

Abstract
Pd(II)-catalyzed enantioselective γ-C(sp3)–H cross-coupling of alkyl amines via desymmetrization and kinetic resolution has been realized for the first time using chiral acetyl-protected aminomethyl oxazoline ligands (APAO). A diverse range of aryl- and vinyl-boron reagents can be used as coupling partners. The chiral γ-arylated alkylamine products are further transformed into chiral 2-substituted 1,2,3,4-tetra-hydroquinolines and spiro-pyrrolidines as important structural motifs in natural products and biologically active molecules.
Graphical Abstract
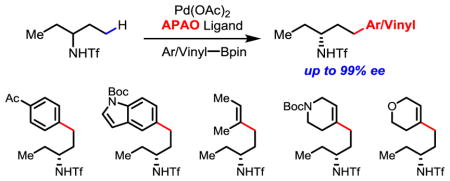
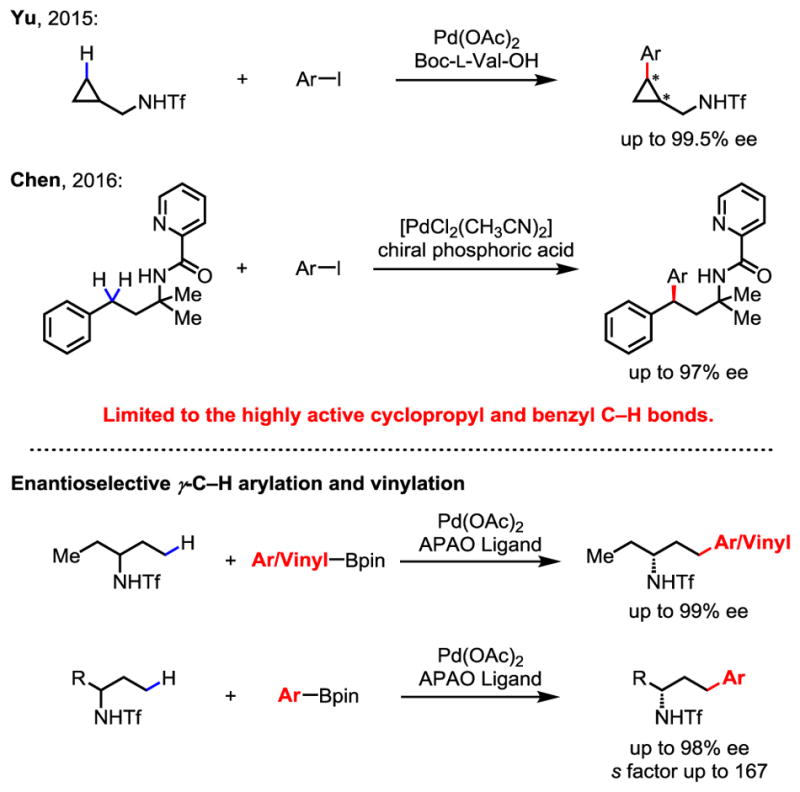
While Pd(OAc)2 has been long known to activate both sp2 and sp3 C–H bonds,1 such reactivity driven by strong coordination from substrates can also lead to persistent background reactions, which presents a fundamental challenge for rendering Pd(II)-catalyzed racemic C–H activation reactions2 enantioselective. Despite recent development of chiral Pd(II) catalysts for enantioselective intermolecular C–H activation reactions through weak coordination and ligand acceleration,3–5 as well as Pd(0)-catalyzed intramolecular enantioselective C–H arylation,6 scope and efficiency remains limited. For example, protocols for enantioselective C–H activation reactions of carboxylic acids and amide substrates are not applicable to alkyl amine substrates. To date, there are only two examples of enantioselective intermolecular γ-C(sp3)–H activation of amine substrates involving cyclopropyl and benzylic C–H bonds (Scheme 1).7, 8 In addition, only arylation via a Pd(II)/Pd(IV) catalytic cycle is compatible with these chiral catalysts. Considering the broad utility of chiral amines in organic synthesis and drug discovery,9 it is of great importance to develop enantioselective C–H activation reactions of simple alkyl amines using other catalytic cycles. Herein, we report the first chiral catalyst for enantioselective γ-C(sp3)–H cross-coupling of aliphatic amines with both aryl and vinyl boron reagents through desymmetrization. Kinetic resolution10 of alkyl amines bearing various α-substituents via γ-C(sp3)–H cross-coupling is also made possible through catalyst modification, allowing the access to a broader range of chiral amines.
