

Abstract
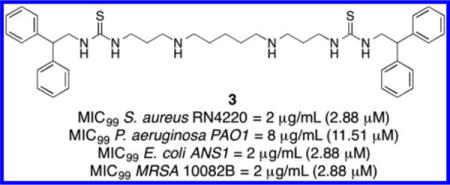
Antibiotic resistance is a growing threat to human health exacerbated by a lack of new antibiotics. We now describe a series of substituted diamines that produce rapid bactericidal activity against both Gram-positive and Gram-negative bacteria, including methicillin-resistant Staphylococcus aureus and stationary-phase bacteria. These compounds reduce biofilm formation and promote biofilm dispersal in Pseudomonas aeruginosa. The most potent analogue, 3 (1,13-bis{[(2,2-diphenyl)-1-ethyl]thioureido}-4,10-diazatridecane), primarily acts by depolarization of the cytoplasmic membrane and permeabilization of the bacterial outer membrane. Transmission electron microscopy confirmed that 3 disrupts membrane integrity rapidly. Compound 3 is also synergistic with kanamycin, demonstrated by the checkerboard method and by time-kill kinetic experiments. In human cell toxicity assays, 3 showed limited adverse effects against the HEK293T human kidney embryonic cells and A549 human adenocarcinoma cells. In addition, 3 produced no adverse effects on Caenorhabditis elegans development, survival, and reproduction. Collectively, diamines related to 3 represent a new class of broad-spectrum antibacterials against drug-resistant pathogens.
Graphical abstract
INTRODUCTION
Infections caused by drug-resistant bacteria have become an increasing threat to human health. Despite the rapidly increasing incidence of antibacterial resistance, few new antibiotics are currently being developed.1 Thus, there is an urgent need to identify new antibiotics from distinct chemical classes and with novel mechanisms of action. Unlike conventional antibiotics, cationic antimicrobial peptides are known to act primarily by disrupting the bacterial cell membrane. Moreover, the charge– charge interaction between antibacterial peptides and bacterial cell membrane does not target intracellular bacterial proteins. Thus, bacteria find it difficult to develop resistance against antibacterial peptides.2 However, antibacterial peptides are mainly limited by their potential for toxicity, high susceptibility to proteolytic degradation, and the high cost of manufacture. In the recent past, several classes of cationic small molecules that mimic the antibacterial peptides have shown some promise as potential antibiotics, and some of them are undergoing clinical trials.3–7
By virtue of the cationic groups in their structure, natural polyamines exhibit a broad range of biological functions that promote bacterial fitness and pathogenesis.8 Recent studies have shown that natural polyamines and their analogues can impair membrane integrity and biofilm formation9–11 without affecting planktonic bacterial growth,12 and also increase antibiotic susceptibility.13,14 Synthetic polyamine-containing sterol analogues related to squalamine also exhibit antimicrobial activities.15–17 These data suggest that there is considerable potential for the use of synthetic diamines as potential antibacterial drug candidates.
Herein, we report the design and development of synthetic diamines 1–16 (Figure 1); compounds 1–12 are bactericidal against Staphylococcus aureus, Pseudomonas aeruginosa, Escher-ichia coli, and methicillin-resistant Staphylococcus aureus (MRSA), while 13–16 were designed to evaluate the requirement for the central charged nitrogens. Importantly, these compounds also strongly inhibit biofilm formation and promote biofilm dispersal in the Gram-negative pathogen P. aeruginosa. The most effective of these analogues, compound 3, was selected for use as a chemical tool to elucidate the exact mechanism of the bactericidal activity for these agents, which until now was unclear. We hypothesize that positively charged diamines such as 1–12 can interact with the negatively charged bacterial cell membrane. Therefore, we employed multiple methods to elucidate the effects of synthetic diamines on membrane homeostasis. We also determined the effect of 3 in combination with selected commercial antibiotics to determine whether they act synergistically. Finally, we determined the effect of 3 against nondividing bacterial cells and evaluated its protease tolerance and toxicity in human cells and Caenorhabditis elegans. Findings from this study provide evidence that diamines are membrane-targeting inhibitors that are active against drug-resistant pathogens.
Figure 1.
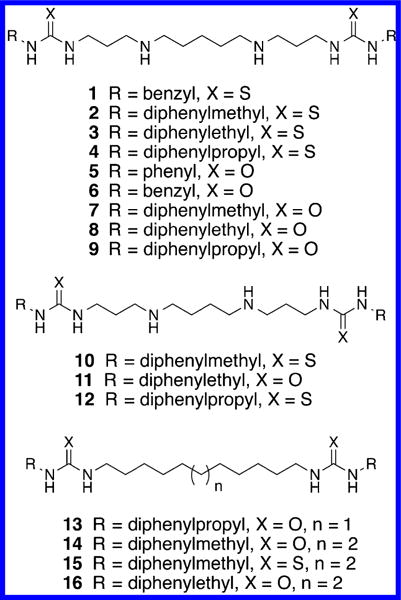
Structures of (bis)ureido or (bis)thioureido(N,N′-alkyl)-diamines 1–10 and (bis)ureido or (bis)thioureidoalkanes 10–16.
CHEMISTRY
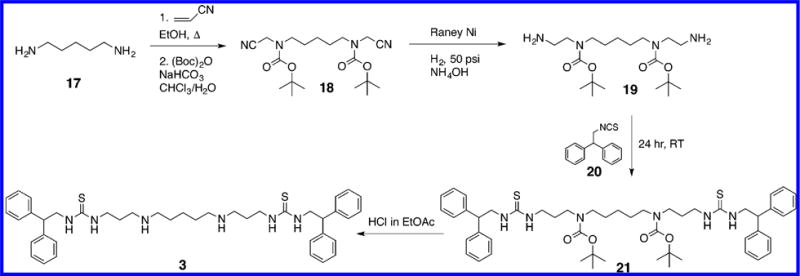
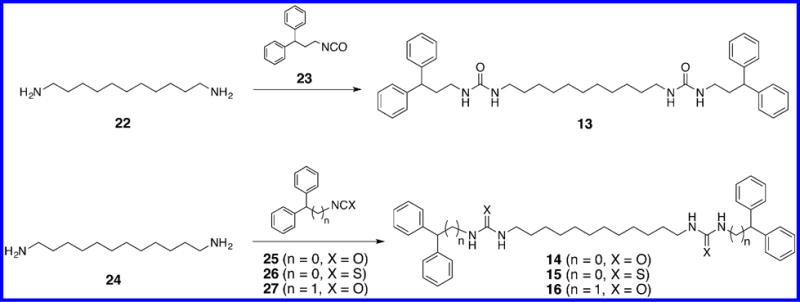
The (bis)ureido- and (bis)thioureido diamines 1–12 described in this article were synthesized according to our recently published pathway,18 as shown in Scheme 1. The synthesis of compound 3 is described below, and all other compounds were synthesized in exactly the same manner. 1,5-Diaminopentane 17 was (bis)cyanoethylated (acrylonitrile, ethanol, reflux), and the central nitrogens were protected [(Boc)2O, NaHCO3, and CHCl3/H2O] to afford the previously described (bis)-N-Boc protected intermediate 18.19,20 Compound 18 was reduced in the presence of Raney nickel (H2, 50 psi, NH4OH) to form the (bis)-N-Boc protected diamine 19. Intermediate 19 was then reacted with the appropriate isocyanate or isothiocyanate (in this case 2,2-diphenylethyl isothiocyanate 20) to yield the protected precursor 21. Removal of the N-Boc protecting groups with mild acid (HCl in EtOAc) then afforded the desired target compound 3. Compounds 13–16, which lack the two internal secondary nitrogens found in 1–12, were synthesized as described in Scheme 2. Briefly, 1,11-diaminoundecane 22 was allowed to react with 2 equiv of 3,3-diphenylpropyl isocyanate 23 to afford the corresponding target compound 13. Likewise, 1,12-diaminodo-decane 24 was coupled to the appropriate isocyanate or isothiocyanate 25–27 to afford the corresponding analogues 14–16.
Scheme 1.
Scheme 2.
