefore, it could be theorized that in this particular model the use of therapeutic anti-PD-1 antibodies might provide higher benefit than others (e.g. anti-CTLA-4, anti-TIM-3). Others studied different combinations of checkpoint inhibitors with virotherapy with somewhat similar28,30,31,47 or opposite outcomes,29 which depicts the need for further understanding of these inhibitory pathways.
Depending on the particular expression levels of the different inhibitory routes, the achievement of a clear antitumor immune response might be heavily dependent on the blockade of a specific route. For example, in one of the conditions tested in this study (Fig 2) the group treated with virotherapy, checkpoint inhibitors and ACT did not perform better than groups with fewer therapies, hypothetically due to high levels of non-blocked PD-1 receptors on the surface of the TILs (Fig 3). Similarly, the group treated with the three therapies (i.e. virotherapy, anti-PD-1 and ACT) might have upregulated secondary inhibitory routes such as CTLA-4 (Supplementary Figure 2B) as a counter response of the stronger initial response.
To add more uncertainty to the understanding of the presence of checkpoint markers, some of them as PD-1 and CTLA-4 are expressed on the surface of cytotoxic lymphocytes after their activation.6 This phenomenon, might lead to ambiguous interpretation of the data as the mere presence of the proteins is not automatically a detrimental feature when analyzing biological samples and actually might mean a higher antitumor efficacy48,49 if the potential inhibitory effect coming after they bind their ligands is avoided (e.g. by the use of an inhibitory antibody).
When studying the tumor microenvironment, it has been claimed that intratumoral injection of cytokines can result in tumor control or beneficial effects on the tumor microenvironment23,50,51 with no need of specific cell populations able to produce those cytokines. In this regard, viruses are a convenient tool to deliver cytokines locally, resulting in extended high level expression without significant systemic exposure.51,52 Also, the innate proinflammatory effects of the virus are amplified in the tumor microenvironment by viral transgene expression (TNFa and IL-2). This is a likely reason why there is an increase (Fig 5H) in Th1 cytokines which may contribute to a favorable inflammatory status with subsequent antitumor effects.53,54 Due to the immune homeostasis,55 a Th2 response typically follows any Th1 response. However, when the upregulation of Th1 and Th2 cytokines in the tumor are compared, the former were expressed 16.8-fold higher on average while the latter were increased only 3.4-fold in virus treated groups. Regarding Th17 signals, their role is more controversial as they could lead to both antitumor (e.g. CD8+ cell activation, intratumoral immune cell recruitment) and protumor (e.g. angiogenesis, myeloid-derived suppressor cells recruitment) changes.43,56,57
The field of immunotherapy has evolved steadily in the last decade but the excitement caused by long-term responses in some patients is frequently coupled with frustrating ignorance regarding optimal combinations, dosing and administration regimes. In this regard, we addressed the sequencing of virotherapy with checkpoint blockade. A better outcome was seen when checkpoint blockade was started after a priming period of virotherapy given as monotherapy. We hypothesized that the initial virus injections alone (i.e. without concomitant anti-PD-1 administration) might allow the virus to “exploit” tumor immunosuppression for avoiding anti-viral immunity while generating anti-tumor immunity. Further studies are needed in this regard
Our results provide the rationale to study the same question with other viruses – it is possible that priming before initiation of anti-PD1 is useful for many types of viruses. It is pertinent to note that the phase Ib trial of T-VEC in combination with pembrolizumab featured a priming period with virus injections before anti-PD-1 was started.22 In contrast, the phase III trial which followed featured simultaneous administration of both drugs without priming. If there is an efficacy difference between these trials, this difference in administration could be an important reason. Our data suggests that it is not trivial how and when the drugs are given.
In summary, we report 100% cure rates and 100% survival when mice with aggressive immunosuppressive melanoma (similar to many human melanoma patients) were injected with adenoviruses coding for TNFa and IL-2, with subsequent initiation of anti-PD-1 therapy in a prime and boost manner. Mechanistic clues suggest that this regime induced proinflammatory danger signals in the tumor microenvironment and led to effective recruitment and stimulation of anti-tumor T cells, whose exhaustion was prevented by the anti-PD-1 antibody. These results set the stage for clinical evaluation of an oncolytic adenovirus coding for TNFa and IL-2 (TILT-123)25 in melanoma patients receiving anti-PD-1 antibody.
