

ABSTRACT
Lung-specific overexpression of prostacyclin synthase (PGIS) decreases tumor initiation in murine lung cancer models. Prostacyclin analogs prevent lung tumor formation in mice and reverse bronchial dysplasia in former smokers. However, the effect of prostacyclin on lung cancer progression has not been well studied. We investigated the effects of pulmonary PGIS overexpression in an orthotopic immunocompetent mouse model of lung cancer using two murine lung cancer cell lines. Pulmonary PGIS overexpression significantly inhibited CMT167 lung tumor growth, increased CXCL9 expression, and increased CD4+ tumor-infiltrating lymphocytes. Immunodepletion of CD4+ T cells abolished the inhibitory effect of pulmonary PGIS overexpression on CMT167 lung tumor growth. In contrast, pulmonary PGIS overexpression failed to inhibit growth of a second murine lung cancer cell line, Lewis Lung Carcinoma (LLC) cells, and failed to increase CXCL9 expression or CD4+ T lymphocytes in LLC lung tumors. Transcriptome profiling of CMT167 cells and LLC cells recovered from tumor-bearing mice demonstrated that in vivo, CMT167 cells but not LLC cells express MHC class II genes and cofactors necessary for MHC class II processing and presentation. These data demonstrate that prostacyclin can inhibit lung cancer progression and suggest that prostacyclin analogs may serve as novel immunomodulatory agents in a subset of lung cancer patients. Moreover, expression of MHC Class II by lung cancer cells may represent a biomarker for response to prostacyclin.
KEYWORDS: Lung cancer, lymphocytes, prostacyclin, prostaglandins, tumor immunology
Introduction
A major breakthrough in lung cancer therapy is the development of immunotherapy. Among the most promising approaches to activating therapeutic antitumor immunity is the blockade of immune checkpoints, such as Programmed Death Receptor 1 (PD-1). PD-L1, which is a ligand for PD-1, is expressed on cancer cells and stromal cells in the tumor microenvironment (TME). Binding of PD-L1 to PD-1 on activated T lymphocytes inhibits T cell function and prevents immune attack on tumors. Disrupting these interactions using monoclonal antibodies against PD-1 or PD-L1 reactivates T cells and can generate long-lasting clinical responses in multiple cancer types, leading to approval of these agents for the treatment of multiple malignancies including non-small cell lung cancer (NSCLC). However, response rates in unselected NSCLC patients across clinical trials of PD-1/PD-L1 checkpoint inhibitors have ranged from 14.5%-20%.1-5 Thus, additional strategies to unleash the immune system in patients with NSCLC are urgently needed.
Eicosanoids represent a family of active signaling molecules that act as autocrine and paracrine factors. These products are metabolites of arachidonic acid, which is released from cells, primarily through the action of cytosolic phospholipase A2. Three major downstream pathways have been defined: the cyclooxygenase pathway, which produces prostaglandins and thromboxanes; the 5-lipoxygenase pathway, which produces leukotrienes, lipoxins and hydroxyeicosatetraenoic acids; and the cytochrome P450 pathway, which produces epoxygenated fatty acids. Increases in prostaglandins have been demonstrated in a subset of NSCLC,6, 7 and elevated prostaglandin E2 (PGE2) production has been associated with lung cancer cell lines expressing oncogenic K-Ras.8 Elevated tumor PGE2 levels have been implicated in angiogenesis, tumor growth and invasion, apoptosis resistance, and suppression of anti-tumor immunity.9 Whereas PGE2 promotes lung cancer progression, prostacyclin (PGI2), which is produced by prostacyclin synthase (PGIS), has been shown to inhibit lung cancer initiation. Targeted overexpression of PGIS to the distal lung epithelia in mice and the stable PGI2 analog iloprost decreased lung tumor formation in chemical carcinogenesis and cigarette smoking models.10-12 These findings led to a phase II/III clinical chemoprevention trial, in which iloprost improved endobronchial dysplasia in former smokers.13
Although these studies have demonstrated a chemopreventive role for PGI2 and iloprost, little is known about the potential therapeutic effects of PGI2 on established lung tumors. Interestingly, pulmonary tumors regressed in a patient with systemic sclerosis and metastatic lung adenocarcinoma that was treated with inhaled iloprost for pulmonary arterial hypertension,14 suggesting PGI2 analogs may have antitumor effects. In this study, we have examined the effects of targeted pulmonary PGIS overexpression on lung cancer progression in an orthotopic immunocompetent mouse model using two murine lung cancer cell lines implanted into the lungs of syngeneic mice. These cells form a primary tumor, which subsequently metastasizes to the other lobes of the lung, and to distant organs including the liver and the brain. We report here that PGI2 selectively inhibited CMT167 primary tumor growth and distant organ metastasis through a CD4+ T cell dependent pathway, but had minimal effects of progression of LLC lung tumors. This study suggests that PGI2 can inhibit progression of a subset of lung cancers and that PGI2 analogs may have a novel role as immunomodulatory agents in the treatment of lung cancer.
Results
Pulmonary PGIS overexpression inhibits CMT167 orthotopic lung tumor progression and metastasis
We have previously shown that CMT167 murine lung cancer cells injected into the lungs of syngeneic C57BL/6 mice form a primary tumor, which subsequently forms secondary pulmonary tumors and metastasizes to the liver and brain.15,16 Since production of orally active iloprost was discontinued by the manufacturer, we employed mice with lung-specific overexpression of PGIS (PGIS OE) to test the role of this eicosanoid. Previous studies have shown that these mice have constitutively elevated levels of PGI2, with no significant change in other eicosanoids.10 CMT167 cells stably expressing luciferase were injected into the left lung of mice with targeted pulmonary PGIS OE or wild-type littermate controls (WT). Animals were sacrificed after four weeks, and primary tumors in the left lung were measured with digital calipers. Pulmonary PGIS overexpression significantly decreased primary tumor size (WT 27.2 ± 5.2 mm3 vs PGIS OE 5.9 ± 2.6 mm3, p = 0.0021; Fig. 1A). The other lung lobes, livers, and brains were harvested from tumor-bearing animals, and metastases were measured by ex vivo bioluminescence. The incidence of secondary pulmonary tumors in PGIS OE mice was half the incidence in control animals (WT 66% versus PGIS OE 33%, p = 0.0895; Fig. 1B). Similarly, pulmonary PGIS overexpression significantly decreased the incidence of liver metastases (WT 47.6% versus PGIS OE 13.3%, p = 0.0399; Fig. 1B). There was no difference in the number or size of secondary pulmonary tumors or liver metastases between PGIS OE and control mice with metastases (Figs. 1C-1E). No brain metastases were detected in either group.
Figure 1.
