

Abstract
The synthesis and redox properties are presented for the electron-rich bis(monothiolate)s Fe2(SR)2-(CO)2(dppv)2 for R = Me ([1]0), Ph ([2]0), CH2Ph ([3]0). Whereas related derivatives adopt C2-symmetric Fe2(CO)2P4 cores, [1]0–[3]0 have Cs symmetry resulting from the unsymmetrical steric properties of the axial vs equatorial R groups. Complexes [1]0–[3]0 undergo 1e− oxidation upon treatment with ferrocenium salts to give the mixed valence cations [Fe2(SR)2(CO)2(dppv)2]+. As established crystallographically, [3]+ adopts a rotated structure, characteristic of related mixed valence diiron complexes. Unlike [1]+ and [2]+ and many other [Fe2(SR)2L6]+ derivatives, [3]+ undergoes C–S bond homolysis, affording the diferrous sulfido-thiolate [Fe2(SCH2Ph)(S)(CO)2(dppv)2]+ ([4]+). According to X-ray crystallography, the first coordination spheres of [3]+ and [4]+ are similar, but the Fe–sulfido bonds are short in [4]+. The conversion of [3]+ to [4]+ follows first-order kinetics, with k = 2.3 × 10−6 s−1 (30 °C). When the conversion is conducted in THF, the organic products are toluene and dibenzyl. In the presence of TEMPO, the conversion of [3]+ to [4]+ is accelerated about 10×, the main organic product being TEMPO-CH2Ph. DFT calculations predict that the homolysis of a C–S bond is exergonic for [Fe2(SCH2Ph)2(CO)2(PR3)4]+ but endergonic for the neutral complex as well as less substituted cations. The unsaturated character of [4]+ is indicated by its double carbonylation to give [Fe2(SCH2Ph)(S)(CO)4(dppv)2]+ ([5]+), which adopts a bioctahedral structure.
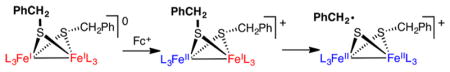
Graphical Abstract
INTRODUCTION
Organometallic radicals have received continuous attention for decades.1–3 Within the area of enzymology, a prominent organometallic radical is the Hox resting state in the [FeFe]-hydrogenases. Featuring an S = 1/2 Fe(II)Fe(I) center, this state is exceptionally well characterized,4 as are low molecular weight synthetic models.5 Briefly, one Fe center is pentacoordinate, typically assigned as Fe(I), with a “rotated” geometry, whereas the other Fe center, assigned as Fe(II), is octahedral. The strikingly unsymmetrical structure presents a vacant coordination site adjacent to the amine cofactor substrate activation.
The Hox active site and its models exhibit three kinds of Fe-centered reactions: reduction to Fe(I)Fe(I) derivatives, binding of CO, and activation of dihydrogen in the presence of a second redox agent6,7 (Scheme 1). This report describes a new kind of reaction of the Hox-like center: their fragmentation with release of organic radicals.
Scheme 1.

Selected Reactions of [Fe2(SR)2(CO)L5]+ (L = CO, PR3)
This report begins with experiments probing the reactivity of bis(monothiolate)s [Fe2(SR)2(CO)2(dppv)2]n+ (dppv = cis-1,2-bis(diphenylphosphino)ethylene). The properties of the related dithiolates have been exhaustively studied,5 but the substituted bis(monothiolate)s have been rarely described,8,9 and mixed valence derivatives have not been characterized.
The radical reactions of iron monothiolates are potentially relevant to the biosynthesis of the [2Fe]H center (the active site) in the [FeFe]-hydrogenases.10–12 The pathway proceeds via a ferrous cysteinate-cyanide “Complex B” assembled within the multifunctional enzyme HydG. Complex B is transferred to HydF, a scaffold where the enzyme HydE probably operates on cysteinate to produce the adt cofactor (Scheme 2). Since HydE is a radical-SAM enzyme, radical reactions of diiron monothiolates are implicated.
Scheme 2.

Radical Reactions Implicated in the Biosynthesis of the [2Fe]H Center
RESULTS
Preparative Background
The displacement of four CO ligands by a pair of dppv groups has been applied to the preparation of chelating dithiolato complexes, e.g., Fe2[S2-(CH2)n](CO)2(dppv)2. In this work, the two dppv were installed for the first time on bis(monothiolate) complexes Fe2(SMe)2(CO)2(dppv)2 ([1]0), Fe2(SPh)2(CO)2(dppv)2 ([2]0), and Fe2(SCH2Ph)2(CO)2(dppv)2 ([3]0). For the preparation of alkylthiolato complexes [1]0 and [3]0 from the corresponding Fe2(SR)2(CO)6 derivatives, either of two methods proved suitable: one-pot UV-irradiation or a two-pot process that involves isolation of the intermediate Fe2(SR)2-(CO)4(dppv) by a thermal reaction, which is converted to the dicarbonyl in a separate photochemical reaction.
The preparation of [2]0 proved more complicated as indicated by a recent report.13 The intermediate Fe2(SPh)2-(CO)4(dppv) could be efficiently prepared, but its reaction with dppv produced significant amounts of Fe(SPh)2(CO)2-(dppv) and Fe(CO)3(dppv). Phenylthiolate is a more weakly bridging ligand than alkylthiolates; hence Fe2(SPh)2(CO)4-(dppv) is more susceptible to rupture of the diiron unit. The preparation of Fe2(SPh)2(CO)2(dppv)2 was achieved by an inefficient photochemical route.
The Fe(I)Fe(I) complexes [1]0, [2]0, and [3]0 are green-brown solids with good solubility in dichloromethane. Solutions appear completely stable at room temperature. The 31P NMR spectra, consisting of two signals, remain unchanged over a broad range of temperatures, indicating relatively rigid structures. Two CH2R singlets are also observed in the 1H NMR spectrum recorded on CD2Cl2 (but not C6D6) solutions (R = Ph, H). The observation of methylene singlets for [3]0 is indicative of a plane of symmetry, since alternative geometries would result in diastereotopic signals. The rigidity reflects the high barriers for the axial-equatorial inversion of the μ-SR groups, which are known to be slow to invert on NMR time scales.14 The unsymmetrical disposition of the thiolate substituents induces the two dppv ligands to adopt a symmetrical arrangement. Thus, [1]0–[3]0 have a rigid Cs-symmetric Fe2(SR)2(CO)2P4 core as the result of the presence of axial and equatorial thiolates.14 By contrast, for the complexes Fe2[(SCH2)2X](CO)2(dppv)2 (X = CH2, O, NH, nothing), the 31P NMR spectra exhibit only a single signal near room temperature. Such compounds adopt a C2-symmetric Fe2(SR)2 core wherein the two dppv ligands undergo a degenerate oscillatory motion (Scheme 3).15
Scheme 3.

Bis(monothiolates) Fe2(SR)2(CO)2(dppv)2 Are More Rigid Stereochemically (Top) than the Related Chelating Dithiolates Fe2[(SCH2)2X](CO)2(dppv)2, Which Undergo Degenerate Racemizationa
aPh groups on phosphorus omitted for clarity.
Stereorigidity was also evident for Fe2(SR)2(CO)4(dppv), intermediates in the syntheses of [1]0–[3]0. Specifically, room temperature NMR spectra of the Fe2(SR)2(CO)4(dppv) (R = Me, CH2Ph, Ph) also indicate that the Fe(CO)(dppv) center is rigid on the NMR time scales. The 31P NMR spectra consist of two doublets. The 1H NMR spectrum of Fe2(SCH2Ph)2-(CO)4(dppv) exhibits four equally intense AB quartets, indicative of the diastereotopicity of the methylene protons. By contrast, only singlets are observed for each of these methylene groups in the 1H NMR spectrum of [3]0 (Scheme 4).
Scheme 4.

