

Abstract
Malarial infection in naïve individuals induces a robust innate immune response. In the recently described model of innate immune memory, an initial stimulus primes the innate immune system to either hyperrespond (termed “training”) or hyporespond (“tolerance”) to subsequent immune challenge. Previous work in both mice and humans demonstrated that infection with malaria can both serve as a priming stimulus and promote tolerance to subsequent infection. In this study, we demonstrate that initial stimulation with Plasmodium falciparum infected red blood cells (iRBCs) or the malaria crystal hemozoin (Hz) induced human adherent PBMCs to hyperrespond to subsequent ligation of TLR2. This hyperresponsiveness correlated with increased H3K4me3 at important immunometabolic promoters, and these epigenetic modifications were also seen in Kenyan children naturally infected with malaria. However, the use of epigenetic and metabolic inhibitors indicated that the induction of trained immunity by malaria and its ligands may occur via previously unrecognized mechanism(s).
Introduction
A hallmark of malaria is robust proinflammatory cytokine production induced by widespread innate immune activation. Multiple innate immune receptors are involved in the recognition of Plasmodium-infected red blood cells (iRBCs) and other malarial ligands [reviewed in (1)]. For example, the malaria crystal hemozoin (Hz), which becomes coated with Plasmodium-derived pathogen-associated molecular patterns (PAMPs) including genomic DNA, activates TLR9 in the phagolysosome (2). Hz crystals can also induce phagolysosomal rupture resulting in the activation of the nucleotide-binding domain, leucine-rich repeat–containing, pyrin domain containing 3 (NLRP3) inflammasome as well as deliver Plasmodium DNA to the cytosol, where it is recognized by cytosolic DNA receptors including those that sense AT-rich stem-loop structures in Plasmodium genomes (3, 4). This innate immune response, although beneficial through limiting parasitemia and assisting in the activation of adaptive immunity, induces the systemic symptoms of fever, nausea, and malaise. Proinflammatory cytokinemia has been implicated in the development of cerebral malaria (5).
Multiple studies have demonstrated memory phenotypes in innate immune cells (6–8). In the prevailing model of innate immune memory, an initial stimulus primes the innate immune system, which induces epigenetic and metabolic changes that result in an increased or decreased response—termed training or tolerance, respectively—to a subsequent challenge occurring days to months later (9). Malarial infection serves as a robust priming stimulus, as whole blood samples from experimentally infected individuals and PBMCs from patients with acute febrile disease are hyperresponsive to ex vivo TLR ligand stimulation—a phenotype that can be recapitulated in vitro (10, 11).
Malaria can induce tolerance to subsequent infection or other immune challenge [reviewed in (12)]. The pyrogenic threshold, i.e., the level of parasitemia required to provoke fever, was higher for individuals after reinfection compared to initial infection (13). In an area of Mali with seasonal malaria transmission, nearly 50% of healthy individuals had detectable parasitemia at the end of the dry season in the absence of symptoms (14). Individuals infected with malaria as fever therapy for neurosyphilis and then challenged 2-3 days post final defervescence with heat-killed Salmonella exhibited depressed febrile responses (15). Tolerance and training appear to be two ends of the same spectrum, as LPS and other ligands induce tolerance at higher concentrations but produce training at much lower concentrations (16). We hypothesized that malarial stimulation would also induce trained immunity and set about to evaluate this possibility directly using human PBMCs.
Materials and Methods
Malaria cultures and iRBC/hemozoin isolation
Plasmodium falciparum clone 3D7 iRBCs were passed through a magnetic field resulting in enrichment consistently ≥90% iRBCs. Hz was isolated by passing malaria culture supernatants through a magnetic field as described previously (2).
Human subject use
Human subject use was approved by the UMMS IRB (H-10368), University Hospitals Cleveland Medical Center IRB (06-11-22), and the KEMRI Ethical Review Committee (SSC No: 2207). Samples from Kenyan children aged 1-10 years with febrile malaria were obtained from Chulaimbo Sub-County Hospital Kisumu. Venous blood was obtained at presentation and 6 weeks after curative treatment. Individuals with submicroscopic infections detected at recovery visits by PCR (17) were excluded from further analysis. PBMCs from Kenyan children and healthy adult North American controls (3 male, 3 female, aged 33-68 years) were cryopreserved (18). Monocytes were negatively selected from thawed PBMCs using a Pan Monocyte Isolation Kit (Miltenyi Biotec).