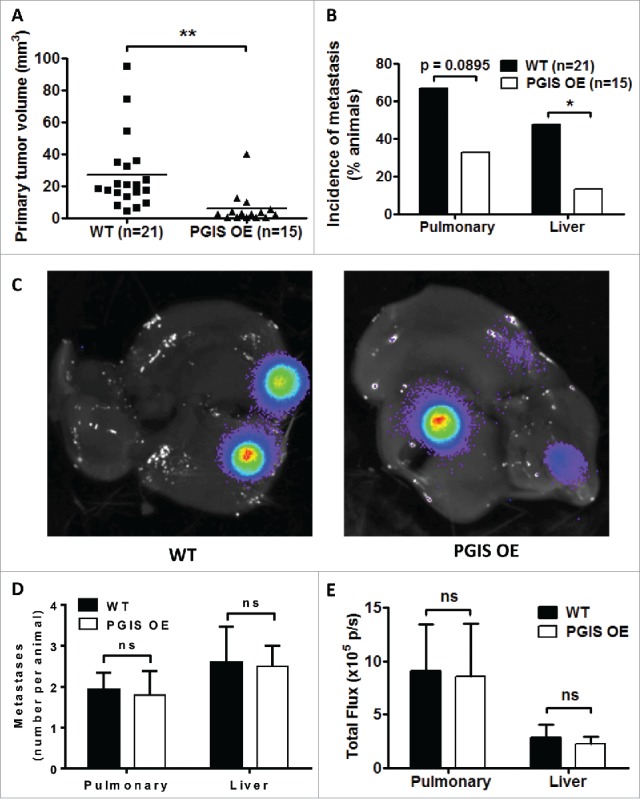
Pulmonary PGIS overexpression inhibits CMT167 tumor progression and metastasis. CMT167 tumors were implanted in pulmonary PGIS overexpressing transgenic mice (PGIS OE, n = 15) or littermate controls (WT, n = 21). After four weeks, lungs and livers were harvested and imaged ex vivo with an IVIS 50 imaging system. (A) Primary lung tumor volumes were measured; each point represents a single tumor; ** = p < 0.01. (B) The incidence of secondary pulmonary tumors and liver metastases was quantified by ex vivo bioluminescence (percent of total mice per group; * = p < 0.05). (C) Representative ex vivo bioluminescent images of livers with metastases from a control mouse (left panel) and a PGIS overexpressor (right panel). The number (D) and signal intensity (E) of pulmonary and liver metastases were quantified by ex vivo bioluminescence imaging.
Prostacyclin analogs do not affect CMT167 cells in vitro
To determine whether decreased lung tumor growth in vivo is mediated through direct effects of PGI2 on cancer cells, we examined the effects of PGI2 analogs on CMT167 cells in vitro. Exposure of CMT167 cells to the stable PGI2 analogs iloprost or treprostinil had no effect on proliferation (Supplemental Fig. S1A). Stimulating CMT167 cells with TGF-β1 resulted in morphological alterations consistent with epithelial-mesenchymal transition (EMT), in which cells changed from a cuboidal morphology to an extended spindle-like morphology (Supplemental Fig. S1B). However, PGI2 analogs did not prevent TGF-β1-induced EMT in CMT167 cells. EMT is associated with decreased expression of the epithelial marker E-cadherin; exposure to TGF-β1 decreased E-cadherin levels in CMT167 cells, but this was not affected by either PGI2 analog (Supplemental Fig. S1C-S1D). Finally, our previous studies indicated that the chemopreventive effects of PGI2 may be mediated through activation of PPARγ.12 PPARγ activation in CMT167 cells was assessed using a PPAR-response element luciferase reporter (PPAR-RE).17 Whereas the specific PPARγ activator pioglitazone induced PPAR-RE activity in CMT167 cells, neither iloprost nor treprostinil increased PPAR-RE activity (Supplemental Fig. S1E). Together these data indicate that PGI2 has minimal effects on CMT167 cells in vitro.
CMT167 tumor growth inhibition by pulmonary PGIS overexpression is mediated through CD4+ T lymphocytes
PGI2 has previously been shown to regulate CD4+ T cell differentiation and function in other disease models.18-20 As a result, we investigated whether pulmonary PGIS overexpression affects recruitment of tumor-infiltrating lymphocytes (TILs). CMT167 lung tumor sections were stained for expression of tumor-infiltrating CD3+, CD4+, and CD8+ T cells (Figs. 2–3). Pulmonary PGIS overexpression significantly increased the density of tumor infiltrating CD3+ T cells (Fig. 3A: WT 49.59 ± 6.73 vs PGIS OE 80.76 ± 10.18 cells/hpf, p = 0.02). Subset analysis revealed that pulmonary PGIS overexpression increased tumor-infiltrating CD4+ T cells compared to control animals (Fig. 3B: WT 23.21 ± 5.70 vs PGIS OE 59.29 ± 11.48 cells/hpf, p = 0.01), but not tumor-infiltrating CD8+ T cells (Fig. 3C: WT 25.79 ± 5.40 vs PGIS OE 21.48 ± 6.25 cells/hpf, n.s.). By flow cytometry, similar levels of CD44 and CD69 expression on CD4+ and CD8+ T cells were found in PGIS overexpressing transgenic mice and wild type littermates implanted with CMT167 orthotopic lung tumors (Supplemental Fig. S2).
Figure 2.
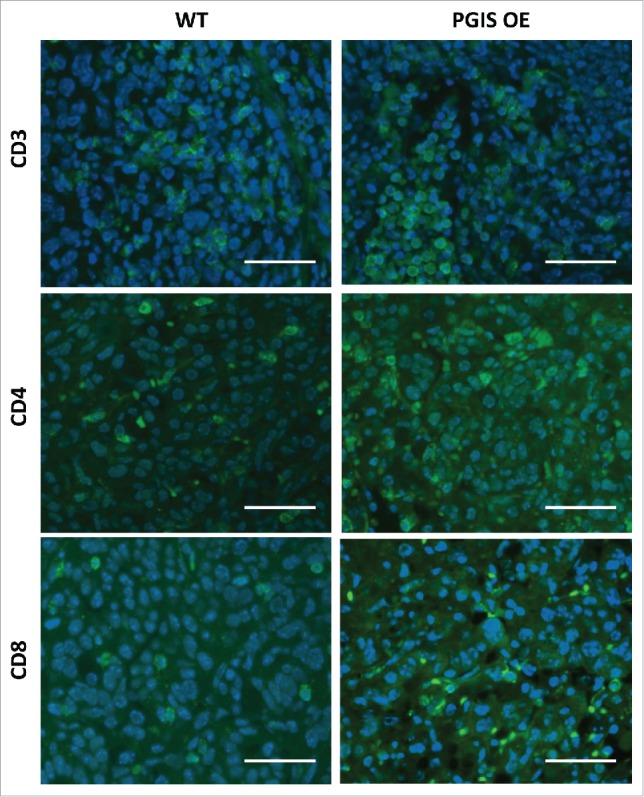
Pulmonary PGIS overexpression increases lymphocyte infiltration in CMT167 tumors. Representative CD3 (top panels), CD4 (middle panels), and CD8 (bottom panels) immunostaining (AF488, green) of sections from PGIS overexpressing mice (right panels) and wildtype littermates (left panels). Nuclei were stained with DAPI (blue); scale bar = 40 μm.
Figure 3.
