

Abstract
We rationally designed allosteric compounds that stimulate Hsp90 ATPase activity and show anticancer potencies in the low micromolar to nanomolar range. In parallel, we clarified their mode of action and developed a quantitative model that links the dynamic ligand-protein cross-talk to observed cellular and in vitro activities. Our results support the potential of using dynamics-based approaches to develop original mechanism-based cancer therapeutics.
Heat Shock Protein 90 (Hsp90) is a chaperone that controls the folding of more than 200 client proteins and constitutes a central node in many signaling pathways [1]. Overexpression and dysregulation of Hsp90 have been linked with cancer and neurodegeneration. It is thus not surprising that this chaperone has become an important drug-target: in principle its inhibition can result in the simultaneous degradation of multiple clients associated with different pathological hallmarks [2]. Inhibitors targeting the N-terminal ATP-binding site have been developed and some reached clinical trials [3]. However, they all showed problems due to the induction of the HSF1 mediated heat shock response and of Hsp70 overexpression, leading to drug resistance and toxicity [3a–c, 4].
A viable alternative to interfering with Hsp90 is represented by allosteric ligands, which perturb the chaperone by targeting sites alternative to the ATP-site. Novobiocin was shown to inhibit Hsp90 by binding the C-terminal region without inducing the heat shock response. Based on this observation, a number of new derivatives with promising activities against a variety of cancers were developed [5].
In this context, we have developed a method for the identification of allosteric pockets via the analysis of residue-pair distance fluctuations in the structural ensemble around the active state of the chaperone. Such analysis unveiled an allosteric pocket at 65Å from the active site, located at the MD:CTD border in a region overlapping with the client binding site [6]. This facilitated the design of modulators showing promising anticancer activities and a novel molecular mechanism of perturbation of Hsp90 functions: the ligands in fact proved to be activators of closure kinetics and ATPase of the chaperone in vitro, induce cancer cell death, and interfere with client maturation. We developed a first Quantitative-Structure-Dynamics-Activity-Relationship (QSDAR) model correlating the structures of an initial set of modulators to observed activation effects [6c].
Here, based on this initial model, we report the rational design of new allosteric ligands, reaching low micromolar to nanomolar anticancer activities, which supports their potential in the development of anticancer therapeutics. On the computational side, we further develop a model to evaluate the potency of allosteric modulators by taking into account the dynamic cross-talk that exists between the protein and the ligand.
The conformational properties of the allosteric site were previously described [6–7]. Docking, MD refinement and analysis of the allosteric effects of the initial O-aryl rhamnoside benzofuran 1 revealed that the glycosidic moiety preferentially interacted with E477 and D503 of one protomer, while the propenyl substituent on the benzofuran could contact a hydrophobic pocket lined by R591 of the other protomer. This model led to redesign 1 into 18 and 19 (Table 1) where the propenyl was substituted by a Cl atom and the sugar moiety with an ethylamine or a propylamine group. Such modifications resulted in an increase of Hsp90 ATPase activity, paralleled by an improvement in the anticancer cytotoxicity of the ligands. A new round of MD simulations and allosteric coordination analysis supported the importance of interactions with E477 and D503 [6b, c]. Here, to establish structure-dynamics-activity relationships we directed design efforts towards obtaining different amine substitutions on the phenyl-benzofuran scaffold. The structures of the compounds are reported in Table 1, their best poses in the Hsp90 pocket are in Figure 1A, and the details of their synthesis in [8].
Table 1.
Structures, stimulatory potencies and cytotoxic activities of designed compounds. ATPase stimulation and cytotoxicity were measured as described in Materials and Methods.
| Compound | Normalized ATPase rate | Toxicity in STO (IC50 uM) | |
|---|---|---|---|
| 1 |
|
1.1a | 57.0 ± 2.1a |
| 18 |
|
4.8a | 8.9 ± 1.1a |
| 19 |
|
6a | 9.9 ± 1.1a |
| 20 |
|
1.5 | 16 ± 1 |
| 21 |
|
2.2 | 8.2 ± 2.7 |
| 22 |
|
1.9 | 11.7 ± 2.4 |
| 23 |
|
2.4 | 3.9 ± 1.8 |
| 24 |
|
4.1 | 8.6 ±2.1 |
| 25 |
|
4.5 | 22.1 ± 1.1 |
| 26 |
|
4.2 | 4.1 ± 0.4 |
| 27 |
|
1.4 | not soluble |
| 28 |
|
1.3 | 36 ± 6 |
| 29 |
|
1.17 | > 75 |
| 30 |
|
0.98 | > 100 |
| 31 |
|
0.57±0.04 |
Data reported in [6b]
Figure 1.