Stereochemistry of Methylene Groups in Two Isomers of Fe2(SCH2R)2(CO)2(dppv)2 (the Isomer on the Left Is Not Observed) and Fe2(SCH2R)2(CO)4(dppv)a
aPh groups on phosphorus omitted for clarity.
Crystallographic analysis confirmed the distinctive stereochemistry of [3]0. The thiolate substituents are indeed axial-equatorial (Figure 1). The two dppv ligands are apical-basal as is normal, but they are eclipsed, which is rarely observed. Correspondingly the CO ligands are cis-dibasal. The observed isomer appears to be stabilized by avoidance of a steric clash between the equatorial μ-SR group with the arylphosphine ligands.16
Figure 1.

Structure of Fe2(SCH2Ph)2(CO)2(dppv)2 ([3]0) with 50% probability ellipsoids and hydrogen atoms removed for clarity. Selected bond distances (Å): Fe1–C1, 1.7500(15), 1.7381(15); Fe–P, 2.1735(4)–2.2144(4); Fe–S, 2.2566(4)–2.2933(4); Fe1–Fe2, 2.5840(3).
Oxidation of [1]0 and [3]0 with FcBF4 afforded [1]BF4 and [3]BF4 (Fc+ = ferrocenium). Samples of [3]BF4 were obtained in analytical purity, while [1]+ was examined spectroscopically. FT-IR spectra of [1]BF4 and [3]BF4 are very similar. The oxidations were accompanied by a color change from red-brown to green. The IR spectrum also changed significantly, including the appearance of a lower energy band near 1904 cm−1 (CH2Cl2 solution) assigned to the semibridging CO ligand. This value is typical for Fe(II)Fe(I)/Fe(I)Fe(I) couples for complexes of the type Fe2(dithiolate)(CO)2(dppv)2.5
The salt [3]BF4 was characterized by X-ray crystallography (Figure 2). The geometry of the diiron center is similar to those of related 33e− [Fe2(SR)2(CO)6–xLx]+ complexes,17–19 which are often referred to as “Hox models”.5 Thus, one Fe center adopts a “rotated geometry” with an open apical coordination site trans to the Fe–Fe vector. One CO ligand is semibridging (Fe(2)–C–O angle = 171.8(2)°). This ligand gives rise to the νCO band observed at 1904 cm−1 in the FT-IR spectrum. The dppv on the nonrotated Fe center spans apical-basal sites, retaining the stereochemistry (relative to the axial-equatorial SCH2Ph groups) seen in [3]0. The average C–S distances are relatively unaffected by the oxidation, being 1.855(2) vs 1.847(2) Å for the neutral and cation, respectively.
Figure 2.

Structure of [Fe2(SCH2Ph)2(CO)2(dppv)2]+ ([3]+) with 50% probability ellipsoids and hydrogen atoms removed for clarity. Selected bond distances (Å): Fe1–C1, 1.752(2); Fe1–P2, 2.2035(6); Fe1–P1, 2.2487(6); Fe1–S2, 2.2504(6); Fe1–S1, 2.2894(5); Fe1– Fe2, 2.6012(4); Fe2–C2, 1.775(2); Fe2–P4, 2.2232(6); Fe2–P3, 2.2263(6); Fe2–S2, 2.2319(5); Fe2–S1, 2.2895(5).
Conversion of [Fe2(SCH2Ph)2(CO)2(dppv)2]+ to [Fe2(SCH2Ph)(S)(CO)2(dppv)2]+
When monitored by FT-IR spectroscopy, solutions of [1]BF4 are stable for days at room temperature. In contrast, C6D6 solutions of [3]BF4 decompose under the same conditions. The principal product is [Fe2-(SCH2Ph)(S)(CO)2(dppv)2]+ ([4]+, Scheme 5). ESI-MS measurements support the formula. The complex is diamagnetic, judging from its well-resolved 1H and 31P NMR spectra. The 1H spectrum is simple, consistent with a single isomer. The 31P NMR spectrum shows signals at δ101.7 and 85.6, also consistent with a single isomer. In situ examination of the conversion of [3]+ into [4]+ by 31P NMR spectroscopy revealed that CH2Cl2, BF4−, and traces of water interfere with the reaction. These contaminants give [Fe2(SCH2Ph)2(X)(CO)2-(dppv)2]+ (X = OH, Cl, F), as further indicated by ESI-MS analysis (the complex [Fe2(SCH2Ph)2(Cl)(CO)2(dppv)2]+ was further identified by X-ray crystallography as its BF4− salt). Complexes of the type [Fe2(SR)2(Cl)(CO)6–x(PR3)x]+ are well-known.20
Scheme 5.

Oxidation of [1]0–[3]0 and Debenzylation of [3]+
To minimize side reactions involving counterions and chlorinated solvent, the decomposition of [3]+ was examined as its BArF4− salt in THF and in benzene. IR spectra for [3]BF4 and [3]BArF4 are identical in the νCO region. The decomposition in dry benzene-d6 gave principally [4]+, obtained in 55% yield over the course of 3 days at room temperature. Some insoluble material is observed, pointing to other degradation pathways that parallel the [3]+ → [4]+ conversion. Solutions of [4]BArF4 are stable in benzene and CH2Cl2, so the modest yields are not attributable to the instability of [4]+.
The debenzylation of [3]+ appears to follow a radical pathway. Together with [4]+, dibenzyl and toluene were observed as coproducts when the reaction was conducted in THF (THF-h8 as well as THF-d8). Using an internal integration standard, the combined yields of dibenzyl and toluene were approximately 60%, which is consistent with the yield of [4]+. When conducted in C6D6, the reaction is slower and toluene was not observed, only dibenzyl (58% yield). When measured by 31P NMR spectroscopy, the appearance of [4]+ followed first-order kinetics with a half-life of about 80 h.
Further support for the radical pathway was provided by the finding that the conversion of [3]+ to [4]+ was accelerated 10× in the presence of 1 equiv of TEMPO. The yield of [4]+, evaluated by in situ 1H NMR analysis vs an internal integration standard, improved to almost 80%. This finding suggests that the diminished yield in the TEMPO-free [3]+ → [4]+ reaction results from attack of •CH2Ph on [3]+ or on [4]+. From the TEMPO-induced reaction, TEMPO-CH2Ph was isolated in 61% yield.
In control tests, solutions of [1]+, [3]0, and [4]+ were unreactive toward TEMPO over the course of days at room temperature. Qualitatively, the rate of the [3]+ + TEMPO reaction is unaffected by excess TEMPO. This finding is consistent with a rate-limiting homolysis reaction, followed by efficient trapping (Scheme 6).
Scheme 6.

Reaction of [3]+ with TEMPO
Crystallographic Characterization of [Fe2(SCH2Ph)(S)-(CO)2(dppv)2]+
X-ray crystallographic analysis of [4]BF4 confirmed the presence of only one benzyl group, which takes an equatorial orientation (Figure 3). In terms of its Fe2S2L6 core, [4]+ resembles [3]0. The Fe–S distances are disparate: Fe–sulfido distances are 2.1643(7) and 2.1275(7) Å, 0.1 Å shorter than the Fe–SCH2Ph distances, which are 0.1 Å longer. The Fe–S(R) bond lengths in complexes of the type [Fe2(SR)2(CO)6–xLx]z are relatively insensitive to oxidation state, as shown by comparison of [3]0 and [3]+. The short Fe–sulfide distance is attributed to steric effects as well as Fe–S π-bonding. The S–Fe–S angles (85.24, 85.53°) are more open than in [3]0 and [3]+, which average 80.5° and 78.7°, respectively.
Figure 3.

Structure of the cation in the salt [Fe2(SCH2Ph)(S)-(CO)2(dppv)2]BF4 ([4]BF4) with 50% probability ellipsoids. Hydrogen atoms are omitted for clarity. Selected bond distances (Å): Fe–C, 1.769(3), 1.766(3); Fe1–S2, 2.1643(7); Fe2–S2, 2.1275(7); Fe1–S1, 2.2476(7); Fe2–S1, 2.2707(7); Fe1–P1, 2.1656(7); Fe1–P2, 2.2356(7); Fe2–P3, 2.2078(7); Fe2–P4, 2.2249(7); Fe1–Fe2, 2.7453(5).
Cyclic Voltammetry of [Fe2(SR)(S)(CO)2(dppv)2]+
The cyclic voltammogram of [4]+ in CH2Cl2 solution exhibits one reversible one-electron reduction wave at −1.1 V (Figure 4). For all couples, the current ratios, ipc/ipa, were >0.9. When compared to the [3]+/0 couple, this value indicates that S2− stabilizes the oxidized state by 300 mV vs PhCH2S−. The data show that SCH2Ph substituents are nearly 100 mV less reducing that the analogous SMe complex (Table 1).
Figure 4.