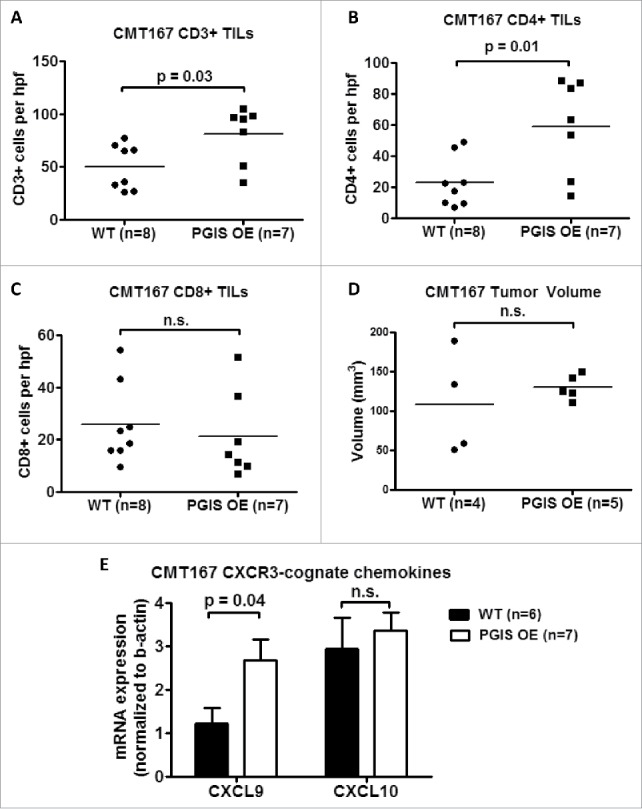
Pulmonary PGIS overexpression increases CD4+ T cell infiltration in CMT167 tumors. Three sections from each animal were stained for (A) CD3+, (B) CD4+, and (C) CD8+ TILs and three random fields (20x) per section were quantified. The mean was used for analysis; each point represents a single mouse. (D) Mice were injected with either anti-CD4 monoclonal antibody or control IgG antibody starting 1 day prior to tumor implantation and throughout the course of the tumor progression experiment. CMT167 tumors were implanted in either PGIS OE (n = 5) or WT littermates (n = 4). After four weeks, primary lung tumors were isolated and tumor volumes were measured by digital calipers. Each point represents a single tumor. (E) Two weeks after CMT167 tumor implantation, total RNA was isolated from tumor-bearing left lungs. CXCL9 and CXCL10 mRNA expression was analyzed by quantitative RT-PCR and normalized to β-actin. Results represent the mean with the S.E.M. indicated. Statistically significant differences are indicated as calculated by unpaired t test, n.s. = not significant.
To determine whether the anti-tumorigenic effects of pulmonary PGIS overexpression are mediated by CD4+ tumor infiltrating lymphocytes, we depleted CD4+ T cells and examined the effects on CMT167 orthotopic lung tumor growth. As shown in Supplemental Fig. S3, mice injected with anti-CD4 antibody had few detectable CD4 T cells in peripheral blood 10 days and 17 days after CD4 depletion was started. Immunodepletion of CD4+ T cells in pulmonary PGIS OE mice restored CMT167 primary tumor size to the level seen in control mice (Fig. 3D: WT 108.3 ± 32.92 vs PGIS OE 130.2 ± 7.023 mm3, n.s.). Similarly, there were no differences in the incidence of pulmonary, mediastinal, liver, or brain metastases in pulmonary PGIS OE mice compared to wildtype littermate control mice (data not shown).
Expression of CXCR3-cognate chemokines (i.e., CXCL9 and CXCL10) is correlated with T cell infiltration status in metastatic melanoma.21 To evaluate whether the increase in tumor-infiltrating CD4+ T cells observed in pulmonary PGIS overexpressing mice is mediated by these cytokines, total RNA was extracted from tumor-bearing lungs and expression of CXCL9 and CXCL10 was determined by quantitative RT-PCR (Fig. 3E). The level of CXCL9 mRNA was significantly higher in pulmonary PGIS overexpressing mice than control mice. However, pulmonary PGIS overexpression had no effect on CXCL10 mRNA expression.
Pulmonary PGIS overexpression has no effect on LLC orthotopic lung tumor progression and metastasis
Tumor progression and metastasis experiments were repeated using a second murine lung carcinoma cell line, LLC cells, on a smaller scale (5 animals per group). There was no significant difference in LLC primary tumor size with pulmonary PGIS overexpression (Fig. 4A, WT 733.1 ± 158.2 mm3 vs PGIS OE 611.5 ± 109.0 mm3, n.s.). Every mouse in both PGIS OE and control groups developed liver metastases, and there was no significant difference in the number of liver metastases per animal in PGIS OE compared to littermate controls (Figs. 4B and 4C). However, quantification by ex vivo bioluminescence imaging showed a trend toward smaller liver metastases in PGIS OE mice (Fig. 4D), suggesting decreased metastatic burden. Compared to littermate controls, PGIS OE mice that developed metastasis to the brain had a trend toward fewer metastases (Fig. 4C, n.s.) and decreased signal intensity by ex vivo bioluminescence (Fig. 4D, n.s.). Moreover, pulmonary PGIS overexpression decreased the incidence of brain metastasis (20% in PGIS OE vs 60% in WT, n.s.; Fig. 4B).
Figure 4.
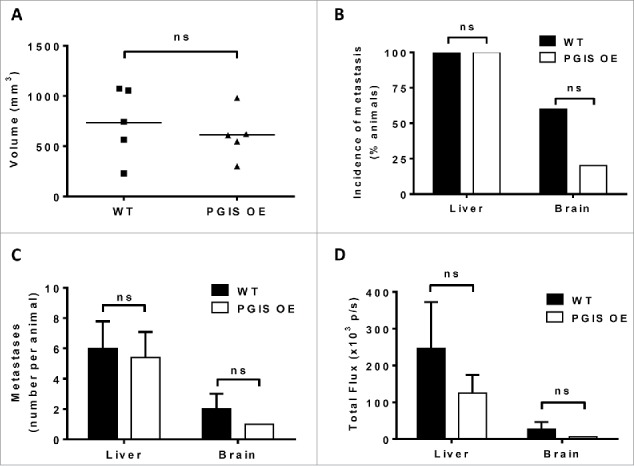
Pulmonary PGIS overexpression has no effect on LLC orthotopic lung tumor progression and metastasis. LLC tumors were implanted in pulmonary PGIS overexpressing transgenic mice (PGIS OE, n = 5) or littermate controls (WT, n = 5). After 3 weeks, lungs, livers, and brains were harvested and imaged ex vivo with an IVIS 50 imaging system. (A) Primary tumor volumes were measured by digital calipers. Each point represents a single tumor. The incidence (B), number (C) and signal intensity (D) of liver and brain metastases were quantified by ex vivo bioluminescent imaging, ns = not significant.
Pulmonary PGIS overexpression has no effect on LLC tumor-infiltrating lymphocytes
As shown in Figs. 5-6, LLC lung tumor sections were stained for expression of tumor infiltrating CD3+, CD4+, and CD8+ T cells. In contrast with CMT167 tumors, LLC tumors had fewer tumor-infiltrating T lymphocytes (LLC 20.40 ± 4.70 vs CMT 49.59 ± 6.73 cells/hpf), and pulmonary PGIS overexpression failed to affect the density of CD3+ T cells (Fig. 6A: 20.40 ± 4.70 vs 16.13 ± 3.35 cells/hpf, n.s.; control vs PGIS OE, respectively). Subset analysis revealed no difference between PGIS OE and control mice in either CD4+ T cell density (Fig. 6B: WT 9.2 ± 2.33 vs PGIS OE 7.2 ± 1.59 cells/hpf, n.s.) or CD8+ T cell density (Fig. 6C: WT 10.8 ± 2.40 vs PGIS OE 8.8 ± 1.59 cells/hpf, n.s.). In contrast to CMT167 tumors, pulmonary PGIS overexpression had no effect on levels of CXCL9 and CXCL10 mRNA expression in mice implanted with LLC tumors (Fig. 6D).