RESULTS
Antibacterial Screen
In order to determine whether synthetic diamines 1–16 possessed an acceptable minimum inhibitory concentration (MIC) in multiple organisms, the MIC99 of compounds 1–16 was determined against a Gram-positive organism (Staphylococcus aureus strain RN4220) and 2 Gram-negative organisms (Pseudomonas aeruginosa strain PAO1 and Escherichia coli strain ANS1). The MIC99 assay for the antibacterial activity of each test compound was performed by the microbroth dilution method of the Clinical and Laboratory Standards Institute of America. MIC99 was defined as the lowest compound concentration at which bacterial growth was completely inhibited after overnight incubation in a 96-well plate (Nunc) at 37 °C without shaking. The results of these studies are summarized in Table 1. Commercially available antibiotics from 4 different classes were used as positive controls: the penicillin derivative ampicillin (AMP), the aminoglycoside kanamycin (KAN), the fluoroquinoline norfloxacin (NOR), and the polymixin antibiotic colistin. The natural polyamines spermidine and spermine were also assayed as negative control compounds. Synthetic diamines 1–4 and 7–12 produced significant antibacterial activity, with MIC99 values between 2 and 32 μg/mL (with one exception: compound 1 had an MIC99 value of 256 μg/mL against P. aeruginosa). The most effective of these analogues, 3, exhibited MIC99 values of 2.0, 8.1, and 2.2 μg/mL against S. aureus RN4220, P. aeruginosa PAO1, and E. coli ANS1, respectively. Compounds 13–16 were synthesized to determine whether the presence of the two central nitrogens was required for activity. Importantly, compounds 13–16 produced no detectable inhibitory effect against any of the 3 tested strains. A direct comparison could be made between active compounds 10–12 and inactive (control) analogues 14–16 since each of these compounds possessed a chain length of 12 atoms between the terminal substituted ureas or thioureas. Substitution of carbon for the internal nitrogens in 10–12 produced a complete abolition of antibacterial activity. On the basis of the fact that simple polyamine analogues such as spermidine and spermine do not produce antibacterial effects, it would appear that the presence of both the central charged nitrogens and the terminal diaryl moieties are required for activity. It may be that the aromatic ring structures produce additional affinity for the bacterial membrane due to a hydrophobic effect. However, this effect in itself in not sufficient to produce the observed antibacterial activity since 13–16 are inactive. Interestingly, it has been shown that the natural polyamines spermidine and spermine, as well as simple diamines like putrescine and cadaverine, have no antibacterial effects, no effect on outer membrane permeability, and did not cause membrane rupture.13 These amine derivatives have no inherent antibacterial activity in pseudomonas and are in fact biosynthesized and imported by numerous bacteria, including Pseudomonas aeruginosa,21 Staphylococcus aureus,22 and Escherichia coli.22 Although diamines such as those mentioned above have no inherent antibacterial activity, addition of exogenous spermidine (1.0 mM) or other polyamines to the pseudomonas growth medium increased the MIC values of multiple antibiotics, including polycationic antibiotics, amino-glycosides, quinolones, and fluorescent dyes.23 In a subsequent study by the same group, spermine was shown to decrease the MIC values for chloramphenicol and β-lactam antibiotics.14 Taken together, these data support the contention that both the central charged nitrogens and the terminal diaryl moieties in 1– 12 are required for the observed antibacterial activity.
Table 1.
In Vitro Antibacterial Activity (MIC99) of Diamines Tested against S. aureus RN4220, P. aeruginosa PAO1, and E. coli ANS1a
| Compound | Chemical Structure | MIC99 (μg/ml) | ||
|---|---|---|---|---|
| S. aureus RN4220 | P. aeruginosa PAOI | E. coli ANSI | ||
| 1 |
|
32 ± 2.4 | 256 ±6.3 | 32 ±3.3 |
| 2 |
|
6.7 ±2.3 | 13.3 ±4.6 | 6 ±2.8 |
| 3 |
|
2 ±0.8 | 8 ± 1.6 | 2± 1.1 |
| 4 |
|
9.3±6.1 | 32 ±5.2 | 10 ±8.5 |
| 5 |
|
>256 | >256 | >256 |
| 6 |
|
106.7±37.3 | 213.3±73.9 | 64 ±8.6 |
| 7 |
|
10.7±4.6 | 13.3±4.6 | 6 ±2.8 |
| 8 |
|
18.7±12.2 | 26.7+9.2 | 4 ±0.8 |
| 9 |
|
13.3±4.6 | 26.7±9.2 | 6 ±2.8 |
| 10 |
|
3.8 ±2.9 | 10.4 ±3.3 | 6.2 ± 1.8 |
| 11 |
|
11.3 ±3.7 | 18.6 ±5.8 | 4.4 ±2.6 |
| 12 |
|
4.9 ± 1.3 | 29.2 ±2.2 | 10.4 ±3.7 |
| 13 |
|
>256 | >256 | >256 |
| 14 |
|
>256 | >256 | >256 |
| 15 |
|
>256 | >256 | >256 |
| 16 |
|
>256 | >256 | >256 |
| Spermine | >512 | >512 | >512 | |
| Spermidine | >512 | >512 | >512 | |
| Ampicillin | 0.125 ±0.7 | 128 ±4.3 | - | |
| Kanamycin | 8 ± 1.1 | 32 ±3.8 | 8 ±3.3 | |
| Norfloxacin | 0.5 ±0.6 | 0.5 ± 02 | 0.25 ± 0.8 | |
| Colistin | >16 | <0.125 | ND | |
MIC99 values were determined three times independently and are presented as averages ± standard deviation (SD). ND = not determined.
A clear structure/activity trend was also observed by comparing the MIC99 values of compounds 1–12. Compounds with (bis)aryl substitutions on the terminal alkyl group were significantly more potent than those with monoaryl substituents, as reflected in MIC99 values. Also, the (bis)arylthioureido analogues were slightly more effective than the corresponding (bis)arylureido analogues, although this difference may not be indicative of a trend. Also, as outlined above, the central charged nitrogen species in 1–12 are required for activity. Because compound 3 was very effective in all three organisms, it was chosen for the experiments described below.
Antibacterial Activity of Compound 3 against Meth-acillin-Resistant Staphylococcus aureus
To further probe the activity of antimicrobial diamines 1–12, compound 3 was evaluated for the ability to kill methacillin-resistant Staphylococcus aureus (MRSA) in vitro. Five MRSA isolates from the Medical University of South Carolina collection were selected for antibacterial activity testing. These isolates represented the most commonly found SPA types in the MUSC collection. Strain 10082 B is SPA type 1, 10076 B is SPA type 2, 30253 CA is SPA type 7, 20225 B is SPA type 15, and 20467 BA is SPA type 59. Compound 3 exhibited potent anti-MRSA activity, exhibiting MIC99 values of 1–2 μg/mL against each isolate (Table 2). These MIC99 values are comparable to the MIC value of 3 against S. aureus RN4220 (2 μg/mL, see Table 1).
Table 2.
Effect of Compound 3 on Five Methacillin-Resistant S. aureus Clinical Isolatesa
| isolate ID | SPA type | MIC99 (μg/mL) |
|---|---|---|
| 10082B | 1 | 1 |
| 10076B | 2 | 1 |
| 30253CA | 7 | 1 |
| 20225B | 15 | 1 |
| 20467BA | 59 | 1 |
MIC99 values are the average of 3 determinations that in each case differed by 5% or less. MIC99 average values were then rounded to the nearest whole number μg value.
Effect of Compound 3 on Biofilm Formation in Pseudomonas aeruginosa
As shown in Figure 2, compound 3 inhibited P. aeruginosa biofilm formation at 64 μg/mL, which was comparable to the commercial antibiotics tetracycline (TET) and NOR, whereas KAN displayed no inhibitory effect below 1024 μg/mL (Figure 2A). Interestingly, 3 had a significantly stronger effect on the dispersal of the formed biofilm (Figure 2B) when compared to that of TET, NOR, and KAN. The term drug tolerance refers to the noninherited drug resistance that develops against antibiotics in nondividing cells such as bacteria in the stationary phase.24 Because we suspected that diamines target the bacterial membrane (see below), we hypothesized that they would be bactericidal against nondividing cells. Thus, the effect of compound 3 on drug tolerant cells was evaluated using bacteria in stationary phase. Actively dividing cells in exponential phase were used for the purpose of comparison. An in vitro time-kill assay for 3 against stationary phase S. aureus and P. aeruginosa revealed the remarkable and rapid bactericidal activity against both multiplying bacteria and nondividing stationary cells (Figure 3). By contrast, the traditional antibiotics AMP, KAN, and NOR displayed strong bactericidal activity against dividing bacteria but did not kill stationary phase bacteria efficiently. When tested at bactericidal concentrations (4× MIC99), AMP, KAN, NOR, and compound 3 showed similar bactericidal activity against log phase S. aureus cells (Figure 3A). However, stationary-phase S. aureus were resistant to AMP, KAN, and NOR, while remaining susceptible to 3 (Figure 3B). Following 4 h of treatment, NOR reduced the viable cell numbers only by 2 logs, while 3 eliminated all viable cells. Log phase P. aeruginosa responded weakly to AMP and KAN, while NOR and 3 both produced a 4 log reduction in viable bacteria (Figure 3C). P. aeruginosa in stationary phase was completely resistant to AMP and KAN, and displayed increased resistance to NOR compared to log phase cells. However, compound 3 elicited potent bactericidal activity against stationary phase P. aeruginosa (Figure 3D) and eliminated all bacteria after 4 h. These data demonstrate that the membrane-targeting diamine 3 can circumvent drug tolerance in non-dividing cells since both dividing log phase and nondividing stationary phase bacteria require intact membranes to survive.
Figure 2.
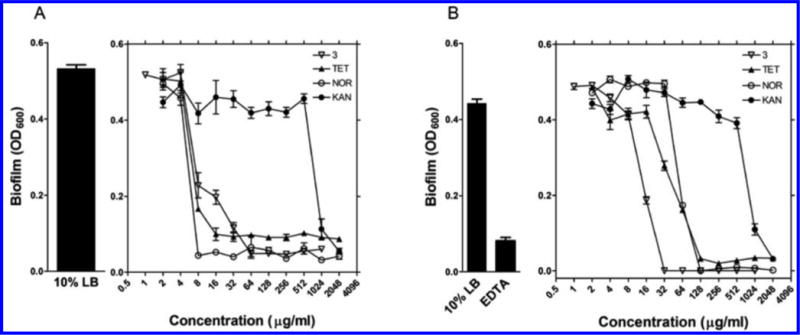
Effect of 3 on biofilm formation (A) and dispersal (B). The pegged-lid method was used to grow the P. aeruginosa biofilm, and the effect of 3 was compared with tetracycline (TET), norfloxacin (NOR), and kanamycin (KAN). EDTA was used as a positive control for biofilm dispersal. Each data point is the average of 3 determinations ± SEM.
Figure 3.
