

Abstract
Gliomas are the most common primary brain tumor and high-grade gliomas the leading cause of brain tumor-related death in both children and adults. An appreciation for the crucial role of the nervous system in the tumor microenvironment is emerging for cancers in general, and the neural regulation of glioma progression has come into sharp focus. Here, we review what is known about the influence of active neurons on glioma pathobiology.
Gliomas, central nervous system cancers that resemble glial cells molecularly and morphologically, are among the most common primary brain tumors in adults and children. High-grade gliomas, a histopathologic class that encompasses devastating tumors such as glioblastoma, anaplastic astrocytoma, anaplastic oligodendroglioma and midline H3K27M mutant gliomas of childhood (Louis et al., 2016) such as diffuse intrinsic pontine glioma (DIPG), have a particularly poor prognosis and remain the primary cause of mortality from brain tumors in patients of all ages. Distinctive properties of the central nervous system during the periods of both postnatal neurodevelopment and adult neural plasticity establish a unique tumor microenvironment for these cancers. Understanding microenvironmental determinants of glioma growth and progression is thus a focus of current lines of research that aim to uncover new targets and strategies for treating the disease and addressing its high burden of morbidity and mortality. Prior work has endeavored to describe interactions of glioma cells with astrocytes, immune cells, and cells of the vascular system (Charles et al., 2011; Pyonteck et al., 2013; Quail and Joyce, 2017; Silver et al., 2013). Emerging research now suggests that interactions with neurons, and the direct and indirect consequences of neuronal activity, represent critically important determinants of glioma cell behavior, as well.
The concept of active neurons as important components of the tumor microenvironment recalls our understanding of neuronal activity as a key regulator of central nervous system development and plasticity. While the glioma cell of origin remains unconfirmed and openly debated, accumulating research suggests that glioma may arise from neural stem or precursor cells of the oligodendroglial lineage, specifically oligodendrocyte precursor cells (OPCs), pre-OPCs or earlier neural precursor cells (NPCs) (Galvao et al., 2014; Liu et al., 2011; Monje et al., 2011; Wang et al., 2009; Nagaraja et al., 2017). The known influence of active neurons on the proliferation, differentiation, and/or function of the cells from which glioma is thought to arise suggest that parallel mechanisms could play a role in glial cancers if co-opted for the promotion of tumor growth and progression.
Activity-Dependent Glioma Growth
Electrical activity of neurons is known to locally and specifically influence the proliferation of myelinating cell precursors, as well as the promotion of circuit myelination by functionally mature oligodendrocytes generated downstream. This was first suggested by early studies demonstrating that OPC proliferation could be suppressed by silencing neuronal activity in the rat optic nerve, either surgically via nerve transection or chemically via exposure to tetrodotoxin (Barres and Raff, 1993). Using optogenetic control of premotor cortical neural activity in awake, behaving mammalian models, NPCs, pre-OPCs and OPCs were found to exhibit a brisk mitogenic response to optogenetically increased cortical activity, leading to downstream differentiation to functionally mature oligodendrocytes and myelination of the active circuit in an adaptive manner (Gibson et al., 2014). Similarly, optogenetic manipulation of cortical neuronal activity also leads to an increased rate of proliferation of primary patient-derived pediatric cortical glioblastoma cells xenografted into the cortex of a mammalian model (Venkatesh et al., 2015). This occurs in a specific manner proximal to the stimulated circuit, and leads to increased tumor burden when optogenetic stimulation is performed repeatedly over time.
Activity-Regulated Secretion of Neuroligin-3
While the mechanism by which neuronal activity leads to increased proliferation of OPCs remains to be determined, an unexpected mechanism was implicated in the observed mitogenic effect of neuronal activity on glioma in vivo. Optogenetic stimulation of cortical slices resulted in the activity-dependent secretion of factors into conditioned medium that, when exposed to glioma cells, demonstrated a broad mitogenic effect on a nine out of ten patient-derived high-grade glioma cultures tested, including diffuse intrinsic pontine glioma, adult and pediatric glioblastoma, and anaplastic oligodendroglioma (Venkatesh et al., 2015). Biochemical and proteomic analyses revealed that the synaptic protein neuroligin-3 (NLGN3), secreted in an activity-regulated manner, was the primary factor responsible for the observed mitogenic effect, along with lesser contributions from known glioma mitogens, the neurotrophin BDNF (brain-derived neurotrophic factor) and GRP78 (78-kDa glucose-regulated protein) (Venkatesh et al., 2015). Secreted NLGN3 activates the PI3K-mTOR signaling pathway to increase glioma cell proliferation, and also leads to the expression of NLGN3 by glioma cells in a feed-forward, potentially autocrine/paracrine loop. Furthermore, NLGN3 expression was found to correlate inversely with overall survival in adult patients with glioblastoma, emphasizing the clinical significance of this mechanism in the human disease. This study provided compelling evidence that active neurons are an important component of the glioma microenvironment, introducing a new potential approach to glioma therapeutics.