Human adherent PBMC isolation and stimulation
PBMCs from healthy donors were plated at 5×105 cells/well (in 96-well round-bottom plates) or 10×106 cells/well (in 6- or 12-well flat-bottom plates) and incubated at 37°C for ≥1 hr. Non-adherent cells were removed by washing 3× with PBS. Adherent PBMCs were then incubated in RPMI supplemented with 10% human serum (RPMI+) and stimulated for 24 hr. Stimulation with iRBCs or Hz did not decrease cell number or viability (data not shown). Cells were washed with PBS and allowed to rest in RPMI+ for 3 days. Cells were then harvested for ChIP analysis or stimulated with Pam3CSK4 (Invivogen) for 4-24 hr. Stimulated cells were harvested for mRNA analysis or supernatants were frozen at −20°C for subsequent cytokine measurement.
Cytokine measurement
TNFα and IL-6 ELISA kits were from R&D. A MTS tetrazolium dye assay [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt] (Promega) was used to determine relative cell number post stimulation. Cytokine values were normalized to cell number.
mRNA expression
Total RNA was extracted using the RNeasy Mini Kit (Qiagen). cDNA synthesis and qPCR were performed using an iScript cDNA synthesis kit and iQ SYBR Green supermix (both Bio-Rad). The following primer pairs were used: ACTB, FW 5′-TGAAGTCTGACGTGGACATC-3′, RV 5′-ACTCGTCATACTCCTGCTTG-3′; GAPDH, FW 5′-GAGTCAACGGATTTGGTCGT-3′, RV 5′-TTGATTTTGGAGGGATCTCG-3′; IL6, FW 5′-ACTCACCTCTTCAGAACGAATTG-3′, RV 5′-CCATCTTTGGAAGGTTCAGGTTG-3′; TNF, FW 5′-TGCTTGTTCCTCAGCCTCTT-3′, RV 5′- GGTTTGCTACAACATGGGCT-3′. Ct values were normalized to ACTB and GAPDH expression and analyzed using Bio-Rad software. NanoString analysis was performed using the nCounter Human Inflammation v2 Codeset, which simultaneously measures mRNA expression levels of 249 inflammation-related genes, according to the manufacturer’s instructions (NanoString Technologies).
Neutralizing antibodies and inhibitors
Mouse anti-human IFNγ IgG1 and IgG1 isotype control were from Biolegend. 5′-Deoxy-5′-(methylthio)adenosine (MTA) and rapamycin were from Sigma.
ChIP analysis
Briefly, cells were fixed in 1% formaldehyde for 10 min and quenched with glycine. Chromatin was sonicated from these cells using a Bioruptor UCD-300 (Diagenode) for four cycles of 10× (30 sec ON, 30 sec OFF) on the HIGH setting. Chromatin precipitation was performed using rabbit anti-human H3K4me3 IgG antibody (Diagenode) as described previously (7). DNA was then quantified using qPCR with the following primer pairs: GAPDH, FW 5′-ATCCAAGCGTGTAAGGGTCC-3′, RV 5′-GACTGAGATTGGCCCGATGG-3′; IL6, FW 5′-AGCTCTATCTCCCCTCCAGG-3′, RV 5′-ACACCCCTCCCTCACACAG-3′; MTOR, FW 5′-ATAAAGAGCGCTAGCCCGAA-3′, RV 5′-GACCCCTCCCGGTGTAATTC-3′; TNF, FW 5′-CAGGCAGGTTCTCTTCCTCT-3′, RV 5′-GCTTTCAGTGCTCATGGTGT-3′. For all ChIP experiments, qPCR values were normalized as percent recovery of the input DNA.
Statistical analysis
Statistical comparisons were made between the indicated condition and control-trained cells (RPMI+). Multiple analyses were carried out for experiments with multiple replicates for an individual donor performed on different days; for the experiments represented in Fig. 1B–C: a mixed model regression analysis, a Wilcoxon signed-rank test with clustering (analysis at the replicate level accounting for correlation of data within donor), a Wilcoxon signed-rank test using one averaged value per donor (mean of all replicates), and a paired t-test using one averaged value per donor. All four tests provided nearly identical hypothesis decisions with an α=0.05, so paired t-tests using one value per donor was used for all other experiments. When ratios to controls were compared, one sample t-tests (H0 log(ratio)=0) were performed on log-transformed ratio values.
Figure 1.
