

ABSTRACT
We advocate for a tRNA- rather than an mRNA-centric model for evolution of the genetic code. The mechanism for evolution of cloverleaf tRNA provides a root sequence for radiation of tRNAs and suggests a simplified understanding of code evolution. To analyze code sectoring, rooted tRNAomes were compared for several archaeal and one bacterial species. Rooting of tRNAome trees reveals conserved structures, indicating how the code was shaped during evolution and suggesting a model for evolution of a LUCA tRNAome tree. We propose the polyglycine hypothesis that the initial product of the genetic code may have been short chain polyglycine to stabilize protocells. In order to describe how anticodons were allotted in evolution, the sectoring-degeneracy hypothesis is proposed. Based on sectoring, a simple stepwise model is developed, in which the code sectors from a 1→4→8→∼16 letter code. At initial stages of code evolution, we posit strong positive selection for wobble base ambiguity, supporting convergence to 4-codon sectors and ∼16 letters. In a later stage, ∼5–6 letters, including stops, were added through innovating at the anticodon wobble position. In archaea and bacteria, tRNA wobble adenine is negatively selected, shrinking the maximum size of the primordial genetic code to 48 anticodons. Because 64 codons are recognized in mRNA, tRNA-mRNA coevolution requires tRNA wobble position ambiguity leading to degeneracy of the code.
KEYWORDS: The last universal common cellular ancestor, tRNA, genetic code, aminoacyl tRNA synthetases
Abbreviations
- LUCA
The last universal common cellular ancestor
- aaRS
aminoacyl tRNA synthetases
- GlyRS
i.e. glycine aminoacyl tRNA synthetase
- DNA tRNAome
the DNA encoding tRNA for an organism
Introduction
We posit that cloverleaf tRNA is the molecular archetype around which translation systems and the genetic code evolved. Evolution of the genetic code was recently comprehensively reviewed, but issues remain unresolved [1,2]. We posit that to grasp code evolution requires a focus on tRNA evolution. To make sense of translation systems, for instance, start with tRNA and work out [3,4]. Translation systems evolved around cloverleaf tRNA, which has not changed very much since LUCA (the last universal common cellular ancestor), and a tRNA-centric view renders translation a much simpler conceptual problem [3,4]. In order to understand evolution of the genetic code, moreover, start with tRNA and work out. As one obvious example, the genetic code is a triplet code because the structure of the tRNA anticodon loop forces a triplet register for two adjacent tRNAs paired to mRNA bound in the ribosome A and P sites at the decoding center. Genetic code evolution, therefore, must have tracked tRNA evolution more closely than mRNA or ribosome evolution. Furthermore, because of unique features of the tight tRNA 7 nt anticodon loop structure with its specialized U turn [5], the anticodon loop of tRNA constrained code evolution much more than the mRNA, which has an extended and partly relaxed conformation on the ribosome. Because of physical limitations of tRNA anticodon wobble sequences immediately following the U turn, the initial expansion of the genetic code in tRNA was limited to 48 anticodons, even though in mRNA all 64 codons are utilized.
Based mostly on analyses of archaeal tRNAs, which are more faithfully conserved from LUCA, a model for evolution of cloverleaf tRNA was proposed [3,4]. According to the model, acceptor stems are derived from a GCG repeat and its CGC complement [3]. The anticodon stem is flanked on both sides by 5 nt relics of acceptor stems, also derived from GCG and CGC repeats. Although largely unpaired in the cloverleaf, in the anticodon loop minihelix, the last 5 nt of the D loop and the 5 nt V loop were paired as acceptor stems [3]. The D loop is derived from a UAGCC repeat. The anticodon stem and loop and the T stem and loop are homologous by sequence and structure. Both loops have a U turn (a U-shaped turn) after a U (in the anticodon loop) or a pseudouridine (in the T loop) between loop positions 2 and 3 out of 7 total [3]. Anticodon loop bases 3–7 are tightly stacked, as if in a helix connecting with the 3’-anticodon stem, making cloverleaf tRNA a relatively stiff adapter to specify contacts to mRNA in the decoding center of the ribosome smaller subunit. The T loop of tRNA is very similar in structure to the anticodon loop, but differs slightly because of T loop interactions with the D loop. Specifically, intercalation of D loop G19 (G18 in historic numbering), between T loop bases 4 and 5, lifts T loop base 5 to contact the T loop stem and flips loop bases 6 and 7 out of the T loop [3]. Based on the tRNA-centric view and the tRNA evolution model, we advocate reassessment of the evolution of the genetic code.