Given the diversity of microenvironmental and cell-intrinsic mechanisms that may promote glioma growth, what is the relative contribution of activity-regulated NLGN3? To answer this question, patient-derived high-grade glioma cells were orthotopically xenografted to neuroligin-3 knock out mice or littermate neuroligin-3 WT controls. Strikingly, glioma xenografts fail to grow in the neuroligin-3 deficient brain, indicating an unexpected dependency on this molecule. Neuroligin-3 dependency is conserved across molecularly and clinically distinct glioma types including adult glioblastoma, pediatric glioblastoma and DIPG (Venkatesh et al, 2017). Detailed phosphoproteomic studies reveal that neuroligin-3 stimulates numerous oncogenic signaling cascades in the glioma cell, with early activation of focal adhesion kinase and downstream activation not only of PI3K-mTOR but also SRC and RAS pathways (Venkatesh et al, 2017). In addition to these signaling consequences, neuroligin-3 also induces numerous gene expression changes in the glioma cell. The most intriguing changes include up-regulated expression of numerous synapse-associated genes. In addition to the previously described feed-forward expression of NLGN3, several glutamate receptor subunit genes and the BDNF receptor gene NTRK2 increase expression following NLGN3 exposure in glioma (Venkatesh et al, 2017). As well, NLGN3 induces tweety homologue-1 (TTHY1) expression, a protein that regulates tumor microtube network formation in glioma (Osswald et al., 2015; Jung et al, 2017). While the functional significance of these gene expression changes remain to be clarified, the complex downstream consequences of activity-regulated neuroligin-3 release in the tumor microenvironment indicate that our understanding is still nascent regarding this crucial molecule and its pathological roles in glioma.
Although there is much to learn about the mechanisms that account for the observed neuroligin-3 dependency in glioma, this activity-regulated molecule represents an important therapeutic target. Neuroligin-3 is cleaved at the membrane by the ADAM10 protease resulting in ectodomain release into the microenvironment (Venkatesh et al., 2017). Inhibiting ADAM10 prevents neuroligin-3 release and dramatically reduces the growth of patient-derived high-grade glioma orthotopic xenografts (Venkatesh et al., 2017), suggesting a new therapeutic strategy targeting this key neuron-glioma interaction.
Neurotrophins in the Glioma Microenvironment
The identification of BDNF as a contributor to activity-dependent glioma proliferation (Venkatesh et al., 2015) is consistent with prior work suggesting a role for neurotrophins in glioma cell survival and growth. Neurotrophins are a family of growth factor molecules in the nervous system that act as major regulators of neuronal function, survival, and maturation, both in development and plasticity; they also govern the proliferation, differentiation and function of OPCs (Tsiperson et al.; VonDran et al.; Wong et al.). This role is coupled to neuronal activity, as BDNF is synthesized in an activity-dependent manner (Hong et al., 2008; Lindholm et al., 1994) and secreted in response to depolarization (Androutsellis-Theotokis et al., 1996; Goggi et al., 2003). BDNF mediates its effects by signaling via the high-affinity TrkB (NTRK2) receptor (Chao et al., 2003; Klein et al., 1991) and indeed, this pathway is also implicated in the mechanism by which BDNF promotes proliferation, survival, and migration of high-grade glioma cells in studies in vitro (Lawn et al., 2015; Xiong et al., 2013). In further support of a role of BDNF-TrkB signaling in the glioma microenvironment, many human glioma cells, particularly astrocytomas, express neurotrophins and their receptors (Assimakopoulou et al., 2007; Lawn et al., 2015; Wadhwa et al., 2003; Wang et al., 1998) and exhibit mutations in Trk genes, including frequently activating fusions of NTRK1, NTRK2, and NTRK3 in pediatric high-grade glioma (Wu et al., 2014), pilocytic astrocytoma (Jones et al., 2013) and less commonly in adult glioblastoma (Frattini et al., 2013), as well as NTRK1 and/or NTRK2 amplifications in about half of diffuse intrinsic pontine gliomas (DIPG) (Grasso et al., 2015). Whether gliomas exhibiting NTRK fusions or amplifications are differentially dependent on or responsive to activity-regulated neurotophins in the microenvironment is unknown and represents an area for further research. These early findings certainly suggest a potentially targetable role of activity-regulated neurotrophin signaling in glioma progression, although the therapeutic potential of disrupting BDNF-TrkB signaling in glioma remains to defined in the literature.