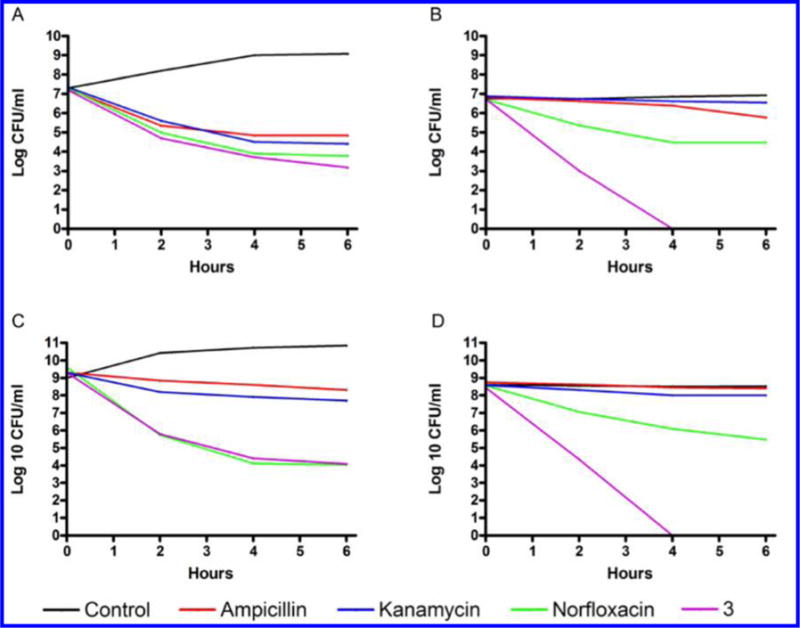
Viability of exponential-phase and stationary-phase bacteria in the presence of 3 or the indicated antibiotics. (A) Exponential-phase S. aureus and (B) stationary-phase S. aureus were treated with antibiotics at the following concentrations: 0.5 μg/mL ampicillin sodium (1.35 μM), 32 μg/mL kanamycin sulfate (54.9 μM), 2 μg/mL norfloxacin (6.26 μM), and 8 μg/mL 3 (11.5 μM). (C) Exponential-phase P. aeruginosa and (D) stationaryphase P. aeruginosa were treated with 512 μg/mL ampicillin sodium (1.38 mM), 128 μg/mL kanamycin sulfate (219.6 μM), 2 μg/mL norfloxacin (6.26 μM), and 32 μg/mL 3 (41.6 μM).
Antibacterial Activity in Plasma
One of the major disadvantages of antibacterial peptides is their potential for proteolytic inactivation. This is reflected in the subsequent loss of antibacterial activity of antibacterial peptides in the presence of plasma.25 To determine whether antibacterial diamines are resistant to hydrolysis by proteases, the MIC99 value of 3 in the presence of plasma was measured. A stock solution was prepared containing 10% fetal bovine serum (FBS), and the MIC99 assay was performed by the microbroth dilution method as described above. Following 12 h of preincubation in the presence of plasma, 3 displayed no loss of activity when cultured in the presence of 10% FBS (data not shown).
Hemolytic Activity and Cytotoxicity
The toxicity of compound 3 in mammalian cells was measured by its ability to lyse human red blood cells using the method of Kustanovich,26 and the results were represented as the HC50 value (the concentration at which 50% of the red blood cells are lysed). The HC50 of compound 3 was 64 μg/mL, indicating that the compound produced selective toxicity toward bacterial cells. The cytotoxicity of compound 3 was also monitored in two additional mammalian cell lines, the HEK293T human kidney embryonic cell line and the A549 human alveolar basal epithelial adenocarcinoma cell line. The results of these studies are shown in Table 3. The selectivity index (SI) in each cell line was the ratio of the CC50 value of cytotoxicity against a given human cell line divided by the MIC99 value for a given bacterial strain. Compound 3 possessed a favorable SI (defined as an SI ≥ 10) between 16 and 64 against S. aureus, E. coli and MRSA clinical isolates. The SI against P. aeruginosa was lower (either 4 or 8), suggesting that additional structure optimization is required to identify a compound that kills P. aeruginosa with an acceptable SI.
Table 3.
Cytotoxicity of Compound 3 in HEK293T Human Kidney Embrionic Cells and A549 Human Alveolar Basal Epithelial Adenocarcinoma Cells
| MIC99a | selectivity indexb
|
||
|---|---|---|---|
| HEK293T cells | A549 cells | ||
| S. aureus RN4220 | 2 | 16 | 32 |
| P. aeruginosa PAO1 | 8 | 4 | 8 |
| E. coli ANSI | 2 | 16 | 32 |
| MRSA | 1 | 32 | 64 |
MIC99 = the concentration that inhibited bacterial growth by 99%.
Selectivity index = CC50/MIC99. The CC50 value of compound 3 against HEK293T cells was 32 μg/mL. The CC50 value of compound 3 against A549 cells was 64 μg/mL. CC50 = the concentration that reduced HEK293T or A549 cell viability by 50%.
Toxicity against Caenorhabditis elegans
C. elegans has been successfully used to study acute bacterial infections27–29 and has also been employed to assess toxicity.30 For our purposes, we incubated C. elegans with high concentrations of 3, gentamicin, and 5-fluorodideoxyuridine (5FU). Gentamicin and 5FU are both toxic toward eukaryotic cells at high concentrations and were used as controls throughout the assay.31,32 Prior to this study, we evaluated the effect of lysed E. coli as a food source for nematodes because 3 has demonstrated the ability to lyse bacterial membranes. C. elegans incubated with mechanically lysed E. coli displayed no alterations in development and procreation (data not shown). C. elegans eggs, worms at L1 and L4 development stages, were incubated with 64 μg/mL of 3 for 96 h, which provided enough time to assess the complete life cycle. We observed no changes in C. elegans egg development, and larvae that hatched produced viable eggs and displayed no deleterious genotypic effects. Juvenile and adult nematodes were also not affected as evidenced by 100% survival, no changes in locomotion, and maintenance of egg production. High concentrations of gentamicin or 5FU were toxic toward C. elegans, specifically affecting the viability of eggs produced and killing juvenile nematodes. Collectively, the assessment of the toxicity of 3 in C. elegans suggests that the compound does not produce overt toxicity in vivo and should be further evaluated in a higher organism. These data also indicate that the toxicity of 3 is low, an observation that is in agreement with our results from toxicity testing in human cells.
Mechanism of Action
The mechanism of action of compound 3 was investigated using various spectroscopic methods that revealed that the diamines primarily acted by targeting the bacterial cell membrane. As described below, our results show that the diamines exhibit rapid and potent bactericidal activity. This fast killing activity was indicative of membrane-targeting activities, which could be mediated by an electrostatic interaction between the positively charged central nitrogens in 3 and its analogues, and the negatively charged bacterial membrane. This hypothesis is consistent with the fact that positively charged antibacterial peptides can disrupt bacterial membrane integrity and depolarize bacterial membranes.33 To test this hypothesis, we first measured the effect of 3 on bacterial membrane potential using a membrane-potential-sensitive fluorescent probe (DiSC3-5). In buffer-treated control cells, the fluorescent signal of the probe was quenched by cell membranes with intact membrane potential (Figure 4A and B). The addition of compound 3 at 4× MIC99 (8 μg/mL) and 1× MIC99 (2 μg/mL) concentrations against S. aureus caused a rapid increase in the probe’s fluorescence intensity (Figure 4A), indicating that the membrane potential was dissipated by the diamine. Treatment of P. aeruginosa by compound 3 led to a similar abrupt increase in DiSC3-5 fluorescence (Figure 4B), demonstrating that the cell membrane of both Gram-positive and Gram-negative bacteria can be depolarized. There is now increasing evidence that cationic antimicrobial agents can depolarize the cytoplasmic membrane. However, it is unclear if depolarization is a consequence of the dying bacteria or is truly tied to the mechanism. To this end, we performed simultaneous testing of bactericidal kinetics and depolarization (Figure 5). Following a 5 min treatment with 3 at 4× MIC99 (8 μg/mL), colony-forming unit (CFU) titers of S. aureus and P. aeruginosa decreased more than 2 log10 units, an effect that was equal to the positive control compound dinitrophenol (DNP). Colistin, a cationic antimicrobial peptide that targets the membrane of Gram-negative bacteria, produced a similar effect in P. aeruginosa. The time-kill kinetics produced by 3 paralleled the observed membrane depolarization, suggesting a causal relationship between the rapid bactericidal effect of 3 and membrane depolarization. Bacterial outer membrane permeabilization of P. aeruginosa was evaluated using another fluorescent probe, 1-N-phenylnaphthylamine (NPN). Intact outer membranes exclude the hydrophobic NPN, while membranes with increased fluidity and permeability allow entry of the probe, resulting in a higher fluorescent signal.34 The aminoglycoside antibiotic gentamicin increases the outer membrane permeability of Gram-negative bacteria35 and was thus included as a positive control. As shown in Figure 4C, the NPN fluorescence signal was increased rapidly in P. aeruginosa treated with 3 at 4× MIC99 (32 μg/mL) and 1× MIC99 (8 μg/mL) in a manner similar to that of gentamicin. These results suggest that 3 increases the permeability of the outer bacterial membrane. Transmission electron microscopy (TEM) was subsequently used to visualize bacterial cell morphology at high resolution and provide direct evidence of the membrane effects caused by 3. Untreated S. aureus (Figure 6A) and P. aeruginosa (Figure 6D) possessed an intact cell envelope with both a well-defined peptidoglycan cell wall and a lipid bilayer membrane. Following treatment with 3 at 2× MIC99 for 30–60 min, S. aureus formed numerous spherical, double layered mesosome-like structures (Figure 6B). Cellular debris of completely lysed cells was also observed after the treatment with 3 (Figure 6C). Similarly, P. aeruginosa treated with 3 at 2× MIC99 (15 min) exhibited extensive membrane damage, as illustrated by the appearance of lysed cells and the release of cell contents (Figure 6E and F).
Figure 4.