It has been suggested that glycine may be a founding amino acid for the genetic code [6–9]. Here, we show that archaeal tRNAGly is very close to the posited root of the tRNA evolutionary tree. We propose therefore the polyglycine hypothesis, that the primordial cloverleaf tRNA (tRNAPri), which most strongly resembles archaeal tRNAGly, diversified by mutation to include all permitted anticodons. The initial purpose of the 3 nt code may have been, therefore, to synthesize short chains of polyglycine, used to stabilize protocells for energy generation. Gly5 is the typical length for polyglycine cross-linking in bacterial cell walls [10]. In the primitive system, polyglycine chains may have been short in length, because of weak translational processivity and/or mRNA codons lacking a corresponding tRNA anticodon. The polyglycine hypothesis provides a functional root, and the tRNA evolutionary model provides a sequence root to the genetic code. Di Giulio has argued against polyglycine as a founding product of the code, but his arguments are centered on proto-mRNAs encoding multiple peptide products [11]. Whether or not Di Giulio is correct about ancient mRNA coding, the genetic code that exists today appears to be evolved around a single primordial cloverleaf tRNA that recruits mRNA [3]. The nearly universal genetic code, therefore, may be a reinvention of coding that surpassed and suppressed older mRNA-centered systems.
In human tRNAGly, adenine in the wobble position was shown to be destabilizing for the anticodon loop [12]. In bacteria and eukaryotes, adenine in the tRNA wobble position is converted to inosine by a tRNA adenosine deaminase, conferring greater loop stability [12,13]. In archaea, adenine is rarely or never found in the tRNA wobble position, indicating that, in the RNA-protein world and at LUCA, adenine was negatively selected at the wobble position. This observation shrinks the maximum size of the initial genetic code in tRNA from 64 anticodons to 48 anticodons.
A hierarchy is observed for the importance of the three tRNA anticodon positions for translation [2,14]. The middle (second) position of the anticodon is most important for translational fidelity, followed by the third position and then followed by the wobble (first) position. Ambiguity of the wobble position, therefore, describes degeneracy of the code and why only ∼20 amino acids are specified rather than a potentially much larger number (i.e. up to 48). From a structural perspective, when a tRNA binds in the ribosome A site (addition site), the wobble position is ambiguous because the tRNA wobble anticodon base is not fully restrained and can make multiple types of contacts (i.e. Watson-Crick pairs and various non Watson-Crick wobble pairs) to mRNA. The middle and third positions of the tRNA anticodon, by contrast, are constrained to form accurate Watson-Crick base pairs, and even G∼U wobble pairs, commonly found in RNA stems, are strongly disfavored at these positions [15]. At the second and third anticodon positions but not the wobble position, the specificity of contacts is checked by a proofreading conformational change in the decoding center of the smaller subunit of the ribosome involving EF-TU and GTP hydrolysis [16]. Because pairing of the wobble base involves multiple types of contacts, the ribosome conformational change cannot now be extended to proofread wobble position contacts. For one thing, multiple essential wobble contacts that rely on non-Watson-Crick base pairs would be disallowed. Also, ambiguity at the wobble anticodon position was likely necessary for early stage evolution of the genetic code, and wobble ambiguity remains positively selected [17]. To our knowledge, the structural explanations for degrees of freedom in pairing at the wobble position are not fully known. Also, the central importance of the middle anticodon position has not been completely elucidated. Considering these issues, however, we discuss the evolution and the degeneracy of the code. We argue that, at earlier stages of evolution, as the code grew toward ∼16 letters, wobble position ambiguity was positively selected.