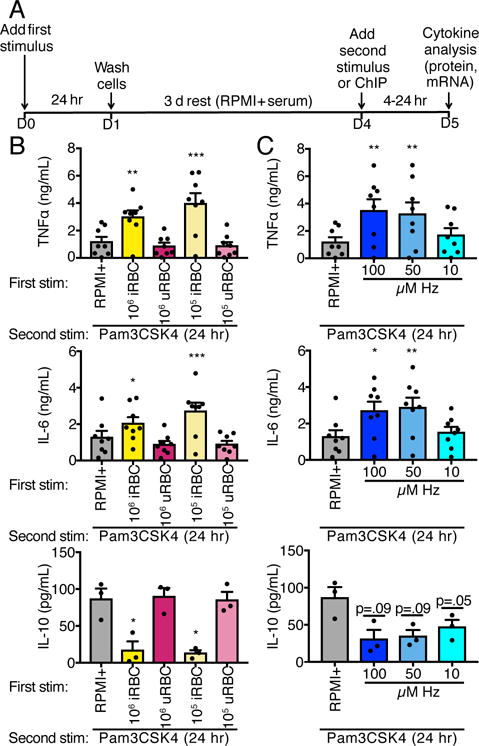
P. falciparum iRBCs and Hz induce trained immunity. (A) Schematic of trained immunity assay. Adherent PBMCs were treated with media (RPMI+), iRBCs, uRBCs, or Hz for 24 hr and then washed. After 3 d rest, cells were harvested for ChIP or challenged with Pam3CSK4 (10 μg/mL) for 4-24 hr. (B and C). TNFα, IL-6, and IL-10 ELISA measurements of supernatants from adherent PBMCs trained with iRBCs (B) or Hz (C), rested for 3 d, then challenged with Pam3CSK4 for 24 hr. Bars represent mean ± SEM for three (IL-10) or eight (TNFα, IL-6) donors (all comparisons to RPMI+ trained, *p<0.05, **p<0.01, ***p<0.001, paired t-test).
Results
Training with malaria parasites or ligands induces increased proinflammatory and decreased anti-inflammatory cytokine production in response to secondary TLR stimulation
Adherent PBMCs from healthy donors were stimulated with RPMI+ alone, P. falciparum iRBCs, uninfected red blood cells (uRBCs), or Hz for 24 hr. The cells were washed and allowed to rest in RPMI+ for three days. At this point, the cells were either harvested for ChIP analysis or stimulated with the TLR2 ligand Pam3CSK4 for 4-24 hr to measure cytokine production (Fig. 1A). Training with either 1×105 or 1×106 iRBCs—but not uRBCs—induced increased production of proinflammatory cytokines TNFα and IL-6 but decreased production of the anti-inflammatory cytokine IL-10 24 hr after second stimulation with Pam3CSK4 as compared to controls (Fig. 1B and Supplemental Fig. 1). Similarly, primary stimulation with Hz induced a similar pattern of training following Pam3CSK4 stimulation (Fig. 1C).
Malarial training has wide-ranging effects on the inflammatory transcriptome post-TLR second stimulus
The prevailing model of trained immunity is that increased responsiveness to secondary stimuli results from epigenetic remodeling that increases chromatin accessibility and that increased proinflammatory gene transcription follows secondary stimulation. In our in vitro model, stimulation of iRBC- or Hz-trained adherent PBMCs with Pam3CSK4 increased transcription at the TNF locus as compared to cells trained with media alone (Fig. 2A). Surprisingly, malarial training did not lead to increased IL6 transcription after 4 hr Pam3CSK4 second stimulation, although there was a trend towards increased IL6 transcription in iRBC- and Hz-trained cells after 12 hr Pam3CSK4 stimulation (Fig. 2B). To determine if malarial training has a more global effect on inflammatory gene expression, we performed a NanoString analysis on RNA harvested from trained cells given Pam3CSK4 as a secondary stimulus. For each gene, we normalized the iRBC-, uRBC-, or Hz-trained transcript count to the control-trained transcript count at each secondary stimulus time point. These normalized ratios are displayed as a heatmap (Fig. 2C), demonstrating that while uRBC training had little effect on the inflammatory transcriptome, both iRBC and Hz training had wide-ranging and similar effects. The majority of transcripts could be classified into the following cohorts: globally upregulated (C1), upregulated after 4 hr Pam3CSK4 stimulation (C2), upregulated after 12 hr Pam3CSK4 stimulation (C3), downregulated after 12 hr Pam3CSK4 stimulation (C4), downregulated after 4 hr Pam3CSK4 stimulation (C5), or globally downregulated (C6). A subset of genes previously implicated in malarial pathogenesis and/or trained immunity are listed for each cohort (19–23), and a complete list of transcripts in each cohort can be found in Supplementary Table I.
Figure 2.