Scheme 1.
Enantioselective γ-C(sp3)–H Activation of Alkyl Amines
Based on our previously developed γ-C(sp3)–H arylation7 and cross coupling11 of alkyl amines, we initiated our investigation to develop γ-C(sp3)–H cross-coupling of amine 1a using various types of bidentate chiral ligands (Table 1). While no C(sp3)–H arylation occurred in the absence of ligand, we were delighted to observe that the acetyl-protected aminoethyl quinoline (APAQ) ligand (L1) provided the desired product in 30% yield with 97% ee, and Ac-L-Val-OH (L2) provided product in 38% yield with 26% ee. However, the efforts to further improve the yield and enantioselectivity failed after an extensive screening of APAQ and mono-N-protected amino acid (MPAA) ligands. Considering the fact that acetyl protected amino oxazoline (APAO) chiral ligands can facilitate carboxylic amide-directed enantioselective C(sp3)–H arylation (Pd(II)/Pd(IV)) and borylation (Pd(II)/Pd(0)),5e, 5f we began to test this ligand for alkyl amine derived substrates 1a. Encouragingly, the use of (S,S)-L3 furnished the cross-coupling product in 54% yield with 98% ee. We then systematically modified APAO ligands at both the side chain and the oxazoline ring to further optimize this transformation (L4–L13). We found that the reactivity of this cross-coupling reaction is highly sensitive to the steric hindrance of the ligand side chain (L4–L7). Reaction with ligand bearing a bulky t-Bu group improved the yield from 54% to 65% (L7). Fine tuning of other substituents on the side chain did not provide better results. The use of (S,R)-L7, the other diastereomer of the optimal ligand, provided the enantiomer of product with 32% yield and 98% ee. Para-trifluoromethyl substitution on the phenyl ring gave slightly lower yield (L8). Increasing the steric hindrance on the oxazoline ring was detrimental to the reaction, presumably due to the ineffective binding of APAO ligand to Pd(II) species (L9–L11). The chiral oxazoline ligands lacking a stereocenter on either the side chain or the oxazoline moiety gave poor yields and enantioselectivities (L12 and L13), which speaks to the importance of both chiral centers on the APAO ligand backbones for chiral induction. Finally, control experiments revealed that the Pd catalyst, silver source, BQ, base, and H2O were all crucial for the C(sp3)–H cross-coupling reaction (see Supporting Information for the detail optimization of these parameters).
Table 1.

|
Reaction conditions: substrate 1a (0.1 mmol), 2a (2.0 equiv.), Pd(OAc)2 (10 mol%), ligand (15 mol%), Ag2CO3 (2.0 equiv.), Na2CO3 (2.0 equiv.), 1,4-benzoquinone (BQ) (0.5 equiv.), H2O (5.5 equiv.), t-AmylOH (1.0 mL), 80 °C, 12 h, N2.
The yields were determined by 1H NMR analysis of the crude product using CH2Br2 as the internal standard. The ee values were determined by HPLC analysis on a chiral stationary phase.
With the optimal reaction conditions established, we set out to explore this enantioselective γ-C(sp3)–H cross-coupling reaction of amine derivative 1a with a wide range of arylboron reagents in the presence of (S,S)-L7 as ligand (Table 2). Reaction with simple phenylboronic acid pinacol ester provided 3b in good yield and excellent enantioselectivity (64%, 97% ee). Arylboron reagents with both electron-donating and electron-withdrawing substituents at the para and meta positions are well tolerated, affording the desired products in good yields with excellent enantioselectivities (>96% ee in all cases). Coupling partners bearing electron-withdrawing ester, acetyl, trifluoromethyl, and fluoro groups gave moderate to good yields (52%–65%) with excellent enantioselectivities (3a, 3c–3e). Coupling partners bearing electron-donating substituents such as methyl and methoxyl groups at the para position also performed well (3f and 3g). Reactions with biphenyl, 1-naphthyl, and 2-naphthyl boronic acid pinacol ester furnished the desired products in good yields with excellent enantioselectivities (3h–3j). Furthermore, meta-substituted and disubstituted arylboron reagents were all well tolerated (3k–3o). Heteroaryl boronate ester was also compatible with this reaction, albeit giving lower yield (3p). The Tf group of 3b could be readily removed in a two-step procedure to yield free amine without loss of optical activity (see Supporting Information for the deprotected reaction). The free amine can also direct γ-C(sp3)–H arylation of the remaining ethyl group using our transient directing group to access a wider range of amines.12
Table 2.