Figure 5.
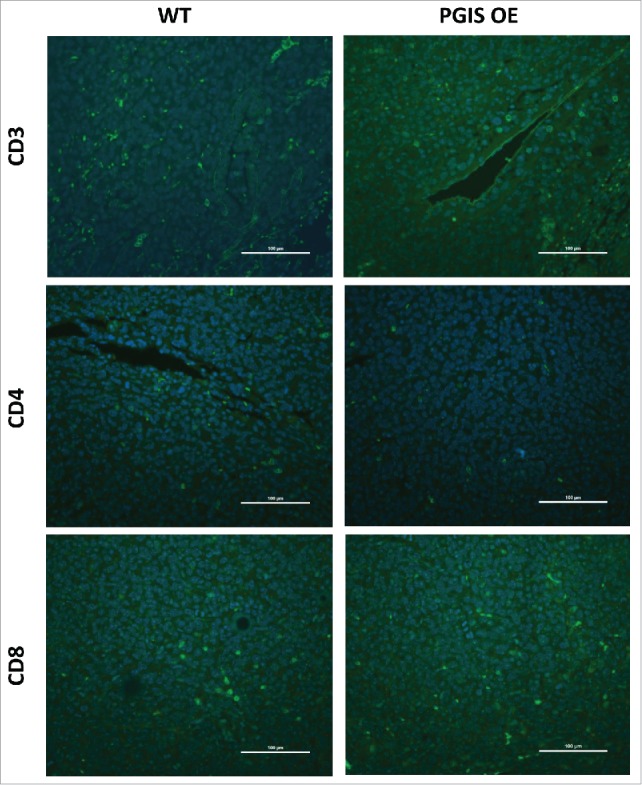
Pulmonary PGIS overexpression has no effect on lymphocyte infiltration in LLCtumors. Representative CD3 (top panels), CD4 (middle panels), and CD8 (bottom panels) immunostaining (FITC, green) of sections from PGIS overexpressing mice (right panels) and wildtype littermates (left panels). Nuclei were stained with DAPI (blue); scale bar = 100 μm.
Figure 6.
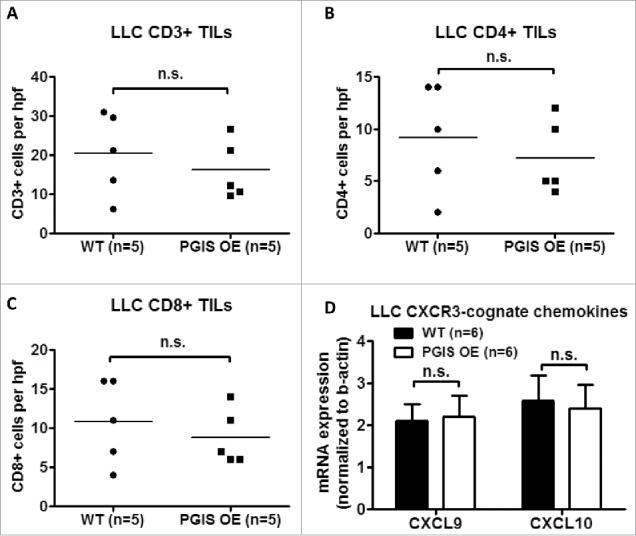
Pulmonary PGIS overexpression has no effect on LLC tumor-infiltrating lymphocytes. Three sections from each animal were stained for (A) CD3+, (B) CD4+, and (C) CD8+ TILs, and three random fields (20x) per section were quantified. The mean was used for analysis; each point represents a single mouse. (D) Two weeks after LLC tumor implantation, total RNA was isolated from tumor-bearing left lungs. CXCL9 and CXCL10 mRNA expression was analyzed by quantitative RT-PCR and normalized to β-actin. Results represent the mean with the S.E.M. indicated. Differences between groups were compared using unpaired t test, n.s. = not significant.
In vivo, CMT167 cells but not LLC cells express MHC class II genes and cofactors necessary for MHC class II processing and presentation
We hypothesized that differences in responses to pulmonary PGIS overexpression between CMT167 and LLC tumors were mediated in part by their ability to interact directly with CD4+ T lymphocytes. RNA was extracted from CMT167 cells or LLC cells recovered from orthotopic tumors implanted in green fluorescence protein (GFP)-expressing transgenic mice using flow cytometry (Supplemental Fig. S4) and was subjected to RNA sequencing. Analysis of MHC genes and associated cofactors demonstrated that in vivo, CMT167 cells but not LLC cells express MHC class II genes H2-Aa and H2-Ab1 and cofactors necessary for MHC class II processing and presentation, such as H2-DM, CIITA, and CD74 (Fig. 7A). Cell surface MHC class II expression was evaluated by flow cytometry on CMT167 and LLC tumor cells, either in vitro from cultured cells or in vivo from tumors implanted in GFP-expressing transgenic mice (Figs. 7B-C). As predicted by the RNA-seq data, CMT167 cells expressed higher levels of MHC class II than LLC cells, both in vitro and in vivo.
Figure 7.
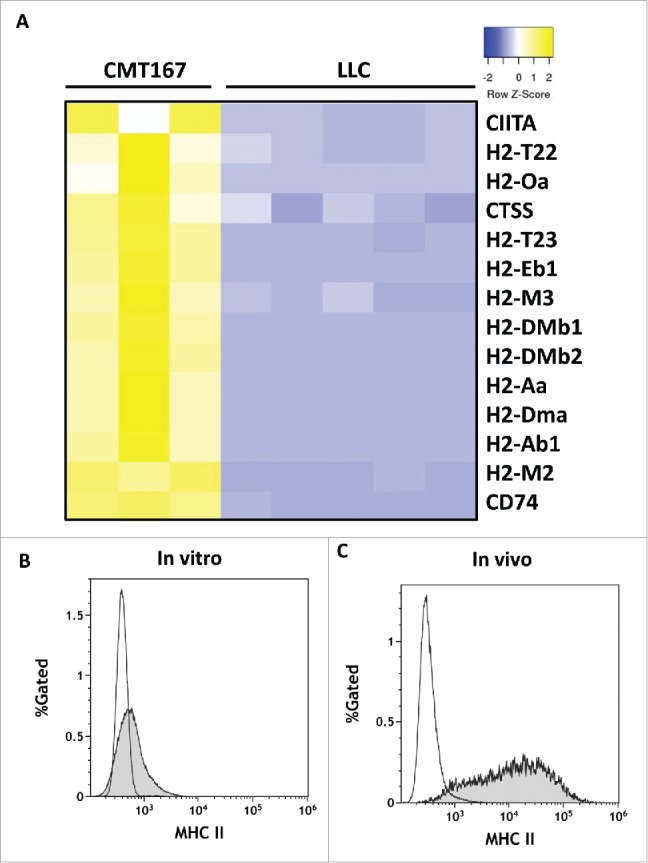
In vivo, CMT167 cells but not LLC cells express MHC class II genes and cofactors necessary for MHC class II processing and presentation. CMT167 tumors and LLC tumors were implanted in GFP-expressing transgenic mice. (A) After several weeks, cancer cells were recovered by sorting for GFP-negative cells using flow cytometry. Total RNA was isolated, analyzed by RNA-seq, and compared with total RNA collected from cancer cells grown in vitro. CMT167 and LLC cancer cells were analyzed for surface expression of MHC II by flow cytometry. The histograms show overlays of MHC II expressed by CMT167 cells (shaded) and LLC cells (white) in vitro (B) and in vivo (C). Data are representative of six independent in vitro experiments and three independent in vivo experiments (n = 2 mice per group).