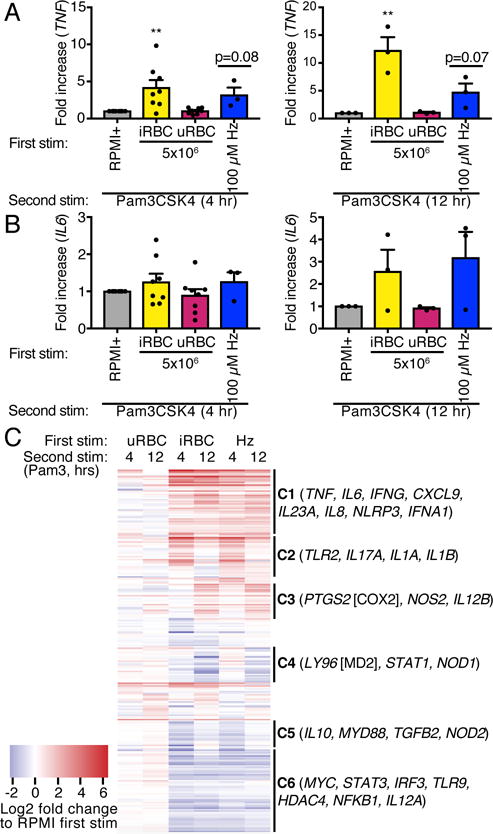
Malaria-induced training has wide-range effects on the transcriptional response to subsequent challenge. (A and B) mRNA was harvested from adherent PBMCs trained with iRBCs or Hz, rested for 3 d, and challenged with Pam3CSK4 for 4 or 12 hr. Levels of TNF (A) and IL6 (B) relative to RPMI+ trained cells are shown. Bars represent mean ± SEM for eight (4 hr Pam3CSK4) or three (12 hr Pam3CSK4) donors (**p<0.01, paired t-test). (C) Gene expression from cells treated as in (A) and (B) was quantified using NanoString technology. Expression counts are represented as log2 fold change compared to control-trained cells using hierarchical clustering. Each row represents one gene. A subset of the total genes measured is listed after each cohort. Values shown are the means of two independent experiments from one of the donors in (A) and (B).
Malarial training does not depend on IFNγ signaling
IFNγ was one of the cytokines highly upregulated by malarial training. NK cells produce IFNγ starting ~6 hr post-iRBC stimulation (24, 25). In a murine malaria model, malarial priming was IFNγ-dependent (11), and IFNγ was required for C. albicans-induced trained immunity (26). We used a neutralizing antibody against IFNγ to test the role of this cytokine in malaria-induced training. Treatment of adherent PBMCs with anti-IFNγ IgG during the 24 hr first stimulation did not inhibit iRBC- or Hz-induced training (Fig. 3). We conclude that IFNγ signaling is dispensable for malarial training.
Figure 3.
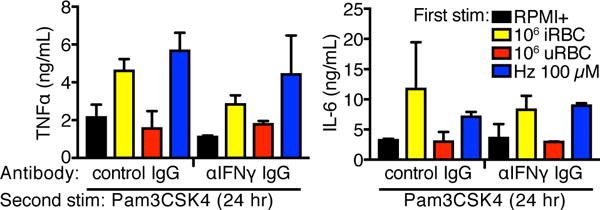
Malaria-induced training is not inhibited by co-treatment with anti-IFNγ neutralizing mAb. TNFα and IL-6 ELISA measurements of supernatants from adherent PBMCs trained with iRBCs or Hz for 24 hr, rested for 3 d, and challenged with Pam3CSK4 for 24 hr. Cells were treated with control IgG or neutralizing antibody during the 24 hr training. Bars represent mean ± SEM for two donors (all comparisons to control IgG conditions were not significant, paired t-test).
Differential epigenetic and metabolic regulation of malaria-induced trained immunity
Trained immunity is believed to be the result of intracellular metabolic shifts leading to changes in epigenetic regulation of immune-related genetic loci (9). To determine the role of naturally-acquired malaria on epigenetic remodeling, we performed H3K4me3 ChIP on monocytes isolated from Kenyan children with acute febrile malaria, the same children 6 weeks after curative antimalarial treatment, and malaria naïve adult North American controls (Fig. 4A). The Pan Monocyte Isolation Kit (Miltenyi) utilized for this monocyte isolation enriches for both CD14++CD16− and CD14+CD16++ monocytes. Increased H3K4me3 was seen at the TNF, IL6, MTOR, and GAPDH promoters during acute malaria compared to controls. Increased H3K4me3 levels were unchanged 6 weeks after antimalarial treatment. We performed a similar experiment using our in vitro model on adherent PBMCs given a 24 hr first stimulation and then rested for three days. Increased H3K4me3 was seen at the TNF, PTGS2, and IL6 promoters after iRBC (but not Hz) training compared to RPMI+ training (Fig. 4B).
Figure 4.