Neurotransmitters and Glioma Growth and Progression
Neuronal activity could potentially also influence glioma growth and progression via neurotransmitter release. Several studies to date have begun to investigate glioma cell response to neurotransmitters in order to identify potential new therapeutic targets. Glioma incidence is reduced in patients with history of long-term therapy with tricyclic antidepressants, which have broad neurotransmitter effects and are thought to act primarily via reuptake inhibition of serotonin and norepinephrine (Walker et al., 2011). Low-grade glioma-bearing mice treated with the tricyclic antidepressant imipramine exhibited prolonged survival, with decreased rate of tumor cell proliferation and reduced progression to high grade lesions; the mechanism of action appears to involve induction of autophagy, leading to apoptosis (Shchors et al., 2015). Glioblastoma cells express dopamine receptors (Dolma et al., 2016; Li et al., 2014), and in a proliferation screen performed on three patient-derived glioma cell cultures exposed to a panel of neurotransmitter agonists, antagonists, and reuptake inhibitors, pharmacologic blockade of dopamine receptor D4 emerged as an effective and selective inhibitor of glioma cell proliferation via disruption of autophagy and downstream induction of apoptosis (Dolma et al., 2016). Glioma cells also express functional serotonin receptors (Mahe et al., 2004), but whether serotonergic signaling influences glioma growth or progression is less clear, as increased serotonin levels as a result of selective serotonin reuptake inhibitor (SSRI) therapy have not been shown to have a survival benefit in retrospective studies of patients with glioblastoma and comorbid depression (Caudill et al., 2011). It should be noted in all glioma studies pharmacologically manipulating neurotransmitter signaling in vivo that it is difficult to de-convolute the cell autonomous effects on glioma from the non-cell autonomous effects on neuronal activity, and these dissecting the various possible effects of neurotransmitter signaling blockade in the glioma ecosystem should be an area of dedicated study.
It also remains to be seen whether GABA, the primary inhibitory neurotransmitter in the mature CNS, regulates glioma growth; low-grade astrocytomas and oligodendrogliomas cells do express functional GABAA receptors, and while the function of such signaling is largely unclear, it is notable that higher grade gliomas did not express GABA receptors, and glioma cell lines without GABA receptor expression exhibited unlimited proliferation in culture (Labrakakis et al., 1998). Indeed, GABAergic signaling to OPCs inhibits their proliferation and promotes differentiation to mature oligodendrocytes (Zonouzi et al., 2015), but it is yet unclear whether glioma cells respond to GABA in a similar manner. This raises the interesting unanswered question regarding the influence GABAergic interneurons may exert on glioma. Understanding the direct role that GABA signaling plays in glioma growth may elucidate additional therapeutic avenues to control disease progression.
Both glial progenitor cells and glioma cells express glutamate receptors (Gallo et al., 1994; Ishiuchi et al., 2002; Labrakakis et al., 1998), and furthermore, non-synaptic secretion of glutamate by glioblastoma cells has been directly observed in vitro (Ye and Sontheimer, 1999) and supported by in vivo studies describing increased extracellular glutamate levels in brain tissue proximal to glioblastomas (Behrens et al., 2000). Glutamate has been found to promote glioblastoma cell survival, growth and migration via calcium influx-mediated activation of PI3K-Akt signaling through AMPA receptors (Ishiuchi et al., 2002; Ishiuchi et al., 2007; Takano et al., 2001). Accordingly, gliomas that secrete greater levels of glutamate also exhibit increased tumor growth in mammalian models (Takano et al., 2001). Thus, nonsynaptic secretion of glutamate by glioma cells appears to have an autocrine/paracrine function in enhancing the tumor growth, survival and progression. Neuronal glutamate release could further contribute to glioma progression, although the extent to which neuronal glutamate release contributes to glioma progression – and whether neuronal glutamate release would signal in the same way as glutamate secreted form glioma cells - has not yet been explored.