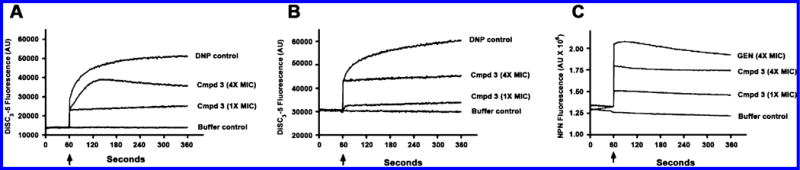
(A) S. aureus cytoplasmic membrane depolarization promoted by 3. (B) P. aeruginosa cytoplasmic membrane depolarization promoted by 3. (C) P. aeruginosa outer membrane permeabilization promoted by 3. The arrow indicates the time point at which the inhibitor was added. Experiments were performed three times independently, and data from a representative experiment are shown.
Figure 5.
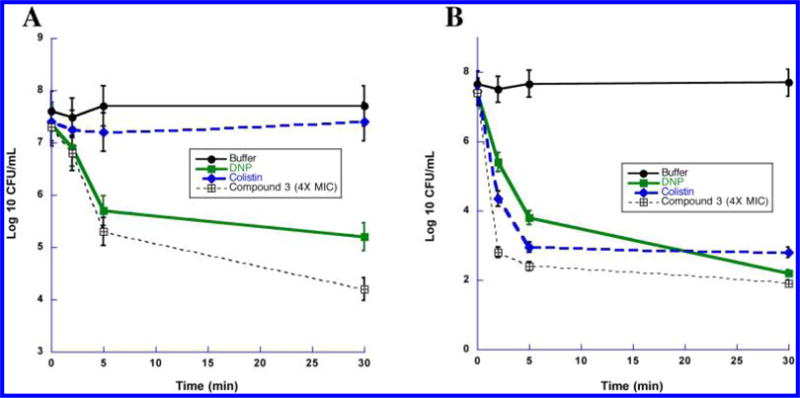
(A) Bactericidal kinetics resulting from cytoplasmic membrane depolarization of S. aureus by 3. (B) Bactericidal kinetics resulting from cytoplasmic membrane depolarization of P. aeruginosa by 3. Each data point is the average of 3 determinations ± SEM.
Figure 6.
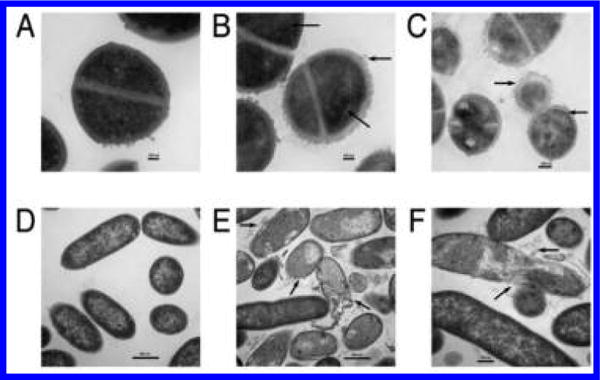
Transmission electron micrographs of (A) untreated S. aureus; (B,C) S. aureus treated with 3 at 2× its MIC99; (D) untreated P. aeruginosa; (E,F) P. aeruginosa treated with 3 at 2× its MIC99. The arrows mark areas of membrane damage and cell content release.
Interaction between Compound 3 and Antibiotics
The interactions between the membrane-targeting diamines and antibiotics with different cellular targets were investigated using checkerboard assays. The checkerboard fractional inhibitory concentration (FIC) for S. aureus and P. aeruginosa treated with 3 was determined in combination with AMP, NOR, or KAN. On the basis of the resulting ΣFIC (Table 4) values and MIC99 isobolograms (Figure 7), compound 3 at 0.5× MIC99 was synergistic with KAN, causing an 8-fold reduction in the MIC99 for KAN against S. aureus (from 16 to 2 μg/mL) and P. aeruginosa (from 64 to 8 μg/mL). By contrast, 3 was not synergistic with AMP or NOR. The synergistic interaction between 3 and KAN is consistent with previous reports that aminoglycosides interact with the bacterial cell envelope and cause membrane damage.36,37 Because the checkerboard method has been reported to be error prone, the synergy between 3 and KAN was verified using a time-kill kinetic assay. Eliopoulos and Eliopoulos have suggested that an interpretation of “synergistic” requires a ≥ 2 log10 decrease in CFU/mL by the combination compared with the most active constituent after 24 h and a ≥ 2 log10 decrease in CFU/mL below the starting inoculum.38 Our results show that the interaction between 3 and KAN meet these criteria (Figure 8) and confirm the synergistic relationship. S. aureus treated with 0.5× the MIC99 of compound 3 combined with 0.5× the MIC99 of KAN reached more than 2 log10 decrease in viable cell count compared with the treatment with 1× the MIC99 of KAN alone, and to treatment at 1× the MIC99 of 3 alone (Figure 8A). Similarly, treatment of P. aeruginosa with 0.5× the MIC99 of 3 plus 0.5× the MIC99 of KAN produced the same effect (Figure 8B).
Table 4.
FIC Values of 3 and Antibiotics AMP, NOR, and KAN As Determined by the Checkerboard Method
| ΣFICa
|
||
|---|---|---|
| S. aureus RN4220 | P. aeruginosa PAO1 | |
| ampicillin | 0.75 | 2.0 |
| norfloxacin | 0.75 | 0.75 |
| kanamycin | 0.25 | 0.75 |
ΣFIC, combined fractional inhibitory concentration. The formula for calculating the ΣFIC is described in the Experimental Section.
Figure 7.
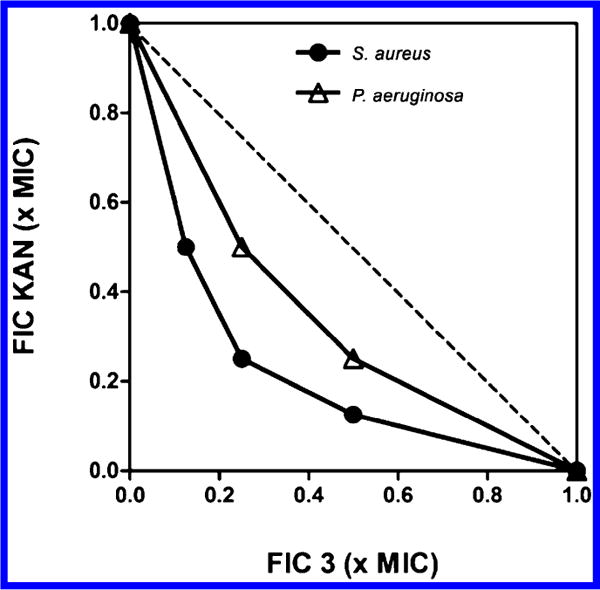
Isobolograms showing the synergistic effects of 3 with kanamycin against S. aureus and P. aeruginosa.
Figure 8.
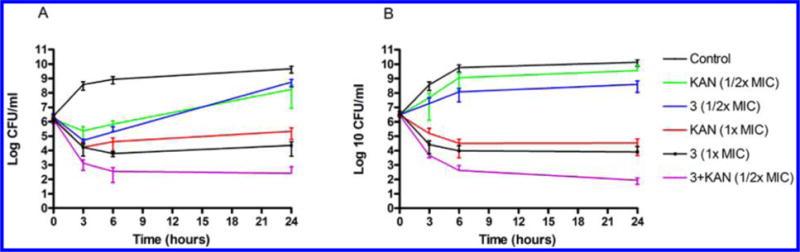
Interaction of 3 and kanamycin (KAN) against (A) S. aureus and (B) P. aeruginosa, determined by time-kill experiments that were performed three times independently. Each data point is the average of 3 determinations ± SEM.
DISCUSSION
In order to discover antibacterial agents with novel mechanisms of action, many recent studies have focused on identifying new bacterial targets or metabolic pathways. A relatively smaller number of studies have described compounds targeted to the bacterial membrane and the formation and disruption of biofilms. The cationic antimicrobial antibacterial peptides have shown promise as antibiotics that are potentially able to overcome bacterial resistance.2 The antimicrobial action of antibacterial peptides is mediated through interaction of their positively charged structures and hydrophobic moieties with the bacterial cell membrane. In this study, we investigated the antibacterial properties of functionalized diamines containing positive moieties that can mimic the antibacterial peptides and confirmed that these compounds have potent bactericidal activity by depolarizing membrane potential and disrupting bacterial membrane integrity. The importance of the positive charges in mediating this activity is underscored by the fact that compounds 13–16, which lack the central nitrogen moieties and thus are uncharged at physiological pH, have no detectable activity as compared to diamines with the same internal chain length. Both Gram positive S. aureus and Gram negative P. aeruginosa and E. coli bacteria were susceptible to the bactericidal effect of the synthetic diamine 3, which was chosen for further investigation. Importantly, compound 3 showed impressive activity against drug-resistant MRSA, dispersed biofilm in log phase and stationary-phase bacteria, and produced a synergistic interaction with the aminoglycoside antibiotic KAN. These effects were accompanied by a desirable selectivity index between the human cells evaluated in this study and bacteria.