|
Reaction conditions: substrate 1a (0.1 mmol), 2 (2.0 equiv.), Pd(OAc)2 (10 mol%), (S,S)-L7 (15 mol%), Ag2CO3 (2.0 equiv.), Na2CO3 (2.0 equiv.), 1,4-benzoquinone (BQ) (0.5 equiv.), H2O (5.5 equiv.), t-AmylOH (1.0 mL), 80 °C, 12 h, N2.
Isolated yields. The ee values were determined by HPLC analysis on a chiral stationary phase.
Extending this enantioselective C–H activation method to γ-C(sp3)–H coupling with vinylboron reagents will allow the access to a broader range of chiral amines. Hence, we next investigated the coupling of Tf-protected amines with various vinylboron reagents, which has not been reported to date. γ-Styrenylation of amine 1a using chiral ligand (S,S)-L7 afforded the product in 50% yield and 96% ee. The use of (S,S,S)-L14, containing an additional chiral center at C-5, improved the yield to 56% and enantioselectivity to 99% ee (Table 3). A number of disubstituted (Z)-vinylboron reagents (5a–5e), as well as trisubstituted vinylboron reagent (5f) were compatible with this reaction. Coupling partners bearing 1-aryl-2-alkyl, diaryl, dialkyl, and trialkyl groups gave moderate yields and excellent enantioselectivities. 1-Cyclohexenyl-Bpin and 1-cyclopentyl-Bpin were both well tolerated as coupling partners in this reaction (5g, 5h). Hetero-cyclohexenyl boronate esters also afforded desired products in good enantioselectivities, albeit in lower yields (5i, 5j). These heterocycle-containing chiral amines are useful intermediates in drug discovery.
Table 3.

|
Reaction conditions: substrate 1a (0.1 mmol), 4 (3.0 equiv.), Pd(OAc)2 (10 mol%), ligand (15 mol%), Ag2CO3 (2.0 equiv.), Na2CO3 (2.0 equiv.), 1,4-benzoquinone (BQ) (0.5 equiv.), H2O (5.5 equiv.), THF (1.0 mL), 70 °C, 24 h, N2.
Isolated yields. The ee values were determined by HPLC analysis on a chiral stationary phase.
Next, we began to develop chiral ligands for enantioselective γ-C(sp3)–H coupling of unsymmetric amines through kinetic resolution (Table 4). Guided by the established enantioselective desymmetrization C–H coupling (Table 1), the reaction of racemic 1b with para-methoxycarbonyl-phenylboronic acid pinacol ester was carried out in the presence of 10 mol% Pd(OAc)2, 15 mol% (S,S)-L7, 2.0 equiv. of Ag2CO3, 2.0 equiv. of Na2CO3, 0.5 equiv. of BQ, and 5.5 equiv. of H2O in t-AmylOH at 80 °C. We were pleased to find that the desired cross-coupling product 6a was obtained with 40% yield and 84% ee.
Table 4.

|
Reaction conditions: substrate 1 (0.1 mmol), 2 (2.0 equiv.), Pd(OAc)2 (10 mol%), ligand (15 mol%), Ag2CO3 (2.0 equiv.), Na2CO3 (2.0 equiv.), 1,4-benzoquinone (BQ) (0.5 equiv.), H2O (5.5 equiv.), t-AmylOH (1.0 mL), 80 °C, 24 h, N2.
Isolated yields. The ee values were determined by HPLC analysis on a chiral stationary phase.
Encouraged by this result, a variety of APAO ligands with different side chains were screened and (S,S)-L15 was found to give the best s-factor of 167 (6a, 34% yield, 98% ee) (see Supporting Information for detail optimization of the reaction). With the optimized reaction conditions in hand, we next investigated different amines and arylboron coupling partners. Notably, differentiation of a hydrogen atom and a relatively small methyl group in 2-butylamine is highly challenging. We were pleased to find that cross-coupling of 2-butylamine derived substrate gave the arylated product in 32% yield and 96% ee (6b). This enantioselective C–H activation method is also compatible with amino alcohol derived substrate (6c, s = 67).
