

ABSTRACT
Tissue transglutaminase (TG2) is the ubiquitously expressed member of transglutaminase family and shown to play a critical role in the development and progression of drug resistance malignancies. We have previously showed the association of TG2 upregulation with progression and metastasis of renal cell carcinoma (RCC) and low disease-free survival. In the present study we further investigate the role of TG2 in cell adhesion, migration and invasion of RCC by silencing TG2 expression in Caki-2 and A-498 primary site and Caki-1 and ACHN metastatic site RCC cell lines. Downregulation of TG2 expression led up to a 60% decrease in actin stress fiber formation and adhesion to β 1 integrin (ITGB1) substrates fibronectin, collagen type I and laminin in both primary and metastatic site RCC cell lines. In addition, treatment with siRNAs against TG2 impaired the migration capacity and cellular invasiveness of ITGB1 substrates in all 4 RCC cell lines. Lastly, the knockdown of TG2 in metastatic Caki-1 cells diminished the expression of CD44, CD73-and CD105 cancer stem cell-like markers. We conclude, for the first time, that TG2 expression is critical for cancer cell adhesion, migration, invasiveness and cancer cell-stemness during RCC progression and dissemination. Therefore, combined targeting of TG2 with drugs widely used in the treatment of RCC may be a promising therapeutic strategy for RCC.
KEYWORDS: cell adhesion, cancer cell stemness, invasion, renal cell carcinoma, tissue transglutaminase
Introduction
Tumor cell migration and invasion can transform a primary tumor into a metastatic and life-threatening disease, which is mostly resistant to therapeutic agents. Development of metastasis is considered as a heterogeneous and adaptive process that is controlled by signaling cascades responsible for the epithelial mesenchymal transition (EMT) and changes in cytoskeletal dynamics.1 During the initial steps of metastasis, cancer cells change their morphology and cytoskeletal organization to facilitate cell locomotion and generate mechanical forces necessary for the extracellular matrix (ECM) remodeling by differentially modulating their cell surface receptors, mainly integrins, and their matrix metalloproteinases profile.2
With their noncovalently linked heterodimers of α and β subunits, integrins are one of the main cell adhesion proteins responsible from the regulation of adhesion, migration, and invasion of ECM proteins including fibronectin (FN), collagen, and laminin.3 In return, due to the association of the integrin β cytoplasmic domain with the cytoskeleton and signaling complexes, integrins can contribute to ECM remodeling and cell survival.4 Reduction in the integrin-ECM interaction triggers apoptosis (“anoikis”), which can be suppressed by tissue transglutaminase (TG2) in complex with FN serving as a cell adhesion protein.5 Of the transglutaminases, TG2 is ubiquitously expressed in mammalian tissues and takes a role in biological processes such as cell adhesion, survival and apoptosis.6 The most well-known function of TG2 is the Ca2+ dependent catalysis of non-degradable Nε(γ-glutamyl)lysine isopeptide bonds between proteins.7 TG2s other pleiotropic functions include GTPase/ATPase, protein disulfide isomerase, and protein kinase activity.6,8 Moreover, independent from its catalytic activity, TG2 once bound to FN can act as a ligand for the heparan sulfate chains of syndecan-4 (SDC4) and lead to the activation of β1 integrin (ITGB1) through inside-out signaling.9
While decreased levels of cell surface TG2 has been thought to induce the migration potential of cancer cells at the early stages of tumor progression,10 the upregulation of TG2 has been associated with the late-stage aggressive metastatic cancer phenotype and increased drug resistance in cancers with epithelial origin such as breast,11-13 ovarian,14 pancreatic cancer15 and most recently renal cell carcinoma.16-19 Additionally it was shown that the aberrant expression of TG2 might induce stem cell properties in cancer cells of breast, ovarian and skin origin by favoring the transition from the epithelial characteristics to the mesenchymal phenotype.20-22
Renal Cell Carcinoma (RCC) is the most common type of kidney carcinoma that comprises 90% of kidney cancer cases. Mortality to incidence ratio of RCC is higher than any other urological cancer and RCC is more resistant to radiotherapy and chemotherapy.23 By analyzing gene expression levels in 95 primary tumor samples, our recent study showed, for the first time, that TG2 together with the cell adhesion receptors ITGB1 and SDC4 drives the development and progression of metastatic RCC.16 Findings from several other research groups corroborated our findings and demonstrated a strong association of increased TG2 expression with poor RCC prognosis and metastatic disease in addition to the reduced disease outcome and low cancer-specific survival.17-19
Given that TG2 cooperatively operates with β 1 integrins in the organization of cell adhesion and migration,6,9 here we have further investigated the main role of TG2 in cell adhesion, migration and invasion of RCC using primary site A-498 and Caki-2 and metastatic site Caki-1 and ACHN RCC cell lines. siRNA downregulation of TG2 was performed to assess the significance of TG2 in cell adhesion, migration and invasion of all 4 RCC cell lines on β 1 integrin substrates of FN, collagen type I (Col1) and laminin (LM). To analyze the long-term inhibitory effect of TG2 on the expression of cancer stem cell (CSC) antigenic markers, metastatic Caki-1 cells possessing high CSC surface markers was transduced with TG2-specific shRNA-expressing lentivirus. Our data suggest for the first time that the silencing of TG2 impaired the cellular adhesion, migration, and invasiveness of all 4 RCC cell lines on ITGB1 substrates and decreased the expression RCC CSC markers.
Results
Importance of TG2 for actin stress fiber assembly in RCC cell lines
Detection of low TG2 activity levels in the metastatic RCC tumors that are characterized with high co-expression levels of TG2 with ITGB1 and SDC4 suggested that the interaction of TG2 with cell surface adhesion receptors may result in the loss of enzyme activity and force TG2 to act as a cell adhesion protein.16 To investigate the possible role of TG2 in the regulation of RCC cell adhesion, actin stress fiber formation was investigated in the primary site originated Caki-2 and A-498, and metastatic Caki-1 and ACHN RCC cell lines. For these experiments, endogenous TG2 was silenced in these cell lines using targeted siRNAs (siR1 and siR6) and the formation of actin stress fibers in TG2 knockdown cells were analyzed by fluorescence microscopy using FITC-phalloidin staining.
To eliminate the possible siR off-target effects and achieve a high silencing efficacy 2 individual siRs were used against the TGM2 gene. Densitometric analysis of TG2 protein levels with Western blots showed that protein levels of TG2 were decreased to 43% of the non-treated control values by siR1 and 6 in Caki-2 cells, while a 63% and 43% decrease was recorded for that of TG2 in A-498 cells following siR1 and 6 treatment, respectively (Fig. 1A top). When compared with the control, specific silencing of TG2 by siR1 and siR6 led to a more than 90% decrease in the protein levels of TG2 in Caki-1 cells, while an approximate 60% reduction was evident in the protein expression of TG2 in ACHN cells after siR1 and siR6 treatment (Fig. 1A bottom). There was no statistically significant difference detected between NS siR treated and non-treated control cells for the protein (Fig. 1A). Similar results were obtained for mRNA expression levels of TG2 following siR treatment (Supplementary Figure 1A). The expression levels of SCD4 and ITGB1, recently identified cell adhesion partners of TG2, were not significantly affected by the siR silencing of TG2 (Supplementary Figure 1B & C).
Figure 1.