Cyclic voltammograms of 2. 7 mM o f [Fe2(SBn)2(CO)2(dppv)2] ([3]0, black), [3]+ (blue), and [4]0 (red) in CH2Cl2 solutions with 0.125 M [Bu4N]PF6 electrolyte (scan rate = 100 mV/s). The presence of a small amount of [4]+ is evident in the sample of [3]+.
Table 1.
Reduction Potentials for [4]+, [3]+, [2]+, and [1]+ as Well as the Related 1,3-Propanedithiolate (pdt2−) Complex
| couple | potential, V vs Fc+/0 | ipa/ipc |
|---|---|---|
| [Fe2(SMe)2(CO)2(dppv)2]+/0 | −0.90 | 1 |
| [Fe2(SPh)2(CO)2(dppv)2]+/0 | −0.85 | 1 |
| [Fe2(SCH2Ph)2(CO)2(dppv)2]+/0 | −0.81 | 1 |
| [Fe2(pdt)(CO)2(dppv)2]+/0 | −0.83 | 1 |
| [Fe2(SCH2Ph)(S)(CO2(dppv)2])+/0 | −1.1 | 0.95 |
DFT Calculations on C–S Homolysis in [Fe2(SCH2Ph)2-(CO)6–x(PMe3)x]]0/+
Method Validation
The pure GGA functional BP86 has been widely adopted when dealing with hydrogenase-inspired Fe2S2 compounds, since it has proven to reliably reproduce their structural, spectroscopic, and redox parameters.21–26 Further calibration of the method was performed taking into account the reference compound 3. The potential of the couple [3]+/0 (in CH2Cl2) has been estimated at the BP86 level to be −0.800 V. The close match of this value to the experimental value of 0.81 V suggests that the pure functional BP86 can accurately describe the electronic structure of both S = 0 and 1/2 spin states of this class of compounds. Moreover, FT-IR νCO bands of both [3]0 and [3]+ simulated at the BP86 level are predicted to be 1895, 1855 cm−1 (exp. 1899, 1855 cm−1) and 1950, 1899 cm−1 (exp. 1942, 1904 cm−1).
To further validate our choice of functional, since we are dealing with chemical processes involving changes of spin multiplicity, we also tested the performance of a hybrid functional (namely, the GGA functional B3LYP). For the [3]+/0 redox potential, the calculated value is −1.231 V, which is far from the experimental value. In addition, to account for the effect of dispersive forces on the homolysis process, we completed the DFT picture by including results obtained at the B97-D/TZVP level. The B97-D functional, which has been developed to a priori include noncovalent interaction effects, has provided the best match with experimental data among “dispersion-based” approaches such as BP86-D3/TZVP and M06-L/TZVP schemes (for further details, see the DFT Methods section and the Supporting Information). Fe–Fe and Fe–S(avg) distances for 3 (eclipsed geometry structure) are very similar when optimized with BP86 vs B97-D (respectively, Fe–Fe: 2.665 and 2.625 Å and Fe–S(avg): 2.296 and 2.287 Å), although this distance is quite elongated with respect to the crystallographic result.
Results
To systematically clarify the effect of redox potential on the homolysis of the S–C bonds in diiron(I) μ-benzylthiolates, calculations examined the impact of replacing pairs of CO ligands with pairs of PMe3 ligands. In the parent complex Fe2(SCH2Ph)2(CO)6, the C–S bond dissociation free energy (BDFE) is only 27.3 kcal/mol at the BP86/TZVP level, and 29.6 kcal/mol at the B97-D/TZVP one (Figure 5). For comparison, the BDEs for HS-CH2Ph and HS-CH3 are 61.7(1.5) and 74.7(1.5) kcal/mol (298 K), respectively.27
Figure 5.

Relative free energies (kcal/mol at 298 K) associated with the homolytic cleavage of the S–C bond in the series of [Fe2(SCH2Ph)2(CO)6–x(PMe3)x]z (x = 0, 2, 4) as well as [Fe2(SCH2Ph)2(CO)2(dppv)2]z ([3]z). Both Fe(I)Fe(I) (z = 0) and Fe(II)Fe(I) (z = 1) redox states are considered.
The C–S bond in [Fe2(SCH2Ph)2(CO)6–x(PMe3)x]0 is weakened upon replacement of CO ligands by PMe3. Each PR3-for-CO substitution weakens the C–S bond by about 4–7 kcal/mol, depending on the level of theory. One-electron oxidation of the diiron complexes further weakens the C–S bond. At the BP86 level, the effect of oxidation is predicted to be greatest for the hexacarbonyl (23.7 vs 7.4 kcal/mol) and smallest for the tetraphosphine derivatives Fe2(SCH2Ph)2-(CO)6–x(PMe3)x (5.2 vs −2.5 kcal/mol) and Fe2(SCH2Ph)2-(CO)2(dppv)2 (1.2 vs −8.4 kcal/mol). When dispersion forces are considered (B97-D), the effect of oxidation is predicted to be almost constant upon CO vs P substitutions (about 16–18 kcal/mol). DFT results are qualitatively consistent with experimental observations. Indeed, the overall scenario remains unchanged when switching from BP86 (without dispersion) to B97-D (dispersion including) methods: the C–S bond is weakened by increasing the number of phosphine ligands and upon oxidation, becoming thus very labile for cationic tetra-substituted species, which is experimentally observed to spontaneously undergo C–S homolysis. In contrast, the complexes Fe2(SCH2Ph)2(CO)6, Fe2(SCH2Ph)2(CO)4(dppv), and Fe2(SCH2Ph)2(CO)2(dppv)2 were thermally stable. Attempts to prepare Fe2(SCH2Ph)2(CO)2(PMe3)4 were unsuccessful.
Calculations were also performed to gain mechanistic insight. Information on spin density distribution and SOMO is presented in Tables 2 and 3 for S = 1/2 FeIFeII species [5]0 and the rotated and unrotated isomers of [4]+. For [5]0, the inorganic sulfur carries most of the unpaired electron fraction.
Table 2.
Spin Densities (at the BP86/TZVP Level) for Selected Atoms in [Fe2(SCH2Ph)(S)L6]0, Rotated-[Fe2(SCH2Ph)2L6]+, and Unrotated-[Fe2(SCH2Ph)2L6]+a
| L6 | [Fe2(SCH2Ph)(S)L6] 0 | [Fe2(SCH2Ph)2L6] + (rot.) | [Fe2(SCH2Ph)2L6] + (unrot.) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|||||||||
| Fe1 | Fe2 | S2 | Fe1 | Fe2 | S1 | S2 | Fe1 | Fe2 | S1 | S2 | |
| (CO)6 | 0.26 | 0.26 | 0.47 | 0.14 | 0.70 | 0.08 | −0.01 | 0.20 | 0.53 | 0.12 | −0.01 |
| (CO)4(PMe3)2 | 0.34 | 0.28 | 0.35 | 0.12 | 0.85 | 0.02 | −0.01 | 0.41 | 0.42 | 0.06 | −0.01 |
| (CO)2(PMe3)4 | 0.40 | 0.34 | 0.27 | 0.09 | 0.89 | 0.03 | −0.02 | 0.38 | 0.54 | 0.07 | −0.01 |
| (CO)2(dppv)2 | 0.53 | 0.21 | 0.23 | 0.09 | 0.86 | 0.04 | −0.01 | 0.29 | 0.50 | 0.07 | −0.01 |
For [Fe2(SCH2Ph)(S)L6]0, values are shown for the inorganic sulfur. For rotated-[Fe2(SCH2Ph)2L6]+, Fe2 has the rotated geometry.
Table 3.
Single Atom Contributions to the SOMO (at the BP86/TZVP Level) for Selected Atoms in [Fe2(SCH2Ph)(S)L6]0, Rotated-[Fe2(SCH2Ph)2L6]+, and Unrotated-[Fe2(SCH2Ph)2L6]+a
| L6 | [Fe2(SCH2Ph)(S)L6]0 | [Fe2(SCH2Ph)2L6]+ (rot.) | [Fe2(SCH2Ph)2L6] + (unrot.) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
|
|
|
|||||||||
| Fe1 | Fe2 | S2 | Fe1 | Fe2 | S1 | S2 | Fe1 | Fe2 | S1 | S2 | |
| (CO)6 | 0.18 | 0.18 | 0.47 | 0.07 | 0.15 | 0.06 | 0.03 | 0.09 | 0.17 | 0.09 | 0.03 |
| (CO)4(PMe3)2 | 0.23 | 0.21 | 0.36 | 0.13 | 0.45 | 0.11 | 0.03 | 0.24 | 0.26 | 0.10 | 0.03 |
| (CO)2(PMe3)4 | 0.26 | 0.24 | 0.28 | 0.23 | 0.41 | 0.14 | 0.01 | 0.26 | 0.29 | 0.08 | 0.01 |
| (CO)2(dppv)2 | 0.30 | 0.19 | 0.27 | 0.17 | 0.43 | 0.12 | 0.02 | 0.21 | 0.25 | 0.14 | 0.03 |
For [Fe2(SCH2Ph)(S)L6]0, values are shown for the inorganic sulfur. For rotated-[Fe2(SCH2Ph)2L6]+, Fe2 has the rotated geometry.
Focusing on the mixed valence species related to [3]+, going from electron-poor to electron-rich derivatives does not affect the spin density distribution, which remains heavily localized on the rotated Fe. In fact, in the more electron-rich complexes, the unpaired electron is even more localized (the rotated Fe). The contribution of the rotated Fe to the SOMO increases when going from (CO)6 to (CO)4(PMe3)2, but little effect is observed in going from (CO)4(PMe3)2 to (CO)2(PMe3)4. We note that a small fraction of the SOMO in [Fe2(SCH2Ph)2-(CO)2(PMe3)4]+ is localized on the sulfur of the equatorial thiolate, but the one that does not undergo homolysis (Figure 6).
Figure 6.