Archaeal species generally have one tRNA species per permitted anticodon (excepting adenine in the anticodon wobble position), but there are a few common exceptions. Many archaea have multiple (generally three) tRNAMet (anticodon CAU), including initiator and elongator tRNAMet. Pyrococcus furiosis (archaea), as a typical example, has two elongator tRNAMet (CAU) and one initiator tRNAiMet (CAU). Interestingly, Pyrococcus furiosis has only one tRNAIle (GAU; in some archaea, a single anticodon UAU or CAU may be utilized). Pyrococcus tRNAMet, with three tRNAs (CAU), and tRNAIle, with only one tRNAIle (generally anticodon GAU), share the same 4-codon sector of the codon-anticodon table (anticodon NAU). From analysis of rooted tRNAomes for ancient archaeal species, it appears that tRNAMet may have been derived from tRNAIle.
Results
Lineages in tRNAs
A DNA tRNAome is defined here as the set of all available coding tRNA DNA sequences from a single strain of a species of organism. Sequences of tRNAs were collected from tRNA databases [18, 19]. Others have used tRNA sequences to indicate phylogenies of species [20]. To improve these comparisons, we root tRNAome trees to tRNAPri and compare tree structures among species. In Supplementary Figures S1–S8, we compare evolutionary trees of rooted DNA tRNAomes from Pyrococcus furiosis DSM3638, Pyrococcus abyssi GE5, Pyrococcus horikoshii OT3, Staphylothermus marinus F1, Pyrobaculum aerophilum str. IM2, Aeropyrum pernix K1, Sulfolobus solfataricus P2 (archaea) and Thermus thermophilus HB27 (bacteria). Pyrococcus, Staphylothermus, Pyrobaculum, Aeropyrum and Sulfolobus species were selected because their tRNAomes appear to be very similar to the LUCA tRNAome. A Pyrococcus typical tRNA (Figure S9), for instance, shows much stronger conservation than a broader archaeal or bacterial typical tRNA, indicating proximity of Pyrococcus to LUCA [3]. Very strong GCG/CGC (acceptor stem) and UAGCC (D loop) repeats are preserved in Pyrococcus tRNAs, indicating that these tRNAomes remain close to the primordial cloverleaf. The Pyrococcus typical tRNA sequence is very close to tRNAPri (the proposed primordial tRNA cloverleaf) sequence (60/79 in-phase identities). Of course, these observations also support our assignment of the tRNAPri sequence. For consistency of interpretation, tRNAome evolutionary trees were rooted to tRNAPri (Figures S1–S8). Thermus was selected based on inspection of typical tRNA diagrams, which indicated Thermus was a bacterial family that was more similar to archaea than others (Figure S8).
In Figure 1, a qualitative interpretation of the evolutionary trees (Figures S1–S8) is shown. With some differences, apparent lineages of tRNAs are maintained in different archaeal species and some tRNA lineages are preserved among both archaeal and bacterial species. A sophisticated bioinformatics analysis (not yet complete), therefore, can trace tRNAomes for many species through the archaea and into the bacteria and eukarya. One useful comparison would be an evolutionary tree of tRNAome trees, comparing intact genetic code structures, organism to organism. Rooting trees to tRNAPri helps to interpret the comparisons.
Figure 1.
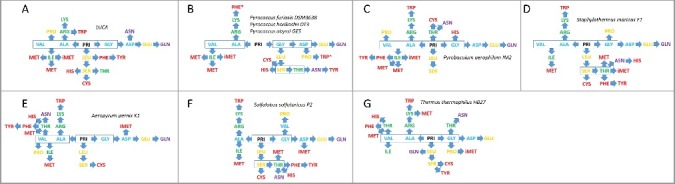
Qualitative maps of the radiations of tRNAomes for various species based on interpretation of evolutionary trees (Figures S1–S8). A) Model for LUCA; B) Three Pyrococcus species; C) Pyrobaculum aerophilium str. IM2; D) Staphylothermus marinus F1; E) Aeropyrum pernix K1; F) Sulfolobus solfataricus P2 (archaea); and G) Thermus thermophilus HB27 (bacteria). We posit that tRNAs are added to the code in the approximate order cyan→orange→green→purple→red. Asterisks (Pyrococcus; Figure 1B) indicate two tRNAs that appear to be reassigned to encode distinct amino acids compared to LUCA and other archaea.