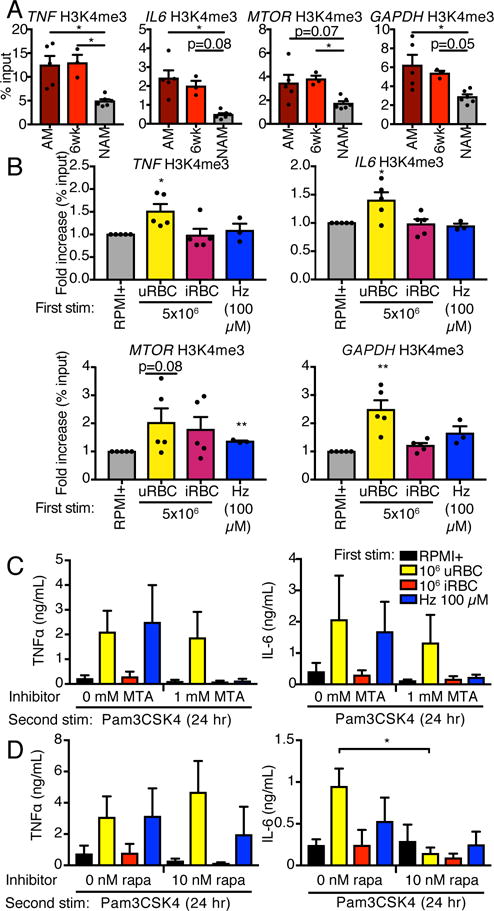
Epigenetic and metabolic regulation of malaria-induced training. (A) H3K4me3 ChIP was performed on monocytes from Kenyan children with acute malaria (AM), the same children 6 weeks after therapy (6wk), or adult North American controls (NAM). Bars represent mean ± SEM for five (AM), three (6wk), or six (NAM) donors (*p<0.05, Kruskal-Wallis test). (B) H3K4me3 ChIP was performed on adherent PBMCs trained for 24 hr and rested for 3 d. Bars represent mean ± SEM for five (RPMI+, iRBC, uRBC) or three (Hz) donors (all comparisons to RPMI+ trained, *p<0.05, **p<0.01, paired t-test). (C and D) Adherent PBMCs were co-treated with MTA (C), rapamycin (D) or relevant vehicle controls during the 24 hr training stimulation. Cells were rested for 3 d and challenged with Pam3CSK4 for 24 hr. Cytokine concentrations in supernatants were assessed by ELISA. Bars represent mean ± SEM for three or four donors (all comparisons to no inhibitor conditions and were not significant unless indicated, paired t-test).
We utilized the methyltransferase inhibitor MTA as well as the mechanistic target of rapamycin (mTOR) inhibitor rapamycin, which have been demonstrated to inhibit BCG-induced training (7) and C. albicans β-glucan-induced training (27), respectively, to investigate the importance of these mechanisms in malaria-induced training. Treatment with 1 mM MTA for the 24 hr first stimulation had no effect on iRBC-induced training (Fig. 4C). MTA treatment appeared to inhibit Hz-induced training, although this did not achieve statistical significance due to the relatively small sample size. Treatment with 10 nM rapamycin during the 24 hr first stimulation blocked iRBC-induced training as measured by IL-6 protein production but had no effect on malarial training as measured by TNFα protein levels (Fig. 4D).
Discussion
Malaria is an extraordinarily complicated disease. Individuals from endemic regions of the world have a clinical syndrome that is influenced by age and previous exposure. Children with little or no immunity to malaria are at highest risk for acute febrile disease that may be complicated by severe sequelae such as cerebral disease (28). This corresponds with our findings that Kenyan children have increased H3K4me3 at promoters relevant to inflammation and immunometabolism both during acute malaria and after curative therapy as compared to healthy North American adult controls. This is especially striking as baseline monocyte H3K4me3 levels are higher in otherwise healthy adults than neonates at both proinflammatory and metabolic promoters (29). Monocytes were chosen for ChIP analysis because enriching for the population of cells that produce the cytokines we measured in our in vitro studies seemed logical. It is worth noting that the t1/2 of monocytes is 1-3 days, suggesting that training almost certainly occurred at the level of myeloid progenitor cells (9). We recognize that this early data using patient specimens was necessary to show, but not conclusive. Long term approaches to definitively identifying training would require following individuals over the course of years (as has been done for BCG vaccine). These data support our hypothesis that malarial infection serves as a robust priming and training stimulus. Indeed, we predict that malaria-infected children are at risk to hyperrespond to superinfection in ways that might be predicted based on the in vitro responses we observed.
Older children and adults from endemic areas are likely to have experienced multiple episodes of malaria. These individuals will often have detectable parasitemia without symptoms (14). While the level of parasitemia in these asymptomatic malaria cases are often quite low, some individuals have levels of parasites in their blood that would be incapacitating in the immunologically naïve. It is our hypothesis that the differences in outcome in disease between young individuals or even immunologically mature adults who become infected for the first time later in life (e.g., travelers) compared to multiply infected asymptomatic individuals, are due to epigenetic changes at proinflammatory and immunometabolic promoters.