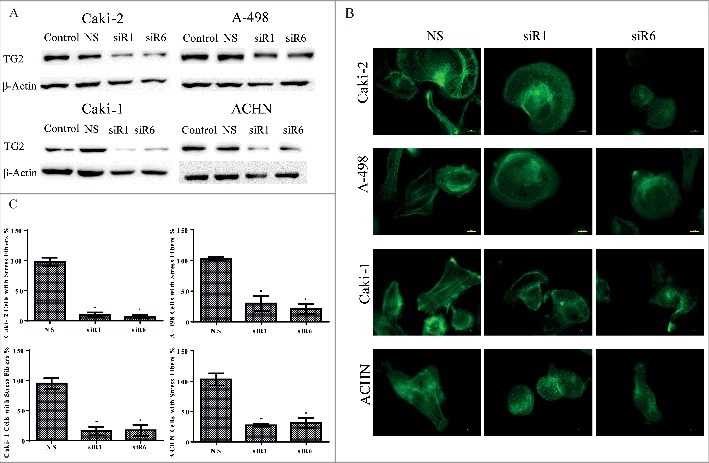
Down-regulation of TG2 reduces actin stress fiber formation in the primary site and metastatic RCC cell lines. (A) Efficiency of siRNA transfection on downregulation of TG2 in Caki-2 and A-498 primary RCC line lines and Caki-1 and ACHN metastatic RCC lines was analyzed by Western blot using actin as the control loading protein. (B) Changes in actin cytoskeleton organization in RCC cell lines in response to TG2 silencing was examined under florescence microscopy using an FITC filter. Bars, 10 µm. Following the treatment of RCC cell lines with NS siRNA and siRNAs against TG2 (siR1 and siR6), cells were seeded on tissue culture plastic for 60 mins and actin stress fibers were stained using FITC-phalloidin as described in Experimental Procedures. The downregulation level of TG2 was determined with gene expression analysis run in parallel with each experiment. (C) Non-overlapping images of 10 random fields/sample were acquired and the number of cells with actin stress fibers were scored. Data values represent the mean percentage of cells with formed actin stress fibers from 3 independent experiments, which were expressed as the percentage of control values. Mean percentage value of stress fiber positive cells treated with NS was used as 100% for each cell line.
A notable alteration in means of stress fiber formation was examined between the non-targeted control siR and TG2 targeted siR treated cells after 60 minutes of cell seeding (Fig. 1B & C). Similar to the non-treated cells, NS siR treated cells displayed well-developed stress fibers formed by dense actin networks in all 4 RCC cell lines. However, TG2 silenced cells failed to form a fibrillar appearance made up by dense stress fibers.
NS siR treated control Caki-2 cells exhibited peripheral actin polymerization with few cortical stress fibers, while only 7–10% of cells displayed actin stress fiber formation after TG2 siR1 and 6 treatment (Fig. 1B & C). A-498 cells treated with NS siR demonstrated mostly longitudinal stress fibers forming medium-thick bundles (Fig. 1B). Following the treatment of A-498 cells with TG2 targeted siRs, a 70% decrease for siR1 and a 79% decrease for siR6 was observed in the number of cells with actin stress formation (Fig. 1C). Compared to the primary site originated RCC cell lines Caki-2 and A-498, the metastatic RCC cell lines Caki-1 and ACHN exhibited longer actin stress fibers mostly stretching across the cell in a thick and condensed fibrillary form (Fig. 1B). siR1 and siR6 treatment strongly inhibited the actin stress formation in Caki-1 cells, by 84%, compared with NS siR treated cells (Fig. 1C). Likewise, a respective 73% and 69% decrease in actin stress fiber assembly was seen in siR1 and 6 treated ACHN cells (Fig. 1C). Taken together, these results suggest that TG2 is an essential component for the cell machinery controlling the formation of organized actin stress fibers in all 4 RCC cell lines.
Role of TG2 in RCC cell adhesion onto β1 integrin matrix substrates
Based on the accumulated findings, TG2 interaction with β1 and β3 integrins or TG2-FN heterocomplex recognized by SDC4 could mediate cell adhesion and migration by inducing the integrin survival signaling.5,9,11,24,25 Given that elevated expression of TG2 along with ITGB1 upregulation increased the risk of RCC metastasis,16,17 using siR treated and non-treated control cells, adhesion assays were undertaken to highlight the potential importance of TG2 in RCC cell attachment on β1 integrin-binding matrix substrates including FN, Col1 and LM. No statistically significant difference was detected between the cell adhesion of non-treated and NS siR treated RCC cells on β1 integrin matrix substrates, therefore only the results for the cell adhesion (attachment and spreading) for NS siR treated cells together with siR1 and siR6 treated cells were used in the construction of graphs (Fig. 2 and 3).
Figure 2.
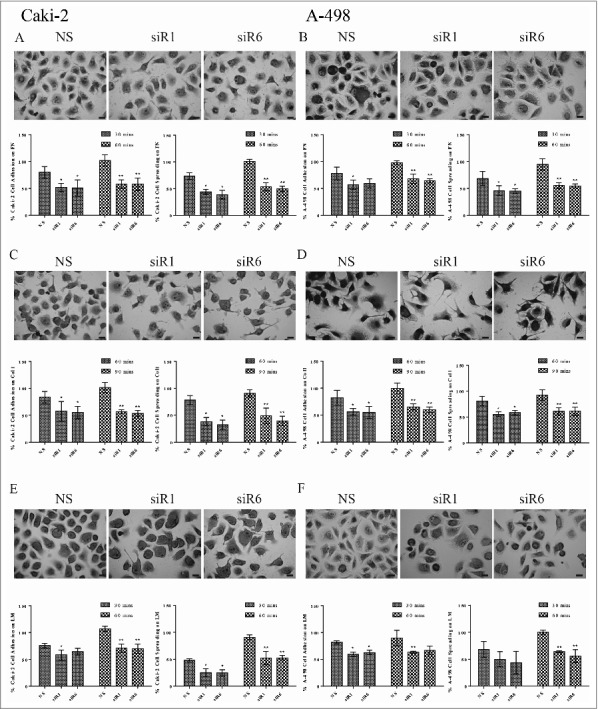
TG2 is required to support cell adhesion and spreading of the primary site RCC cell lines Caki-2 and A-498 on integrin β1 substrates FN, Col 1, LM. The percentage of attached cells or spread cells ± _SD shown were from the mean of at least 3 independent experiments, performed in triplicate. As described in Experimental Procedures, NS siRNA, siRNA 1 (siR1) and siRNA 6 (siR6) treated Caki-2 cells seeded on (A) FN, (C) Col1, (E) LM and A-498 cells seeded on (B) FN, (D) Col1, (F) LM were allowed to attach for the indicated early (30 mins for FN and LM, and 60 mins for Col1) and the late (60 mins for FN and LM, and 90 mins for Col1) time points. Cell attachment or spreading of control cells (cells treated with NS siRNA) on integrin β1 substrates recorded for the late time point represents 100%. Bars, 20 µm.
Figure 3.
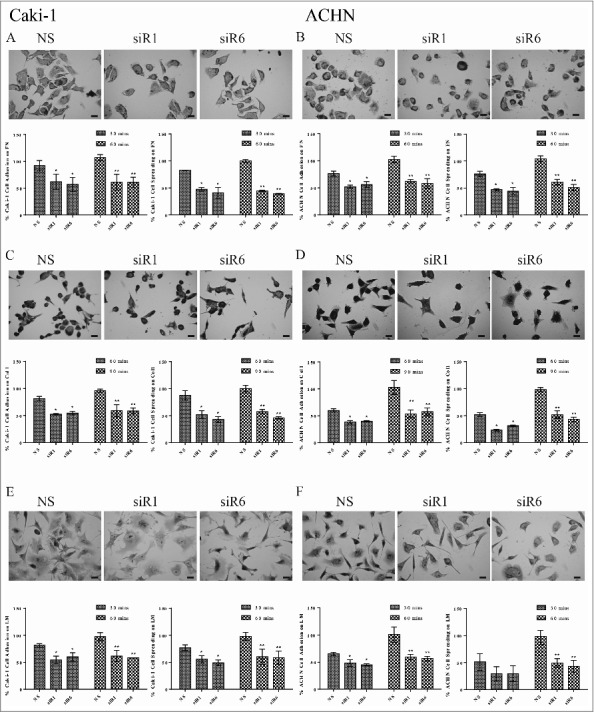
TG2 is also necessary for the cell adhesion and spreading of the metastatic RCC cell lines Caki-1 and ACHN on integrin β1 substrates FN, Col 1, LM. Means of at least 3 independent experiments, performed in triplicate was used to calculate the percentage of attached cells or spread cells ± _SD. NS siRNA, siRNA 1 (siR1) and siRNA 6 (siR6) treated Caki-1 cells seeded on (A) FN, (C) Col1, (E) LM and ACHN cells seeded on (B) FN, (D) Col1, (F) LM were allowed to attach for indicated early (30 mins for FN and LM, and 60 mins for Col1) and late (60 mins for FN and LM, and 90 mins for Col 1) time points as described in the Experimental Procedures. Cell attachment or spreading obtained at the late time point for cells treated with NS siRNA on integrin β1 substrates was taken as 100%. Bars, 20 µm.