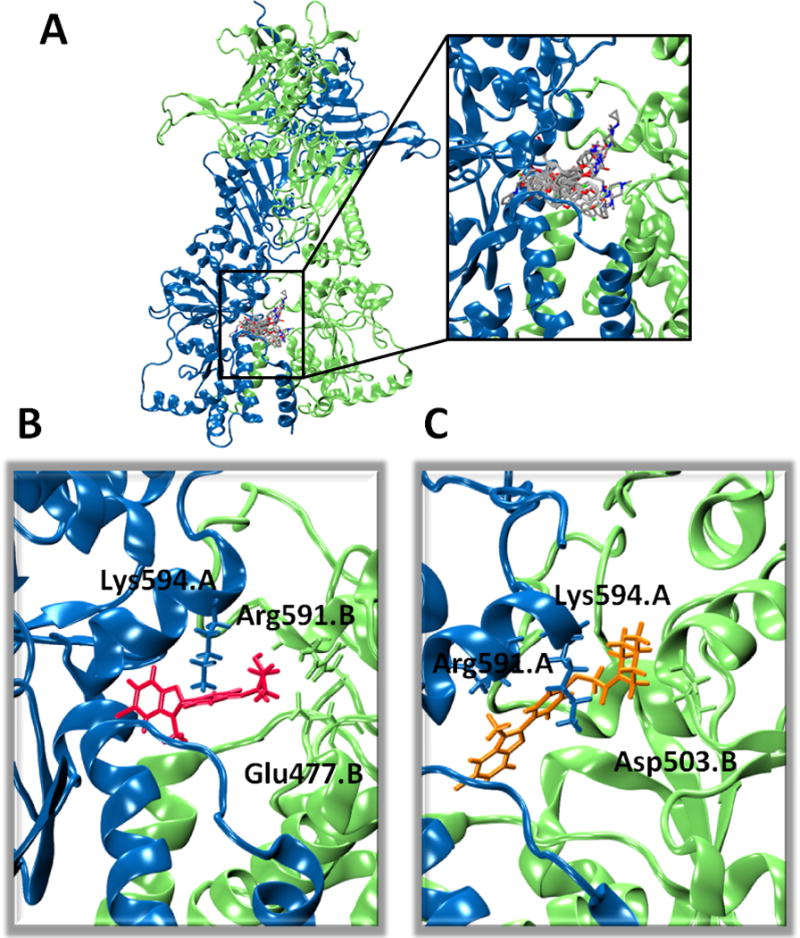
(A) Overall view and detailed superposition of the best pose for each compound in the allosteric site. Color code: Protomers A and B, light blue and green respectively; compounds: C=grey, O=red, N=blue, Cl=green, F=dark yellow. (B) Best pose obtained for compound 25, in red, showing the highest ATPase stimulation. The sidechains of residues interacting with 25 are shown with the color code reported in (A). (C) Same as (B), reporting compound 22 in orange, displaying lowest stimulation.
All compounds resulted in activation of ATP hydrolysis, with the activity depending on the nature of the substituent linked to the O-aryl moiety (Table 1). Compounds 20, 21, 22, and 23, characterized by the presence of a bulky substituent on the amino group, display the lowest activity. The compounds with N-methyl piperazinic group on the hydroxyphenyl functionality, stimulate ATPase to a degree comparable to that induced by the glycosidic derivatives previously tested [6b, c], suggesting that the 6-membered ring actually mimics sugar substituents. Maximal stimulatory effects were observed for 24, 25 and 26 indicating that minimizing the steric hindrance on the amine-pharmacophore, while maintaining its alkylation, is beneficial to ATPase stimulation, resulting in a 4-5 fold increase [6–7]. Finally, compounds 27, 28, 29, and 30, carrying a carboxylate group at the terminal of the propenyl group on the benzofuran scaffold, while conserving the N,N-dimethyl amino pharmacophore on the phenol ring, showed a decreased stimulation compared to 24, 25, and 26. The structural reason is that the carboxylate group points to the negatively charged area defined by E477 and D503 on the protomer alternative to the one where the amino group is anchored: despite the presence of K and R residues in this region, repulsive interactions are detrimental to the activity of these derivatives.