Although the precise mechanism of action of antibacterial diamines such as 3 has not been fully elucidated, our findings strongly suggest that the Gram positive and Gram negative bacterial membrane is the primary target for these diamines. The interaction between diamines related to 3 and the bacterial membrane is likely mediated by an electrostatic interaction between the negatively charged bacterial membrane and the positively charged compounds. Binding of 3 to the membrane dissipated cell membrane potential, increased membrane permeability, and led to cell content release and lysis. Transmission electron microcopy provided the most direct evidence of the membrane disrupting action of 3; cells treated with 3 formed abnormal membranous structures and exhibited extensive cell membrane damage. Preliminary structure–activity relationship experiments demonstrate that analogues of 3 lacking charged central nitrogens were not effective against S. aureus and P. aeruginosa. While the positive charge of the compound is required for activity, it is also likely that this charge contributes to the cytotoxicity observed with these analogues at higher concentrations.20,39 However, this toxicity can be effectively managed when compounds related to 3 are used at lower doses as antimicrobial agents.40,41 Hit-to-lead optimization of antimicrobial diamines such as 3 is an ongoing concern in our laboratories.
Classical antibiotics are effective in killing actively dividing cells, but they are ineffective against bacteria that are not dividing, a phenomenon known as noninherited antibiotic resistance.24 Membrane-active compounds can overcome this problem and are promising antibacterial drug candidates since both actively dividing cells and drug-resistant dormant cells require an intact membrane to stay viable.42 Consistent with its membrane-targeting activity, compound 3 exhibited bactericidal activity against both exponential phase and stationary phase bacteria. Interestingly, the stationary phase cells were more susceptible to 3 than the exponential phase cells (Figure 3). The increased sensitivity of stationary phase cells may be due to changes in bacterial surface charges between the two growth phases. For example, the increased negative charge in the stationary-phase S. aureus membrane43 may enhance the attraction of 3, causing increased sensitivity. However, the precise mechanism of this increased sensitivity remains to be fully elucidated. The Gram-negative outer membrane is also highly negatively charged. Stationary-phase P. aeruginosa secretes membrane vesicles with more negative surface charges than log phase cells,44 suggesting increased negative charges on the outer membrane. Finally, E. coli MG1655 also shows increased negative charge on the outer membrane as cells enter stationary phase.45
As outlined above, results from checkerboard assays demonstrated a synergistic interaction between 3 and KAN (Table 4 and Figures 7 and 8). The molecular basis for synergistic interactions is complex; however, synergy often occurs between compounds targeting overlapping pathways.46 In addition to inhibiting protein synthesis, cationic aminoglycosides interact with the outer membrane of Gram-negative bacteria by displacing essential metal cations.37 Aminoglycosides bind to outer membrane lipopolysaccharides, form transient small holes, and increase outer membrane permeability.35–37 The cationic diamine 3 also increased outer membrane permeability (Figure 4), suggesting that it may have an effect on the outer membrane permeability similar to that of the aminoglycosides.
CONCLUSIONS
In conclusion, this article illustrates that the synthetic diamines, which contain positively charged structures, not only hold promise as potent broad-spectrum bactericidal antibiotics but also can be used in combination to increase the efficacy of other antibiotics by increasing outer membrane permeability.
EXPERIMENTAL SECTION
Synthesis
All reagents and dry solvents were purchased from Aldrich Chemical Co. (Milwaukee, WI), Sigma Chemical Co. (St. Louis, MO), VWR (Radnor, PA), or Fisher Scientific (Chicago, IL) and were used without further purification except as noted below. Pyridine was dried by passing it through an aluminum oxide column and then stored over KOH. Triethylamine was distilled from potassium hydroxide and stored in a nitrogen atmosphere. Dry methanol, ethyl acetate, tetrahydrofuran, dimethylformamide, and hexane were prepared using a Glass Contour Solvent Purification System (Pure Process Technology, LLC, Nashua, NH). Routine chromatographic purification on silica gel was performed on a Teledyne Isco CombiFlash Rf200. Preparative scale chromatographic procedures were carried out using a CombiFlash Rf200 chromatography system (Teledyne-Isco, Lincoln, NE) fitted with silica gel 60 cartridges (230–440 mesh). Thin layer chromatography was conducted on Merck precoated silica gel 60 F-254. Ion exchange chromatography was conducted on Dowex1X8-200 anion exchange resin.
All 1H- and 13C NMR spectra were recorded on a Varian Mercury 400 mHz spectrometer, and all chemical shifts are reported as δ values referenced to TMS or DSS. Splitting patterns are indicated as follows: s, singlet; d, doublet; t, triplet; m, multiplet; and br, broad peak. In all cases, 1H NMR, 13C NMR, and MS spectra were consistent with assigned structures. Mass spectra were recorded by LC/MS on a Waters autopurification liquid chromatography with a model 3100 mass spectrometer detector. Prior to biological testing procedures, all compounds were determined to be >95% pure by UPLC chromatography (95% H2O/5% acetonitrile to 20% H2O/80% acetonitrile over 10 min) using a Waters Acquity H-series ultrahigh-performance liquid chromatography fitted with a C18 reversed-phase column (Acquity UPLC BEH C18 1.7 M, 2.1 × 50 mm). Experimental details and analytical data for compounds 1–12 appear in previous publications.18,40
1,11-Bis-[(3,3-diphenylpropyl)ureido]undecane (13)
A 0.255 g (1.07 mmol) portion of 3,3-diphenylpropyl-1-isocyanate 23 in 10 mL of dichloromethane was slowly added to 0.100 g (0.537 mmol) of 1,11-diaminoundecane 22 in 10 mL of dichloromethane with stirring at 0 °C. The reaction mixture was stirred for 15 min at 0 °C, warmed to room temperature, and allowed to stir overnight. Completion of the reaction was verified by TLC, the solvent was evaporated, and the crude product was chromatographed on silica gel 5% ethyl acetate in hexane to afford 0.634 g of pure 13 (89.6%) as a white, crystalline solid. 1H NMR (CDCl3): δ 7.23 (m, 20H), 4.71 (s, 4H), 3.92 (t, 2H, J = 19 Hz), 3.12 (t, 4H, J = 17 Hz), 3.04 (t, 4H, J = 19 Hz), 2.24 (q, 4H, J = 19 Hz), 1.40 (m, 4H, J = 15 Hz), 1.24 (m, 14H, J = 12 Hz). 13C NMR (CDCl3): δ 162.1, 147.3, 131.7, 128.6, 125.3, 42.8, 36.6, 29.7, 28.2, 26.4; MS calculated 660.44; found, 661.89 ([M+1]+).
1,12-Bis-[(1,1-diphenylmethyl)ureido]dodecane (14)
Compound 14 was prepared exactly as described for compound 13 above from 1,12-diaminododecane 24 and 1,1-(diphenyl)methyl-1-isocyanate 25 (0.978 g, 73.2%) as a white amorphous solid. 1H NMR (CDCl3): δ 7.73 (m, 20H), 6.41 (s, 2H), 5.85 (s, 2H), 5.80 (s, 2H), 3.83 (t, 4H, J = 17 Hz), 1.90 (m, 4H, J = 14 Hz), 1.75 (m, 16H, J = 19 Hz). 13C NMR (CDCl3): δ 163.4, 143.3, 130.1, 127.9, 124.3, 62.2, 39.7, 30.5, 29.6, 29.1, 26.6; MS calculated 618.39; found, 619.77 ([M+1]+).
1,12-Bis-[(1,1-diphenylmethyl)thioureido]dodecane (15)
Compound 15 was prepared exactly as described for compound 13 above from 1,12-diaminododecane 24 and 1,1-(diphenyl)methyl-1-isothiocyanate 26 (0.980 g, 68.4%) as a white amorphous solid. 1H NMR (CDCl3): δ 7.31 (m, 20H), 6.40 (s, 2H), 5.61 (s, 2H), 5.30 (s, 2H), 3.44 (t, 4H, J = 19 Hz), 3.41 (m, 4H, J = 13 Hz), 1.52 (m, 4H), 1.40 (m, 4H), 0.85–1.28 (m, 8H). 13C NMR (CDCl3): δ 183.2, 142.6, 129.7, 128.8, 125.0, 63.7, 46.6, 31.2, 28.4, 28.5; MS calculated 650.35; found, 651.78 ([M+1]+)
1,12-Bis-[(2,2-diphenylethyl)ureido]dodecane (16)
Compound 16 was prepared exactly as described for compound 13 above from 1,12-diaminododecane 24 and 2,2-(diphenyl)ethyl-1-isocyanate 27 (1.1 g, 81.2%) as a white amorphous solid. 1H NMR (CDCl3): δ 7.28 (m, 20H), 5.36 (s, 4H), 4.21 (m, 2H, J = 14 Hz), 3.75 (d, 4H, J = 17 Hz), 3.59 (m, 4H, J = 12 Hz), 1.41 (m, 4H, J = 19 Hz), 1.24 (m, 16H). 13C NMR (CDCl3): δ 159.9, 140.8, 128.7, 128.4, 125.5, 44.2, 40.7, 39.3, 28.5, 27.9, 26.4; MS calculated 646.42; found, 647.85 ([M+1]+)
Strains and Growth Medium
Staphylococcus aureus strain RN4220 (ATCC No. 35556), Pseudomonas aeruginosa strain PAO1 (ATCC No. 47085), and Escherichia coli strain ANS147 were grown in Luria–Bertani (LB) medium. Methicillin-resistant Staphylococcus aureus (MRSA) clinic isolates were grown in Mueller-Hinton (MH) medium. Human kidney cells HEK293T/17 (ATCC CRL11268) and lung cells A549 (ATCC CCL-185) were grown in Dulbecco’s minimum essential medium (DMEM) with 10% heat inactivated fetal bovine serum (FBS). Nematode Caenorhabditis elegans strain N2 was obtained from the Caenorhabditis Genetics Center at University of Minnesota, and worms were grown in nematode growth medium (NGM) for toxicity experiments.