Primary site originated cancer cells, Caki-2 and A-498, exhibited a higher adhesion potential on Col1 than on FN and LM, consecutively (Fig. 2). NS siR treated Caki-2 cells displayed 80% cell attachment and 74% spreading on FN at the early time point of 30 mins, when compared with the cell adhesion recorded for the late time point of 60 mins (Fig. 2A). Similar values were obtained for the NS siR treated A-498 cells with 78% and 69% FN-cell attachment and spreading at the early time point (Fig. 2B). TG2 downregulation in Caki-2 cells led to a 45% loss of cell attachment and an average 54% decrease in cell spreading on FN at both time points (Fig. 2A). At the early time point, TG2 silencing led to a respective reduction in the cell attachment and spreading of A-498 cells by 42% and 54%, which was increased further by 8% in the late time point (Fig. 2B). NS siR treated Caki-2 and A-498 cell lines displayed an ∼83% cell attachment and ∼80% cell spreading on Col1 at the early time point of 60 mins, while an average of 44% decrease was recorded in cell attachment of siR1and6 treated Caki-2 and A-498 cells on Col1 at 60 mins (Fig. 2C and D). Silencing of TG2 resulted in a 45% and 56% reduction in Caki-2 cell attachment and spreading, respectively (Fig. 2C), when an average of 62% cell adhesion was evident in siR1and6 treated A-498 cells at the late time point of 90 mins (Fig. 2D). In contrast to FN and Col1, a lower cell adhesion on LM was observed for Caki-2 and A-498 cells, yet TG2 downregulation still significantly decreased the cell attachment and spreading rates on this matrix (Fig. 2E & F). TG2 silencing reduced the cell attachment levels of Caki-2 cells on LM to 61% and 70% of cell attachment recorded for the control non-treated cells at the early 30min- and late 60 min-time points, respectively (Fig. 2E). TG2 downregulation in Caki-2 cells respectively decreased the spreading levels on LM to 25% and 52% at 30 and 60 mins. NS siR treated A-498 cells displayed a higher adhesion potential on LM when compared with NS siR treated Caki-2 cells, where the cell attachment and spreading was respectively recorded as 82% and 68% in the early time point. TG2 silencing led to an average of 37% decrease in the attachment of A-498 cells on LM at both time points (Fig. 2F). Interestingly, the treatment with siRs did not lead to a significant decrease in the cell spreading of A-498 cells on LM at the early time point, however, a significant 40% decrease was measured in the spreading of A-498 cells on LM at the late time point.
Similar to Caki-2 and A498 cells, TG2 was shown to be necessary for the cell adhesion machinery of Caki-1 and ACHN metastatic RCC cell lines (Fig. 3). When compared with the other 3 RCC cell lines, Caki-1 cells displayed the highest cell attachment and spreading level on FN with 92% and 82%, respectively, for the early time point of 30 mins (Fig. 3A). In this cell line, TG2 downregulation reduced the degree of cell attachment and spreading on FN by ∼40% in early and an average of 57% in the late time points. Analysis of NS siR treated ACHN cells showed a 76% cell attachment and spreading on FN at 30 mins, while TG2 downregulation decreased the potential of cell attachment to 57% and spreading to 50% at the early and late time points (Fig. 3B). Caki-1 attachment and spreading on Col1 was determined as an average of 85% at the early time point whereas TG2 downregulation led to a 47% and 53% decrease in cell adhesion at 60 mins with no significant increase recorded for the late time point of 90 mins (Fig. 3C). At the early time point, ACHN cells exhibited a 60% cell attachment and 52% spreading on Col1, which were brought down to 40% and 30% respectively, with siR1and6 treatment. A 16% increase in cell attachment and 20% increase in cell spreading on Col1 was evident for the siR1and6 treated ACHN cells for 90 mins (Fig. 3D). NS siR treated Caki-1 cells showed a 82% attachment and 77% spreading on LM whereas TG2 downregulation led to an approximate 45% decrease both in attachment and spreading at the early time point. Once more, a further 30 mins-incubation only led to an insignificant 5% increase in the cell adhesion potential of TG2 silenced Caki-1 cells on LM substrate (Fig. 3E). ACHN cells treated with NS siR, demonstrated 65% cell attachment and 51% cell spreading on LM at the early time point, while TG2 silencing led to a 54% and 42% decrease in cell attachment at 30 mins. Although no significant difference was detected between the NS-treated and siR1and6 treated cell spreading on LM at the early time point, TG2 downregulation led to a 54% decrease in the ACHN cell spreading on LM at the late time point of 60 mins (Fig. 3F). Taken together, our data suggested that TG2 plays an important role in the cell attachment and spreading of primary site-originated and metastatic RCC cell lines on β1 integrin-binding matrix substrates.
Effect of TG2 downregulation in the migration potential of RCC cells
To determine whether TG2 plays an essential role in the primary and metastatic RCC cell migration, wound scratch assays were performed using siR treated and non-treated RCC cells under low serum concentrations to limit cell proliferation during the experiment. Quantitative analysis of images taken from the scratched area revealed that TG2 silencing reduced cell motility in each of the 4 RCC cell lines compared with control NS siR treated cells, which showed no detectable difference from the migration potential of the non-treated cells (Fig. 4). It was found that all of the RCC cells totally closed the scratch area after 48 hours, therefore the experiment was performed at 24 and 48 hours, which were defined as early and late time points.
Figure 4.
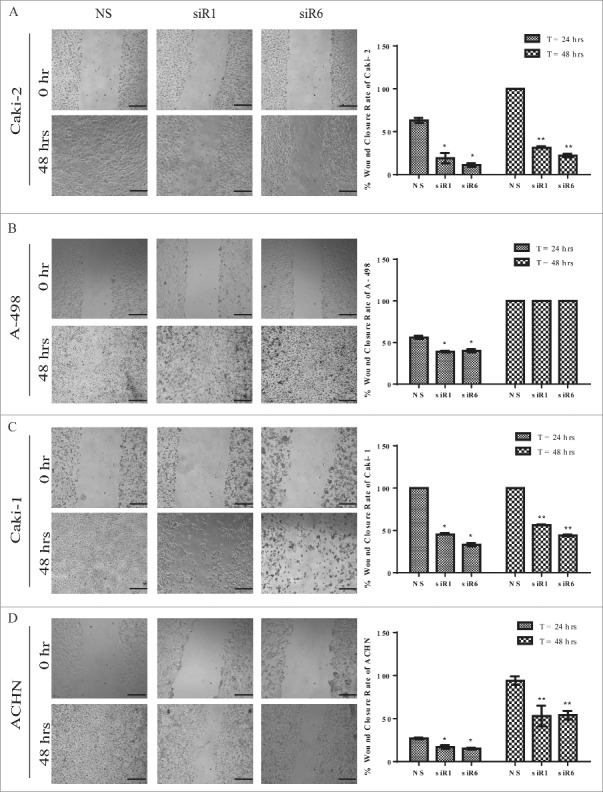
Silencing of TG2 delays or impairs the migration of RCC cell lines (A) Caki-2 (B) A-498, (C) Caki-1, and (D) ACHN in wound scratch assay. Percentage wound closure rate was expressed as the percentage of non-silencing RNA (NS) treated cells for each RCC cell line. Data values represent average of 3 independent experiments. Non-overlapping images of the whole wound area for each well were captured at the time of 0, 24 and 48 hours, as described in Experimental Procedures. A complete wound closure was detected for each cell line at 48 hours. Representative images were shown for each RCC cell line at the time of 0 and 48 hours after wounding. Bar, 500 µm.