SOMOs for [Fe2(SCH2Ph)2(CO)4(PMe3)2]+ and [Fe2(SCH2Ph)2(CO)2(PMe3)4]+ (BP86/TZVP optimized structures). Isosurface boundary = 0.05 au.
Carbonylation of [4]+
With 32 valence electrons, [4]+ is highly coordinatively unsaturated. Indeed, in solution, [4]+ binds not 1, but 2 equiv of CO (Scheme 7). The reaction occurs rapidly at room temperature at 1 atm. Previous examples of double carbonylation of metal complexes (without ligand displacement) are invariably associated with insertion of one CO into a metal–carbon bond.28 The dicarbonylation of [5]+ is reversible. When the decarbonylation was monitored by FT-IR spectroscopy, intermediates were not detected. The 31P NMR and IR spectra of [5]+ indicate multiple isomers.
Scheme 7.

Double Carbonylation of [4]+. Only the Crystallographically Verified Isomer of [5]+ Is Showna
aPh groups omitted for clarity.
In terms of its structure, [5]+ consists of an edge-shared bioctahedron (Figure 7). The Fe⋯Fe distance elongates from 2.7453(5) Å in [4]+ to 3.588(3) Å in [5]+. The Fe–S distances, both to the thiolate and especially the sulfide, are strongly affected, elongating to ca. 2.32 Å from 2.26 and 2.14 Å, respectively. Two octahedral Fe(II) centers are bridged by the dithiolate. For one Fe center, the two CO ligands are trans, and for the other center, the CO ligands are cis. Compounds of the type Fe(pdt)(CO)2(diphosphine) often exist as mixtures of isomers.29
Figure 7.

Structure of [Fe2(SBn)(S)(CO)4(dppv)2]+ ([5]+) with 50% probability ellipsoids and hydrogen atoms omitted for clarity. Selected bond distances (Å): Fe1–C27, 1.8125(16); Fe1–C28, 1.8171(16); Fe1–P1, 2.2263(4); Fe1–P2, 2.2521(4); Fe1–S2, 2.3256(4); Fe1– S1, 2.3432(4); Fe2–C37, 1.7916(15); Fe2–C36, 1.7985(16); Fe2– P4, 2.2494(4); Fe2–P3, 2.3059(4); Fe2–S1, 2.3336(4); Fe2–S2, 2.3398(4).
DISCUSSION
Hundreds of bis(monothiolate) complexes are known of the type Fe2(SR)2(CO)6;30 however, their substituted derivatives Fe2(SR)2(CO)6–xLx have received little attention. These substituted complexes are shown to adopt novel stereochemistry, a consequence of the axial-equatorial disposition of the organic substituents on sulfur. These substituted derivatives oxidize at mild potentials, which led to the discovery that the benzyl derivative [3]+ has a labile C–S bond.
Mechanism and Implications of the Dealkylation of [3]+
The finding that the benzyl derivative [3]+ is more labile than the methyl and phenylthiolates is reasonable in view of the relative bond dissociation energies of the HS–CH2Ph, HS– CH3, and HS–Ph bonds, which are 258.2 (6.3), 312.5 (4.2), 367.8 (6.3) kJ/mol.27
Regarding the mechanism of desulfurization, a free-radical pathway is indicated. The evidence includes diagnostic products (dibenzyl, toluene), the accelerating effect of TEMPO, and the nonreactivity of the SMe and SPh derivatives. The pathway for formation of CH2Ph radicals from [3]+ involves two geometric processes: (i) a small twisting of the “rotated” Fe(dppv)(CO) center to an octahedral geometry observed in [4]+ and (ii) breaking of the C–S bond (Scheme 8). The unrotation of mixed valence Fe(II)Fe(I) dithiolates is nearly barrierless in the few cases that have been investigated.31 When monitored by FT-IR and NMR spectroscopies, the conversion of [3]+ into [4]+ produced no detected intermediates.
Scheme 8.

Two Pathways for Debenzylation of [Fe2(SCH2Ph)2(CO)2(PMe3)4]+
The conversion of [3]+ → [4]+ is an example of a well-defined desulfurization of a thiolate by a metal complex. In this case, well-defined means that the precursor and products are well characterized and the reaction proceeds in good yields with good stoichiometry. The homolysis of C–S bonds is implicated in hydrodesulfurization (HDS) catalysis. The closest model for HDS of a thiol involves the reaction of an organoMo-Co cluster (eq 1).32,33
| (1) |
For eq 1 and related reactions,32–34 the discrete desulfurization step has not been observed. With regard to the conversion [3]+ → [4]+, our results point to two aspects of complexes and catalysts that facilitate C–S homolysis: (i) the significantly weakened C–S bond in bridging thiolate ligands and (ii) the requirement for redox-active metals that can accommodate the conversion Mn(μ-SR) → Mn(μ-S) + R•. The qualitative weakening of C–S bonds is implicated for a variety of tri-and tetrametallic complexes.32,33,35
New Members of [Fe2(SR)2–x(S)xL6]z Series
With [Fe2(SR)2(CO)6]0 as the best known members, organometallic 2Fe-2S complexes can be organized according to their oxidation states (Figure 8). The anions [Fe2(S)2–x(SH)x(CO)6](2–x)− are equivalent to [Fe2(SR)2(CO)6]0 with regards to the oxidation state of Fe.30,36–38 A mixed valence subset of this family take the form [Fe2(SR)2L6]+, manifested in synthetic models for the Hox state of the [FeFe]-hydrogenases.17–19 The diferrous members, i.e., [Fe2(SR)2–x(S)xL6](2–x)+, have not been observed previously but are represented by [4]+.
Figure 8.