Given the importance of Hsp90 in determining cancer maintenance and development, we tested the antiproliferative activity of the new compounds in an experimental model of diffuse malignant peritoneal mesothelioma (STO), an uncommon and locally aggressive tumor, poorly responsive to conventional therapies [9].
Cells were exposed to increasing concentrations (0.01-100 μM) of each compound for 72 hours, and the effect on cell proliferation was assessed by MTS assay. STO cells were highly responsive to compounds 23, 24, and 26, as indicated by the IC50 of 3.9±1.8, 8.6±2.1, and 4.1±0.4μM, respectively. A slightly less pronounced effect was observed after exposure of STO cells to compounds 22 and 25, highlighting the relevance of alkyl substitutions on the amino groups to increase the antiproliferative activity. By contrast, the presence of the negatively charged carboxylate group led to a decreased (28) or null (29 and 30) activity (Table 1).
Interestingly, a remarkable antiproliferative effect (0.57±0.04μM) was observed for compound 31 (Table 1), obtained by the introduction of a phosphonium group to improve cell and mitochondrial penetration. In this context, we measured the activity of succinate dehydrogenase in human cervix carcinoma HeLa cells to test the activation of Hsp90 mitochondrial homologue TRAP1. Indeed, compound 31 decreases the activity of the enzyme in a dose-dependent way (Figure 2) as expected for a potential TRAP1 activator [10]. As a possible caveat, it is worth noting that we are assuming that TRAP1 is targeted by the compound in a similar way to Hsp90.
Figure 2.
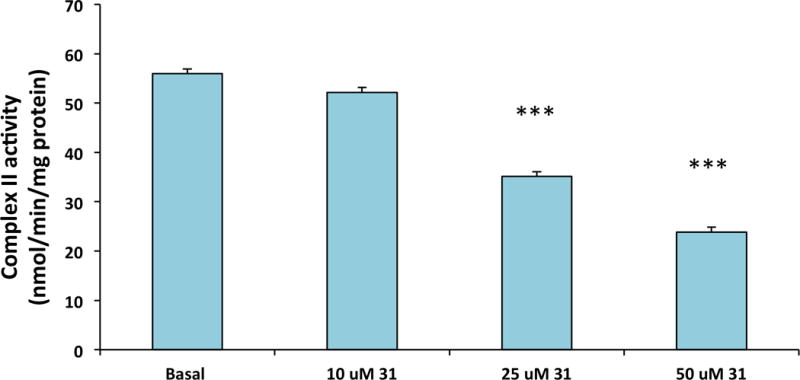
Analysis of the SQR enzymatic activity of succinate dehydrogenase in human cervix carcinoma HeLa cells pretreated for 30 minutes with compound 31 at the reported concentrations. Data are reported as mean ± SD values (n = 3); asterisks indicate significant differences (*** for a p<0.01 with a Student’s t test analysis)
Because the designed compounds target a dynamic allosteric site on Hsp90 that does not overlap with and is far removed from the ATP binding site, it is expectedly hard to obtain correlations between computationally determined affinities and effects on chaperone functions or cellular activities [11].