While 63% of the wound area was closed by NS siR treated Caki-2 cells at 24 hours, only 19% and 11% of wound closure was measured for Caki-2 cells treated with siR1 and siR6, respectively. At the end of 48 hours, while the control cells completely closed the scratch, siR1 and siR6 treated Caki-2 cells had only covered the wound area by 31% and 22%, respectively (Fig. 4A). In A-498 cells the downregulation of TG2 resulted in a delayed response in cell migration, in that, at the end of 48 hours both TG2 targeted and NS siR treated A-498 cells re-covered 100% of the wound area. At the early time point, siR1 and siR6 treatment led to a significant 17% reduction in the migration rate of A-498 cells into the wound area when compared with the NS siR treated control cells (Fig. 4B). Caki-1 cells displayed an accelerated wound closure rate in comparison to the other 3 RCC cell lines, such that, NS siR treated Caki-1 cells totally closed the wound area even at the early time point of 24 hours. The siR1 and siR6 treated Caki-1 cells only recovered 42% and 50% of wound area at 24 and 48 hours, respectively (Fig 4C). ACHN exhibited the slowest wound closure rate among the other RCC cells by only closing 27% of the wound area at 24 hours (Fig. 4D). TG2 downregulation affected ACHN cell migration approximately 50% at both time points when compared with the control NS siR treated cells. The siR1 and siR6 treatment led to a 10% reduction in the migratory potential of ACHN cells at 24 hours, which was increased to 47% at the end of 48 hours. These results suggest that downregulation of TG2 is associated with the repression of cell motility of RCC cell lines.
Regulation of RCC cell invasion and migration in β1 integrin matrix substrates by TG2
Recent studies indicated that an increase in the expression of TG2 is associated with accelerated cancer cell migration and invasion during metastasis.26 Given that TG2 silencing significantly suppressed the actin stress fiber formation and markedly reduced the cell adhesion of RCC cell lines on β1 integrin substrates, we next explored the effect of TG2 downregulation on cellular invasion of RCC cell lines through FN, Col1 and LM. For this purpose, TG2 silenced and non-silenced control cells were seeded on the upper chamber of transwells coated with these β1 integrin matrix substrates and allowed to transmigrate at the predetermined time points, which were chosen according to the time when the initial invasion (early time point) has started and completed (late time point). For the evaluation of invasion potential, percentage values were determined by considering the number of control NS cell migration recorded at the late time point as 100%.
Among all 4 RCC cell lines, NS siR treated Caki-2 cells exhibited the highest invasion potential on FN by 341 cells, while an average of 236 cells was recorded for A-498, Caki-1 and ACHN at the early time point (Fig. 5). Treatment of Caki-2 cells with siR1 and siR6 inhibited FN invasion at a level of 63% and 46% in the early and late time point, respectively. TG2 targeted siR treatment led to an 80% reduction in the FN invasion potential of A-498 and Caki-1 cells at the end of 3 hours, which was increased by 15% for A-498 and by 10% for Caki-1 at 6 hours. Invasion thorough the FN matrix was inhibited by 60% at 3 hours and by 50% at 6 hours by the downregulation of TG2 in ACHN cells (Fig 5A).
Figure 5.
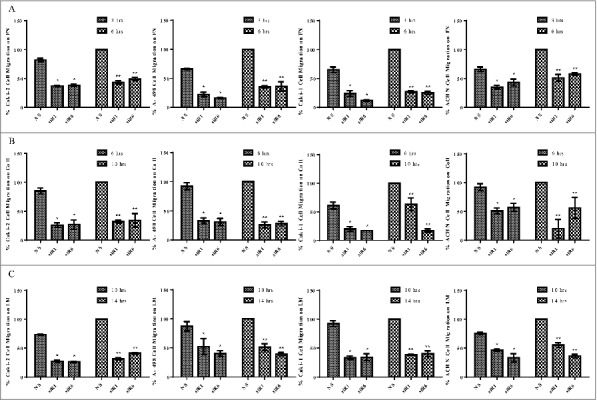
TG2 downregulation reduces the invasion of β1 integrin substrates by RCC cell lines. NS siRNA, siRNA 1 (siR1) and siRNA 6 (siR6) treated RCC cell lines seeded in AIM-V medium on (A) FN, (B) Col 1, and (C) LM coated transwells and allowed to migrate through β1 integrin substrates for indicated early (3 hours for FN, 6 hours for Col1 and 10 hours for LM) and late (6 hours for FN, 10 hours for Col1 and 14 hours for LM) time points as described under “Experimental Procedures.” Images of migrated cells were captured from 10 different non-overlapping areas for each point and analyzed using Scion Image analysis. Percentage of migrated cells was calculated by considering the number of control NS cells migrated at the late time point as 100%. The percentage of migrated cells ± _SD shown were from the mean of at least 3 independent experiments.
Time-dependent analysis of RCC cell invasion in Col1 matrix showed that ACHN cells treated with NS siR displayed a higher level invasion of Col1, which was recorded as 608 cells, compared with the other 3 RCC cell lines (Fig. 5B). TG2 silencing led to a 74% decrease in the number of Caki-2 cells invaded through Col1 at the early time point and only a 7% increase in the migration of siR treated Caki-2 cells (siR1 and siR6) was recorded at the late time point. Down-regulation of TG2 by siRs led to a significant 70% reduction in Col1 invasion rate of A-498, when compared with the NS siR treated control A-498 at the both time points. Caki-1 cells exhibited the slowest migration capability on Col1 by 61% while an average of 82% decline was recorded for siR1 and siR6 treated Caki-1 cells at the indicated early time point. At the late time point, the reduction in the migratory cell number remained the same for the siR6 treated Caki-1 cells, while siR1 treatment led to a 37% decrease in that of Caki-1 cells. Analysis of siR1 treated ACHN invasion of Col1 revealed an 80% reduction at the early time point, which was increased by 30% at the late time point. The number of siR6 treated cells invaded through Col1 matrix remained the same at 57% of NS siR treated control ACHN for both time points (Fig 5B).
When the invasion capacity of the 4 RCC cell lines on LM was compared, Caki-1 cells displayed the highest invasive potential. The number of RCC cells that invaded through LM matrix was recorded as 588 cells for Caki-2, 434 cells for A-498, 681 cells for Caki-1 and 397 cells for ACHN at the early time point. NS siR treated Caki-2 cells invaded through LM by 73%, while TG2 silencing led to a respective 73% and 64% decrease in the number of migrated siR1 and siR6 treated cells at the early and late time points. TG2 downregulation in A-498 cells led to a 54% reduction in the number of LM invaded cells for both early and late time points. siR treatment against TG2 significantly decreased the invasion capacity of Caki-1 cells by an average of 65% at each time point. Finally, siR1 and siR6 treated ACHN cells exhibited a 61% and 55% reduction in their invasion on LM at the early and late time points, respectively (Fig 5C). Overall, migration experiments performed on β1 integrin matrix substrate-coated transwells suggested that the downregulation of TG2 in RCC cell lines leads to an impediment in the invasion potential of RCC cells through these matrices.