Discussion
Lung cancer is the leading cause of cancer deaths worldwide, and novel therapeutic approaches are urgently needed. Based on preclinical and clinical studies, manipulation of the PGI2 pathway prevents lung cancer.10-13 Early studies also demonstrated that PGI2 inhibits metastasis using an experimental tail vein injection mouse model.22 However, the role of PGI2 in more relevant models of lung cancer metastasis has not been explored. Three PGI2 analogs have been approved by the FDA: epoprostenol, treprostinil, and iloprost. Unfortunately, the short half-lives of these drugs necessitate delivery by continuous intravenous infusion, or frequent subcutaneous injections or inhalations. Although iloprost was previously available in an oral formulation, this formulation has been discontinued by the manufacturer. Thus, we used targeted PGIS overexpression in mice to increase pulmonary PGI2 levels.
Targeted PGIS overexpression significantly inhibited tumor progression of CMT167 tumors in our immunocompetent orthotopic mouse model. Pulmonary PGIS overexpression significantly decreased primary tumor growth and the incidence of both secondary pulmonary and liver metastases. However, there was no difference in the number of secondary liver tumors per mouse or in the size of the metastases. Since PGI2 levels are elevated in the lung only, we conclude that pulmonary PGI2 affects the ability of CMT167 cells to leave the primary tumor. However, metastatic CMT167 cells from different animals grow at the same rate once they seed another organ and are no longer exposed to elevated PGI2 levels. Unfortunately, with pulmonary PGIS overexpressing transgenic mice and our orthotopic model, we are unable to determine the effect PGI2 has on the growth rate of metastatic lesions.
Treating CMT167 cells in vitro with stable PGI2 analogs failed to affect proliferation or TGFβ1-mediated EMT, suggesting that the inhibitory effects of PGIS overexpression in vivo are not due to direct effects on cancer cells, but are likely mediated through alterations in the TME. In pulmonary PGIS overexpressing transgenic mice, we observed increases in CMT167 tumor-infiltrating CD4+ T cells, but no difference in CD8+ T cells. Using an anti-CD4 depleting antibody, we demonstrated a critical role for CD4+ T cells in mediating the effects of pulmonary PGIS overexpression. We attempted to characterize the activation status of CD4 and CD8 T cells infiltrating the tumors. By flow cytometry, we find similar levels of CD44 and CD69 expression on CD4+ and CD8+ T cells in both PGIS overexpressing transgenic mice and wild type littermates, with both CMT167 and LLC orthotopic lung tumors. Thus, we are unable to find significant differences between CD4+ and CD8+ T cells by flow cytometry. In our opinion, this reflects the limitations of flow cytometry in analyzing tumor-infiltrating lymphocytes in our orthotopic model of lung cancer. Flow cytometry will define the populations in the entire tumor-bearing lung, which comprises both tumor and surrounding normal lung tissue. Since the tumors comprise a relatively small proportion of the lung, especially in the setting of CMT167 tumors, we are unable to distinguish between T cells that infiltrate the tumors and T cells in normal regions of the lung.
PGI2 has previously been shown to regulate CD4+ T cell subset differentiation and function, but its exact role is unclear. Whereas some studies have demonstrated that PGI2 inhibits Th1 and Th2 effector cytokine production and drives Th17 differentiation,18, 19 other studies have suggested that PGI2 promotes Th1 differentiation.20 We propose that in CMT167 tumors, PGI2 increases the recruitment and proliferation of antitumor CD4+ T cells, which leads to tumor inhibition. Our data suggest that one mechanism whereby PGI2 increases tumor-infiltrating CD4+ T cells is through expression of key chemokines such as CXCL9. Several cell types within the TME could potentially produce chemokines important for recruitment of T lymphocytes, including lung cancer cells themselves, macrophages, endothelial cells, and even other recruited T cells. Further work will be required to identify the cell types that produce CXCL9 and how PGI2 upregulates CXCL9 expression.
To extend our findings with CMT167 cells, we repeated the experiment with a second murine Kras mutant lung cancer cell line, LLC cells,23 which are more aggressive and metastasize more quickly than CMT167 cells in our orthotopic mouse model.15 Surprisingly, pulmonary PGIS overexpression had no effect on LLC primary tumor growth, CXCL9 mRNA expression, or tumor-infiltrating CD4+ T cells. We hypothesized that differences in responses to pulmonary PGIS overexpression between CMT167 and LLC tumors were mediated in part by their ability to interact directly with CD4+ T cells. Using RNA-seq and flow cytometry, we analyzed CMT167 and LLC cancer cells recovered from in vivo tumors. Interestingly, CMT167 cells but not LLC cells express MHC class II genes and cofactors necessary for MHC class II processing and presentation. This suggests that CMT167 cells may function as antigen presenting cells and interact directly with CD4+ T cells, whereas LLC cells will not. We hypothesize that LLC cells are less dependent on changes in CD4+ T cells, and alterations in the phenotype of these cells induced by PGI2 therefore has a limited effect on LLC tumor progression.
In summary, pulmonary-specific PGIS overexpression inhibits lung cancer progression in an immunocompetent orthotopic mouse model. This effect is mediated by increases in tumor-infiltrating CD4+ T cells and decreased levels of pro-tumorigenic prostaglandins. However, the antitumorigenic effects of PGI2 may be limited to tumors that express MHC class II genes on the cancer cells. Thus, in addition to their chemopreventive role, PGI2 analogs may represent novel therapeutic agents for the treatment of lung cancer.
Materials and methods
Culture of murine lung cancer cell lines and in vitro assays
CMT167 cells,24 originally provided by Dr. Alvin Malkinson (University of Colorado), were stably transfected with firefly luciferase as previously described15 and maintained in DMEM containing 10% FBS, 100 U/mL penicillin, 100 μg/ml streptomycin, MEM Non-Essential Amino Acids, 10 mM HEPES, and 500 μg/mL G418 at 37°C in a humidified 5% CO2 atmosphere. DNA sequencing of Kras confirmed that CMT167 cells harbor an activating KrasG12V mutation, as has been previously reported.25 Luciferase-expressing Lewis Lung Carcinoma cells were purchased from Caliper Life Sciences (Bioware LL/2-luc-M38) and maintained in DMEM containing 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 500 μg/mL G418 at 370C in a humidified 5% CO2 atmosphere. Both cell lines were periodically tested for mycoplasma infection and were last retested in November 2017. To avoid cross-contamination and phenotypic changes, these cell lines have not been maintained in long-term cultures. All cells used in this study were maintained as frozen stocks and cultured only for two to four weeks before use in the experiments. Authentication of these cell lines based on morphology, growth curve analysis, and metastatic phenotype was performed regularly, and no phenotypic changes were observed through the duration of the study.