In Figure 1(A), a model for a LUCA tRNAome is shown based on comparison of maps and the relatedness of encoded amino acids. Some of the LUCA tRNAome assignments are based on information in an accompanying paper [17]. The model for LUCA gives an indication of lineages that appear most conserved. Lineages often connect tRNAs for related amino acids, indicating the likelihood of the lineage. A strongly conserved lineage appears to be tRNAPri→tRNAGly→tRNAAsp→tRNAGlu→tRNAGln (Figure 1(A–F)). Another apparent conserved lineage is tRNAPri/tRNAGly→tRNALeu→tRNASer (Figure 1(A–G)). Because V loop inserts were deleted from tRNA alignments before generating trees, the strong similarity of tRNALeu and tRNASer is due to the tRNA cloverleaf core sequence (1–75) and is not due to alignments of tRNALeu and tRNASer extended V loops. The archaeal species that are most similar to LUCA connect tRNASer→tRNAThr, which appear to radiate to tRNACys, tRNAHis, possibly to tRNAAsn and possibly to tRNAPhe→tRNATyr. Because Phe and Tyr are related amino acids, a tRNAPhe→tRNATyr lineage seems reasonable, whether or not this lineage roots properly to tRNASer/tRNAThr. In Pyrobaculum aerophilum str. IM2, for instance, the lineage tRNAAla/tRNAVal→tRNAIle→tRNAPhe→tRNATyr is indicated. Based on amino acid relatedness and positions in the codon-anticodon table, however, tRNALeu→tRNAPhe→tRNATyr might be a more reasonable model [17]. We posit that tRNAAsn may originally be derived from tRNAAsp, similar to tRNAGln being apparently derived from tRNAGlu, as might be expected based on amino acid similarity. If this surmise is correct, tRNAAsn was forced to diverge further from tRNAAsp to maintain accuracy of AsnRS and AspRS charging and translation. Another apparent conserved lineage is tRNAAla→tRNAArg→tRNALys→tRNATrp (Figure 1(A,C–G)). In the archaea that appear most closely related to LUCA, tRNAIle→tRNAMet (one initiator and two elongator) is likely (Figure 1(A–C)). In more derived species, one tRNAMet and tRNAiMet appear to have specialized and further diverged from tRNAIle (Figure 1(D–F)). Some conserved lineage structures appear to extend from archaea→bacteria.
In three Pyrococcus species, it appears that two tRNAs may have been reassigned to attach a different amino acid compared to LUCA tRNAs. Partly because of its placement in the map, Pyrococcus tRNAPhe appears to be a reassigned tRNATrp. Also, Pyrococcus tRNATrp appears to be a reassigned tRNAPro (Figure 1(B)). Because three Pyrococcus species are considered, it is difficult to attribute these apparent tRNA reassignments based on sequencing errors. We posit that these two tRNAs duplicated and evolved to assume new identities. So far as we can judge, the other tRNAs considered for the 8 organisms analyzed here may have maintained their original identities, although divergent and convergent evolution of tRNAs causes tRNAs to move in the lineage maps. Evolution of tRNAs, therefore, can suppress evidence of tRNA reassignments. Migration of tRNAs in the maps tends to make rooting of tRNA lineages ambiguous and causes lineages to appear more shallow (i.e. in bacteria) than in species with tRNAs that are more similar to LUCA tRNAs (i.e. Pyrococcus and Pyrobaculum).