Characterization of TG2 silenced Caki-1 RCC cell lines for stem cell markers
Previous reports implicated a role for TG2 in the induction of stem cell character in mammary epithelial cells21 and the maintenance of cancer stem cell (CSC) markers in metastatic ovarian and glioma cell lines20,27 prompted us to quantify the previously identified RCC CSC surface marker CD105 in the metastatic site RCC cell lines.28 Since, between the 2 metastatic cell lines, only Caki-1 tested positive for CD105 antigen expression, it was used as the cell model. To observe the long-term effects of TG2 silencing, Caki-1 cells were stably transduced with lentiviral particles carrying 3 different shRNA products targeting the TGM2 gene (shTG2) and scrambled control (Scr).
The shRNA targeting of TG2 led to an 83% decrease in the TG2 protein levels in these cells (Fig 6A). Scr control Caki-1 cells were found to be negative for the haematopoietic markers CD14, CD31, CD34, CD45 but displayed high mesenchymal CD marker profile of CD44 (99.8%), CD 73 (99.35%) and CD 105 (99.54%) except for the mesenchymal stem cell marker CD90. While the knockdown of TG2 did not lead to any significant change in the haematopoietic CD markers, a more than 90% decrease was detected in the mesenchymal stem cell markers CD44, CD73 and CD105. Given that CD105 positive renal CSCs isolated from human renal carcinomas display high tumor initiating potential,28 our results showing that TG2 knockdown mediates the loss of mesenchymal CD markers suggests that TG2 expression is associated with the maintenance of cancer cells with CSC antigens, which may be related to CSC state and tumor dissemination.
Figure 6.
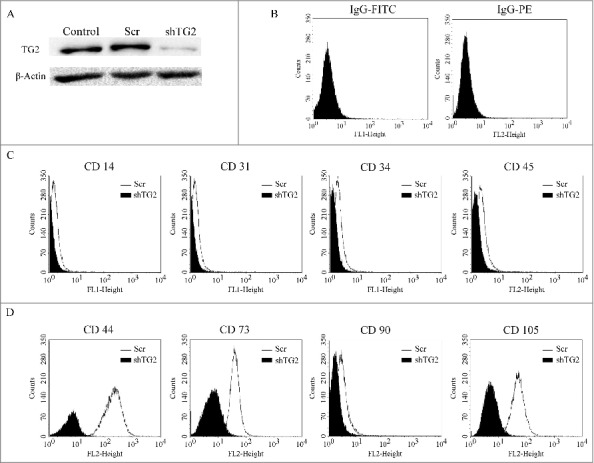
Decrease in TG2 expression in Caki-1 cells results in the loss of cell surface CSC markers. (A) TG2 gene knockdown was achieved by using shRNA lentiviral particles and silencing efficiency was determined by Western bot using actin as the loading control protein. Flow cytometric analysis of CD markers on Caki-1 scrambled control (light-shaded histograms) and TG2-shRNA transduced cells (dark-shaded histograms). (B) Caki-1 scrambled cells incubated with isotype-matched FITC- and PE-IgG were used as negative control. Cells were either stained with FITC- or PE- conjugated (C) haematopoietic and (D) mesenchmal markers represented below x-axis as FL1 and FL2, respectively. Background fluorescence was detected with FITC- or PE-conjugated isotype control mouse IgG antibodies.
Discussion
We previously showed that regardless from its enzymatic activity, the upregulation of TG2 along with ITGβ1 and SDC-4 facilitates the progression and metastasis of RCC in 95 primary tumor samples.16 In a follow-up study performed between metastatic and primary RCC tumor tissue samples, an increase in TG2 protein levels in the primary tumors of metastatic patients was shown along with a significantly lower 5-year progression free and cancer-specific survival rates suggesting that the initial increment of TG2 expression in primary site tumor cells could be a predictive marker during the diagnosis of advanced RCC.17 While these findings pointed to a critical role for TG2 in advanced RCC, the molecular identity of its contribution to the progression of RCC remained to be specified. In the current study, our goal was to use primary and metastatic site RCC cell lines to gain insight into the actions of TG2 in cell adhesion, migration and invasiveness as well as the maintenance of CSC markers in RCC. To our knowledge, this is the first investigation in which the role of TG2 in RCC is characterized either in terms of cell adhesion and migration or assessing the cell invasiveness and CSC surface marker phenotype.
Emerging evidence suggested that the upregulation of TG2 provided a marked survival advantage to various cancer types by rendering cells resistant to serum starvation,29 apoptosis,18,30,31 anoikis,32,33 and hypoxic conditions.34 In this context, the silencing of TG2 expression or activity has been shown to halt the tumor growth and dissemination and increase the tumor sensitivity to chemotherapeutic agents in several cancers.11,35-39 TG2 may attend to the orchestration of cancer phenotype as a main player or a pawn to drive the transformed cells to acquire metastatic and chemo resistance features.
Aside from the implicated functions of TG2 in various signaling pathways responsible for cancer cell maintenance,26 TG2 plays a central role in the regulation of cytoskeletal organization, cell adhesion, migration and invasion of cancer cells.11,40 Thus, we sought to examine the impact of TG2 downregulation in the adherence and migration potential of RCC cells, isolated both from primary and metastatic sites of the tumor, so as to gain a broad insight into RCC progression and dissemination. On the cell surface, TG2 potentiates integrin clustering and activates the modulator of actin stress fibers ROCK kinase through a signaling pathway mediated by FAK, Src, and p190RhoGAP in the upstream.41 Intracellularly, TG2 was co-localized with actin in the protruding ends of migrating cancer cells possibly serving as a binding partner for the recruitment of necessary proteins to drive actin polymerization.40 On the basis of these observations, we investigated the effect of TG2 downregulation on actin stress fiber formation of the 4 RCC cell lines using 2 different siR constructs targeting different regions of TGM2 mRNA. Because reduction of TG2 levels in RCC cell lines induces p53-mediated cellular apoptosis under amino acid starvation conditions,18,42 cells were kept in serum containing media throughout the siR transfection protocol. Silencing of TG2 expression caused a significant disruption in the actin cytoskeleton organization of all 4 RCC cell lines, although Caki-2 exhibited the greatest reduction in the number and size of stress fibers, followed by its metastatic counterpart Caki-1.
Given that in RCC cell lines TG2 was necessary in potentiating the actin stress fiber formation which regulates cells' ability to adhere and migrate, the next step was to exploit the effect of TG2 downregulation both on the cell adhesion potential on ITGB1 substrates and the migration profile. Indeed, our results from cell adhesion experiments demonstrated that once TG2 expression was downregulated, the attachment and spreading of primary and metastatic RCC cells on FN was significantly reduced. Our results suggesting TG2 as an important carcinogenic protein necessary for RCC cell adhesion were consistent with the previous reports showing that the silencing of TG2 in breast cancer cell line with high TG2 expression11 or targeting TG2 binding to FN with synthetic compounds37,43 in ovarian cancer cells impaired the cells attachment on FN. In addition, here we have for the first time demonstrated that the inhibition of TG2 expression also blocked RCC cell adhesion onto other β1 integrin-binding matrix substrates such as Col1, the main component of connective tissue ECM, and LM, the structural glycoprotein of basement membrane. We also showed that the silencing of TG2 by siR significantly hindered the migration potential of Caki-2 primary site and Caki-1 and ACHN metastastic site cell lines both at early and late time points. Interestingly, a decrease in the migratory ability in siR treated A-498 cells was compensated by 48 hours suggesting that there are either other mechanisms underlying the cell migration in this particular cell line or the downregulation of TG2 was compensated by a cell-specific compensation mechanism.44
Recent studies suggested that TG2 powers cancer metastasis by enhancing the invasive potential and stem cell phenotype through the induction of epithelial mesenchymal transition, a process in which epithelial cells adopt mesenchymal like properties and gain motility and invasive capacity.26 We observed that the number of cells that invaded through β1 integrin matrix substrates FN, Col1 and LM was significantly lower for TG2 targeting siR treated RCC cell lines suggesting that TG2 is critical for the invasive behavior of both primary and metastatic site RCC cell lines. Consistently, the knockdown of TG2 in breast11 and ovarian cancer37 and squamous carcinoma33,45 cell lines abrogated their invasive activity suggesting that TG2 induction was required for cell migration and invasiveness. In addition, studies on these cancer types suggested that TG2 fosters stem cell phenotype by facilitating cellular anchorage-independent growth in soft agar or spheroid assays and inducing the stem cell CD marker expression profiles.20,21,26,27 On the basis of a previous report showing that a subpopulation of CD105+ cells also expressing CD44, CD90, and CD73 stem cell markers in renal carcinomas, possess the ability to initiate tumor formation and maintain their stem cell marker through animal passaging,28 we set out on a quest to determine whether silencing of TG2 in metastatic RCC cell line Caki-1 eliminates these renal CSC markers. Since the previous literature reported a CD105− phenotype for ACHN, these cell lines were not used in the study.46 Our data show that the inhibition of TG2 expression eliminates CD105+/ CD44+/CD73 phenotype in Caki-1 cells suggesting that TG2 is necessary for the expression of these CSC surface antigens and therefore may be important for the mesenchymal phenotype associated with EMT. Indeed, our preliminary results suggest that TG2 expression potentiates the expression of EMT inducing transcription factors Zeb1, Snail, Slug and Twist 1 and 2 as well as EMT markers N-cadherin and vimentin in RCC (unpublished).