Supplementary Material
Conflict of interest
A.H. is shareholder in Targovax ASA (Oslo, Norway) and in TILT Biotherapeutics Ltd. (Helsinki, Finland). A.H., M.S., R.H., S.P., J.M.S., and V.CC. are employees of TILT Biotherapeutics Ltd. Other coauthors declare no potential conflict of interest.
Acknowledgments
We thank Eleonora Munaro, Susanna Grönberg-Vähä-Koskela and Minna Oksanen for their assistance during the study. The Biomedicum Flow Cytometry Core Facility (University of Helsinki) as well as the Laboratory Animal Centre (University of Helsinki) are appreciated for technical support. This study was supported by Marie Skłodowska-Curie Innovative Training Networks (ITN-EID VIRION H2020-MSCA-ITN-2014 project number 643130), Jane and Aatos Erkko Foundation, HUCH Research Funds (EVO), Sigrid Juselius Foundation, Finnish Cancer Organizations, University of Helsinki and TILT Biotherapeutics Ltd.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. PMID:21376230 [DOI] [PubMed] [Google Scholar]
- 2.Puzanov I, Milhem MM, Minor D, Hamid O, Li A, Chen L, Chastain M, Gorski KS, Anderson A, Chou J, et al.. Talimogene Laherparepvec in Combination With Ipilimumab in Previously Untreated, Unresectable Stage IIIB-IV Melanoma. J Clin Oncol. 2016;34:2619–26. doi: 10.1200/JCO.2016.67.1529. PMID:27298410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Guarinos C, Juarez M, Egoavil C, Rodríguez-Soler M, Pérez-Carbonell L, Salas R, Cubiella J, Rodríguez-Moranta F, de-Castro L, Bujanda L, et al.. Prevalence and characteristics of MUTYH-associated polyposis in patients with multiple adenomatous and serrated polyps. Clin Cancer Res. 2014;20:1158–68. doi: 10.1158/1078-0432.CCR-13-1490. PMID:24470512 [DOI] [PubMed] [Google Scholar]
- 4.Jiang Y, Li Y, Zhu B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015;6:e1792. doi: 10.1038/cddis.2015.162. PMID:26086965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Quereux G, Pandolfino MC, Knol AC, Khammari A, Volteau C, Nguyen JM, Dreno B. Tissue prognostic markers for adoptive immunotherapy in melanoma. Eur J Dermatol. 2007;17:295–301. PMID:17540635 [DOI] [PubMed] [Google Scholar]
- 6.Baumeister SH, Freeman GJ, Dranoff G, Sharpe AH. Coinhibitory Pathways in Immunotherapy for Cancer. Annu Rev Immunol. 2016;34:539–573. doi: 10.1146/annurev-immunol-032414-112049. PMID:26927206 [DOI] [PubMed] [Google Scholar]
- 7.Kyi C, Postow MA. Immune checkpoint inhibitor combinations in solid tumors: opportunities and challenges. Immunotherapy. 2016;8:821–37. doi: 10.2217/imt-2016-0002. PMID:27349981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teng MW, Ngiow SF, Ribas A, Smyth MJ. Classifying Cancers Based on T-cell Infiltration and PD-L1. Cancer Res. 2015;75:2139–45. doi: 10.1158/0008-5472.CAN-15-0255. PMID:25977340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mellman I, Coukos G, Dranoff G. Cancer immunotherapy comes of age. Nature. 2011;480:480–89. doi: 10.1038/nature10673. PMID:22193102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen DS, Mellman I. Elements of cancer immunity and the cancer-immune set point. Nature. 2017;541:321–30. doi: 10.1038/nature21349. PMID:28102259 [DOI] [PubMed] [Google Scholar]
- 11.Breitbach CJ, Lichty BD, Bell JC. Oncolytic Viruses: Therapeutics With an Identity Crisis. EBioMedicine. 2016;9:31–36. doi: 10.1016/j.ebiom.2016.06.046. PMID:27407036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Niemann J, Kuhnel F. Oncolytic viruses: adenoviruses. Virus Genes. 2017;53(5):700–06. doi: 10.1007/s11262-017-1488-1. PMID:28702840 [DOI] [PubMed] [Google Scholar]