Influence of Glioma on Cortical Excitability
Emerging work also suggests that the influence of active neurons on glioma cells may actually represent one facet of a bidirectional relationship (Figure 1). Of note, glutamate secretion by glioma cells contributes to hyperexcitability of neural circuits in the glioma microenvironment, including promotion of seizure activity (Buckingham et al., 2011; Campbell et al., 2012). This suggests that gliomas may actually enhance their own growth and progression not only via direct autocrine/paracrine effects of nonsynaptic secreted glutamate, but also by enhancing local neural activity, inducing an increase in subsequent release of activity-dependent mitogens by other cell types in the microenvironment. This may be a factor underlying the well-known clinical feature of seizure activity among human glioma patients, particularly in those with glioblastoma. Increased cortical excitability may also occur as gliomas progress and tumor cell behavior evolves. A recent study identified functionally diverse subgroups of astrocytes whose gene expression profiles match with those of apparently analogous populations in glioma (John Lin et al., 2017). In a transgenic mouse model of glioma, the emergence of a subpopulation of glioma cells with particularly synaptogenic properties was observed as tumors progressed, which correlated with the onset of clinical seizure activity in the tumor-bearing mice, suggesting that increased and/or altered synaptogenesis due to shifting predominance of tumor cell phenotypes may underlie the cortical hyperexcitability seen in later-stage glioma in this model (John Lin et al., 2017). Interestingly, this increased excitability also correlated with enhanced migratory behavior of the glioma cells, suggesting a possible link between synaptic neuronal activity and promotion of infiltration, though this association has yet to be directly tested. Additional work is also needed to demonstrate whether seizure activity as well as physiologic cortical activity similarly promotes glioma growth. Furthermore, as clinically apparent seizures are more common in individuals with low-grade gliomas and oligodendrogliomas than those with astrocytomas, it will be important to determine if these glioma types promote cortical hyperexcitability via similar mechanisms.
Figure 1. Bidirectional signaling between neurons and gliomas promote glioma growth.
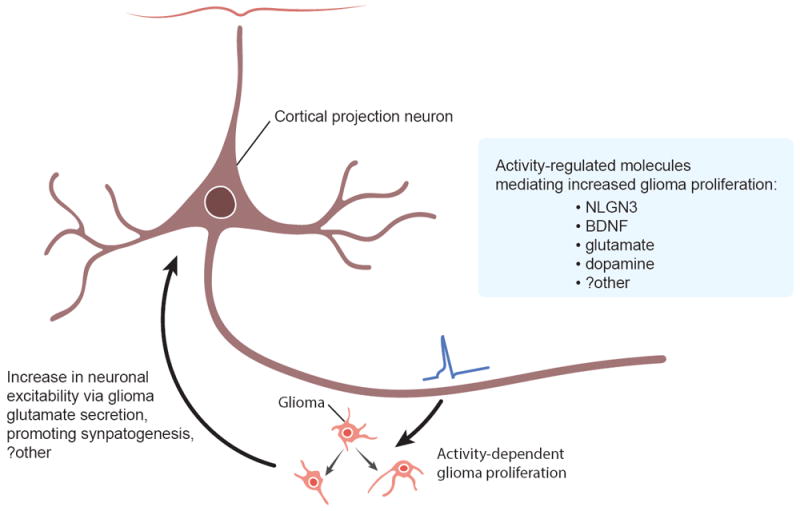
Neuronal activity promotes glioma proliferation and growth through activity-regulated secretion of brain-derived neurotrophic factor (BDNF), soluble neuroligin-3 (NLGN3), glutamate, dopamine and likely other factors. In turn, gliomas encourage neuronal activity through glutamate release, promoting synaptogenesis, and possibly additional mechanisms.
Conclusion
Gliomas thus join an ever-increasing number of cancers for which neural regulation of the tumor microenvironment plays a key role in malignancy (for review, please see (Venkatesh and Monje, 2017). A fascinating model is emerging from the literature to date, suggesting multiple mechanisms by which active neurons may promote the growth and progression of glioma, including activity-regulated neurotransmitter, growth factor, and NLGN3 release into the glioma microenvironment. Furthermore, as glioma remodels its microenvironment in order to promote hyperexcitability of local circuits, the resultant increase in neuronal activity may contribute even further to various activity-dependent mechanisms of tumor growth and progression. While additional work is needed to elucidate the nature of many of these mechanisms, the important influence of neuronal activity on glial precursor cell growth and behavior during development and plasticity may suggest parallel interactions between active neurons and glioma cells. A better understanding of this underrecognized aspect of the glioma microenvironment, and the degree to which glioma cells may co-opt or diverge from physiological activity-dependent processes, may reveal promising new approaches to developing therapeutics for the treatment of this devastating group of neural cancers.
Highlights.
-
*
Neuronal activity promotes glioma growth
-
*
Gliomas increase neuronal excitability and promote seizures
-
*
Bidirectional communication between gliomas and neurons in the tumor microenvironment drives a cycle of malignancy
Acknowledgments
The authors gratefully acknowledge support from the National Institute of Neurological Disorders and Stroke (R01NS092597 to M.M.) and California Institute for Regenerative Medicine (CIRM RN3-06510 to M.M.). Special thanks to Sarah Chen for figure illustration.