Structure of known and hypothetical Fe2(S)2L6]z complexes in various oxidation states and degrees of alkylation.
SUMMARY
This report describes the properties of the bis(monothiolate)s [Fe2(SR)2(CO)2(dppv)2]n+, rare complexes of the type [Fe2(SR)2(CO)6–xLx]n+. The related chelating dithiolates have been heavily studied.5 Studies on the redox properties of bis(monothiolate)s are rare. Similarities obviously exist between the bis(monothiolate)s and the chelating dithiolates: the νCO band positions in the IR spectra and the E1/2 values are comparable. The distinguishing features of the bis-(monothiolate)s arise from the unsymmetrical disposition of the SR groups. The two Fe(CO)(PR3)2 centers are related by mirror symmetry. This unsymmetrical steric field rigidifies these [Fe(I)]2 complexes.
The second advance in this work is the discovery that [Fe2(SCH2Ph)2(CO)2(dppv)2]+ is prone to homolysis of one C–S bond. According to DFT analysis, the C–S bond in these μ-thiolate complexes is weakened, especially in more electron-rich derivatives. Oxidation of these complexes further weakens the C–S bond. According to our calculations, C–S bond strength follows the order HS-CH2Ph (61.7 kcal/mol) < Fe2(SCH2Ph)2(CO)6 < Fe2(SCH2Ph)2(CO)2(PMe3)4 < [Fe2-(SCH2Ph)2(CO)2(PMe3)4]+ or [Fe2(SCH2Ph)2(CO)2-(dppv)2]+, independent of the level of theory.
The third area of discovery involves the product of the C–S homolysis, the 32 e complex [Fe2(SCH2Ph)(S)(CO)2- (dppv)2]+. This complex, which is of the type [Fe2(SR)(S)L6]+, is unprecedented within the otherwise well-studied realm of low-spin 2Fe-2S compounds.30 The unusual character of the new sulfido-thiolate is shown by its unique ability to undergo reversible double decarbonylation.
EXPERIMENTAL SECTION
Materials and Methods
General procedures have been described previously.21 Reagents were used as received.39 Literature routes were followed to prepare Fe2(SMe)2(CO)6, Fe2(SCH2Ph)2(CO)6,40,41 and Fe2(SPh)2(CO)6.42 Photochemical reactions were conducted in Pyrex flasks; the light source was an array of 100 W LEDs emitting at 365 nm.
Cyclic voltammograms were recorded using a CH Instruments 760D Electrochemical workstation (Austin, TX). A standard three electrodes configuration was employed using glassy carbon (3 mm diameter) as the working electrode, a Pt wire as a counter electrode, and a “no leak” Ag/AgCl reference electrode (Warner Instruments, Hamden, CN). These reference electrodes have been calibrated after each experiment by adding ferrocene to the solution and recording its half-wave potential.
Fe2(SMe)2(CO)2(dppv)2 ([1]0)
A mixture of 0.10 g (0.27 mmol) of Fe2(SMe)2(CO)6 and 0.106 g (0.27 mmol) of dppv in 10 mL of benzene was heated at reflux for 2 h. The solvent was removed in vacuum; the residue was extracted into 2 mL of CH2Cl2. The product, Fe2(SMe)2(CO)4(dppv), precipitated as a light-brown solid upon the addition of 20 mL of pentane. Yield: 0.168 g (88%). A mixture of 0.10 g (0.14 mmol) of Fe2(SMe)2(CO)4(dppv) and 0.055 g (0.14 mmol) of dppv in 90 mL of toluene was irradiated at 365 nm until the conversion was complete (~2 h) as indicated by IR spectroscopy. Solvent was removed under vacuum, and the resulting residue was extracted into 2 mL of CH2CI2. Upon addition of 30 mL of pentane, the product precipitated as a green-brown solid. Yield: 0.125 g (85%). Anal. Calcd for C56H50Fe2O2P4S2·1.5CH2Cl2: C, 58.15; H, 4.32. Found: C, 58.34; H, 4.42. 1H NMR (500 MHz, CD2Cl2): δ 7.88–6.89, (m, 44H, 8C6H5, 2CH═CH), 0.26, 0.86 (s, 6H, 2CH3). 31P NMR (202 MHz, CD2Cl2): δ 89.2(d, Jp-p = 20 Hz), 85.6 (d, Jp-p = 20 Hz). IR (CH2Cl2): νCO = 1896, 1861 cm−1.
Fe2(SPh)2(CO)2(dppv)2 ([2]0)
A solution of 0.10 g (0.2 mmol) of Fe2(SPh)2(CO)6 in 10 mL of toluene was treated with a solution of 0.015 g (0.2 mmol) of Me3NO in 2 mL of CH3CN. After 15 min, 0.056 g (0.2 mmol) of dppv was added to the mixture, which was then stirred for 2 h. Solvent was removed under vacuum, and the resulting solid residue was extracted into 2 mL of CH2CI2. Upon addition of 30 mL of pentane to the reaction mixture, the product precipitated as a green-brown solid. Yield: 0.145 g (86%). A solution of 0.10 g (0.12 mmol) of Fe2(SPh)2(CO)4(dppv) and 0.047 g (0.12 mmol) of dppv in 90 mL of toluene was irradiated at 365 nm until the conversion was complete (~2 h) as indicated by IR spectroscopy. The solvent was removed under vacuum, and the resulting dark residue was washed with 10 mL of Et2O to remove other organoiron compounds. The residue was extracted into 2 mL of CH2CI2. Upon dilution of this extract with 30 mL of pentane, the product precipitated as a green-brown solid. Yield: 0.049 g (35%). Anal. Calcd for C66H54Fe2O2P4S2·CH2Cl2: C, 63.68; H, 4.47. Found: C, 63.53; H, 4.32. 1H NMR (500 MHz, CD2Cl2): δ 8.15–6.23, (m, 10C6H5, 2CH═CH). 31P NMR (202 MHz, CD2Cl2): δ 94.0(d, Jp-p = 20 Hz), 81.3 (d, Jp-p = 20 Hz). IR (CH2Cl2): νCO = 1900, 1851 cm−1.
Fe2(SCH2Ph)2(CO)2(dppv)2 ([3]0)
A mixture of 0.10 g (0.19 mmol) of Fe2(SCH2Ph)2(CO)6 and 0.15 g (0.38 mmol) of dppv in 90 mL of toluene was irradiated at 365 nm until the conversion was complete (~2 h) as indicated by IR spectroscopy. Solvent was removed under vacuum, and the resulting solid residue was extracted into 2 mL of CH2CI2. Addition of 30 mL of pentane precipitated a green-brown solid. Yield: 0.12 g (52%). Syntheses of related Fe2(SR)2(CO)2(dppv)2 complexes typically proceed in yields near 80%. We verified that Fe2(SR)2(CO)2(dppv)2 is sensitive to UV irradiation, which may explain the modest yields. 1H NMR (500 MHz, CD2Cl2): δ 7.86–5.97, (m, 54H, C6H5, CH═CH), 2.14, 2.23 (s, 4H, 2CH2). 31P NMR (202 MHz, CD2Cl2): δ 85.7(d, Jp-p = 20 Hz), 80.3 (d, Jp-p = 20 Hz). IR (CH2Cl2): νCO = 1899, 1855 cm−1. Single crystals were grown by slow diffusion of hexane into a CH2CI2 solution.
[Fe2(SCH2Ph)2(CO)2(dppv)2]BF4 ([3]BF4)
A stirred solution of 75 mg (0.06 mmol) of [3]0 in 3 mL of CH2CI2 at −40 °C was treated dropwise with a solution of 17 mg (0.06 mmol) of FcBF4 in 3 mL of CH2CI2. Within a few min., the solution color changed from green brown to dark green. The solution was concentrated to ~1 mL. A dark-green solid precipitated upon addition of 20 mL of pentane to this solution. Yield: 72 mg (90%). IR (CH2Cl2): νCO = 1942, 1904 cm−1. Anal. Calcd for C68H58Fe2O2P4S2BF4·CH2Cl2: C, 60.11; H, 4.39. Found: C, 60.48; H, 4.42. Crystals were grown by slow diffusion of hexane into a CH2CI2 solution.
[Fe2(SCH2Ph)(S)(CO)2(dppv)2]BF4 ([4]BF4)