For effects of PGI2 analogs on proliferation, cells were treated with iloprost (10 μM, Cayman 18215) or treprostinil (10 μM, Cayman 10162), and proliferation was determined by hemocytometer counting at 24, 48, and 72 hours. For assessment of epithelial-to-mesenchymal transition, cells were treated with recombinant mouse TGF-β1 (5 ng/mL, R&D Systems 7666-MB) in the presence or absence of iloprost or treprostinil (10 μM) for 72 hours. Cells were lysed in ice-cold MAPK lysis buffer [0.5% Triton X-100, 50 mM β-glycerophosphate (pH 7.2), 0.1 mM sodium vanadate, 2 mM MgCl2, 1 mM EGTA, 1 mM dithiothreitol, 2 μg/ml leupeptin, and 4 μg/ml aprotinin) containing protease inhibitors and centrifuged at 10,000 xg for 5 minutes. Supernatants were immunoblotted with mouse monoclonal anti-E-cadherin (BD Transduction 610182) and mouse monoclonal anti-β-actin (Sigma-Aldrich A5441). For luciferase reporter assays, cells were co-transfected with 1 μg of a promoter construct encoding three consensus peroxisome proliferator-activated receptor-γ (PPARγ)-response elements ligated to a luciferase reporter (PPARγ-RE) and 1 μg CMV-β-galactosidase using Lipofectamine 2000 (Invitrogen 11668019). After an overnight incubation, cells were stimulated for 24 hours with pioglitazone (10 μM, Cayman 71745), iloprost (10 μM), or treprostinil (10 μM). Extracts were prepared in Reporter Lysis Buffer (Promega E3971), and PPARγ-RE activity normalized to β-galactosidase was determined.
Animals
FVB mice with targeted overexpression of PGIS, consisting of the human surfactant protein-C (SP-C) promoter driving expression of full-length rat PGIS cDNA,26 were transferred to the C57BL/6 background by completing 10 generations of backcrosses. Transgenic mice were genotyped by PCR on genomic DNA as previously described.26 GFP-expressing mice [C57BL/6-Tg(UBC-GFP)30Scha/J] were obtained from Jackson Laboratory. Animals were bred and maintained in the Center for Comparative Medicine at the University of Colorado Anschutz Medical Campus. Experiments were performed on 8–12 week old male mice. This study was carried out with the recommendations in the U.S. Department of Health and Human Services Guide for the Care and Use of Laboratory Animals. All procedures were performed under protocols approved by the Institutional Animal Care and Use Committee at the University of Colorado Anschutz Medical Campus and Veterans Affairs Eastern Colorado Health Care System.
Orthotopic mouse model
Surgeries were performed under inhaled isoflurane anesthesia, and all efforts were made to minimize suffering. As previously described,27 a transverse skin incision was made along the left lateral axillary line at the level of the xyphoid process, and subcutaneous fat was dissected away to expose the left lung. Luciferase-expressing CMT167 or LLC cells (105) were suspended in Hank's Buffered Salt Solution containing 1.35 mg/mL Matrigel Basement Membrane Matrix (Corning 354234) and injected into the left lung using a 30 g needle. Following injection, the incision was closed using veterinary skin adhesive. Four weeks after CMT167 tumor implantation or three weeks after LLC tumor implantation, mice were injected intraperitoneally with 300 mg/kg body weight D-Luciferin (PerkinElmer 122799) before euthanasia with inhaled carbon dioxide. The circulation was perfused and the lungs were inflated with heparinized PBS. The heart, lungs, liver, and brain were removed for ex vivo bioluminescence imaging. The left lung was isolated from the remaining lung lobes and imaged for bioluminescence separately; remaining lung lobes, liver, and brain were also imaged using an IVIS 50 Imaging System (Xenogen). Primary tumor size was measured using digital calipers. Incidence of metastases to the other lung lobes and liver was quantified by ex vivo bioluminescence using Living Image software (PerkinElmer).
CD4+ T cell immunodepletion
To deplete CD4+ T cells, rat anti-mouse CD4 monoclonal antibody (clone GK1.5, BE0003-1) and rat IgG2b (clone LTF-2, BE0090) were purchased from BioXCell. Mice were injected intraperitoneally with antibody [200 μg in PBS per dose (8-10mg/kg)] twice weekly starting 1 day prior to tumor implantation and throughout the course of the tumor progression experiment. Depletion was assessed 10 days and 17 days after starting treatment by collecting whole blood (100-150 μL) from the submandibular plexus of mice (two mice per group). Red blood cell lysis was performed using RBC lysis buffer [0.15 M NH4Cl, 0.1mM KHCO3, 0.1mM Na2EDTA (pH 7.2)]. Whole blood leukocytes were analyzed by flow cytometry as described below.
Evaluation of TILs
Sections (5 μm thick) were cut from formalin fixed, paraffin embedded (FFPE) tissue blocks. Tissue sections were deparaffinized, hydrated, and heated for 20 minutes at 100ºC in Antigen Unmasking Solution (Vector Labs H-3300). Slides were incubated with 3% normal goat serum (Vector Labs S-1000) for one hour. Serial sections were incubated at 4ºC overnight with rabbit monoclonal antibody to CD3ε (Invitrogen clone SP7, MA5-14524), rat monoclonal antibody to CD4 (eBioscience clone 4SM95, 14-9766-80), or rat monoclonal antibody to CD8α (eBioscience clone 4SM15, 14-0808-82) to stain for T lymphocytes. After washing with 0.05M Tris-based solution in 0.15M NaCl with 0.1% v/v Triton-X-100 (pH 7.6), slides were incubated with corresponding Alexa Fluor 488 conjugated secondary antibody (Invitrogen A-11006) for 30 minutes at room temperature. Slides were coverslipped with VECTASHIELD Mounting Medium with DAPI (Vector Labs H-1200). Three sections from each animal were stained with each antibody, and three random fields (40x) per section were quantified independently by two blinded observers (HYL and MM); the mean was used for analysis.
Quantitative real-time PCR of tumor-bearing lungs
Two weeks after tumor implantation, left lobes of tumor-bearing mice were frozen in liquid nitrogen and homogenized in 350 μL of RLT Plus buffer (Qiagen). Total RNA was isolated using an RNeasy Plus Mini Kit (Qiagen 74136). Reverse transcription was performed on 1 μg of total RNA per sample using qScript cDNA SuperMix (Quanta BioSciences 95048-025). Real-time PCR analysis was conducted in triplicate in a C1000 Touch Thermal Cycler (Bio-Rad) using Power SYBR Green PCR Master Mix (Applied Biosystems 4367659). The relative message levels of each target gene were normalized to β-actin. Sequences of primers used were: CXCL9 (For: 5′-GAGCAGTGTGGAGTTCGAGG-3′, Rev: 5′-TCCGGATCTAGGCAGGTTTG-3′), CXCL10 (For: 5′-GGATGGCTGTCCTAGCTCTG-3′, Rev: 5′-TGAGCTAGGGAGGACAAGGA-3′), β-actin (For: 5′-GGCTGTATTCCCCTCCATCG-3′, Rev: 5′-CCAGTTGGTAACAATGCCATGT-3′).