Footnotes
Conflict of Interest Statement
Stanford University has filed a patent application relevant to our work on the neuronal regulation of glioma growth. We have no other conflicts of interest to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Androutsellis-Theotokis A, McCormack WJ, Bradford HF, Stern GM, Pliego-Rivero FB. The depolarisation-induced release of [125I]BDNF from brain tissue. Brain Res. 1996;743:40–48. doi: 10.1016/s0006-8993(96)00981-x. [DOI] [PubMed] [Google Scholar]
- Assimakopoulou M, Kondyli M, Gatzounis G, Maraziotis T, Varakis J. Neurotrophin receptors expression and JNK pathway activation in human astrocytomas. BMC cancer. 2007;7:202. doi: 10.1186/1471-2407-7-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Barres BA, Raff MC. Proliferation of oligodendrocyte precursor cells depends on electrical activity in axons. Nature. 1993;361:258–260. doi: 10.1038/361258a0. This seminal paper demonstrated the first link between neuronal activity and the behavior of myelin-forming cells. [DOI] [PubMed] [Google Scholar]
- Behrens PF, Langemann H, Strohschein R, Draeger J, Hennig J. Extracellular glutamate and other metabolites in and around RG2 rat glioma: an intracerebral microdialysis study. J Neurooncol. 2000;47:11–22. doi: 10.1023/a:1006426917654. [DOI] [PubMed] [Google Scholar]
- **.Buckingham SC, Campbell SL, Haas BR, Montana V, Robel S, Ogunrinu T, Sontheimer H. Glutamate release by primary brain tumors induces epileptic activity. Nat Med. 2011;17:1269–1274. doi: 10.1038/nm.2453. This foundational paper showed that glioma cell glutamate secretion influences neuronal activity and promotes tumor-associated seizures. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **.Campbell SL, Buckingham SC, Sontheimer H. Human glioma cells induce hyperexcitability in cortical networks. Epilepsia. 2012;53:1360–1370. doi: 10.1111/j.1528-1167.2012.03557.x. Related to the Buckingham paper above, this study showed that glioma cells increase neuronal excitability and promotes neuronal firing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caudill JS, Brown PD, Cerhan JH, Rummans TA. Selective serotonin reuptake inhibitors, glioblastoma multiforme, and impact on toxicities and overall survival: the mayo clinic experience. American journal of clinical oncology. 2011;34:385–387. doi: 10.1097/COC.0b013e3181e8461a. [DOI] [PubMed] [Google Scholar]
- Chao MV. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nature reviews Neuroscience. 2003;4:299–309. doi: 10.1038/nrn1078. [DOI] [PubMed] [Google Scholar]
- Charles NA, Holland EC, Gilbertson R, Glass R, Kettenmann H. The brain tumor microenvironment. Glia. 2011;59:1169–1180. doi: 10.1002/glia.21136. [DOI] [PubMed] [Google Scholar]
- **.Dolma S, Selvadurai HJ, Lan X, Lee L, Kushida M, Voisin V, Whetstone H, So M, Aviv T, Park N, et al. Inhibition of Dopamine Receptor D4 Impedes Autophagic Flux, Proliferation, and Survival of Glioblastoma Stem Cells. Cancer Cell. 2016;29:859–873. doi: 10.1016/j.ccell.2016.05.002. This elegant study demonstrated that dopaminergic signaling in glioblastoma promotes autophagy and growth, identifying a new therapeutic target. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frattini V, Trifonov V, Chan JM, Castano A, Lia M, Abate F, Keir ST, Ji AX, Zoppoli P, Niola F, et al. The integrated landscape of driver genomic alterations in glioblastoma. Nat Genet. 2013;45:1141–1149. doi: 10.1038/ng.2734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo V, Wright P, McKinnon RD. Expression and regulation of a glutamate receptor subunit by bFGF in oligodendrocyte progenitors. Glia. 1994;10:149–153. doi: 10.1002/glia.440100209. [DOI] [PubMed] [Google Scholar]