MIC99 Assay for S. aureus Strain RN4220, P. aeruginosa Strain PAO1, and E. coli Strain ANS1
The MIC99 assay for the antibacterial activity of each test compound was performed by the microbroth dilution method of the Clinical and Laboratory Standards Institute of America. MIC99 was defined as the lowest compound concentration at which bacterial growth was completely inhibited after overnight incubation in a 96-well plate (Nunc) at 37 °C without shaking. MIC99s were tested three times independently and were presented as averages ± standard deviation (SD).
MIC Assay for Methacillin-Resistant S. aureus (MRSA)
Strains were isolated from Medical University of South Carolina ICU rooms with high touch objects fabricated using US-EPA registered antimicrobial copper alloys in concert with the studies described.48,49 Briefly, surfaces of 100 cm2 were sampled once a week with wipes using uniform pressure and motion, 5 strokes horizontally and 5 strokes vertically, for a total of 10 strokes. MRSA isolates were recovered from MRSA-Chromagar and were then subjected to PFGE with subsequent SPA typing.50 The isolates were stored at −80 °C until they were reanimated via cultivation on MH broth for antimicrobial susceptibility testing. MIC99 values of test compounds against MRSA strains were determined using the microbroth dilution method described above. Overnight cultures of five MRSA strains in MH broth were diluted in fresh medium. Ten microliters of diluted cell suspension was used to inoculate 96-well plates containing 100 μL of serially diluted compounds to yield ∼105 CFU/mL. The plates were incubated at 37 °C for 24 h before cell density was measured by absorbance at 620 nm using a Thermo MultiSkan FC plate reader.
Time-Kill Assay against Exponential Phase and Stationary Phase Cells
Ten milliliter overnight cultures of strain RN4220 or PAO1 were harvested, then washed and resuspended in PBS to their initial volume, and diluted 100 fold in LB or 1% PBS to mimic the exponential phase or stationary phase cells, respectively.27 Antibiotics AMP, KAN, and NOR or compound 3 was added to 4× MIC99. The cultures were incubated at 37 °C with shaking at 200 rpm. At 0, 2, 4, and 6 h time points, viable bacterial numbers were estimated by diluting, plating, and counting the colony-forming unit (CFU) on LB agar plates.
Biofilm Inhibition Assay
A biofilm inhibition experiment was performed with a slight modification as mentioned before.51 P. aeruginosa PAO1 was grown in LB for 2 h with shaking at 37 °C and then diluted 1:1000 in 10% LB-PBS to reach a concentration of 3 × 105 CFU/mL. The first step of the assay was to fill each well of the clear-bottomed 96-well plate (Nunc) with 25 μL of medium containing the culture inoculums. Then, 50 μL of serial diluted compounds was added into each well, and 10% LB-PBS was tested as the control. The 96-well plate was covered with transferable solid-phase (TSP) pegged-lids (Nunc), and the plate was incubated at 37 °C for 24 h without shaking. After 24 h of incubation, biofilms formed on the pegged lid were washed twice by submerging the lid into a basin of sterilized water and then stained with 0.1% crystal violet (CV) in a new 96-well plate for 15 min with gently shaking. After this, the excess CV was washed off with sterilized water. The CV maintained in the biofilms was eluted into 150 μL of 95% ethanol, and the solublized CV was quantitated spectrophotometrically at 600 nm using a Biotek Synergy HT plate reader.
To investigate the dispersal effect of the diamines, we grew the biofilms on the pegged lid without test compounds for 24 h as described above, and biofilms formed on the pegged lid were washed twice by submerging the lid into a basin of sterilized water. Then, 100 μL of serial diluted compounds or 10% LB-PBS was added into a new 96-well plate. Then, the pegged lid with biofilms was transferred into the 96-well plate and incubated at 37 °C for another 24 h. The biofilms maintained in the pegged lid were measured as described above. Three hundred micromolar EDTA was used as the positive control for biofilm dispersal.
Cytoplasmic Membrane Depolarization Assay
The membrane depolarization activity of compound 3 was determined using a fluorescent probe 3,3′-dipropylthiacarbocyanine DiSC3-5 (Sigma), which is sensitive to membrane potential.52 Briefly, cells from log phase culture (OD600 ∼ 0.7) were harvested, washed, and resuspended in 5 mM HEPES buffer at pH 7.2 containing 5 mM glucose and 100 mM KCl to an OD600 of 0.05. The cell suspension was incubated with 0.4 μM DiSC3-5 in darkness, until a stable and approximately 90% reduction in fluorescence signal was reached. The reduction of the probe signal was due to the quenching in the cell with an intact membrane potential. After compound 3 was added (at 4× or 1× MIC99 concentration), changes in fluorescence due to the collapse of the cytoplasmic membrane potential were continuously recorded at 25 °C using a fluorophotometer (Photon Technology), with an excitation wavelength of 622 nm and an emission wavelength of 670 nm. Compound 3 exhibited no fluorescence disturbance of the probe signal. 2,4-Dinitrophenol (DNP) (Acros), which is a known inhibitor that depolarized the membrane potential, was used as a positive control.53
Outer Membrane Permeabilization Assay
The effect of compound 3 on the permeability of the outer membrane of P. aeruginosa strain PAO1 was determined by the 1-N-phenylnaphthylamine (NPN) (Acros) assay described previously.35,54 Cells from strain PAO1 culture at OD600 ∼ 0.5 were harvested, washed, and resuspended in 5 mM HEPES buffer at pH 7.2, NPN was added to the cell suspension to reach 10 μM final concentration, and sodium azide was added to reach 10 mM final concentration. After gently mixing, the cell suspension was incubated at room temperature for 30 min in darkness. Compound 3 at 4× or 1× MIC99 concentration was added, and changes of NPN fluorescence were continuously recorded at the excitation wavelength of 350 nm and emission wavelength of 420 nm. Gentamicin is a known inhibitor that increases outer membrane permeability35 and was used as a positive control at 4× MIC99 concentration.
Transmission Electron Microscopy
Membrane damage caused by compound 3 was visualized with transmission electron microscopy. Bacteria were grown to the early exponential phase in LB (OD600 ∼ 0.4) and treated with compound 3 at 2× MIC99 concentration for 15–60 min. Cells were harvested, washed twice with phosphate buffer, and then fixed with 2.5% glutaraldehyde. Samples were prepared and analyzed by transmission electron microscopy as described in ref 24.
Drug Interaction by the Checkerboard MIC99 Assay
The interactions between compound 3 and three different classes of antibiotics, AMP, KAN, and NOR, were determined by the checker-board MIC99 method as previously described.25,26 Briefly, compound 3 was serially diluted along the ordinate of a 96-well plate, while the antibiotic was serially diluted along the abscissa in 100 μL of LB. Each well was inoculated with 2 × 104 CFU in 100 μL of LB, and the plates were incubated at 37 °C for 24 h. The combined fractional inhibitory concentration (ΣFIC) was calculated as follows: ΣFIC = FIC A + FIC B, where FIC A is the MIC99 of drug A in the combination/MIC99 of drug A alone, and FIC B is the MIC99 of drug B in the combination/MIC99 of drug B alone. The drug combination is considered synergistic when the ΣFIC is less than 0.5, indifferent when the ΣFIC is between 0.5 to 4, and antagonistic when the ΣFIC is greater than 4.26
Drug Interaction by the Time-Kill Assay
The synergistic interaction between compound 3 and kanamycin was further confirmed by the time-kill assay. The test groups were compound 3 or KAN alone at 1× and 0.5× MIC99 and in combination at 0.5× MIC99. Strain RN4220 or PAO1 was used to inoculate the culture at 1 × 106 CFU/mL, and all cultures were incubated at 37 °C with shaking at 300 rpm. At 0, 3, 6, and 24 h time points, viable bacterial densities were estimated by diluting, plating, and colony counting on LB agar.
Hemolysis Assay
Hemolytic experiments were performed as previously described.55 Human red blood cells were purchased from Fisher and kept in 4 °C before use. Briefly, in a 96-well plate, 150 μL of red blood cell suspension was added into 50 μL of serially diluted compound 3. The plate was incubated for 1 h at 37 °C without shaking, and then the plate was centrifuged at 3500 rpm for 5 min. One hundred microliters of the supernatant was transferred to a new 96-well plate, and A540 was measured. Triton X-100 was the positive control.
Cytotoxicity in Human Cells
Human embryonic kidney cells HEK293T/17 and human epithelial lung cells A549 were cultured in DMEM supplemented with 10% FBS at 37 °C with 5% CO2. The cells were seeded into tissue-culture treated 96-well plates and at a density of 104 cells per well. Twenty-four hours postseeding, the wells were washed with sterile PBS once to remove the unattached cells, and then 100 μL of serially diluted compound 3 was added into the adherent cells and incubated for 48 h. Cytotoxicity was determined using the MTS conversion assay (Promega) mentioned previously.56 The concentration of the test compound that reduced the cell viability by 50% (CC50) was determined by nonlinear regression analysis using GraphPad Prism4 software.
C. elegans Toxicity Assay
The nematode C. elegans has emerged as a good model for toxicology testing57 and was used to further determine the toxicity of diamines. Strain N2 worms were maintained on NGM agar at 22 °C between passages and synchronization. The effects of the test compound on adult worm survival and reproduction, juvenile worm development, and egg hatching were determined.30 NGM agar containing compound 3 was prepared by mixing the compound with warm molten agar. Adult L4 and juvenile L1 worms were selected and transferred onto the test plate using a platinum wire. Eggs were gathered from maintenance plates using the synchronization protocol and transferred onto the test plate. Worms were incubated at 22 °C and monitored every 12 h for death, development, and egg production.
Statistical Analysis
Values are presented as the means ± standard deviations (SD) and analyzed by one-way ANOVA analysis of variance (Graphpad Prism). For all analyses, the criterion for significance was a P value of <0.05.