Recovery of lung cancer cells from tumor-bearing GFP-expressing transgenic mice
Lung tumors were implanted in GFP-expressing transgenic mice as described above. Three weeks after CMT167 tumor implantation or two weeks after LLC tumor implantation, the pulmonary circulation was perfused with heparinized PBS and tumor-bearing left lungs were isolated. Tumor-bearing lungs were pooled, mechanically dissociated, incubated at 37°C for 45 minutes with Collagenase A (1 mg/mL, Roche 10103578001) and DNAse I (40 μg/mL, Sigma D4527), and processed using a GentleMACS Dissociator (Miltenyi Biotec). Red blood cell lysis was performed as described above. Cell sorting was performed at the University of Colorado Cancer Center Flow Cytometry Shared Resource using an XDP-100 cell sorter (Beckman Coulter). The sorting strategy excluded debris and cell doublets by light scatter and dead cells by DAPI. Lung cancer cells were recovered from tumor-bearing GFP-expressing transgenic mice by sorting for GFP-negative cells.
RNA sequencing (RNA-seq)
CMT167 cells were recovered from three pools of mice consisting of five GFP-expressing mice each, whereas LLC cells were recovered from five pools of mice consisting of four GFP-expressing mice each. Total RNA was isolated using an RNeasy Plus kit (QIAGEN) and each RNA isolate was sequenced separately. Total RNA was also isolated from cells in culture at the time of tumor implantation. RNA quality and quantity were analyzed using an Agilent 2100 electrophoresis bioanalyzer. RNA-seq library preparation and sequencing were conducted at the University of Colorado Cancer Center Genomics and Microarray Shared Resource. RNA libraries were constructed using an Illumina TruSEQ stranded mRNA Sample Prep Kit (Cat# RS-122-2101). Total RNA was combined with RNA purification beads to bind PolyA RNA to oligodT magnetic beads. mRNA was eluted and converted to double stranded DNA. A-tailing, adapter ligation, and PCR amplification using 15 cycles was performed to complete the library construction. Libraries were quantitated via Qubit, analyzed on a Bioanalyzer Tape Station, and diluted to appropriate concentration to run on an Illumina HiSEQ 2500 High Throughput Flow Cell.
RNA-seq reads were obtained using Illumina Hi-seq analysis Pipeline. Reads quality was checked using FastQC (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc). The median number of reads per condition was 24 million. Reads were then processed and aligned to the UCSC Mus musculus reference genome (build mm10) using TopHat v2.28 The pre-built Mus musculus UCSC mm10 index was downloaded from the TopHat homepage and used as the reference genome. The default parameters for TopHat were used. The aligned read files were processed by Cufflinks v2.0.2.29 Reads were assembled into transcripts and their abundance was estimated. Cufflinks uses the normalized RNA-Seq fragment counts to measure the relative abundances of transcripts. The unit of measurement is Fragments Per Kilobase of exon per Million fragments mapped (FPKM). Confidence intervals for FPKM estimates were calculated using a Bayesian inference method.29
Flow cytometry
Peripheral blood leukocytes and CMT167 and LLC cancer cells from tumors implanted in GFP-expressing transgenic mice were stained for 1 hour at 4°C with CD3ε (clone 145-2C11, eBioscience), CD4 (clone RM4-5, eBioscience), CD8 (clone 53–6.7, eBioscience), CD44 (clone IM7, eBioscience), CD45 (clone 30-F11, eBioscience), CD69 (clone H1.2F3, eBioscience), LIVE/DEAD Fixable Dead Cell Stain (Invitrogen), and MHC class II (clone M5/114.15.2, BioLegend). Samples were acquired at the University of Colorado Cancer Center Flow Cytometry Shared Resource using a Gallios flow cytometer (Beckman Coulter) and data were analyzed using Kaluza Software (Beckman Coulter).
Statistical analysis
Data are presented as mean ± SEM. Differences between groups were identified using the Student unpaired t test. One-way ANOVA was used to compare more than two groups. Fisher's exact test was used to compare incidence of metastases between groups. Under all circumstances, P < 0.05 was considered to be significant.
Supplementary Material
Funding Statement
This work was supported by the U.S. Department of Veterans Affairs Career Development Program under Grant IK2BX001282 to H. Li; the National Cancer Institute of the National Institutes of Health under Grants R01 CA162226 and R01 CA108610 to R.A. Nemenoff, and Colorado Lung SPORE Grant P50 CA058187 to H. Li and R.A. Nemenoff; and the Cancer League of Colorado Grant AWD-162952 to H. Li. The University of Colorado Cancer Center Flow Cytometry Shared Resource and Genomics and Microarray Shared Resource receive direct funding support from the National Cancer Institute of the National Institutes of Health through the University of Colorado Cancer Center Support Grant (P30 CA046934). The content is solely the responsibility of the authors and does not necessarily represent the official views of the U.S. Department of Veterans Affairs, the National Institutes of Health, or the Cancer League of Colorado.
Acknowledgments
We thank Mark Geraci for developing and providing the pulmonary PGIS overexpressing transgenic mice, originally on the FVB background.
Disclosure statement
The authors disclose no potential conflicts of interest.
References
- 1.Borghaei H, Paz-Ares L, Horn L, Spigel DR, Steins M, Ready NE, Chow LQ, Vokes EE, Felip E, Holgado E, et al.. Nivolumab versus Docetaxel in Advanced Nonsquamous Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373(17):1627–39. doi: 10.1056/NEJMoa1507643. PMID:26412456 PMCID: . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brahmer J, Reckamp KL, Baas P, Crino L, Eberhardt WE, Poddubskaya E, Antonia S, Pluzanski A, Vokes EE, Holgado E, et al.. Nivolumab versus Docetaxel in Advanced Squamous-Cell Non-Small-Cell Lung Cancer. N Engl J Med. 2015;373:123–35. doi: 10.1056/NEJMoa1504627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gettinger SN, Horn L, Gandhi L, Spigel DR, Antonia SJ, Rizvi NA, Powderly JD, Heist RS, Carvajal RD, Jackman DM, et al.. Overall Survival and Long-Term Safety of Nivolumab (Anti-Programmed Death 1 Antibody, BMS-936558, ONO-4538) in Patients With Previously Treated Advanced Non-Small-Cell Lung Cancer. J Clin Oncol: official journal of the American Society of Clinical Oncology. 2015;33:2004–12. doi: 10.1200/JCO.2014.58.3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rizvi NA, Mazieres J, Planchard D, Stinchcombe TE, Dy GK, Antonia SJ, Horn L, Lena H, Minenza E, Mennecier B, et al.. Activity and safety of nivolumab, an anti-PD-1 immune checkpoint inhibitor, for patients with advanced, refractory squamous non-small-cell lung cancer (CheckMate 063): a phase 2, single-arm trial. Lancet Oncol. 2015;16:257–65. doi: 10.1016/S1470-2045(15)70054-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garon EB, Rizvi NA, Hui R, Leighl N, Balmanoukian AS, Eder JP, Patnaik A, Aggarwal C, Gubens M, Horn L, et al.. Pembrolizumab for the treatment of non-small-cell lung cancer. N Engl J Med. 2015;372:2018–28. doi: 10.1056/NEJMoa1501824. [DOI] [PubMed] [Google Scholar]