In this work, we report that the malaria parasite and its crystal Hz can induce trained immunity as measured by inflammatory gene expression after a secondary TLR ligand challenge. Although these two stimuli have similar effects on the inflammatory transcriptome post-Pam3CSK4 challenge, the differential regulation of iRBC- and Hz-induced training by known trained immunity inhibitors and differing levels of H3K4me3 at immunometabolic promoters suggest divergent training mechanisms for the two stimuli. One potential explanation for this divergence is that the parasite contains multiple additional ligands with innate immune stimulatory capacity. Although these TLRs utilize the same signaling cascades and transcription factors as the receptors stimulated by Hz, the balance in signaling between the cascades may be sufficiently different to explain these differences.
Rapamycin treatment only inhibited training as measured by IL-6 and not TNFα, which differs from previous studies on other training stimuli (27, 30). This discrepancy in rapamycin inhibition is not completely surprising given that the TNF and IL6 loci have different initiation kinetics and epigenetic requirements for transcription (31). However, along with the apparent dispensability of IFNγ signaling for malaria training, this provides further evidence that both malaria parasites and Hz induce innate immune training by mechanisms that differ from other infectious stimuli. Any hope for immunomodulation of malaria, either for the purposes of improving outcome in the critically ill or enhancing acquired immunity, requires a more thorough understanding of the basic pathogenesis of the disease. The methods for acquiring this understanding are at hand.
Supplementary Material
Acknowledgments
Footnotes
This work was funded by NIH grants R01AI079293 (DTG, KAF, RTG, GWR), R01NS098747 (RTG), U19AI0089681 (DTG, RTG) T32AI095213 (JES), R01AI095192 (JWK, AED, KRD) and R01AI130131 (JWK). MGN was supported by an ERC Consolidator Grant (#310372) and a Spinoza Grant of the Netherlands Organization for Scientific Research.
Abbreviations in this article
- iRBC
Plasmodium falciparum-infected red blood cell
- uRBC
uninfected red blood cell
- Hz
hemozoin
- PAMP
pathogen-associated molecular pattern
- NLRP3
nucleotide-binding domain, leucine-rich repeat–containing, pyrin domain containing 3
- AIM2
absent in melanoma 2
- mTOR
mechanistic target of rapamycin
- BCG
bacille Calmette-Guérin
References
- 1.Gazzinelli RT, Kalantari P, Fitzgerald KA, Golenbock DT. Innate sensing of malaria parasites. Nat Rev Immunol. 2014;14:744–757. doi: 10.1038/nri3742. [DOI] [PubMed] [Google Scholar]
- 2.Parroche P, Lauw FN, Goutagny N, Latz E, Monks BG, Visintin A, Halmen KA, Lamphier M, Olivier M, Bartholomeu DC, Gazzinelli RT, Golenbock DT. Malaria hemozoin is immunologically inert but radically enhances innate responses by presenting malaria DNA to Toll-like receptor 9. Proc Natl Acad Sci USA. 2007;104:1919–1924. doi: 10.1073/pnas.0608745104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kalantari P, DeOliveira RB, Chan J, Corbett Y, Rathinam V, Stutz A, Latz E, Gazzinelli RT, Golenbock DT, Fitzgerald KA. Dual engagement of the NLRP3 and AIM2 inflammasomes by plasmodium-derived hemozoin and DNA during malaria. CellReports. 2014;6:196–210. doi: 10.1016/j.celrep.2013.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sharma S, DeOliveira RB, Kalantari P, Parroche P, Goutagny N, Jiang Z, Chan J, Bartholomeu DC, Lauw F, Hall JP, Barber GN, Gazzinelli RT, Fitzgerald KA, Golenbock DT. Innate immune recognition of an AT-rich stem-loop DNA motif in the Plasmodium falciparum genome. Immunity. 2011;35:194–207. doi: 10.1016/j.immuni.2011.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hunt NH, Grau GE. Cytokines: accelerators and brakes in the pathogenesis of cerebral malaria. Trends in Immunology. 2003;24:491–499. doi: 10.1016/s1471-4906(03)00229-1. [DOI] [PubMed] [Google Scholar]
- 6.Foster SL, Hargreaves DC, Medzhitov R. Gene-specific control of inflammation by TLR-induced chromatin modifications. Nature. 2007;447:972–978. doi: 10.1038/nature05836. [DOI] [PubMed] [Google Scholar]