Acknowledgments
We thank Nancy Smythe for her help with transmission electron microscopy and Dr. Alena Fedarovich for her assistance with the fluorometry experiments. This work was funded in part by the National Institute of Health (NIH) grant 5 RO1 CA149095 (to P.M.W.), Department of Defense (DOD) Grant DM090161 (to Y.M.Z.), and a grant from Progen Pharmaceuticals, Ltd. (to P.M.W.). The project was also supported by a pilot project award from the South Carolina Clinical & Translational Research (SCTR) Institute, with an academic home at the Medical University of South Carolina, NIH/NCATS Grant number UL1TR000062.
ABBREVIATIONS
- MRSA
methacillin-resistant Staphylococcus aureus
- MIC
minimum inhibitory concentration
- AMP
ampicillin
- KAN
kanamycin
- NOR
norfloxacin
- TET
tetracycline
- SI
selectivity index
- CC50
concentration that reduced cell viability by 50%
- 5-FU
5-fluorodideoxyuridine
- NPN
1-N-phenylnaphthylamine
- TEM
transmission electron microscopy
- FIC
fractional inhibitory concentration
- CFU
colony-forming unit
- DNP
dinitrophenol
- TMS
tetramethylsilane
- DSS
dioctyl sodium sulfosuccinate
- LC/MS
liquid chromatography/mass spectroscopy
- UPLC
ultra high performance liquid chromatography
Footnotes
Note
The authors declare no competing financial interest.
References
- 1.Boucher HW, Talbot GH, Bradley JS, Edwards JE, Gilbert D, Rice LB, Scheld M, Spellberg B, Bartlett J. Bad bugs, no drugs: no ESKAPE! An update from the Infectious Diseases Society of America. Clin Infect Dis. 2009;48(1):1–12. doi: 10.1086/595011. [DOI] [PubMed] [Google Scholar]
- 2.Brogden KA. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol. 2005;3(3):238–250. doi: 10.1038/nrmicro1098. [DOI] [PubMed] [Google Scholar]
- 3.Choi S, Isaacs A, Clements D, Liu D, Kim H, Scott RW, Winkler JD, DeGrado WF. De novo design and in vivo activity of conformationally restrained antimicrobial arylamide foldamers. Proc Natl Acad Sci U S A. 2009;106(17):6968–6973. doi: 10.1073/pnas.0811818106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu D, Choi S, Chen B, Doerksen RJ, Clements DJ, Winkler JD, Klein ML, DeGrado WF. Nontoxic membrane-active antimicrobial arylamide oligomers. Angew Chem, Int Ed. 2004;43(9):1158–1162. doi: 10.1002/anie.200352791. [DOI] [PubMed] [Google Scholar]
- 5.Haug BE, Stensen W, Kalaaji M, Rekdal O, Svendsen JS. Synthetic antimicrobial peptidomimetics with therapeutic potential. J Med Chem. 2008;51(14):4306–4314. doi: 10.1021/jm701600a. [DOI] [PubMed] [Google Scholar]
- 6.Svenson J, Stensen W, Brandsdal BO, Haug BE, Monrad J, Svendsen JS. Antimicrobial peptides with stability toward tryptic degradation. Biochemistry. 2008;47(12):3777–3788. doi: 10.1021/bi7019904. [DOI] [PubMed] [Google Scholar]
- 7.Isaksson J, Brandsdal BO, Engqvist M, Flaten GE, Svendsen JS, Stensen W. A synthetic antimicrobial peptidomimetic (LTX 109): stereochemical impact on membrane disruption. J Med Chem. 2011;54(16):5786–5795. doi: 10.1021/jm200450h. [DOI] [PubMed] [Google Scholar]
- 8.Di Martino ML, Campilongo R, Casalino M, Micheli G, Colonna B, Prosseda G. Polyamines: emerging players in bacteria-host interactions. Int J Med Microbiol. 2013;303(8):484–491. doi: 10.1016/j.ijmm.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 9.Bottcher T, Kolodkin-Gal I, Kolter R, Losick R, Clardy J. Synthesis and activity of biomimetic biofilm disruptors. J Am Chem Soc. 2013;135(8):2927–2930. doi: 10.1021/ja3120955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goytia M, Dhulipala VL, Shafer WM. Spermine impairs biofilm formation by Neisseria gonorrhoeae. FEMS Microbiol Lett. 2013;343(1):64–69. doi: 10.1111/1574-6968.12130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balakrishna R, Wood SJ, Nguyen TB, Miller KA, Suresh Kumar EV, Datta A, David SA. Structural correlates of antibacterial and membrane-permeabilizing activities in acylpolyamines. Antimicrob Agents Chemother. 2006;50(3):852–861. doi: 10.1128/AAC.50.3.852-861.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rabin N, Zheng Y, Opoku-Temeng C, Du Y, Bonsu E, Sintim HO. Agents that inhibit bacterial biofilm formation. Future Med Chem. 2015;7(5):647–671. doi: 10.4155/fmc.15.7. [DOI] [PubMed] [Google Scholar]
- 13.Kwon DH, Lu CD. Polyamines increase antibiotic susceptibility in Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2006;50(5):1623–1627. doi: 10.1128/AAC.50.5.1623-1627.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kwon DH, Lu CD. Polyamine effects on antibiotic susceptibility in bacteria. Antimicrob Agents Chemother. 2007;51(6):2070–2077. doi: 10.1128/AAC.01472-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Djouhri-Bouktab L, Vidal N, Rolain JM, Brunel JM. Synthesis of new 3,20-bispolyaminosteroid squalamine analogues and evaluation of their antimicrobial activities. J Med Chem. 2011;54(20):7417–7421. doi: 10.1021/jm200506x. [DOI] [PubMed] [Google Scholar]
- 16.Djouhri-Bouktab L, Alhanout K, Andrieu V, Raoult D, Rolain JM, Brunel JM. Squalamine ointment for Staphylococcus aureus skin decolonization in a mouse model. J Antimicrob Chemother. 2011;66(6):1306–1310. doi: 10.1093/jac/dkr114. [DOI] [PubMed] [Google Scholar]
- 17.Salmi C, Loncle C, Vidal N, Letourneux Y, Brunel JM. New stereoselective titanium reductive amination synthesis of 3-amino and polyaminosterol derivatives possessing antimicrobial activities. Eur J Med Chem. 2008;43(3):540–547. doi: 10.1016/j.ejmech.2007.04.006. [DOI] [PubMed] [Google Scholar]
- 18.Nowotarski SL, Pachaiyappan B, Holshouser SL, Kutz CJ, Li Y, Huang Y, Sharma SK, Casero JRA, Woster PM. Structure-activity study for (bis)ureidopropyl- and (bis)thioureidopropyldiamine LSD1 inhibitors with 3–5-3 and 3–6-3 carbon backbone architectures. Bioorg Med Chem. 2015;23:1601–1612. doi: 10.1016/j.bmc.2015.01.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bellevue FH, III, Boahbedason M, Wu R, Woster PM, Casero JRA, Rattendi D, Lane S, Bacchi CJ. Structural comparison of alkylpolyamine analogues with potent in vitro antitumor or antiparasitic activity. Bioorg Med Chem Lett. 1996;6(22):2765–2770. [Google Scholar]
- 20.Sharma S, Wu Y, Steinbergs N, Crowley M, Hanson A, Casero RAJ, Woster P. Bis)urea and (bis)thiourea inhibitors of lysine-specific demethylase 1 as epigenetic modulators. J Med Chem. 2010;53(14):5197–5212. doi: 10.1021/jm100217a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krossa S, Faust A, Ober D, Scheidig AJ. Comprehensive Structural Characterization of the Bacterial Homospermidine Synthase-an Essential Enzyme of the Polyamine Metabolism. Sci Rep. 2016;6:19501. doi: 10.1038/srep19501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ozogul F, Tabanelli G, Toy N, Gardini F. Impact of Cell-free Supernatant of Lactic Acid Bacteria on Putrescine and Other Polyamine Formation by Foodborne Pathogens in Ornithine Decarboxylase Broth. J Agric Food Chem. 2015;63(24):5828–5835. doi: 10.1021/acs.jafc.5b02410. [DOI] [PubMed] [Google Scholar]
- 23.Kwon DH, Lu CD. Polyamines induce resistance to cationic peptide, aminoglycoside, and quinolone antibiotics in Pseudomonas aeruginosa PAO1. Antimicrob Agents Chemother. 2006;50(5):1615–1622. doi: 10.1128/AAC.50.5.1615-1622.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Levin BR, Rozen DE. Non-inherited antibiotic resistance. Nat Rev Microbiol. 2006;4(7):556–562. doi: 10.1038/nrmicro1445. [DOI] [PubMed] [Google Scholar]
- 25.Radzishevsky IS, Rotem S, Bourdetsky D, Navon-Venezia S, Carmeli Y, Mor A. Improved antimicrobial peptides based on acyl-lysine oligomers. Nat Biotechnol. 2007;25(6):657–659. doi: 10.1038/nbt1309. [DOI] [PubMed] [Google Scholar]
- 26.Kustanovich I, Shalev DE, Mikhlin M, Gaidukov L, Mor A. Structural requirements for potent versus selective cytotoxicity for antimicrobial dermaseptin S4 derivatives. J Biol Chem. 2002;277(19):16941–16951. doi: 10.1074/jbc.M111071200. [DOI] [PubMed] [Google Scholar]
- 27.Mahajan-Miklos S, Tan MW, Rahme LG, Ausubel FM. Molecular mechanisms of bacterial virulence elucidated using a Pseudomonas aeruginosa-Caenorhabditis elegans pathogenesis model. Cell. 1999;96(1):47–56. doi: 10.1016/s0092-8674(00)80958-7. [DOI] [PubMed] [Google Scholar]