To define a quantitative relationship between the ligands’ structures, dynamics cross-talk with the protein and observed activities, we made use of an ensemble approach: we docked each of the ligands into the ten most representative structures extracted upon clusterization of Hsp90 MD trajectories (see Methods). For each ligand, we rank the best pose from the 10 runs, and for each run the pose with the best docking score was selected for further calculations. Superposition of the best pose selected for each ligand is reported in Figure 1A. All the compounds are characterized by interactions with the same set of residues: E477 interacts with the amine in all compounds, but it also binds the OH group in position 2 of the propenyl group present in compounds 24 and 25 (Figure 1B). The O-aryl moiety contacts R591 of the same protomer. Compound 23 interacts with D503 (Figure 1C), as observed previously for O-aryl-sugars [6b, c]. The benzofuran moiety mainly establishes pi-cation interactions with K594 from the other protomer in all the ligands.
Next, we calculated Dynamic Ligand Efficiency (DLE) to correlate predicted docking scores to ATPase stimulation and cellular effects. 31 was not included in this list, as its fluorescence prevented measuring ATPase. In Supplementary Figure 1 (SF1), we report the correlation between the DLE of the set of newly designed compounds together with the ones previously described in [6c]. The model provides a good correlation between DLEs and measured ATPase stimulations (R = −0.66 considering all compounds discussed here and in [6c]; R = −0.71 when considering only co-generic amino-derivatives). This finding supports the validity of our model for the design of allosteric activators of Hsp90. Next, we assessed the capacity of our new DLE descriptor to evaluate the potency of the designed compounds in antiproliferative assays. Importantly, the calculated DLE shows a significant correlation with measured cytotoxicities against the cancer STO cell line, with correlation value of 0.62 when considering the whole series, which raises to 0.67 when considering only amines (see Table 1; SF1) indicating the ability of this very simple model to quantitatively capture the main determinants of cytotoxic activities.
To the best of our knowledge, these results are the first that show the actual feasibility of pushing integrated knowledge of dynamic protein-ligand cross-talk into the design of new Hsp90 allosteric compounds, with novel functional impacts as well as improved antiproliferative activities. Interestingly, our compounds stimulate Hsp90 ATPase activity favoring the active state: we hypothesize that this reverberates in a modification of the population of the chaperone structural ensembles and of the timing with which Hsp90 conformational families are presented to interaction with co-chaperones and clients. Consistent with recent findings based on mutational studies [6b, 12], this novel way of perturbing chaperone populations and kinetics can expectedly be detrimental to cell viability. In conclusion, our compounds may represent a new series of Hsp90 modulators as anticancer drugs with a novel mechanism of action based on the perturbation of the conformational population and kinetics of the Hsp90 machinery.
Supplementary Material
Acknowledgments
This work was supported through funding to G.C. from AIRC (Associazione Italiana Ricerca sul Cancro) grant IG 15420, funding from Fondazione Cariplo Grant 2011.1800, funding from Italian Ministry of Foreign Affairs (MAE) through the project PERTNET, and Università degli Studi di Milano (research fellowship to S.S.).
References
- 1.Krukenberg KA, Street TO, Lavery LA, Agard DA. Q Rev Biophys. 2011;44:229–255. doi: 10.1017/S0033583510000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.a) Whitesell L, Lindquist SL. Nat Rev Cancer. 2005;5:761–772. doi: 10.1038/nrc1716. [DOI] [PubMed] [Google Scholar]; b) Taipale M, Jarosz DF, Lindquist S. Nat Rev Mol Cell Biol. 2010;11:515–528. doi: 10.1038/nrm2918. [DOI] [PubMed] [Google Scholar]