- 7.Kleinnijenhuis J, Quintin J, Preijers F, Joosten LAB, Ifrim DC, Saeed S, Jacobs C, van Loenhout J, de Jong D, Stunnenberg HG, Xavier RJ, van der Meer JWM, van Crevel R, Netea MG. Bacille Calmette-Guerin induces NOD2-dependent nonspecific protection from reinfection via epigenetic reprogramming of monocytes. Proc Natl Acad Sci USA. 2012;109:17537–17542. doi: 10.1073/pnas.1202870109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quintin J, Saeed S, Martens JHA, Giamarellos-Bourboulis EJ, Ifrim DC, Logie C, Jacobs L, Jansen T, Kullberg BJ, Wijmenga C, Joosten LAB, Xavier RJ, van der Meer JWM, Stunnenberg HG, Netea MG. Candida albicans infection affords protection against reinfection via functional reprogramming of monocytes. Cell Host and Microbe. 2012;12:223–232. doi: 10.1016/j.chom.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Netea MG, Joosten LAB, Latz E, Mills KHG, Natoli G, Stunnenberg HG, O’Neill LAJ, Xavier RJ. Trained immunity: A program of innate immune memory in health and disease. Science. 2016;352:aaf1098–aaf1098. doi: 10.1126/science.aaf1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCall MBB, Netea MG, Hermsen CC, Jansen T, Jacobs L, Golenbock D, van der Ven AJAM, Sauerwein RW. Plasmodium falciparum Infection Causes Proinflammatory Priming of Human TLR Responses. The Journal of Immunology. 2007;179:162–171. doi: 10.4049/jimmunol.179.1.162. [DOI] [PubMed] [Google Scholar]
- 11.Franklin BS, Parroche P, Ataíde MA, Lauw F, Ropert C, de Oliveira RB, Pereira D, Tada MS, Nogueira P, da Silva LHP, Bjorkbacka H, Golenbock DT, Gazzinelli RT. Malaria primes the innate immune response due to interferon-gamma induced enhancement of toll-like receptor expression and function. Proc Natl Acad Sci USA. 2009;106:5789–5794. doi: 10.1073/pnas.0809742106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boutlis CS, Yeo TW, Anstey NM. Malaria tolerance – for whom the cell tolls? Trends in Parasitology. 2006;22:371–377. doi: 10.1016/j.pt.2006.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gatton ML, Cheng Q. Evaluation of the pyrogenic threshold for Plasmodium falciparum malaria in naive individuals. Am J Trop Med Hyg. 2002;66:467–473. doi: 10.4269/ajtmh.2002.66.467. [DOI] [PubMed] [Google Scholar]
- 14.Portugal S, Tran TM, Ongoiba A, Bathily A, Li S, Doumbo S, Skinner J, Doumtabe D, Kone Y, Sangala J, Jain A, Davies DH, Hung C, Liang L, Ricklefs S, Homann MV, Felgner PL, Porcella SF, Färnert A, Doumbo OK, Kayentao K, Greenwood BM, Traore B, Crompton PD. Treatment of Chronic Asymptomatic Plasmodium falciparum Infection Does Not Increase the Risk of Clinical Malaria Upon Reinfection. Clin Infect Dis. 2017;64:645–653. doi: 10.1093/cid/ciw849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heyman A, Beeson PB. Influence of various disease states upon the febrile response to intravenous injection of typhoid bacterial pyrogen; with particular reference to malaria and cirrhosis of the liver. J Lab Clin Med. 1949;34:1400–1403. [PubMed] [Google Scholar]
- 16.Ifrim DC, Quintin J, Joosten LAB, Jacobs C, Jansen T, Jacobs L, Gow NAR, Williams DL, van der Meer JWM, Netea MG. Trained immunity or tolerance: opposing functional programs induced in human monocytes after engagement of various pattern recognition receptors. Clin Vaccine Immunol. 2014;21:534–545. doi: 10.1128/CVI.00688-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Singh B, Bobogare A, Cox-Singh J, Snounou G, Abdullah MS, Rahman HA. A genus- and species-specific nested polymerase chain reaction malaria detection assay for epidemiologic studies. Am J Trop Med Hyg. 1999;60:687–692. doi: 10.4269/ajtmh.1999.60.687. [DOI] [PubMed] [Google Scholar]
- 18.Fuss IJ, Kanof ME, Smith PD, Zola H. Isolation of whole mononuclear cells from peripheral blood and cord blood. Curr Protoc Immunol Chapter. 2009;7 doi: 10.1002/0471142735.im0701s85. Unit7.1. [DOI] [PubMed] [Google Scholar]