The polyglycine hypothesis
It appears that the initial purpose of the triplet genetic code may have been to synthesize short chain polyglycine [6–9]. A reason to consider this hypothesis is that tRNAPri most resembles archaeal tRNAGly (Figure 2; Figures S1–S5). The GCGGCGG 5’-acceptor stem GCG repeat of tRNAPri is most similar to an archaeal tRNAGly acceptor stem. Searching the primordial tRNA sequence against genomic DNA sequences (i.e. all archaea) produces tRNAGly (GCC) as the top hit (not shown). Searching against the archaeal Aeropyrum pernix tRNAome also produces tRNAGly (anticodon GCC) as the top hit, with an e-value of 8 × 10−18 and 64/78 in-phase identities (Figure 2). Searching against the bacterial Thermus thermophilus DNA tRNAome gives tRNAGly (anticodon UCC) as a top in-phase hit, with an e-value of 2 × 10−5 and 51/79 identities. There is a 2 nt deletion in the D loop of Thermus tRNAGly (anticodon UCC). D loop deletions are found in archaeal tRNAs but are almost universal for bacterial and eukaryotic tRNAs [3,4]. Analysis of tRNAomes (Figures S1–S5) indicates that tRNAGly is initially most similar to tRNAPri.
Figure 2.
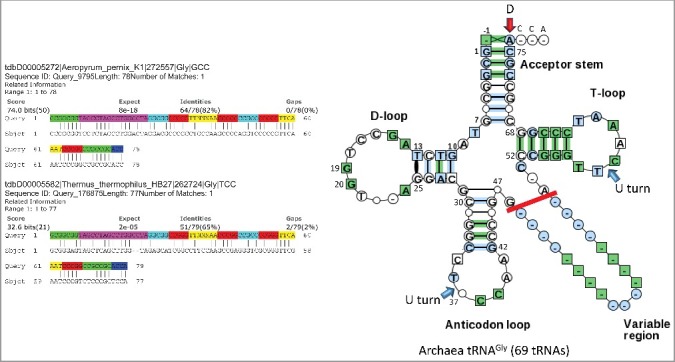
The primordial tRNA cloverleaf is most similar to archaeal tRNAGly. A blast search of the primordial tRNA sequence against the Aeropyrum pernix (archaea) DNA tRNAome and the Thermus thermophilus HB27 DNA tRNAome (bacteria). Coloring of the primordial tRNA sequence: green) acceptor stems; magenta) D loop; cyan) acceptor stem remnants; red) anticodon loop and T loop stems; yellow) anticodon loop and T loop; blue) 3’-ACCA. Right image) A typical tRNA diagram generated from 69 archaeal tRNAGly sequences is shown. Numbering of the tRNA is based on a 75 nt tRNA core sequence. Blue arrows indicate U turns. The red arrow indicates the discriminator (D). Only 5 nt of the V loop are considered in the evolutionary model. Longer V loops include inserts (i.e. tRNALeu and tRNASer).
Radar graphs
Radar graphs (Figure 3) provide a characteristic and identifying tRNAome “fingerprint” that can readily be compared among organisms. Radiations of tRNAome sequences from tRNAPri (at the origin) are compared for Pyrococcus furiosis DSM3638, Pyrococcus abyssi GE5, Pyrococcus horikoshii OT3, Staphylothermus marinus F1, Pyrobaculum aerophilum str. IM2, Aeropyrum pernix K1, Sulfolobus solfataricus P2 (archaea) and Thermus thermophilus HB27 (bacteria). We note that radar graphs provide insight into the evolution of species and their relatedness. From the similarity of graphs, three Pyrococcus species, Pyrobaculum, Staphylothermus and Aeropyrum appear closely related. In particular, compare radar graphs for tRNAs encoding Asn, Asp, Cys, Gln, Glu, Gly and His for these organisms to observe the clear similarities in graph shapes. Based on the extent of radiation from tRNAPri, Pyrobaculum aerophilum may be the closest species of those selected to a LUCA tRNAome. In this comparison, the archaeal species were selected to be similar to LUCA by inspection of typical tRNA sequences. In many archaea, tRNAPri is most similar to tRNAGly, as expected from the polyglycine hypothesis. Some features of radar graph shapes appear to be conserved to Thermus (a bacteria). We conclude that species relatedness and divergence can be determined by analysis of tRNAomes, for instance, as represented in radar graphs. Note that the comparisons shown in radar graphs are also embedded in evolutionary trees although, using trees, the information is more difficult to visually compare species to species (Figures S1–S8).