- Galvao RP, Kasina A, McNeill RS, Harbin JE, Foreman O, Verhaak RG, Nishiyama A, Miller CR, Zong H. Transformation of quiescent adult oligodendrocyte precursor cells into malignant glioma through a multistep reactivation process. Proc Natl Acad Sci U S A. 2014 doi: 10.1073/pnas.1414389111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Gibson EM, Purger D, Mount CW, Goldstein AK, Lin GL, Wood LS, Inema I, Miller SE, Bieri G, Zuchero JB, et al. Neuronal Activity Promotes Oligodendrogenesis and Adaptive Myelination in the Mammalian Brain. Science. 2014;344(6183):1252304. doi: 10.1126/science.1252304. 487; Using in vivo optogenetic stimulation of the premotor circuit in awake, behaving mice, this study demonstrates that cortical projection neuronal activity promotes circuit-specific oligodendrocyte precursor cell proliferation, oligodendrogenesis and myelin microstructural changes consequential to neurological function. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goggi J, Pullar IA, Carney SL, Bradford HF. The control of [125I]BDNF release from striatal rat brain slices. Brain Res. 2003;967:201–209. doi: 10.1016/s0006-8993(03)02225-x. [DOI] [PubMed] [Google Scholar]
- Grasso CS, Tang Y, Truffaux N, Berlow NE, Liu L, Debily MA, Quist MJ, Davis LE, Huang EC, Woo PJ, et al. Functionally defined therapeutic targets in diffuse intrinsic pontine glioma. Nat Med. 2015;21:555–559. doi: 10.1038/nm.3855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong EJ, McCord AE, Greenberg ME. A biological function for the neuronal activity-dependent component of Bdnf transcription in the development of cortical inhibition. Neuron. 2008;60:610–624. doi: 10.1016/j.neuron.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **.Ishiuchi S, Tsuzuki K, Yoshida Y, Yamada N, Hagimura N, Okado H, Miwa A, Kurihara H, Nakazato Y, Tamura M, et al. Blockage of Ca(2+)-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat Med. 2002;8:971–978. doi: 10.1038/nm746. This important study showed the effect of glutamatergic signaling through AMPA receptors on glioma migration and survival. [DOI] [PubMed] [Google Scholar]
- *.Ishiuchi S, Yoshida Y, Sugawara K, Aihara M, Ohtani T, Watanabe T, Saito N, Tsuzuki K, Okado H, Miwa A, et al. Ca2+-permeable AMPA receptors regulate growth of human glioblastoma via Akt activation. J Neurosci. 2007;27:7987–8001. doi: 10.1523/JNEUROSCI.2180-07.2007. This study demonstrates a growth-promoting effect of glutamatergic signaling through AMPA receptors in glioma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **.John Lin CC, Yu K, Hatcher A, Huang TW, Lee HK, Carlson J, Weston MC, Chen F, Zhang Y, Zhu W, et al. Identification of diverse astrocyte populations and their malignant analogs. Nat Neurosci. 2017;20:396–405. doi: 10.1038/nn.4493. This elegant study demonstrates molecularly distinct subpopulations of normal astrocytes and malignant astrocytoma cells. One subpopulation of normal astrocytes and a correlate population in glioma promote synaptogenesis. The emergence of this pro-synaptogenic population in glioma correlates with the onset of seizures in a genetically engineered mouse model of high-grade glioma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT, Hutter B, Jager N, Korshunov A, Kool M, Warnatz HJ, Zichner T, Lambert SR, Ryzhova M, Quang DA, et al. Recurrent somatic alterations of FGFR1 and NTRK2 in pilocytic astrocytoma. Nat Genet. 2013;45:927–932. doi: 10.1038/ng.2682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung E, Osswald M, Blaes J, Wiestler B, Sahm F, et al. Tweety-Homolog 1 Drives Brain Colonization of Gliomas. J Neurosci. 2017;37:6837–50. doi: 10.1523/JNEUROSCI.3532-16.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein R, Nanduri V, Jing SA, Lamballe F, Tapley P, Bryant S, Cordon-Cardo C, Jones KR, Reichardt LF, Barbacid M. The trkB tyrosine protein kinase is a receptor for brain-derived neurotrophic factor and neurotrophin-3. Cell. 1991;66:395–403. doi: 10.1016/0092-8674(91)90628-c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Labrakakis C, Patt S, Hartmann J, Kettenmann H. Functional GABA(A) receptors on human glioma cells. Eur J Neurosci. 1998;10:231–238. doi: 10.1046/j.1460-9568.1998.00036.x. This study demonstrated GABAergic signaling in glioma. [DOI] [PubMed] [Google Scholar]