- 19.Ayimba E, Hegewald J, A. Y. S. gb na. Gantin RG, Lechner CJ, Agosssou A, Banla M, Soboslay PT. Proinflammatory and regulatory cytokines and chemokines in infants with uncomplicated and severe Plasmodium falciparum malaria. Clin Exp Immunol. 2011;166:218–226. doi: 10.1111/j.1365-2249.2011.04474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Prakash D, Fesel C, Jain R, Cazenave PA, Mishra GC, Pied S. Clusters of cytokines determine malaria severity in Plasmodium falciparum-infected patients from endemic areas of Central India. J Infect Dis. 2006;194:198–207. doi: 10.1086/504720. [DOI] [PubMed] [Google Scholar]
- 21.Portugal S, Moebius J, Skinner J, Doumbo S, Doumtabe D, Kone Y, Dia S, Kanakabandi K, Sturdevant DE, Virtaneva K, Porcella SF, Li S, Doumbo OK, Kayentao K, Ongoiba A, Traore B, Crompton PD. Exposure-Dependent Control of Malaria-Induced Inflammation in Children. PLoS Pathog. 2014;10:e1004079–16. doi: 10.1371/journal.ppat.1004079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ishida H, Imai T, Suzue K, Hirai M, Taniguchi T, Yoshimura A, Iwakura Y, Okada H, Suzuki T, Shimokawa C, Hisaeda H. IL-23 protection against Plasmodium bergheiinfection in mice is partially dependent on IL-17 from macrophages. European Journal of Immunology. 2013;43:2696–2706. doi: 10.1002/eji.201343493. [DOI] [PubMed] [Google Scholar]
- 23.Sellau J, Alvarado CF, Hoenow S, Mackroth MS, Kleinschmidt D, Huber S, Jacobs T. IL-22 dampens the T cell response in experimental malaria. Nature Publishing Group. 2016:1–11. doi: 10.1038/srep28058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Artavanis-Tsakonas K, Riley EM. Innate immune response to malaria rapid induction of IFN-gamma from human NK cells by live Plasmodium falciparum-infected erythrocytes. The Journal of Immunology. 2002;169:2956–2963. doi: 10.4049/jimmunol.169.6.2956. [DOI] [PubMed] [Google Scholar]
- 25.Artavanis-Tsakonas K, Eleme K, McQueen KL, Cheng NW, Parham P, Davis DM, Riley EM. Activation of a Subset of Human NK Cells upon Contact with Plasmodium falciparum-Infected Erythrocytes. The Journal of Immunology. 2003;171:5396–5405. doi: 10.4049/jimmunol.171.10.5396. [DOI] [PubMed] [Google Scholar]
- 26.Ifrim DC, Quintin J, Meerstein-Kessel L, Plantinga TS, Joosten LAB, van der Meer JWM, van de Veerdonk FL, Netea MG. Defective trained immunity in patients with STAT-1-dependent chronic mucocutaneaous candidiasis. Clin Exp Immunol. 2015;181:434–440. doi: 10.1111/cei.12642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheng S-C, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, Giamarellos-Bourboulis EJ, Martens JHA, Rao NA, Aghajanirefah A, Manjeri GR, Li Y, Ifrim DC, Arts RJW, van der Veer BMJW, van der Meer BMJW, Deen PMT, Logie C, O’Neill LA, Willems P, van de Veerdonk FL, van der Meer JWM, Ng A, Joosten LAB, Wijmenga C, Stunnenberg HG, Xavier RJ, Netea MG. mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science. 2014;345:1250684–1250684. doi: 10.1126/science.1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marsh K, Kinyanjui S. Immune effector mechanisms in malaria. Parasite Immunol. 2006;28:51–60. doi: 10.1111/j.1365-3024.2006.00808.x. [DOI] [PubMed] [Google Scholar]
- 29.Bermick JR, Lambrecht NJ, denDekker AD, Kunkel SL, Lukacs NW, Hogaboam CM, Schaller MA. Neonatal monocytes exhibit a unique histone modification landscape. Clinical Epigenetics. 2016:1–15. doi: 10.1186/s13148-016-0265-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arts RJW, Carvalho A, La Rocca C, Palma C, Rodrigues F, Silvestre R, Kleinnijenhuis J, Lachmandas E, Gonçalves LG, Belinha A, Cunha C, Oosting M, Joosten LAB, Matarese G, van Crevel R, Netea MG. Immunometabolic Pathways in BCG-Induced Trained Immunity. CellReports. 2016;17:2562–2571. doi: 10.1016/j.celrep.2016.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramirez-Carrozzi VR, Braas D, Bhatt DM, Cheng CS, Hong C, Doty KR, Black JC, Hoffmann A, Carey M, Smale ST. A Unifying Model for the Selective Regulation of Inducible Transcription by CpG Islands and Nucleosome Remodeling. Cell. 2009;138:114–128. doi: 10.1016/j.cell.2009.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.