Figure 3.
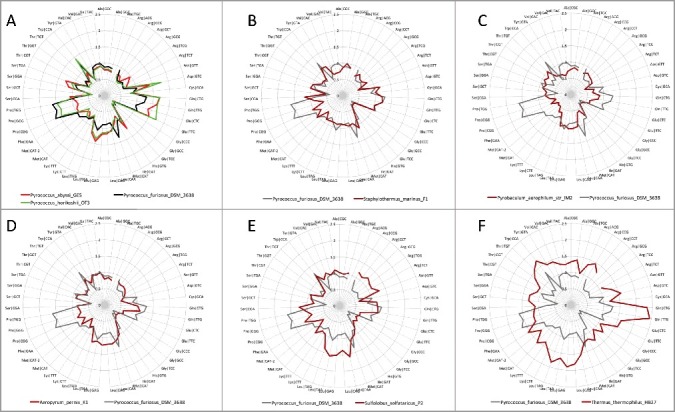
Radar graphs of the radiations of DNA tRNAomes from tRNAPri (at the origin). Evolutionary distances are shown. Comparisons of: A) three Pyrococcus species; B) Staphylothermus marinus F1 versus Pyrococcus furiosis; C) Pyrobaculum aerophilium str. IM2 versus Pyrococcus furiosis D) Aeropyrum pernix versus Pyrococcus furiosis; E) Sulfolobus solfataricus versus Pyrococcus furiosis (archaea); and F) Thermus thermophilus (bacteria) versus Pyrococcus furiosis.
In archaea, tRNAGly is generally most similar to tRNAPri (Figures 2 and 3(A–D)). Although others have also posited that glycine is the founding amino acid for evolution of the code [6–9], we believe we are the first to posit the polyglycine hypothesis, which simplifies the understanding of genetic code evolution. Also, archaeal tRNAs are more closely related to tRNAPri than bacterial tRNAs (Figures 2 and 3(F)), indicating that archaeal tRNAomes are more similar to LUCA tRNAomes than bacterial tRNAomes [3,4]
tRNA mutagenesis and divergence
Anticodon loop positions 1 and 7 weakly interact (i.e. a C∼A wobble pair), restricting the sequences that appear at these positions [21]. Loop position 2 is always U in archaea and bacteria. Loop position 6 is always A or G. Generally, the anticodon of tRNA can mutate without much effect on the cloverleaf fold, so the anticodon is expected to be the fastest sequence in tRNA to change in evolution. Many other substitutions in the cloverleaf, however, are likely to be disruptive and may require a compensatory mutation or a tRNA modification to rescue the fold. Substitutions in stems, for instance, generally require a compensating mutation in the complementary base. We posit, therefore, consistent with the polyglycine hypothesis, that essentially all allowed anticodons became available on tRNAs with acceptor stems that are expected to attach glycine. So populating the codon-anticodon table with a primordial tRNAGly may have occurred before additional amino acids were added to the code. Polyglycine (Gly5) is a component of bacterial cell walls [10]. We imagine that, in the evolving RNA-protein world, polyglycine cross-linking stabilized protocells to facilitate energy generation, which requires a proton gradient across a membrane. Polyglycine, therefore, had evolutionary value from early times, and the genetic code may have initially evolved as an improved mechanism to generate short chain polyglycine to stabilize protocells. If the polyglycine hypothesis is correct, different tRNA species within an organism are expected to have lineages derived from tRNAPri/tRNAGly (Figures 1, 3, and S1–S8). Initially, tRNAPri and its earliest radiations appear to have attached glycine and then evolved to attach other amino acids.