- 3.a) Workman P, Burrows FJ, Neckers L, Rosen N. Ann NY Acad Sci. 2007;1113:202–216. doi: 10.1196/annals.1391.012. [DOI] [PubMed] [Google Scholar]; b) Trepel JB, Mollapour M, Giaccone G, Neckers L. Nat Rev Cancer. 2010;10:537–549. doi: 10.1038/nrc2887. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Neckers L, Workman P. Clin Cancer Res. 2013;18 doi: 10.1158/1078-0432.CCR-11-1000. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Neckers L, Trepel JB. Clin Cancer Res. 2014;20:275–277. doi: 10.1158/1078-0432.CCR-13-2571. [DOI] [PubMed] [Google Scholar]; e) Bracci A, Colombo G, Ronchetti F, Compostella F. European Journal of Organic Chemistry. 2009:5913–5919. [Google Scholar]
- 4.Brandt GEL, Blagg BSJ. Curr Top Med Chem. 2009;9:1447–1461. doi: 10.2174/156802609789895683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Byrd KM, Subramanian C, Sanchez J, Motiwala HF, Liu W, Cohen MS, Holzbeierlein J, Blagg BSJ. Chemistry A European Journal. 2016;22:6921–6931. doi: 10.1002/chem.201504955. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhao HP, Donnelly AC, Kusuma BR, Brandt GEL, Brown D, Rajewski RA, Vielhauer G, Holzbeierlein J, Cohen MS, Blagg BSJ. J Med Chem. 2011;54:3839–3853. doi: 10.1021/jm200148p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Morra G, Neves MAC, Plescia CJ, Tsutsumi S, Neckers L, Verkhivker G, Altieri DC, Colombo G. J Chem Theory Comput. 2010;6:2978–2989. doi: 10.1021/ct100334n. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sattin S, Tao JH, Vettoretti G, Moroni E, Pennati M, Lopergolo A, Morelli L, Bugatti A, Zuehlke A, Moses M, Prince T, Kijima T, Beebe K, Rusnati M, Neckers L, Zaffaroni N, Agard DA, Bernardi A, Colombo G. Chemistry-a European Journal. 2015;21:13598–13608. doi: 10.1002/chem.201502211. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Vettoretti G, Moroni E, Sattin S, Tao J, Agard D, Bernardi A, Colombo G. Sci Rep. 2016;6:23830. doi: 10.1038/srep23830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Morra G, Potestio R, Micheletti C, Colombo G. Plos Comput Biol. 2012;8:e1002433. doi: 10.1371/journal.pcbi.1002433. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Moroni E, Zhao H, Blagg BS, Colombo G. J Chem Inf Model. 2014;54:195–208. doi: 10.1021/ci4005767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sattin S, Panza M, Vasile F, Berni F, Goti G, Tao JH, Moroni E, Agard D, Colombo G, Bernardi A. European Journal of Organic Chemistry. 2016:3349–3364. [Google Scholar]
- 9.a) Zaffaroni N, Costa A, Pennati M, De Marco C, Affini E, Madeo M, Erdas R, Cabras A, Kusamura S, Baratti D, Deraco M, Daidone MG. Cell Oncol. 2007;29:453–466. doi: 10.1155/2007/456839. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) De Cesare M, Cominetti D, Doldi V, Lopergolo A, Deraco M, Gandellini P, Friedlander S, Landesman Y, Kauffman MG, Shacham S, Pennati M, Zaffaroni N. Oncotarget. 2015;6:13119–13132. doi: 10.18632/oncotarget.3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sciacovelli M, Guzzo G, Morello V, Frezza C, Zheng L, Nannini N, Calabrese F, Laudiero G, Esposito F, Landriscina M, Defilippi P, Bernardi P, Rasola A. Cell Metabolism. 2013;17:988–999. doi: 10.1016/j.cmet.2013.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Gestwicki JE, Crabtree GR, Graef IA. Science. 2004;306:865–869. doi: 10.1126/science.1101262. [DOI] [PubMed] [Google Scholar]; b) Li X, Srinivasan SR, Connarn J, Ahmad A, Young ZT, Kabza AM, Zuiderweg ER, Sun D, Gestwicki JE. 2013;4:1042–1047. doi: 10.1021/ml400204n. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Miyata Y, Li X, Lee HF, Jinwal UK, Srinivasan SR, Seguin SP, Young ZT, Brodsky JL, Dickey CA, Sun D, Gestwicki JE. ACS Chem Neurosci. 2013;4:930–939. doi: 10.1021/cn300210g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.a) Zierer BK, Rubbelke M, Tippel F, Madl T, Schopf FH, Rutz DA, Richter K, Sattler M, Buchner J. Nat Struct Mol Biol. 2016;23:1020–1028. doi: 10.1038/nsmb.3305. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Rehn A, Moroni E, Zierer BK, Tippel F, Morra G, John C, Richter K, Colombo G, Buchner J. J Mol Biol. 2016;428:4559–4571. doi: 10.1016/j.jmb.2016.09.014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.