Together, our results provide evidence, for the first time, that TG2 expression is critical for cancer cell adhesion, migration, invasiveness and cancer cell-stemness during RCC progression and dissemination. Our results further support earlier observations implicating TG2 in RCC tumor aggressiveness and higher pathological grades associated with poor prognosis and disease-free survivals.16,17,19 Thus, combined targeting of TG2 with drugs widely used in the treatment of RCC may be a promising therapeutic strategy in RCC.
Materials and methods
Reagents and antibodies
Human placenta LM and Col I, and plasma FN were purchased from Sigma-Aldrich. Antibodies used in Western blot analysis included mouse monoclonal anti-TG2 Cub7402 (Thermosceintific, USA), anti-mouse integrin β1 (BD Bioscience, USA) and mouse anti-actin (Santa Cruz Biotech, USA). For characterization of cell surface markers, anti-CD31-FITC, anti-CD34-FITC, anti-CD45-PE, anti-CD44-PE, anti-CD73-PE, anti-CD90-PE, anti-CD-105-PE and mouse IgG1 control antibodies used in flow cytometry analysis were purchased from Abcam Inc. (USA).
Cell lines
Human primary RCC cell lines Caki-2 and A-498, and metastatic RCC cell lines Caki-1 and ACHN were obtained from the American Type Culture Collection. Caki-2 and Caki-1 were maintained in McCoy's 5A modified medium, whereas ACHN and A-498 were maintained in Dulbecco's modified Eagle's medium, both of which contained 10% (v/v) Fetal bovine serum (FBS) and penicillin/streptomycin (100 units/ml and 100 µg/ml, respectively).16
Quantitative real-time PCR and Western blot Analysis
Total RNA isolation from cultured cells was performed as described previously16 using same primers and PCR conditions. Cells were lysed in homogenization buffer and proteins were separated in SDS-PAGE and transferred to nitrocellulose membranes as stated before.9 Membranes were immunoprobed using anti-TG2 Cub7402 and anti-mouse integrin β1. β-actin was used as an internal control to confirm the equal loading.
siRNA transfection
For in vitro gene knockdown experiments, non-silencing control (NS) siRNA (AllStars SI03650318) human TGM2 targeting siRNAs (Hs_TGM2_1:SI00743715 and Hs_TGM2_6:SI03055465) were purchased from Qiagen. Transfection was performed using RNAiFact transfection reagent (Qiagen) according to the manufacture's protocol, as described previously.24
Cytoskeletal staining and cell adhesion assay
Cells transfected with control NS and TG2-trageted siRNA were dislodged and F-actin stress fiber staining and cell adhesion assay was performed following the protocol of Verderio et al.5 For visualization of actin stress fibers, cells were seeded in 8-well glass permanox-chamber slides at a density of 2 × 105 cells per well, allowed to attach and spread for 60 minutes. Following fixation and permeabilization steps, cells were treated with FITC-labeled phalloidin, mounted in the aqueous mounting media and examined using Nikon ECLIPSE TE200 fluorescence microscopy. The quantification of actin stress fibers was performed on 10 images taken per well using Scion Image Analysis Program. For cell adhesion assay, 96-well plates were coated with FN (5 µg/ml) overnight at 4°C, or with 40µg/ml human placenta Col1 and 10µg/ml human placenta LM for 2 hours at 37°C. Cells were seeded into 96-well plates at 20.000 cells/well density and incubated for 30 and 60 mins for FN and LM and 60 and 90 mins for Col1. Following the fixation and permeabilization, cells were stained and 3 non-overlapping fields per well for samples in triplicate were acquired using a Carl Zeiss Primovert microscope equipped with Camera System Axiocam 105 Digital Color. Number of attached and spread cells were counted and analyzed using Scion Image Analysis Program as described before.5,9
Wound healing and Transwell assay
In vitro scratch assay was performed to analyze the migration potential of RCC cell lines. Control NS cells and TG2 downregulated cells were seeded on 12 well plate, allowed to form a monolayer. Wound area in a cell monolayer was created using a 1 ml pipette tip and cells were allowed to migrate toward the wound area for 48 hours in 2% (v/v) FBS containing media to limit any interference on migration by cell proliferation. 10 non-overlapping digital images covering the central area of each well were captured at 4X magnification at 0 hour, 24 hours and 48 hrs. Wound area after 24 hours and 48 hours was measured and compared with 0 hour and using Image J software. The percentage closure of wound areas was presented as mean ± SD from 3 separate experiments. In vitro cell migration assay was performed using 6.5 mm, 8.0 /∼m pore size Transwells (Costar), which were coated with FN (5µg/ml), Col1 (40µg/ml) and LM (10µg/ml) as described above. Cells in AIM-V medium were seeded on the upper chambers of coated transwell inserts and allowed to migrate at time points of 3 and 6 hours for FN; 6 and 10 hours for Col1 and 10 and 14 hours for LM. At the end of the incubation non-migrant cells were gently removed by a cotton swab from the upper face of the transwell membrane and migrant cells, those attached to the lower face, were fixed and stained using crystal violet. 10 non-overlapping digital images covering the central area of each well were captured at 4X magnification using ECLIPSE TE200 microscope and number of cells was analyzed as described above.
shRNA lentivirus transduction of Caki-1 cells and characterization of cell surface CD markers
Lentiviral constructs expressing TG2 shRNAs and non-target control shRNAs were purchased from Santa Cruz Biotechnology (USA). Caki-1 cells were pre-treated with 8µg/ml polybrene for 10 hours and transduced with lentiviral particles at MOI = 2 for 24 hours. Following the incubation, the media was replaced with fresh McCoy's 5A and antibiotic selection was initiated using puromycin at 3µg/ml for 10 d. Stable silencing of TG2 was analyzed by real-time PCR and Western blot. The analysis of cell surface markers was performed on 5 × 105cells using IgG isotype controls as described by Kumar et al..21
Data analysis
All data were presented as the mean ± standard deviation (SD). Statistical analysis was performed using One-way ANOVA with Tukey post test using GraphPad Prism version 5.03 for Windows (Graphpad Software, San Diego California USA). p values < 0.05 were considered statistically significant.