- 13.Li X, Wang P, Li H, Du X, Liu M, Huang Q, Wang Y, Wang S. The Efficacy of Oncolytic Adenovirus Is Mediated by T-cell Responses against Virus and Tumor in Syrian Hamster Model. Clin Cancer Res. 2017;23:239–49. doi: 10.1158/1078-0432.CCR-16-0477. PMID:27435398 [DOI] [PubMed] [Google Scholar]
- 14.Tahtinen S, Gronberg-Vaha-Koskela S, Lumen D, Merisalo-Soikkeli M, Siurala M, Airaksinen AJ, Vähä-Koskela M, Hemminki A. Adenovirus Improves the Efficacy of Adoptive T-cell Therapy by Recruiting Immune Cells to and Promoting Their Activity at the Tumor. Cancer Immunol Res. 2015;3:915–25. doi: 10.1158/2326-6066.CIR-14-0220-T. PMID:25977260 [DOI] [PubMed] [Google Scholar]
- 15.Woller N, Gurlevik E, Fleischmann-Mundt B, Schumacher A, Knocke S, Kloos AM, Saborowski M, Geffers R, Manns MP, Wirth TC, et al.. Viral Infection of Tumors Overcomes Resistance to PD-1-immunotherapy by Broadening Neoantigenome-directed T-cell Responses. Mol Ther. 2015;23:1630–40. doi: 10.1038/mt.2015.115. PMID:26112079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kanerva A, Nokisalmi P, Diaconu I, Koski A, Cerullo V, Liikanen I, Tähtinen S, Oksanen M, Heiskanen R, Pesonen S, et al.. Antiviral and antitumor T-cell immunity in patients treated with GM-CSF-coding oncolytic adenovirus. Clin Cancer Res. 2013;19:2734–44. doi: 10.1158/1078-0432.CCR-12-2546. PMID:23493351 [DOI] [PubMed] [Google Scholar]
- 17.Lawler SE, Speranza MC, Cho CF, Chiocca EA. Oncolytic Viruses in Cancer Treatment: A Review. JAMA Oncol. 2017;3:841–49. doi: 10.1001/jamaoncol.2016.2064. PMID:27441411 [DOI] [PubMed] [Google Scholar]
- 18.Seymour LW, Fisher KD. Oncolytic viruses: finally delivering. Br J Cancer. 2016;114:357–61. doi: 10.1038/bjc.2015.481. PMID:26766734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koski A, Bramante S, Kipar A, Oksanen M, Juhila J, Vassilev L, Joensuu T, Kanerva A, Hemminki A. Biodistribution Analysis of Oncolytic Adenoviruses in Patient Autopsy Samples Reveals Vascular Transduction of Noninjected Tumors and Tissues. Mol Ther. 2015;23:1641–52. doi: 10.1038/mt.2015.125. PMID:26156245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bommareddy PK, Patel A, Hossain S, Kaufman HL. Talimogene Laherparepvec (T-VEC) and Other Oncolytic Viruses for the Treatment of Melanoma. Am J Clin Dermatol. 2017;18:1–15. doi: 10.1007/s40257-016-0238-9. PMID:27988837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fonteneau JF, Achard C, Zaupa C, Foloppe J, Erbs P. Oncolytic immunotherapy: The new clinical outbreak. Oncoimmunology. 2016;5:e1066961. doi: 10.1080/2162402X.2015.1066961. PMID:26942085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ribas A, Dummer R, Puzanov I, VanderWalde A, Andtbacka RHI, Michielin O, Olszanski AJ, Malvehy J, Cebon J, Fernandez E, et al.. Oncolytic Virotherapy Promotes Intratumoral T Cell Infiltration and Improves Anti-PD-1 Immunotherapy. Cell. 2017;170:1109–19 e1110. doi: 10.1016/j.cell.2017.08.027. PMID:28886381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tahtinen S, Kaikkonen S, Merisalo-Soikkeli M, Grönberg-Vähä-Koskela S, Kanerva A, Parviainen S, Vähä-Koskela M, Hemminki A. Favorable alteration of tumor microenvironment by immunomodulatory cytokines for efficient T-cell therapy in solid tumors. PLoS One. 2015;10:e0131242. doi: 10.1371/journal.pone.0131242. PMID:26107883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Siurala M, Havunen R, Saha D, Lumen D, Airaksinen AJ, Tähtinen S, Cervera-Carrascon V, Bramante S, Parviainen