To showcase the synthetic utility of this enantioselective C–H activation reaction, the cross-coupling products were further transformed into chiral 2-substituted 1,2,3,4-tetra-hydroquinolines without loss of optical activity (Table 5).13 Various substituents such as 2-naphthyl, para- and meta-substituted arylated products were well tolerated in the condition. Treatment of the vinylated product 5j with concentrated sulfuric acid in DCM gave the spiro-pyrrolidine derivative 7g in 79% yield.14 Deprotection was readily accomplished by treating the cyclization product with Red-Al in toluene at 50 °C, affording chiral 2-methyl-1,2,3,4-tetra-hydroquinoline in 99% yield and 96% ee (Scheme 2). It should be noted that 2-substituted 1,2,3,4-tetra-hydroquinolines and spiro-pyrrolidines are important structural motifs in biologically active molecules and natural products.
Table 5.

|
Reaction conditions: substrate 3 or 6 (0.1 mmol), 1,3-diiodo-5,5-dimethylhydantoin (DIH) (2.0 equiv.), DCE (5.0 mL), hv, 60 °C, 6 h, N2.
Isolated yields. The ee values were determined by HPLC analysis on a chiral stationary phase.
Scheme 2. The Removal of Auxiliary a, b.

a Reaction conditions: 7a (0.1 mmol), Red-Al (10.0 equiv.), toluene (1.0 mL), 50 °C, N2, 10 h. b Isolated yield.
In summary, we have developed Pd(II)-catalyzed enantioselective γ-C(sp3)–H cross-coupling of alkyl amines protected by a removable Tf group. C–H arylation and vinylation via desymmetrization or kinetic resolution provide an access to a broad range of chiral alkyl amines.
Supplementary Material
Acknowledgments
We gratefully acknowledge The Scripps Research Institute and the NIH (NIGMS, 2R01GM084019) for financial support. We thank David Hill for chiral separation of compound 7g.
Footnotes
Supporting Information Available: Experimental procedures and spectral data for all new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Constable AG, McDonald WS, Sawkins LC, Shaw BL. J Chem Soc, Chem Commun. 1978;0:1061. [Google Scholar]; (b) Balavoine G, Clinet JC. J Organomet Chem. 1990;390:C84. [Google Scholar]; (c) Carr K, Sutherland JK. J Chem Soc Chem, Commun. :1984, 1227. [Google Scholar]; (d) Baldwin JE, Nájera C, Yus M. J Chem Soc Chem, Commun. 1985:126. [Google Scholar]
- 2.(a) Neufeldt SR, Sanford MS. Acc Chem Res. 2012;45:936. doi: 10.1021/ar300014f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Daugulis O, Do HQ, Shabashov D. Acc Chem Res. 2009;42:1074. doi: 10.1021/ar9000058. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) He J, Wasa M, Chan KSL, Shao Q, Yu JQ. Chem Rev. 2017;117:8754. doi: 10.1021/acs.chemrev.6b00622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.For reviews on enantioselective C–H activation, see: Giri R, Shi BF, Engle KM, Maugel N, Yu JQ. Chem Soc Rev. 2009;38:3242. doi: 10.1039/b816707a.Herrmann P, Bach T. Chem Soc Rev. 2011;40:2022. doi: 10.1039/c0cs00027b.Newton CG, Wang SG, Oliveira CC, Cramer N. Chem Rev. 2017;117:8908. doi: 10.1021/acs.chemrev.6b00692.Baudoin O. Acc Chem Res. 2017;50:1114. doi: 10.1021/acs.accounts.7b00099.
- 4.For intermolecular enantioselective Pd(II)-catalyzed C(sp2)–H activation reactions, see: Shi BF, Maugel N, Zhang YH, Yu JQ. Angew Chem, Int Ed. 2008;47:4882. doi: 10.1002/anie.200801030.Shi BF, Zhang YH, Lam JK, Wang DH, Yu JQ. J Am Chem Soc. 2010;132:460. doi: 10.1021/ja909571z.Gao DW, Shi YC, Gu Q, Zhao ZL, You SL. J Am Chem Soc. 2013;135:86. doi: 10.1021/ja311082u.Pi C, Li Y, Cui X, Zhang H, Han YB, Wu Y. Chem Sci. 2013;4:2675.Chu L, Xiao KJ, Yu JQ. Science. 2014;346:451. doi: 10.1126/science.1258538.Du ZJ, Guan J, Wu GJ, Xu P, Gao LX, Han FS. J Am Chem Soc. 2015;137:632. doi: 10.1021/ja512029x.