Supplementary Material
Abbrevations
- CSC
Cancer stem cell
- Col 1
Collagen type I
- EMT
Epithelial mesenchymal transition
- ECM
Extracellular Matrix
- FN
Fibronectin
- ITGB1
Integrin β−1
- LM
Laminin
- MOI
Multiplicity of infection
- NS siR
Non-silencing siRNA
- Scr
Scrambled control
- siR
Silencing RNA
- shRNA
Short hairpin RNA
- RCC
Renal cell carcinoma
- SDC4
Syndecan-4
- TG2
Tissue-transglutaminase
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Funding
This work was supported in part by EU Marie Curie ITN “TRANSPATH” under Grant number 289964, and by TÜBİTAK under Grant number 109S431.
References
- [1].Valastyan S, Weinberg RA. Tumor metastasis: molecular insights and evolving paradigms. Cell 2011; 147:275-92; PMID:22000009; https://doi.org/ 10.1016/j.cell.2011.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Seguin L, Desgrosellier JS, Weis SM, Cheresh DA. Integrins and cancer: regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol 2015; 25:234-40; PMID:25572304; https://doi.org/ 10.1016/j.tcb.2014.12.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Iwamoto DV, Calderwood DA. Regulation of integrin-mediated adhesions. Curr Opin Cell Biol 2015; 36:41-7; PMID:26189062; https://doi.org/ 10.1016/j.ceb.2015.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Ley K, Rivera-Nieves J, Sandborn WJ, Shattil S. Integrin-based therapeutics: biological basis, clinical use and new drugs. Nat Rev Drug Discov 2016; 15:173-83; PMID:26822833; https://doi.org/ 10.1038/nrd.2015.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Verderio EA, Telci D, Okoye A, Melino G, Griffin M. A novel RGD-independent cel adhesion pathway mediated by fibronectin-bound tissue transglutaminase rescues cells from anoikis. J Biol Chem 2003; 278:42604-14; PMID:12732629; https://doi.org/ 10.1074/jbc.M303303200 [DOI] [PubMed] [Google Scholar]
- [6].Eckert RL, Kaartinen MT, Nurminskaya M, Belkin AM, Colak G, Johnson GV, Mehta K. Transglutaminase regulation of cell function. Physiol Rev 2014; 94:383-417; PMID:24692352; https://doi.org/ 10.1152/physrev.00019.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Folk J. Transglutaminases. Annu Rev Biochem 1980; 49:517-31 [DOI] [PubMed] [Google Scholar]
- [8].Nurminskaya MV, Belkin AM. Cellular functions of tissue transglutaminase. Int Rev Cell Mol Biol 2012; 294:1-97; PMID:22364871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Telci D, Wang Z, Li X, Verderio EA, Humphries MJ, Baccarini M, Basaga H, Griffin M. Fibronectin-tissue transglutaminase matrix rescues RGD-impaired cell adhesion through syndecan-4 and beta1 integrin co-signaling. J Biol Chem 2008; 283:20937-47; PMID:18499669; https://doi.org/ 10.1074/jbc.M801763200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Belkin AM, Zemskov EA, Hang J, Akimov SS, Sikora S, Strongin AY. Cell-surface-associated tissue transglutaminase is a target of MMP-2 proteolysis. Biochem 2004; 43:11760-9; https://doi.org/ 10.1021/bi049266z [DOI] [PubMed] [Google Scholar]
- [11].Mangala LS, Fok JY, Zorrilla-Calancha IR, Verma A, Mehta K. Tissue transglutaminase expression promotes cell attachment, invasion and survival in breast cancer cells. Oncogene 2007; 26:2459-70; PMID:17043648; https://doi.org/ 10.1038/sj.onc.1210035 [DOI] [PubMed] [Google Scholar]
- [12].Herman JF, Mangala LS, Mehta K. Implications of increased tissue transglutaminase (TG2) expression in drug-resistant breast cancer (MCF-7) cells. Oncogene 2006; 25:3049-58; PMID:16449978; https://doi.org/ 10.1038/sj.onc.1209324 [DOI] [PubMed] [Google Scholar]
- [13].Mehta K. Prognostic significance of tissue transglutaminase in drug resistant and metastatic breast cancer. Clin Cancer Res 2004; 10:8068-76; PMID:15585642; https://doi.org/ 10.1158/1078-0432.CCR-04-1107 [DOI] [PubMed] [Google Scholar]
- [14].Satpathy M, Cao L, Pincheira R, Emerson R, Bigsby R, Nakshatri H, Matei D. Enhanced peritoneal ovarian tumor dissemination by tissue transglutaminase. Cancer Res 2007; 67:7194-202; PMID:17671187; https://doi.org/ 10.1158/0008-5472.CAN-07-0307 [DOI] [PubMed] [Google Scholar]
- [15].Verma A, Guha S, Diagaradjane P, Kunnumakkara AB, Sanguino AM, Lopez-Berestein G, Sood AK, Aggarwal BB, Krishnan S, Gelovani JG, et al.. Therapeutic significance of elevated tissue transglutaminase expression in pancreatic cancer. Clin Cancer Res 2008; 14:2476-83; PMID:18413840; https://doi.org/ 10.1158/1078-0432.CCR-07-4529 [DOI] [PubMed] [Google Scholar]
- [16].Erdem M, Erdem S, Sanli O, Sak H, Kilicaslan I, Sahin F, Telci D. Up-regulation of TGM2 with ITGB1 and SDC4 is important in the development and metastasis of renal cell carcinoma. Urol Oncol 2014; 32:25 e13-20; PMID:23499501; https://doi.org/ 10.1016/j.urolonc.2014.03.013 [DOI] [PubMed] [Google Scholar]
- [17].Erdem S, Yegen G, Telci D, Yildiz I, Tefik T, Issever H, Kilicaslan I, Sanli O. The increased transglutaminase 2 expression levels during initial tumorigenesis predict increased risk of metastasis and decreased disease-free and cancer-specific survivals in renal cell carcinoma. World J Urol 2015; 33:1553-60; PMID:25515319; https://doi.org/ 10.1007/s00345-014-1462-7 [DOI] [PubMed] [Google Scholar]
- [18].Ku BM, Kim D-S, Kim KH, Yoo BC, Kim S-H, Gong Y-D, Kim SY. Transglutaminase 2 inhibition found to induce p53 mediated apoptosis in renal cell carcinoma. FASEB J 2013; 27:3487-95; PMID:23704086; https://doi.org/ 10.1096/fj.12-224220 [DOI] [PubMed] [Google Scholar]
- [19].Park MJ, Baek HW, Rhee YY, Lee C, Park JW, Kim HW, Moon KC. Transglutaminase 2 expression and its prognostic significance in clear cell renal cell carcinoma. J Pathol Transl Med 2015; 49(1):37-43; PMID:25812656; https://doi.org/ 10.4132/jptm.2014.10.25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Cao L, Shao M, Schilder J, Guise T, Mohammad KS, Matei D. Tissue transglutaminase links TGF-beta, epithelial to mesenchymal transition and a stem cell phenotype in ovarian cancer. Oncogene 2012; 31:2521-34; PMID:21963846; https://doi.org/ 10.1038/onc.2011.429 [DOI] [PubMed] [Google Scholar]