S, Vähä-Koskela M, et al.. Adenoviral Delivery of Tumor Necrosis Factor-alpha and Interleukin-2 Enables Successful Adoptive Cell Therapy of Immunosuppressive Melanoma. Mol Ther. 2016;24:1435–43. doi: 10.1038/mt.2016.137. PMID:27357626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Havunen R, Siurala M, Sorsa S, Grönberg-Vähä-Koskela S, Behr M, Tähtinen S, Santos JM, Karell P, Rusanen J, Nettelbeck DM, et al.. Oncolytic Adenoviruses Armed with Tumor Necrosis Factor Alpha and Interleukin-2 Enable Successful Adoptive Cell Therapy. Mol Ther Oncolytics. 2017;4:77–86. doi: 10.1016/j.omto.2016.12.004. PMID:28345026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Siurala M, Bramante S, Vassilev L, Hirvinen M, Parviainen S, Tähtinen S, Guse K, Cerullo V, Kanerva A, Kipar A, et al.. Oncolytic adenovirus and doxorubicin-based chemotherapy results in synergistic antitumor activity against soft-tissue sarcoma. Int J Cancer. 2015;136:945–54. doi: 10.1002/ijc.29048. PMID:24975392 [DOI] [PubMed] [Google Scholar]
- 27.Senzer NN, Kaufman HL, Amatruda T, Nemunaitis M, Reid T, Daniels G, Gonzalez R, Glaspy J, Whitman E, Harrington K, et al.. Phase II clinical trial of a granulocyte-macrophage colony-stimulating factor-encoding, second-generation oncolytic herpesvirus in patients with unresectable metastatic melanoma. J Clin Oncol. 2009;27:5763–71. doi: 10.1200/JCO.2009.24.3675. PMID:19884534 [DOI] [PubMed] [Google Scholar]
- 28.Quetglas JI, Labiano S, Aznar MA, Bolaños E, Azpilikueta A, Rodriguez I, Casales E, Sánchez-Paulete AR, Segura V, Smerdou C, et al.. Virotherapy with a Semliki Forest Virus-Based Vector Encoding IL12 Synergizes with PD-1/PD-L1 Blockade. Cancer Immunol Res. 2015;3:449–54. doi: 10.1158/2326-6066.CIR-14-0216. PMID:25691326 [DOI] [PubMed] [Google Scholar]
- 29.Shim KG, Zaidi S, Thompson J, Kottke T, Evgin L, Rajani KR, Schuelke M, Driscoll CB, Huff A, Pulido JS, et al.. Inhibitory Receptors Induced by VSV Viroimmunotherapy Are Not Necessarily Targets for Improving Treatment Efficacy. Mol Ther. 2017;25:962–75. doi: 10.1016/j.ymthe.2017.01.023. PMID:28237836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Engeland CE, Grossardt C, Veinalde R, Bossow S, Lutz D, Kaufmann JK, Shevchenko I, Umansky V, Nettelbeck DM, Weichert W, et al.. CTLA-4 and PD-L1 checkpoint blockade enhances oncolytic measles virus therapy. Mol Ther. 2014;22:1949–1959. doi: 10.1038/mt.2014.160. PMID:25156126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moesta AK, Cooke K, Piasecki J, Mitchell P, Rottman JB, Fitzgerald K, Zhan J, Yang B, Le T Belmontes B, et al.. Local Delivery of OncoVEXmGM-CSF Generates Systemic Antitumor Immune Responses Enhanced by Cytotoxic T-Lymphocyte-Associated Protein Blockade. Clin Cancer Res. 2017;23:6190–202. doi: 10.1158/1078-0432.CCR-17-0681. PMID:28706012 [DOI] [PubMed] [Google Scholar]
- 32.Pennisi M. A mathematical model of immune-system-melanoma competition. Comput Math Methods Med. 2012;2012:850754. doi: 10.1155/2012/850754. PMID:22701144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cerullo V, Diaconu I, Romano V, Hirvinen M, Ugolini M, Escutenaire S, Holm SL, Kipar A, Kanerva A, Hemminki A. An oncolytic adenovirus enhanced for toll-like receptor 9 stimulation increases antitumor immune responses and tumor clearance. Mol Ther. 2012;20:2076–86. doi: 10.1038/mt.2012.137. PMID:22828500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gabrielson A, Wu Y, Wang H, Jiang J, Kallakury B, Gatalica Z, Reddy S, Kleiner D, Fishbein T, Johnson L, et al.. Intratumoral CD3 and CD8 T-cell Densities Associated with Relapse-Free Survival in HCC. Cancer Immunol Res. 2016;4:419–30. doi: 10.1158/2326-6066.CIR-15-0110. PMID:26968206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rathore AS, Kumar S, Konwar R, Makker A, Negi MP, Goel MM. CD3+, CD4+ & CD8+ tumour infiltrating lymphocytes (TILs) are predictors of favourable survival outcome in infiltrating ductal carcinoma of breast. Indian J Med Res. 2014; 140:361–9. PMID:25366203 [PMC free article] [PubMed] [Google Scholar]