- Lawn S, Krishna N, Pisklakova A, Qu X, Fenstermacher DA, Fournier M, Vrionis FD, Tran N, Chan JA, Kenchappa RS, et al. Neurotrophin signaling via TrkB and TrkC receptors promotes the growth of brain tumor-initiating cells. J Biol Chem. 2015;290:3814–3824. doi: 10.1074/jbc.M114.599373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhu S, Kozono D, Ng K, Futalan D, Shen Y, Akers JC, Steed T, Kushwaha D, Schlabach M, et al. Genome-wide shRNA screen revealed integrated mitogenic signaling between dopamine receptor D2 (DRD2) and epidermal growth factor receptor (EGFR) in glioblastoma. Oncotarget. 2014;5:882–893. doi: 10.18632/oncotarget.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindholm D, Castren E, Berzaghi M, Blochl A, Thoenen H. Activity-dependent and hormonal regulation of neurotrophin mRNA levels in the brain--implications for neuronal plasticity. J Neurobiol. 1994;25:1362–1372. doi: 10.1002/neu.480251105. [DOI] [PubMed] [Google Scholar]
- *.Liu C, Sage JC, Miller MR, Verhaak RG, Hippenmeyer S, Vogel H, Foreman O, Bronson RT, Nishiyama A, Luo L, et al. Mosaic analysis with double markers reveals tumor cell of origin in glioma. Cell. 2011;146:209–221. doi: 10.1016/j.cell.2011.06.014. Using elegant genetic mouse modeling, this paper provided direct evidence that oligodendrocyte precursor cells can give rise to malignant astrocytomas and that while the cell of mutation may be a neural stem cell, it is not until the neural stem cell differentiates into an oligodendrocyte precursor cell that gliomas arise. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louis DN, Perry A, Reifenberger G, von Deimling A, Figarella-Branger D, Cavenee WK, Ohgaki H, Wiestler OD, Kleihues P, Ellison DW. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: a summary. Acta Neuropathol. 2016;131:803–820. doi: 10.1007/s00401-016-1545-1. [DOI] [PubMed] [Google Scholar]
- Mahe C, Bernhard M, Bobirnac I, Keser C, Loetscher E, Feuerbach D, Dev KK, Schoeffter P. Functional expression of the serotonin 5-HT7 receptor in human glioblastoma cell lines. British Journal of Pharmacology. 2004;143:404–410. doi: 10.1038/sj.bjp.0705936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monje M, Mitra SS, Freret ME, Raveh TB, Kim J, Masek M, Attema JL, Li G, Haddix T, Edwards MS, et al. Hedgehog-responsive candidate cell of origin for diffuse intrinsic pontine glioma. Proc Natl Acad Sci U S A. 2011;108(11):4453–4458. doi: 10.1073/pnas.1101657108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraja S, Vitanza NA, Woo PJ, Taylor KR, Liu F, et al. Transcriptional Dependencies in Diffuse Intrinsic Pontine Glioma. Cancer Cell. 2017;31:635–52. doi: 10.1016/j.ccell.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osswald M, Jung E, Sahm F, Solecki G, Venkataramani V, et al. Brain tumour cells interconnect to a functional and resistant network. Nature. 2015;528:93–8. doi: 10.1038/nature16071. [DOI] [PubMed] [Google Scholar]
- Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, et al. CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nat Med. 2013;19:1264–1272. doi: 10.1038/nm.3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, Joyce JA. The Microenvironmental Landscape of Brain Tumors. Cancer Cell. 2017;31:326–341. doi: 10.1016/j.ccell.2017.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shchors K, Massaras A, Hanahan D. Dual Targeting of the Autophagic Regulatory Circuitry in Gliomas with Repurposed Drugs Elicits Cell-Lethal Autophagy and Therapeutic Benefit. Cancer Cell. 2015;28:456–471. doi: 10.1016/j.ccell.2015.08.012. [DOI] [PubMed] [Google Scholar]
- Silver DJ, Siebzehnrubl FA, Schildts MJ, Yachnis AT, Smith GM, Smith AA, Scheffler B, Reynolds BA, Silver J, Steindler DA. Chondroitin sulfate proteoglycans potently inhibit invasion and serve as a central organizer of the brain tumor microenvironment. J Neurosci. 2013;33:15603–15617. doi: 10.1523/JNEUROSCI.3004-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takano T, Lin JH, Arcuino G, Gao Q, Yang J, Nedergaard M. Glutamate release promotes growth of malignant gliomas. Nat Med. 2001;7:1010–1015. doi: 10.1038/nm0901-1010. [DOI] [PubMed] [Google Scholar]