- [21].Kumar A, Gao H, Xu J, Reuben J, Yu D, Mehta K. Evidence that aberrant expression of tissue transglutaminase promotes stem cell characteristics in mammary epithelial cells. PLoS One 2011; 6:e20701; PMID:21687668; https://doi.org/ 10.1371/journal.pone.0020701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lin CY, Tsai PH, Kandaswami CC, Chang GD, Cheng CH, Huang CJ, Lee PP, Hwang JJ, Lee MT. Role of tissue transglutaminase 2 in the acquisition of a mesenchymal-like phenotype in highly invasive A431 tumor cells. Mol Cancer 2011; 10:87; PMID:21777419; https://doi.org/ 10.1186/1476-4598-10-87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Greef B, Eisen T. Medical treatment of renal cancer: new horizons. Brit J Cancer 2016; 23:505-16; https://doi.org/ 10.1038/bjc.2016.230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Wang Z, Collighan RJ, Gross SR, Danen EH, Orend G, Telci D, Griffin M. RGD-independent cell adhesion via a tissue transglutaminase-fibronectin matrix promotes fibronectin fibril deposition and requires syndecan-4/2 alpha5beta1 integrin co-signaling. J Biol Chem 2010; 285:40212-29; PMID:20929862; https://doi.org/ 10.1074/jbc.M110.123703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Akimov SS, Krylov D, Fleischman LF, Belkin AM. Tissue transglutaminase is an integrin-binding adhesion coreceptor for fibronectin. J Cell Biol 2000; 148:825-38; PMID:10684262; https://doi.org/ 10.1083/jcb.148.4.825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Eckert RL, Fisher ML, Grun D, Adhikary G, Xu W, Kerr C. Transglutaminase is a tumor cell and cancer stem cell survival factor. Mol Carcinog 2015; 54:947-58; PMID:26258961; https://doi.org/ 10.1002/mc.22375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Fu J, Yang QY, Sai K, Chen FR, Pang JC, Ng HK, Kwan AL, Chen ZP. TGM2 inhibition attenuates ID1 expression in CD44-high glioma-initiating cells. Neuro Oncol 2013; 15:1353-65; PMID:23877317; https://doi.org/ 10.1093/neuonc/not079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Bussolati B, Bruno S, Grange C, Ferrando U, Camussi G. Identification of a tumor-initiating stem cell population in human renal carcinomas. FASEB J 2008; 22:3696-705; PMID:18614581; https://doi.org/ 10.1096/fj.08-102590 [DOI] [PubMed] [Google Scholar]
- [29].Boroughs LK, Antonyak MA, Cerione RA. A novel mechanism by which tissue transglutaminase activates signaling events that promote cell survival. J Biol Chem 2014; 289:10115-25; PMID:24569994; https://doi.org/ 10.1074/jbc.M113.464693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Li Z, Xu X, Bai L, Chen W, Lin Y. Epidermal growth factor receptor-mediated tissue transglutaminase overexpression couples acquired tumor necrosis factor-related apoptosis-inducing ligand resistance and migration through c-FLIP and MMP-9 proteins in lung cancer cells. J Biol Chem 2011; 286:21164-72; PMID:21525012; https://doi.org/ 10.1074/jbc.M110.207571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mehta K, Kumar A, Im Kim H. Transglutaminase 2: a multi-tasking protein in the complex circuitry of inflammation and cancer. Biochem Pharmacol 2010; 80:1921-9; PMID:20599779; https://doi.org/ 10.1016/j.bcp.2010.06.029 [DOI] [PubMed] [Google Scholar]
- [32].Li B, Antonyak MA, Druso JE, Cheng L, Nikitin AY, Cerione RA. EGF potentiated oncogenesis requires a tissue transglutaminase-dependent signaling pathway leading to Src activation. Proc Natl Acad Sci 2010; 107:1408-13; PMID:20080707; https://doi.org/ 10.1073/pnas.0907907107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Fisher ML, Keillor JW, Xu W, Eckert RL, Kerr C. Transglutaminase is required for epidermal squamous cell carcinoma stem cell survival. Mol Cancer Res 2015; 13:1083-94; PMID:25934691; https://doi.org/ 10.1158/1541-7786.MCR-14-0685-T [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kumar S, Donti TR, Agnihotri N, Mehta K. Transglutaminase 2 reprogramming of glucose metabolism in mammary epithelial cells via activation of inflammatory signaling pathways. Int J Cancer 2014; 134:2798-807; PMID:24477458; https://doi.org/ 10.1002/ijc.28623 [DOI] [PubMed] [Google Scholar]
- [35].Verma A, Wang H, Manavathi B, Fok JY, Mann AP, Kumar R, Mehta K. Increased expression of tissue transglutaminase in pancreatic ductal adenocarcinoma and its implications in drug resistance and metastasis. Cancer Res 2006; 66:10525-33; PMID:17079475; https://doi.org/ 10.1158/0008-5472.CAN-06-2387 [DOI] [PubMed] [Google Scholar]
- [36].Cao L, Petrusca DN, Satpathy M, Nakshatri H, Petrache I, Matei D. Tissue transglutaminase protects epithelial ovarian cancer cells from cisplatin-induced apoptosis by promoting cell survival signaling. Carcinogenesis 2008; 29:1893-900; PMID:18667446; https://doi.org/ 10.1093/carcin/bgn158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Yakubov B, Chelladurai B, Schmitt J, Emerson R, Turchi JJ, Matei D. Extracellular tissue transglutaminase activates noncanonical NF-κB signaling and promotes metastasis in ovarian cancer. Neoplasia 2013; 15:609-19; PMID:23730209; https://doi.org/ 10.1593/neo.121878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Han AL, Kumar S, Fok JY, Tyagi AK, Mehta K. Tissue transglutaminase expression promotes castration-resistant phenotype and transcriptional repression of androgen receptor. Eur J Cancer 2014; 50:1685-96; PMID:24656569; https://doi.org/ 10.1016/j.ejca.2014.02.014 [DOI] [PubMed] [Google Scholar]
- [39].Cao J, Huang W. Compensatory increase of Transglutaminase 2 is responsible for resistance to mTOR inhibitor treatment. PloS one 2016; 11:e0149388; PMID:26872016; https://doi.org/ 10.1371/journal.pone.0149388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Antonyak MA, Li B, Regan AD, Feng Q, Dusaban SS, Cerione RA. Tissue transglutaminase is an essential participant in the epidermal growth factor-stimulated signaling pathway leading to cancer cell migration and invasion. J Biol Chem 2009; 284:17914-25; PMID:19403524; https://doi.org/ 10.1074/jbc.M109.013037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Janiak A, Zemskov EA, Belkin AM. Cell surface transglutaminase promotes RhoA activation via integrin clustering and suppression of the Src-p190RhoGAP signaling pathway. Mol Biol Cell 2006; 17:1606-19; PMID:16452636; https://doi.org/ 10.1091/mbc.E05-06-0549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Kang J, Lee J, Hong D, Lee S, Kim N, Lee WK, Sung TW, Gong YD, Kim SY. Renal cell carcinoma escapes death by p53 depletion through transglutaminase 2-chaperoned autophagy. Cell Death Dis 2016; 7:e2163; PMID:27031960; https://doi.org/ 10.1038/cddis.2016.14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Khanna M, Chelladurai B, Gavini A, Li L, Shao M, Courtney D, Turchi JJ, Matei D, Meroueh S. Targeting ovarian tumor cell adhesion mediated by tissue transglutaminase. Mol Cancer Ther 2011; 10:626-36; PMID:21330459; https://doi.org/ 10.1158/1535-7163.MCT-10-0912 [DOI] [PubMed] [Google Scholar]
- [44].Deasey S, Shanmugasundaram S, Nurminskaya M. Tissue-specific responses to loss of transglutaminase 2. Amino Acids 2013; 44:179-87; PMID:22194042; https://doi.org/ 10.1007/s00726-011-1183-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Chen SH, Lin CY, Lee LT, Chang GD, Lee PP, Hung CC, Kao WT, Tsai PH, Schally AV, Hwang JJ, et al.. Up-regulation of fibronectin and tissue transglutaminase promotes cell invasion involving increased association with integrin and MMP expression in A431 cells. Anticancer Res 2010; 30:4177-86; PMID:21036738 [PubMed] [Google Scholar]
- [46].Myszczyszyn A, Czarnecka AM, Matak D, Szymanski L, Lian F, Kornakiewicz A, Bartnik E, Kukwa W, Kieda C, Szczylik C. The role of hypoxia and cancer stem cells in renal cell carcinoma pathogenesis. Stem Cell Rev 2015; 11:919-43; PMID:26210994; https://doi.org/ 10.1007/s12015-015-9611-y [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.