A solution of 26 mg (0.02 mmol) of [3]BF4 in 3 mL of CH2CI2 was treated with a solution of 3 mg (0.02 mmol) of TEMPO in 1 mL of CH2CI2. After 4 h, the solution changed from dark green to dark red. The solution was concentrated to ~1 mL. A dark-red solid precipitated upon the addition of 20 mL of pentane to this concentrated solution. Yield: 18 mg (75%). 1H NMR (500 MHz, CD2Cl2): δ 8.20–6.87, (m, 49H, C6H5, 2CH═CH), 3.67 (s, 2H, CH2). 31P NMR (202 MHz, CD2Cl2): δ 101.7 (t, Jp-p = 20 Hz), 85.6 (t, Jp-p = 20 Hz). IR (CH2Cl2): νCO = 1958, 1936 cm−1. ESI-MS: m/z 1115.2 [M – BF4]+. Crystals were grown by slow diffusion of hexanes into a CH2CI2 solution. Characterization of TEMPO-CH2Ph: Yield: 3 mg (61%). 1H NMR (500 MHz, CD2Cl2): 7.27–7.38 (m, 5H, C6H5), 4.81 (s, 2H, Ph- CH2), 1.42–1.59 (m, 6H, CH2CH2CH2), 1.15, 1.26 (2s, 12H, CH3).43 HR ESI-MS: m/z 248.2003 [M + H] C16H25NO Calcd: 248.2014.
[Fe2(SCH2Ph)(S)(CO)4(dppv)2]BF4 ([5]BF4)
A solution of [4]BF4 (12 mg, 0.01 mmol) in 1 mL of CH2CI2 was purged with CO. After 1 min, the solution color changed from dark red to bright red, and the IR spectrum indicated a complete conversion. Yield: ~9.5 mg (80%). FT-IR (CH2Cl2): νCO = 2016, 1989, 1945, 1898 cm−1. 1H NMR (500 MHz, CD2Cl2): δ 8.05–6.56, (m, 49H, 9C6H5, 2CH═CH); 3.38, 3.35, 3.27, 3.15, 3.13 (m, 2H, CH2). 31P NMR (202 MHz, CD2Cl2): δ 88.6, 85.6, 84.9, 75.9, 74.4, 72.3, 71.8, 55.0. Anal. Calcd for C63H51Fe2O4P4S2BF4.1.5CH2Cl2: C, 56.0 (55.90); H, 4.08 (3.93). Crystals of [5]BF4 were grown by diffusion of CO-saturated hexane into the CH2Cl2 solution at room temperature. Purging a solution of [5]BF4 with N2 results in a color change from bright red to dark red and the appearance of IR bands characteristic of [4]+.
DFT Methods
Density Functional Theory (DFT) computations have been carried out with the TURBOMOLE 7.2 programs suite,44 by using the pure functional BP8645,46 and an all-electron valence triple-ζ basis set with polarization functions on all atoms (TZVP).47 This level of theory, which has proved to reliably reproduce structures, spectroscopic properties, and reactivity of hydrogenase-mimics,21–25 has been further validated by reproducing experimental IR bands and redox potential for the [3]+/0 (vide infra). In addition, computations regarding the S–C bond homolysis process have been also performed with the GGA functional B97-D, developed by Grimme to account for noncovalent interactions.48
Alternative approaches, such as Grimme’s empirical dispersion corrections (BP86-D3)49 and the use of Truhlar’s M06-L functional,50 have been also tested for the prediction of S–C homolysis energies. Results indicate that, while absolute ΔG values are quite sensitive to the functional choice, the overall picture predicted by the BP86 method is retained, independent of the level of theory. Indeed, S–C homolysis is always predicted to be facilitated upon oxidation and by increasing the number of P-ligands. Only results, however, obtained at the BP86/TZVP and B97-D/TZVP levels are indicative for a process that is spontaneous only for FeIIFeI tetra-substituted derivatives, in agreement with experiments. Hybrid functionals have not been used to evaluate homolysis ΔG’s, mainly because of their poor performances in reproducing redox potentials which has been verified in the present investigation (see values obtained with the B3LYP13–15 functional in the SI).45,51,52
The resolution-of-identity (RI)53 technique has been applied to speed up calculations. Geometry optimizations have been performed by means of energy gradient techniques, and full vibrational analysis has been carried out to further characterize each stationary point. The homolysis products [Fe2(SCH2Ph)(S)L6]0 have been treated as unrestricted open-shell doublets (S = 1/2), while [Fe2(SCH2Ph)(S)- L6]0 as overall unrestricted open-shell singlet (S = 0), following the Broken Symmetry (BS) approach.54,55 This approximation allows the treatment of antiferromagnetic spin couplings in the framework of the unrestricted formalism, by localizing opposite spins of the mono-determinant wave function in different parts of the molecule. High spin solutions have not been considered since they correspond to high energy structures. Free energy (ΔG) values have been obtained from the electronic KS-SCF energy considering three contributions to the total partition function (Q), qtranslational, qrotational, qvibrational, assuming that Q can be written as the product of them.56 To evaluate enthalpy and entropy contributions, the values for temperature and pressure have been set to 298.15 K and 1 bar, respectively. The scaling factor for the SCF wavenumbers was set either to 0.9914 (default value in the TURBOMOLE).44 Solvent was modeled according to the conductor-like screening model (COSMO)57,58 by considering a polarizable continuum medium with ε = 8.93 (CH2Cl2). Solvent effects have been included in the evaluation of homolysis for [3]0 and [3]+, their FT-IR νCO bands, and the potential of the [3]+/0 couple. The latter has been computed using the equation ΔG°solv(SCF) = −nFE°, where ΔG°solv refers to the free energy difference (respectively) between the optimized reduced and the oxidized structures (including an implicit solvent model), n is the number of electrons involved in the redox process, F is the Faraday constant, and E° is the standard absolute redox potential (which has subsequently been referred to the Fc+/Fc couple absolute potential, computed at the same level of theory). Both ΔE°solv and ΔG°solv have been used since it has been shown that the inclusion of entropic correction does not necessarily provide a better match of Fe2S2 experimental redox potentials.26 The computed redox potentials for [3]+/0 are E°(ΔE°solv) = −0.800 V and E°(ΔG°solv) = −0.731 V for BP86, E°(ΔE°solv) = −0.867 V and E°(ΔG°solv) = −0.799 V for B97-D, E°(ΔE°solv) = −1.126 V and E°(ΔG°solv) = −1.050 V for M06-L. Computed values at other levels of theory can be found in the Supporting Information.
Supplementary Material
Acknowledgments
This work was supported by GM-61153 from the National Institutes of Health.
Footnotes
Notes
The authors declare no competing financial interest.
Accession Codes
CCDC 1590590–1590593 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.inorgchem. 8b00094.
Spectroscopic data, coordinates for DFT-calculated structures (PDF)
Contributor Information
Qianli Li, School of Chemical Sciences, University of Illinois, Urbana, Illinois 61801, United States.
Noémie Lalaoui, School of Chemical Sciences, University of Illinois, Urbana, Illinois 61801, United States.
Toby J. Woods, School of Chemical Sciences, University of Illinois, Urbana, Illinois 61801, United States
Thomas B. Rauchfuss, School of Chemical Sciences, University of Illinois, Urbana, Illinois 61801, United States.