- 5.For intermolecular enantioselective Pd(II)-catalyzed C(sp3)–H activation reactions, see: Wasa M, Engle KM, Lin DW, Yoo EJ, Yu JQ. J Am Chem Soc. 2011;133:19598. doi: 10.1021/ja207607s.Chen G, Gong W, Zhuang Z, Andra MS, Chen YQ, Höng X, Yang YF, Liu T, Houk KN, Yu JQ. Science. 2016;353:1023. doi: 10.1126/science.aaf4434.Jain P, Verma P, Xia G, Yu JQ. Nat Chem. 2016;353:1023.Smalley AP, Cuthbertson JD, Gaunt MJ. J Am Chem Soc. 2017;139:1412. doi: 10.1021/jacs.6b12234.Wu QF, Shen PX, He J, Wang XB, Zhang F, Shao Q, Zhu RY, Mapelli C, Qiao JX, Poss MA, Yu JQ. Science. 2017;355:499. doi: 10.1126/science.aal5175.He J, Shao Q, Wu Q, Yu JQ. J Am Chem Soc. 2017;139:3344. doi: 10.1021/jacs.6b13389.
- 6.For intramolecular enantioselective Pd(0)-catalyzed C–H activation, see: Nakanishi M, Katayev D, Besnard C, Kündig EP. Angew Chem, Int Ed. 2011;50:7438. doi: 10.1002/anie.201102639.Anas S, Cordi A, Kagan HB. Chem Commun. 2011;47:11483. doi: 10.1039/c1cc14292e.Saget T, Cramer N. Angew Chem, Int Ed. 2012;51:12842. doi: 10.1002/anie.201207959.Shintani R, Otomo H, Ota K, Hayashi T. J Am Chem Soc. 2012;134:7305. doi: 10.1021/ja302278s.Ladd CL, Charette AB. Org Lett. 2016;18:6046. doi: 10.1021/acs.orglett.6b02982.Yang L, Melot R, Neuburger M, Baudoin O. Chem Sci. 2017;8:1344. doi: 10.1039/c6sc04006c.Pedroni J, Cramer N. J Am Chem Soc. 2017;139:12398. doi: 10.1021/jacs.7b07024.Zhu C, Wang D, Zhao Y, Sun WY, Shi Z. J Am Chem Soc. 2017;139:16486. doi: 10.1021/jacs.7b10365.
- 7.Chan KSL, Fu H-Y, Yu J-Q. J Am Chem Soc. 2015;137:2042. doi: 10.1021/ja512529e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang H, Tong H-R, He G, Chen G. Angew Chem, Int Ed. 2016;55:15387. doi: 10.1002/anie.201609337. [DOI] [PubMed] [Google Scholar]
- 9.(a) Zhi L, Tegley CM, Marschke KB, Jones TK. Bioorg Med Chem Lett. 1999;9:1009. doi: 10.1016/s0960-894x(99)00119-5. [DOI] [PubMed] [Google Scholar]; (b) Bohl CE, Chang C, Mohler ML, Chen J, Miller DD, Swaan PW, Dalton JT. J Med Chem. 2004;47:3765. doi: 10.1021/JM0499007. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lindsley SR, Moore KP, Rajapakse HA, Selnick HG, Young MB, Zhu H, Munshi S, Kuo L, McGaughey GB, Colussi D, Crouthamel MC, Lai MT, Pietrak B, Price EA, Sankaranarayanan S, Simon AJ, Seabrook GR, Hazuda DJ, Pudvah NT, Hochman JH, Graham SL, Vacca JP, Nantermet PG. Bioorg Med Chem Lett. 2007;17:4057. doi: 10.1016/j.bmcl.2007.04.072. [DOI] [PubMed] [Google Scholar]
- 10.For a single example of kinetic resolution through a intramolecular C(sp3)–H arylation see: Katayev D, Larionov E, Nakanishi M, Besnard C, Kündig E. Chem - Eur J. 2014;20:15021. doi: 10.1002/chem.201403985.
- 11.Shao Q, He J, Wu Q, Yu J-Q. ACS Catal. 2017;7:7777. [Google Scholar]
- 12.Wu Y, Chen Y-Q, Liu T, Eastgate M-D, Yu J-Q. J Am Chem Soc. 2016;138:14554. doi: 10.1021/jacs.6b09653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moroda A, Furuyama S, Togo H. Synlett. 2009;8:1336. [Google Scholar]
- 14.Henderson L, Knight DW, Williams AC. Synlett. 2012;23:1667. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.