- 28.Tan MW, Mahajan-Miklos S, Ausubel FM. Killing of Caenorhabditis elegans by Pseudomonas aeruginosa used to model mammalian bacterial pathogenesis. Proc Natl Acad Sci U S A. 1999;96(2):715–720. doi: 10.1073/pnas.96.2.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neu AK, Mansson M, Gram L, Prol-Garcia MJ. Toxicity of bioactive and probiotic marine bacteria and their secondary metabolites in Artemia sp. and Caenorhabditis elegans as eukaryotic model organisms. Appl Environ Micro. 2014;80(1):146–153. doi: 10.1128/AEM.02717-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hunt PR, Olejnik N, Sprando RL. Toxicity ranking of heavy metals with screening method using adult Caenorhabditis elegans and propidium iodide replicates toxicity ranking in rat. Food Chem Toxicol. 2012;50(9):3280–3290. doi: 10.1016/j.fct.2012.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smith CR, Lipsky JJ, Laskin OL, Hellmann DB, Mellits ED, Longstreth J, Lietman PS. Double-blind comparison of the nephrotoxicity and auditory toxicity of gentamicin and tobramycin. N Engl J Med. 1980;302(20):1106–1109. doi: 10.1056/NEJM198005153022002. [DOI] [PubMed] [Google Scholar]
- 32.Mojardin L, Botet J, Quintales L, Moreno S, Salas M. New insights into the RNA-based mechanism of action of the anticancer drug 5′-fluorouracil in eukaryotic cells. PLoS One. 2013;8(11):e78172. doi: 10.1371/journal.pone.0078172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lunde CS, Hartouni SR, Janc JW, Mammen M, Humphrey PP, Benton BM. Telavancin disrupts the functional integrity of the bacterial membrane through targeted interaction with the cell wall precursor lipid II. Antimicrob Agents Chemother. 2009;53(8):3375–3383. doi: 10.1128/AAC.01710-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Helander IM, Mattila-Sandholm T. Fluorometric assessment of gram-negative bacterial permeabilization. J Appl Microbiol. 2000;88(2):213–219. doi: 10.1046/j.1365-2672.2000.00971.x. [DOI] [PubMed] [Google Scholar]
- 35.Hancock RE, Farmer SW, Li ZS, Poole K. Interaction of aminoglycosides with the outer membranes and purified lipopolysac-charide and OmpF porin of Escherichia coli. Antimicrob Agents Chemother. 1991;35(7):1309–1314. doi: 10.1128/aac.35.7.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Peterson AA, Hancock RE, McGroarty EJ. Binding of polycationic antibiotics and polyamines to lipopolysaccharides of Pseudomonas aeruginosa. J Bacteriol. 1985;164(3):1256–1261. doi: 10.1128/jb.164.3.1256-1261.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Martin NL, Beveridge TJ. Gentamicin interaction with Pseudomonas aeruginosa cell envelope. Antimicrob Agents Chemother. 1986;29(6):1079–1087. doi: 10.1128/aac.29.6.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eliopoulos GM, Eliopoulos CT. Antibiotic combinations: should they be tested? Clin Microbiol Rev. 1988;1(2):139–156. doi: 10.1128/cmr.1.2.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nowotarski SL, Woster PM, Casero RA., Jr Polyamines and cancer: implications for chemotherapy and chemoprevention. Expert Rev Mol Med. 2013;15:e3. doi: 10.1017/erm.2013.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Verlinden BK, de Beer M, Pachaiyappan B, Besaans E, Andayi WA, Reader J, Niemand J, van Biljon R, Guy K, Egan T, Woster PM, Birkholtz LM. Interrogating alkyl and arylalkylpolya-mino (bis)urea and (bis)thiourea isosteres as potent antimalarial chemotypes against multiple lifecycle forms of Plasmodium falciparum parasites. Bioorg Med Chem. 2015;23(16):5131–5143. doi: 10.1016/j.bmc.2015.01.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verlinden BK, Niemand J, Snyman J, Sharma SK, Beattie RJ, Woster PM, Birkholtz LM. Discovery of novel alkylated (bis)urea and (bis)thiourea polyamine analogues with potent antimalarial activities. J Med Chem. 2011;54(19):6624–6633. doi: 10.1021/jm200463z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hurdle JG, O’Neill AJ, Chopra I, Lee RE. Targeting bacterial membrane function: an underexploited mechanism for treating persistent infections. Nat Rev Microbiol. 2011;9(1):62–75. doi: 10.1038/nrmicro2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Matsuo M, Oogai Y, Kato F, Sugai M, Komatsuzawa H. Growth-phase dependence of susceptibility to antimicrobial peptides in. Staphylococcus aureus Microbiology. 2011;157(Pt 6):1786–1797. doi: 10.1099/mic.0.044727-0. [DOI] [PubMed] [Google Scholar]
- 44.Tashiro Y, Ichikawa S, Shimizu M, Toyofuku M, Takaya N, Nakajima-Kambe T, Uchiyama H, Nomura N. Variation of physiochemical properties and cell association activity of membrane vesicles with growth phase in Pseudomonas aeruginosa. Appl Environ Micro. 2010;76(11):3732–3739. doi: 10.1128/AEM.02794-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eboigbodin KE, Newton JR, Routh AF, Biggs CA. Bacterial quorum sensing and cell surface electrokinetic properties. Appl Microbiol Biotechnol. 2006;73(3):669–675. doi: 10.1007/s00253-006-0505-4. [DOI] [PubMed] [Google Scholar]
- 46.Wolska KI, Grzes K, Kurek A. Synergy between novel antimicrobials and conventional antibiotics or bacteriocins. Polish J Microbiol. 2012;61(2):95–104. [PubMed] [Google Scholar]
- 47.Jackowski S, Zhang YM, Price AC, White SW, Rock CO. A missense mutation in the fabB (beta-ketoacyl-acyl carrier protein synthase I) gene confers tiolactomycin resistance to Escherichia coli. Antimicrob Agents Chemother. 2002;46(5):1246–1252. doi: 10.1128/AAC.46.5.1246-1252.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salgado CD, Sepkowitz KA, John JF, Cantey JR, Attaway HH, Freeman KD, Sharpe PA, Michels HT, Schmidt MG. Copper surfaces reduce the rate of healthcare-acquired infections in the intensive care unit. Infect Control Hosp Epidemiol. 2013;34(5):479–486. doi: 10.1086/670207. [DOI] [PubMed] [Google Scholar]
- 49.Schmidt MG, Attaway HH, Sharpe PA, John J, Jr, Sepkowitz KA, Morgan A, Fairey SE, Singh S, Steed LL, Cantey JR, Freeman KD, Michels HT, Salgado CD. Sustained reduction of microbial burden on common hospital surfaces through introduction of copper. J Clin Microbiol. 2012;50(7):2217–2223. doi: 10.1128/JCM.01032-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shopsin B, Gomez M, Montgomery SO, Smith DH, Waddington M, Dodge DE, Bost DA, Riehman M, Naidich S, Kreiswirth BN. Evaluation of protein A gene polymorphic region DNA sequencing for typing of Staphylococcus aureus strains. J Clin Microbiol. 1999;37(11):3556–3563. doi: 10.1128/jcm.37.11.3556-3563.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Junker LM, Clardy J. High-throughput screens for small-molecule inhibitors of Pseudomonas aeruginosa biofilm development. Antimicrob Agents Chemother. 2007;51(10):3582–3590. doi: 10.1128/AAC.00506-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu M, Maier E, Benz R, Hancock RE. Mechanism of interaction of different classes of cationic antimicrobial peptides with planar bilayers and with the cytoplasmic membrane of Escherichia coli. Biochemistry. 1999;38(22):7235–7242. doi: 10.1021/bi9826299. [DOI] [PubMed] [Google Scholar]
- 53.Shchepina LA, Pletjushkina OY, Avetisyan AV, Bakeeva LE, Fetisova EK, Izyumov DS, Saprunova VB, Vyssokikh MY, Chernyak BV, Skulachev VP. Oligomycin, inhibitor of the F0 part of H+-ATP-synthase, suppresses the TNF-induced apoptosis. Oncogene. 2002;21(53):8149–8157. doi: 10.1038/sj.onc.1206053. [DOI] [PubMed] [Google Scholar]
- 54.Loh B, Grant C, Hancock RE. Use of the fluorescent probe 1-N-phenylnaphthylamine to study the interactions of aminoglycoside antibiotics with the outer membrane of Pseudomonas aeruginosa. Antimicrob Agents Chemother. 1984;26(4):546–551. doi: 10.1128/aac.26.4.546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ghosh C, Manjunath GB, Akkapeddi P, Yarlagadda V, Hoque J, Uppu DS, Konai MM, Haldar J. Small molecular antibacterial peptoid mimics: the simpler the better! J Med Chem. 2014;57(4):1428–1436. doi: 10.1021/jm401680a. [DOI] [PubMed] [Google Scholar]
- 56.Leon CG, Lee J, Bartlett K, Gershkovich P, Wasan EK, Zhao J, Clement JG, Wasan KM. In vitro cytotoxicity of two novel oral formulations of Amphotericin B (iCo-009 and iCo-010) against Candida albicans, human monocytic and kidney cell lines. Lipids Health Dis. 2011;10:144. doi: 10.1186/1476-511X-10-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Leung MC, Williams PL, Benedetto A, Au C, Helmcke KJ, Aschner M, Meyer JN. Caenorhabditis elegans: an emerging model in biomedical and environmental toxicology. Toxicol Sci. 2008;106(1):5–28. doi: 10.1093/toxsci/kfn121. [DOI] [PMC free article] [PubMed] [Google Scholar]