Federica Arrigoni, Department of Biotechnology and Biosciences, University of Milano-Bicocca, Piazza della Scienza 2, 20126-Milan, Italy.
Giuseppe Zampella, Department of Biotechnology and Biosciences, University of Milano-Bicocca, Piazza della Scienza 2, 20126-Milan, Italy.
References
- 1.Tyler DR. Mechanistic Aspects of Organometallic Radical Reactions. Prog Inorg Chem. 1988;36:125–194. [Google Scholar]
- 2.Torraca KE, McElwee-White L. Ligand-Centered Reactivity of Organometallic Radicals. Coord Chem Rev. 2000;206–207:469–491. [Google Scholar]
- 3.Poli R. Relationship Between One-Electron Transition-Metal Reactivity and Radical Polymerization Processes. Angew Chem, Int Ed. 2006;45:5058–5070. doi: 10.1002/anie.200503785. [DOI] [PubMed] [Google Scholar]
- 4.Lubitz W, Ogata H, Rüdiger O, Reijerse E. Hydrogenases. Chem Rev. 2014;114:4081–4148. doi: 10.1021/cr4005814. [DOI] [PubMed] [Google Scholar]
- 5.Schilter D, Camara JM, Huynh MT, Hammes-Schiffer S, Rauchfuss TB. Hydrogenase Enzymes and Their Synthetic Models: The Role of Metal Hydrides. Chem Rev. 2016;116:8693–8749. doi: 10.1021/acs.chemrev.6b00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Camara JM, Rauchfuss TB. Combining Acid–Base, Redox and Substrate Binding Functionalities to Give a Complete Model for the [FeFe]-Hydrogenase. Nat Chem. 2012;4:26–30. doi: 10.1038/nchem.1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Camara JM, Rauchfuss TB. Mild Redox Complementation Enables H2 Activation by [FeFe]-Hydrogenase Models. J Am Chem Soc. 2011;133:8098–8101. doi: 10.1021/ja201731q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ellgen PC, Gerlach JN. Kinetics and Mechanism of Substitution Reactions of Bis(mercaptotricarbonyliron) Complexes. Inorg Chem. 1973;12:2526–2532. [Google Scholar]
- 9.Mathieu R, Poilblanc R, Lemoine P, Gross M. Electrochemical Behavior and Chemical Oxidation Study of Thio- and Phosphido-Bridged Binuclear Iron Complexes. J Organomet Chem. 1979;165:243–252. [Google Scholar]
- 10.Byer AS, Shepard EM, Peters JW, Broderick JB. Radical S-Adenosyl-L-methionine Chemistry in the Synthesis of Hydrogenase and Nitrogenase Metal Cofactors. J Biol Chem. 2015;290:3987– 3994. doi: 10.1074/jbc.R114.578161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shepard EM, Byer AS, Betz JN, Peters JW, Broderick JB. A Redox Active [2Fe-2S] Cluster on the Hydrogenase Maturase HydF. Biochemistry. 2016;55:3514–3527. doi: 10.1021/acs.biochem.6b00528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shepard EM, Byer AS, Aggarwal P, Betz JN, Scott AG, Shisler K, Usselman RJ, Eaton GR, Eaton SS, Broderick JB. Electron Spin Relaxation and Biochemical Characterization of the Hydrogenase Maturase HydF: Insights into [2Fe-2S] and [4Fe-4S] Cluster Communication and Hydrogenase Activation. Biochemistry. 2017;56:3234–3247. doi: 10.1021/acs.biochem.7b00169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ghosh S, Hollingsworth N, Warren M, Holt KB, Hogarth G. Electrocatalytic Proton Reduction by [Fe(CO)2(κ1-dppv)(κ1- SAr)2] (dppv = cis-1,2-bis(diphenylphosphino)ethylene; Ar = C6F5, C6H5, C6H4CH3-p) Polyhedron. 2017;137:140–146. [Google Scholar]
- 14.Adams RD, Cotton FA, Cullen WR, Hunter DL, Mihichuk L. Fluxional Behavior of Some Dinuclear Iron and Cobalt Hexacarbonyl Compounds with Alkylsulfur and Dialkylphosphorus, -arsenic, -germanium, and -tin Bridges. Inorg Chem. 1975;14:1395– 1399. [Google Scholar]
- 15.Carroll ME, Barton BE, Rauchfuss TB, Carroll PJ. Synthetic Models for the Active Site of the [FeFe]-Hydrogenase: Catalytic Proton Reduction and the Structure of the Doubly Protonated Intermediate. J Am Chem Soc. 2012;134:18843–18852. doi: 10.1021/ja309216v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hogarth G, Kabir SE, Richards I. Diphosphine Mobility at a Binuclear Metal Center: A Concerted Double Trigonal-Twist in Bis(dithiolate) Complexes [M2(CO)4(μ-dppm){μ-S(CH2)nS}] (M = Fe, Ru; n = 2, 3) Organometallics. 2010;29:6559–6568. [Google Scholar]
- 17.Justice AK, De Gioia L, Nilges MJ, Rauchfuss TB, Wilson SR, Zampella G. Redox and Structural Properties of Mixed- Valence Models for the Active Site of the [FeFe]-Hydrogenase: Progress and Challenges. Inorg Chem. 2008;47:7405–7414. doi: 10.1021/ic8007552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu T, Darensbourg MY. A Mixed-Valent Fe(II)Fe(I), Diiron Complex Reproduces the Unique Rotated State of the [FeFe]-Hydrogenase Active Site. J Am Chem Soc. 2007;129:7008–7009. doi: 10.1021/ja071851a. [DOI] [PubMed] [Google Scholar]
- 19.Thomas CM, Liu T, Hall MB, Darensbourg MY. Series of Mixed Valent Fe(II)Fe(I) Complexes That Model the Hox State of [FeFe]Hydrogenase: Redox Properties, Density-Functional Theory Investigation, and Reactivities with Extrinsic CO. Inorg Chem. 2008;47:7009–7024. doi: 10.1021/ic800654a. [DOI] [PubMed] [Google Scholar]
- 20.Haines RJ, de Beer JA, Greatrex R. Reactions of Metal Carbonyl Derivatives. Part XIX. Halogenation Studies of Di-μ- alkylthio- and Di-μ-arylthio-bis(tricarbonyliron) and Their Substituted Derivatives. J Chem Soc, Dalton Trans. 1976:1749–1757. [Google Scholar]
- 21.Zhou X, Barton BE, Chambers GM, Rauchfuss TB, Arrigoni F, Zampella G. Preparation and Protonation of Fe2(pdt)- (CNR)6, Electron-Rich Analogues of Fe2(pdt)(CO)6. Inorg Chem. 2016;55:3401–3412. doi: 10.1021/acs.inorgchem.5b02789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zampella G, Bruschi M, Fantucci P, Razavet M, Pickett CJ, De Gioia L. Dissecting the Intimate Mechanism of Cyanation of {2Fe3S} Complexes Related to the Active Site of All-Iron Hydrogenases by DFT Analysis of Energetics, Transition States, Intermediates and Products in the Carbonyl Substitution Pathway. Chem - Eur J. 2005;11:509–520. doi: 10.1002/chem.200400442. [DOI] [PubMed] [Google Scholar]
- 23.Tard C, Liu X, Ibrahim SK, Bruschi M, De Gioia L, Davies SC, Yang X, Wang LS, Sawers G, Pickett CJ. Synthesis of the H-cluster Framework of Iron-Only Hydrogenase. Nature. 2005;433:610–613. doi: 10.1038/nature03298. [DOI] [PubMed] [Google Scholar]
- 24.Boyke CA, van der Vlugt JI, Rauchfuss TB, Wilson SR, Zampella G, De Gioia L. Diferrous Cyanides as Models for the Fe-only Hydrogenases. J Am Chem Soc. 2005;127:11010–11018. doi: 10.1021/ja051584d. [DOI] [PubMed] [Google Scholar]
- 25.Chambers GM, Rauchfuss TB, Arrigoni F, Zampella G. Effect of Pyramidalization of the M2(SR)2 Center: The Case of (C5H5)2Ni2(SR)2. Organometallics. 2016;35:836–846. [Google Scholar]
- 26.Filippi G, Arrigoni F, Bertini L, De Gioia L, Zampella G. DFT Dissection of the Reduction Step in H2 Catalytic Production by [FeFe]-Hydrogenase-Inspired Models: Can the Bridging Hydride Become More Reactive Than the Terminal Isomer? Inorg Chem. 2015;54:9529–9542. doi: 10.1021/acs.inorgchem.5b01495. [DOI] [PubMed] [Google Scholar]