- 36.Strickland KC, Howitt BE, Shukla SA, Rodig S, Ritterhouse LL, Liu JF, Garber JE, Chowdhury D, Wu CJ, D'Andrea AD, et al.. Association and prognostic significance of BRCA1/2-mutation status with neoantigen load, number of tumor-infiltrating lymphocytes and expression of PD-1/PD-L1 in high grade serous ovarian cancer. Oncotarget. 2016;7:13587–98. doi: 10.18632/oncotarget.7277. PMID:26871470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Linardakis E, Bateman A, Phan V, Ahmed A, Gough M, Olivier K, Kennedy R, Errington F, Harrington KJ, Melcher A, et al.. Enhancing the efficacy of a weak allogeneic melanoma vaccine by viral fusogenic membrane glycoprotein-mediated tumor cell-tumor cell fusion. Cancer Res. 2002;62:5495–504. PMID:12359759 [PubMed] [Google Scholar]
- 38.Bianchi V, Bulek A, Fuller A, Lloyd A, Attaf M, Rizkallah PJ, Dolton G, Sewell AK, Cole DK. A Molecular Switch Abrogates Glycoprotein 100 (gp100) T-cell Receptor (TCR) Targeting of a Human Melanoma Antigen. J Biol Chem. 2016;291:8951–59. doi: 10.1074/jbc.M115.707414. PMID:26917722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Capasso C, Magarkar A, Cervera-Carascon V, Fusciello M, Feola S, Muller M, Garofalo M, Kuryk L, Tähtinen S, Pastore L, et al.. A novel in silico framework to improve MHC-I epitopes and break the tolerance to melanoma. Oncoimmunology. 2017;6:e1319028. doi: 10.1080/2162402X.2017.1319028. PMID:28932628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nishikawa H, Sakaguchi S. Regulatory T cells in cancer immunotherapy. Curr Opin Immunol. 2014;27:1–7. doi: 10.1016/j.coi.2013.12.005. PMID:24413387 [DOI] [PubMed] [Google Scholar]
- 41.Aarntzen EH, De Vries IJ Lesterhuis WJ et al.. Targeting CD4(+) T-helper cells improves the induction of antitumor responses in dendritic cell-based vaccination. Cancer Res. 2013;73:19–29. doi: 10.1158/0008-5472.CAN-12-1127. PMID:23087058 [DOI] [PubMed] [Google Scholar]
- 42.Bailey SR, Nelson MH, Himes RA, Li Z, Mehrotra S, Paulos CM. Th17 cells in cancer: the ultimate identity crisis. Front Immunol. 2014;5:276. doi: 10.3389/fimmu.2014.00276. PMID:24987392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Guery L, Hugues S. Th17 Cell Plasticity and Functions in Cancer Immunity. Biomed Res Int. 2015;2015:314620. doi: 10.1155/2015/314620. PMID:26583099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Martinez NE, Sato F, Kawai E, Omura S, Chervenak RP, Tsunoda I. Regulatory T cells and Th17 cells in viral infections: implications for multiple sclerosis and myocarditis. Future Virol. 2012;7:593–608. doi: 10.2217/fvl.12.44. PMID:23024699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.McCarthy MK, Zhu L, Procario MC, Weinberg JB. IL-17 contributes to neutrophil recruitment but not to control of viral replication during acute mouse adenovirus type 1 respiratory infection. Virology. 2014;456–457:259–67. doi: 10.1016/j.virol.2014.04.008. PMID:24889245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Anderson AC, Joller N, Kuchroo VK. Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity. 2016;44:989–1004. doi: 10.1016/j.immuni.2016.05.001. PMID:27192565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Blake SJ, Ching AL, Kenna TJ, Galea R, Large J, Yagita H, Steptoe RJ. Blockade of PD-1/PD-L1 promotes adoptive T-cell immunotherapy in a tolerogenic environment. PLoS One. 2015;10:e0119483. doi: 10.1371/journal.pone.0119483. PMID:25741704 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fernandez-Poma SM, Salas-Benito D, Lozano T, Casares N, Riezu-Boj JI, Mancheño U, Elizalde E, Alignani D, Zubeldia N, Otano I, et al.. Expansion of Tumor-Infiltrating CD8+ T cells Expressing PD-1 Improves the Efficacy of Adoptive T-cell Therapy. Cancer Res. 2017;77:3672–84. doi: 10.1158/0008-5472.CAN-17-0236. PMID:28522749 [DOI] [PubMed] [Google Scholar]
- 49.Tahtinen S, Blattner C, Vaha-Koskela M, Saha D, Siurala M, Parviainen S, Utikal J, Kanerva A, Umansky V, Hemminki A. T-Cell Therapy Enabling Adenoviruses Coding for IL2 and TNFalpha Induce Systemic Immunomodulation in Mice With Spontaneous Melanoma. J Immunother. 2016;39:343–54. doi: 10.1097/CJI.0000000000000144. PMID:27741089 [DOI] [PubMed] [Google Scholar]
- 50.Vacchelli E, Aranda F, Obrist F et al.. Trial watch: Immunostimulatory cytokines in cancer therapy. Oncoimmunology. 2014;3:e29030. doi: 10.4161/onci.29030. PMID:25083328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Santos JM, Havunen R, Siurala M, Cervera-Carrascon V, Tähtinen S, Sorsa S, Anttila M, Karell P, Kanerva A, Hemminki A. Adenoviral production of interleukin-2 at the tumor site removes the need for systemic postconditioning in adoptive cell therapy. Int J Cancer. 2017;141(7):1458–68. doi: 10.1002/ijc.30839. PMID:28614908 [DOI] [PubMed] [Google Scholar]
- 52.Zafar S, Parviainen S, Siurala M, et al.. Intravenously usable fully serotype 3 oncolytic adenovirus coding for CD40 L as an enabler of dendritic cell therapy. Oncoimmunology. 2017;6:e1265717. doi: 10.1080/2162402X.2016.1265717. PMID:28344872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Haabeth OA, Lorvik KB, Hammarstrom C, Donaldson IM, Haraldsen G, Bogen B, Corthay A. Inflammation driven by tumour-specific Th1 cells protects against B-cell cancer. Nat Commun. 2011;2:240. doi: 10.1038/ncomms1239. PMID:21407206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thakur A, Schalk D, Sarkar SH, Al-Khadimi Z, Sarkar FH, Lum LG. A Th1 cytokine-enriched microenvironment enhances tumor killing by activated T cells armed with bispecific antibodies and inhibits the development of myeloid-derived suppressor cells. Cancer Immunol Immunother. 2012;61:497–509. doi: 10.1007/s00262-011-1116-1. PMID:21971587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Muraille E, Leo O, Moser M. TH1/TH2 paradigm extended: macrophage polarization as an unappreciated pathogen-driven escape mechanism? Front Immunol. 2014;5:603. doi: 10.3389/fimmu.2014.00603. PMID:25505468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yamamoto M, Kamigaki T, Yamashita K, Hori Y, Hasegawa H, Kuroda D, Moriyama H, Nagata M, Ku Y, Kuroda Y. Enhancement of anti-tumor immunity by high levels of Th1 and Th17 with a combination of dendritic cell fusion hybrids and regulatory T cell depletion in pancreatic cancer. Oncol Rep. 2009;22:337–43. PMID:19578774 [PubMed] [Google Scholar]
- 57.Murugaiyan G, Saha B. Protumor vs antitumor functions of IL-17. J Immunol. 2009;183:4169–75. doi: 10.4049/jimmunol.0901017. PMID:19767566 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.