- Tsiperson V, Huang Y, Bagayogo I, Song Y, VonDran MW, DiCicco-Bloom E, Dreyfus CF. Brain-derived neurotrophic factor deficiency restricts proliferation of oligodendrocyte progenitors following cuprizone-induced demyelination. ASN neuro. 2015;7 doi: 10.1177/1759091414566878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **.Venkatesh HS, Johung TB, Caretti V, Noll A, Tang Y, Nagaraja S, Gibson EM, Mount CW, Polepalli J, Mitra SS, et al. Neuronal Activity Promotes Glioma Growth through Neuroligin-3 Secretion. Cell. 2015;161:803–816. doi: 10.1016/j.cell.2015.04.012. Using patient-derived cultures and orthotopic xenograft models of high-grade glioma together with in vivo optogenetic techniques, this work demonstrated that neuronal activity promotes the growth a range of pediatric and adult high-grade glioma types. Key mechanisms of activity-regulated growth include secretion of the neurotrophin BDNF and a synaptic protein called neuroligin-3 into the tumor microenvironment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesh HS, Monje M. Neuronal Activity in Ontogeny and Oncology. Trends in Cancer. 2017;3:89–112. doi: 10.1016/j.trecan.2016.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- **.Venkatesh HS, Tam LT, Woo PJ, Nagaraja S, Gillespe SM, Lennon J, Ni J, Duveau DY, Morris PJ, Zhao JJ, Thomas CJ, Monje M. Targeting neuronal activity-regulated neuroligin-3 dependency for high-grade glioma. Nature. 2017;549:533–537. doi: 10.1038/nature24014. Following on the work described in the 2015 paper by the same authors, this study describes an unexpected dependency of gliomas on neuroligin-3 from the brain microenvironment, determines that neuroligin-3 is cleaved and released into the tumor microenvironment by the activity-regulated action of the ADAM10 protease and demonstrated that ADAM10 inhibition represents a promising therapeutic strategy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VonDran MW, Singh H, Honeywell JZ, Dreyfus CF. Levels of BDNF impact oligodendrocyte lineage cells following a cuprizone lesion. J Neurosci. 2011;31:14182–14190. doi: 10.1523/JNEUROSCI.6595-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadhwa S, Nag TC, Jindal A, Kushwaha R, Mahapatra AK, Sarkar C. Expression of the neurotrophin receptors Trk A and Trk B in adult human astrocytoma and glioblastoma. Journal of biosciences. 2003;28:181–188. doi: 10.1007/BF02706217. [DOI] [PubMed] [Google Scholar]
- Walker AJ, Card T, Bates TE, Muir K. Tricyclic antidepressants and the incidence of certain cancers: a study using the GPRD. Br J Cancer. 2011;104:193–197. doi: 10.1038/sj.bjc.6605996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Hagel C, Hamel W, Muller S, Kluwe L, Westphal M. Trk A, B, and C are commonly expressed in human astrocytes and astrocytic gliomas but not by human oligodendrocytes and oligodendroglioma. Acta Neuropathol. 1998;96:357–364. doi: 10.1007/s004010050906. [DOI] [PubMed] [Google Scholar]
- Wang Y, Yang J, Zheng H, Tomasek GJ, Zhang P, McKeever PE, Lee EY, Zhu Y. Expression of mutant p53 proteins implicates a lineage relationship between neural stem cells and malignant astrocytic glioma in a murine model. Cancer Cell. 2009;15:514–526. doi: 10.1016/j.ccr.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AW, Xiao J, Kemper D, Kilpatrick TJ, Murray SS. Oligodendroglial expression of TrkB independently regulates myelination and progenitor cell proliferation. J Neurosci. 2013;33:4947–4957. doi: 10.1523/JNEUROSCI.3990-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, Diaz AK, Paugh BS, Rankin SL, Ju B, Li Y, Zhu X, Qu C, Chen X, Zhang J, et al. The genomic landscape of diffuse intrinsic pontine glioma and pediatric non-brainstem high-grade glioma. Nat Genet. 2014;46:444–450. doi: 10.1038/ng.2938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- *.Xiong J, Zhou L, Lim Y, Yang M, Zhu YH, Li ZW, Zhou FH, Xiao ZC, Zhou XF. Mature BDNF promotes the growth of glioma cells in vitro. Oncology reports. 2013;30:2719–2724. doi: 10.3892/or.2013.2746. This paper provided early evidence that the neurotrophin BDNF can promote glioma growth. [DOI] [PubMed] [Google Scholar]
- Ye ZC, Sontheimer H. Glioma cells release excitotoxic concentrations of glutamate. Cancer Res. 1999;59:4383–4391. [PubMed] [Google Scholar]
- Zonouzi M, Scafidi J, Li P, McEllin B, Edwards J, Dupree JL, Harvey L, Sun D, Hubner CA, Cull-Candy SG, et al. GABAergic regulation of cerebellar NG2 cell development is altered in perinatal white matter injury. Nat Neurosci. 2015;18:674–682. doi: 10.1038/nn.3990. [DOI] [PMC free article] [PubMed] [Google Scholar]