- 27.Luo Y-R, Cheng J-P. In: Handbook of Chemistry and Physics. Rumble JR, editor. CRC Press; New York: 2017. [Google Scholar]
- 28.Atagi LM, Mayer JM. Reactions of the Tungsten-Carbyne Complex W(≡CMe)Cl(PMe3)4 with π-Acceptor Ligands: Carbon Monoxide, Alkynes, and Alkenes. Organometallics. 1994;13:4794– 4803. [Google Scholar]
- 29.Carroll ME, Chen J, Gray DE, Lansing JC, Rauchfuss TB, Schilter D, Volkers PI, Wilson SR. Ferrous Carbonyl Dithiolates as Precursors to FeFe, FeCo, and FeMn Carbonyl Dithiolates. Organometallics. 2014;33:858–867. doi: 10.1021/om400752a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y, Rauchfuss TB. Synthesis of Diiron(I) Dithiolato Carbonyl Complexes. Chem Rev. 2016;116:7043–7077. doi: 10.1021/acs.chemrev.5b00669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bruschi M, Greco C, Fantucci P, De Gioia L. Structural and Electronic Properties of the [FeFe] Hydrogenase H-Cluster in Different Redox and Protonation States. A DFT Investigation. Inorg Chem. 2008;47:6056–6071. doi: 10.1021/ic8006298. [DOI] [PubMed] [Google Scholar]
- 32.Curtis MD, Druker SH. Homolytic C–S Bond Scission in the Desulfurization of Aromatic and Aliphatic Thiols Mediated by a Mo/Co/S Cluster: Mechanistic Aspects Relevant to HDS Catalysis. J Am Chem Soc. 1997;119:1027–1036. [Google Scholar]
- 33.Dungey KE, Curtis MD. Homolytic C–S Bond Cleavage on a Heterogeneous Co/Mo/S Hydrodesulfurization Catalyst. J Am Chem Soc. 1997;119:842–843. [Google Scholar]
- 34.Markö L, Takács J. Trinuclear Metal Complexes. Inorg Synth. 1989;26:243–246. [Google Scholar]
- 35.Adams RD, Horvath IT, Mathur P, Segmueller BE. Cleavage of Carbon-Sulfur Bonds in Thiolato Ligands in Osmium Carbonyl Cluster Compounds. The Synthesis and Structural Characterization of H2Os6(CO)18(μ4-S)(μ3-S) and Two Isomers of H2Os6(CO)17(μ4-S)(μ3-S) Organometallics. 1983;2:996–1005. [Google Scholar]
- 36.Seyferth D, Kiwan AM, Sinn E. Sulfur-bridged Dimers Obtained from μ-Dithiobis(tricarbonyliron) J Organomet Chem. 1985;281:111–118. [Google Scholar]
- 37.Wu X, Bose KS, Sinn E, Averill BA. Isolation and X-Ray Structure of an Intermediate in the Reaction of (μ-S)2Fe2(CO)6 with Thiolates: the [(μ-S)(μ-S2-t-Bu)Fe2(CO)6]− Ion. Organometallics. 1989;8:251–253. [Google Scholar]
- 38.Franz JA, Lee SJ, Bowden TA, Alnajjar MS, Appel AM, Birnbaum JC, Bitterwolf TE, Dupuis M. Activation of the S-H Group in Fe(μ2-SH)Fe Clusters: S-H Bond Strengths and Free Radical Reactivity of the Fe(μ2-SH)Fe Cluster. J Am Chem Soc. 2009;131:15212–15224. doi: 10.1021/ja904602p. [DOI] [PubMed] [Google Scholar]
- 39.Ulloa OA, Huynh MT, Richers CP, Bertke JA, Nilges MJ, Hammes-Schiffer S, Rauchfuss TB. Mechanism of H2 Production by Models for the [NiFe]-Hydrogenases: Role of Reduced Hydrides. J Am Chem Soc. 2016;138:9234–9245. doi: 10.1021/jacs.6b04579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nametkin NS, Tyurin VD, Kukina MA. Synthesis and Some Properties of Sulfur-Containing Iron Tricarbonyl Complexes. J Organomet Chem. 1978;149:355–370. [Google Scholar]
- 41.Haley AL, Broadbent LN, McDaniel LS, Heckman ST, Hinkle CH, Gerasimchuk NN, Hershberger JC, Mebi CA. [Fe–Fe] Hydrogenase Models: Iron(I)-Carbonyl Clusters Coupled to alpha-para-Toluenethiolate Ligands. Polyhedron. 2016;114:218– 224. [Google Scholar]
- 42.Si Y, Hu M, Chen C. Diiron Models for Active Site of FeFe- Hydrogenase with Aromatic Thiolate Bridges: Structures and Electrochemistry. C R Chim. 2008;11:932–937. [Google Scholar]
- 43.Nomura M, Takayama C, Kajitani M. Electrochemical Behavior of Nickeladithiolene S,S′-Dialkyl Adducts: Evidence for the Formation of a Metalladithiolene Radical by Electrochemical Redox Reactions. Inorg Chem. 2003;42:6441–6446. doi: 10.1021/ic034402u. [DOI] [PubMed] [Google Scholar]
- 44.Ahlrichs R, Bär M, Häser M, Horn H, Kölmel C. Electronic structure calculations on workstation computers: The program system turbomole. Chem Phys Lett. 1989;162:165–169. [Google Scholar]
- 45.Becke AD. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behaviour. Phys Rev A: At, Mol, Opt Phys. 1988;38:3098–3100. doi: 10.1103/physreva.38.3098. [DOI] [PubMed] [Google Scholar]
- 46.Perdew JP. Density-Functional Approximation for the Correlation-energy of the Inhomogenous Electron Gas. Phys Rev B: Condens Matter Mater Phys. 1986;33:8822–8824. doi: 10.1103/physrevb.33.8822. [DOI] [PubMed] [Google Scholar]
- 47.Schäfer A, Huber C, Ahlrichs R. Fully Optimized Contracted Gaussian Basis Sets of Triple Zeta Valence Quality for Atoms Li to Kr. J Chem Phys. 1994;100:5829–5835. [Google Scholar]
- 48.Grimme S. Semiempirical GGA-type Density Functional Constructed with a Long-Range Dispersion Correction. J Comput Chem. 2006;27:1787–1799. doi: 10.1002/jcc.20495. [DOI] [PubMed] [Google Scholar]
- 49.Grimme S, Antony J, Ehrlich S, Krieg H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J Chem Phys. 2010;132:154104. doi: 10.1063/1.3382344. [DOI] [PubMed] [Google Scholar]
- 50.Zhao Y, Truhlar DG. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Non-covalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-class Functionals and 12 Other Functionals. Theor Chem Acc. 2008;120:215–241. [Google Scholar]
- 51.Becke AD. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J Chem Phys. 1993;98:5648–5652. [Google Scholar]
- 52.Lee C, Yang W, Parr RG. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys Rev B: Condens Matter Mater Phys. 1988;37:785–789. doi: 10.1103/physrevb.37.785. [DOI] [PubMed] [Google Scholar]
- 53.Eichkorn K, Weigend F, Treutler O, Ahlrichs R. Auxiliary Basis Sets for Main Row Atoms and Transition Metals and Their Use to Approximate Coulomb Potentials. Theor Chem Acc. 1997;97:119–124. [Google Scholar]
- 54.Noodleman L, Norman JG., Jr The Xα Valence Bond Theory of Weak Electronic Coupling. Application to the Low-Lying States of Mo2Cl84−. J Chem Phys. 1979;70:4903–4906. [Google Scholar]
- 55.Noodleman L. Valence Bond Description of Antiferromagnetic Coupling in Transition Metal Dimers. J Chem Phys. 1981;74:5737– 5743. [Google Scholar]
- 56.Jensen F. Introduction to Computational Chemistry. John Wiley & Sons; Chichester, England: 1999. [Google Scholar]
- 57.Klamt A. Conductor-like Screening Model for Real Solvents: A New Approach to the Quantitative Calculation of Solvation Phenomena. J Phys Chem. 1995;99:2224–2235. [Google Scholar]
- 58.Klamt A. Calculation of UV/Vis Spectra in Solution. J Phys Chem. 1996;100:3349–3353. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.