- 6.Hida T, Yatabe Y, Achiwa H, Muramatsu H, Kozaki K, Nakamura S, Ogawa M, Mitsudomi T, Sugiura T, Takahashi T. Increased expression of cyclooxygenase 2 occurs frequently in human lung cancers, specifically in adenocarcinomas. Cancer Res. 1998;58:3761–4. [PubMed] [Google Scholar]
- 7.Wolff H, Saukkonen K, Anttila S, Karjalainen A, Vainio H, Ristimaki A. Expression of cyclooxygenase-2 in human lung carcinoma. Cancer Res. 1998;58:4997–5001. [PubMed] [Google Scholar]
- 8.Heasley LE, Thaler S, Nicks M, Price B, Skorecki K, Nemenoff RA. Induction of cytosolic phospholipase A2 by oncogenic Ras in human non-small cell lung cancer. J Biol Chem. 1997;272:14501–4. doi: 10.1074/jbc.272.23.14501. [DOI] [PubMed] [Google Scholar]
- 9.Nakanishi M, Rosenberg DW. Multifaceted roles of PGE2 in inflammation and cancer. Semin Immunopathol. 2013;35:123–37. doi: 10.1007/s00281-012-0342-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Keith RL, Miller YE, Hoshikawa Y, Moore MD, Gesell TL, Gao B, Malkinson AM, Golpon HA, Nemenoff RA, Geraci MW. Manipulation of pulmonary prostacyclin synthase expression prevents murine lung cancer. Cancer Res. 2002;62:734–40. [PubMed] [Google Scholar]
- 11.Keith RL, Miller YE, Hudish TM, Girod CE, Sotto-Santiago S, Franklin WA, Nemenoff RA, March TH, Nana-Sinkam SP, Geraci MW. Pulmonary prostacyclin synthase overexpression chemoprevents tobacco smoke lung carcinogenesis in mice. Cancer Res. 2004;64:5897–904. doi: 10.1158/0008-5472.CAN-04-1070. [DOI] [PubMed] [Google Scholar]
- 12.Nemenoff R, Meyer AM, Hudish TM, Mozer AB, Snee A, Narumiya S, Stearman RS, Winn RA, Weiser-Evans M, Geraci MW, et al.. Prostacyclin prevents murine lung cancer independent of the membrane receptor by activation of peroxisomal proliferator–activated receptor gamma. Cancer Prev Res. 2008;1:349–56. doi: 10.1158/1940-6207.CAPR-08-0145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Keith RL, Blatchford PJ, Kittelson J, Minna JD, Kelly K, Massion PP, Franklin WA, Mao J, Wilson DO, Merrick DT, et al.. Oral iloprost improves endobronchial dysplasia in former smokers. Cancer Prev Res. 2011;4:793–802. doi: 10.1158/1940-6207.CAPR-11-0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pehlivan Y, Turkbeyler IH, Balakan O, Sevinc A, Yilmaz M, Bakir K, Onat AM. Possible anti-metastatic effect of iloprost in a patient with systemic sclerosis with lung cancer: a case study. Rheumatol Int. 2012;32:1437–41. doi: 10.1007/s00296-011-1848-4. [DOI] [PubMed] [Google Scholar]
- 15. Weiser-Evans MC, Wang XQ, Amin J, Van Putten V Choudhary R, Winn RA, Scheinman R, Simpson P, Geraci MW, Nemenoff RA. Depletion of cytosolic phospholipase A2 in bone marrow-derived macrophages protects against lung cancer progression and metastasis. Cancer Res. 2009;69:1733–8. doi: 10.1158/0008-5472.CAN-08-3766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li H, Sorenson AL, Poczobutt J, Amin J, Joyal T, Sullivan T, Crossno JT, Weiser-Evans MC, Nemenoff RA. Activation of PPARgamma in myeloid cells promotes lung cancer progression and metastasis. PLoS One. 2011;6:e28133. doi: 10.1371/journal.pone.0028133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bren-Mattison Y, Meyer AM, Van Putten V Li H, Kuhn K, Stearman R, Weiser-Evans M, Winn RA, Heasley LE, Nemenoff RA. Antitumorigenic effects of peroxisome proliferator-activated receptor-gamma in non-small-cell lung cancer cells are mediated by suppression of cyclooxygenase-2 via inhibition of nuclear factor-kappaB. Mol Pharmacol. 2008;73:709–17. [DOI] [PubMed] [Google Scholar]
- 18.Zhou W, Blackwell TS, Goleniewska K, O'Neal JF, Fitzgerald GA, Lucitt M, Breyer RM, Jr Peebles RS. Prostaglandin I2 analogs inhibit Th1 and Th2 effector cytokine production by CD4T cells. J Leuk Biol. 2007;81:809–17. doi: 10.1189/jlb.0606375. [DOI] [PubMed] [Google Scholar]
- 19.Zhou W, Dowell DR, Huckabee MM, Newcomb DC, Boswell MG, Goleniewska K, Lotz MT, Toki S, Yin H, Yao S, et al.. Prostaglandin I2 signaling drives Th17 differentiation and exacerbates experimental autoimmune encephalomyelitis. PLoS One. 2012;7:e33518. doi: 10.1371/journal.pone.0033518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakajima S, Honda T, Sakata D, Egawa G, Tanizaki H, Otsuka A, Moniaga CS, Watanabe T, Miyachi Y, Narumiya S, et al.. Prostaglandin I2-IP signaling promotes Th1 differentiation in a mouse model of contact hypersensitivity. J Immunol. 2010;184:5595–603. doi: 10.4049/jimmunol.0903260. [DOI] [PubMed] [Google Scholar]
- 21.Harlin H, Meng Y, Peterson AC, Zha Y, Tretiakova M, Slingluff C, McKee M, Gajewski TF. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res. 2009;69:3077–85. doi: 10.1158/0008-5472.CAN-08-2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Honn KV, Cicone B, Skoff A. Prostacyclin: a potent antimetastatic agent. Science. 1981;212:1270–2. doi: 10.1126/science.7015512. [DOI] [PubMed] [Google Scholar]
- 23.Agalioti T, Giannou AD, Krontira AC, Kanellakis NI, Kati D, Vreka M, Pepe M, Spella M, Lilis I, Zazara DE, et al.. Mutant KRAS promotes malignant pleural effusion formation. Nat Commun. 2017;8:15205. doi: 10.1038/ncomms15205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Layton MG, Franks LM. Heterogeneity in a spontaneous mouse lung carcinoma: selection and characterisation of stable metastatic variants. Br J Cancer. 1984;49:415–21. doi: 10.1038/bjc.1984.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Justilien V, Regala RP, Tseng IC, Walsh MP, Batra J, Radisky ES, Murray NR, Fields AP. Matrix metalloproteinase-10 is required for lung cancer stem cell maintenance, tumor initiation and metastatic potential. PLoS One. 2012;7:e35040. doi: 10.1371/journal.pone.0035040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Geraci MW, Gao B, Shepherd DC, Moore MD, Westcott JY, Fagan KA, Alger LA, Tuder RM, Voelkel NF. Pulmonary prostacyclin synthase overexpression in transgenic mice protects against development of hypoxic pulmonary hypertension. J Clin Invest. 1999;103:1509–15. doi: 10.1172/JCI5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Poczobutt JM, Gijon M, Amin J, Hanson D, Li H, Walker D, Weiser-Evans M, Lu X, Murphy RC, Nemenoff RA. Eicosanoid profiling in an orthotopic model of lung cancer progression by mass spectrometry demonstrates selective production of leukotrienes by inflammatory cells of the microenvironment. PLoS One. 2013;8:e79633. doi: 10.1371/journal.pone.0079633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.