Adenine in the anticodon wobble position
In bacteria and archaea, adenine is rarely found in the anticodon wobble position of tRNAs (Figure 4) [12,13]. In 6368 bacterial tRNAs (shown as DNA), adenine is underrepresented at the wobble position (180/6368 → 2.8%). In most bacterial species, tRNAArg (anticodon ACG) is the only tRNA with adenine encoded in the anticodon wobble position. In 1088 archaeal tRNAs [18], remarkably, adenine is never found at the wobble position. In bacteria and eukaryotes, adenine in the anticodon wobble position is converted to inosine by a tRNA adenosine deaminase, a modification that may stabilize the anticodon loop and that expands wobble position contacts to mRNA [12,13]. Archaea lack the tRNA adenosine deaminase to convert wobble adenine to inosine [12]. We conclude that, at LUCA, adenine in the anticodon wobble position was under strong negative selection, probably, for two reasons. Wobble adenine can have destabilizing effects on the anticodon loop [12]. Also, adenine can only pair strongly with uridine, whereas inosine can pair with adenine, cytidine or uridine [22,23], indicating positive selection for ambiguity at the wobble anticodon position. Unmodified adenine, therefore, in the tRNA anticodon wobble position specifies U in the mRNA codon wobble position. Because of these restrictions, the initial genetic code included at most 48 and not 64 anticodons, as has generally been believed. Interestingly, 46 tRNAs (44 unique anticodons) are found in many archaeal and bacterial species, allowing for 3 stop codons. In prokaryotes, there are generally three tRNAMet (CAU anticodon), including one initiator and two elongator tRNAMet (CAU anticodon), and commonly absent tRNAIle (generally a GAU anticodon is preferred and UAU is not utilized) [18].
Figure 4.
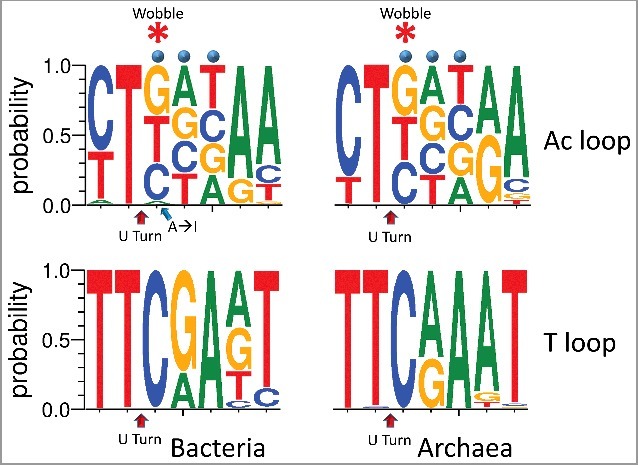
A strong negative selection against adenine in the anticodon wobble position. The homologous T loop is shown below the anticodon (Ac) loop for comparison. DNA sequence logos of the 7 nt anticodon and T loops are shown. Right panels) In archaea (1088 tRNAs), no A is detected at the anticodon wobble position. Left panels) In bacteria (6368 tRNAs), adenine (A) is rarely used, except in tRNAArg (anticodon ACG), and adenine is converted to inosine (I) by tRNA adenosine deaminase [12]. Blue dots indicate the anticodon positions of the loop. The asterisk indicates the wobble position of the anticodon loop.
Evolution of the standard genetic code
Not all of the utilized 48 anticodons specify distinct amino acids, so the genetic code is considered to be degenerate, and structural ambiguity in the reading of the wobble anticodon position causes degeneracy. Because of loop destabilization and the potential for wobble position over-specification, in the initial code, adenine never occupies the anticodon wobble position. Others have noted that the tRNA second (middle) base of the anticodon is most important for translational accuracy, followed by the third base, and then followed by the first (wobble) base, which is recognized with ambiguity [2,14]
These observations suggest an order in which amino acids may have been added to the genetic code (Figure 5) [24,25]. We propose the following approximate pathway for sectoring the code. Initially, essentially all permitted anticodons specify Gly to synthesize polyglycine. Then, the code sectors on the middle position of the anticodon, which is most important for translational accuracy, to specify Val, Ala, Asp and Gly [24]. The second most important position for translational accuracy is the third anticodon position, but this position sectors with difficulty. We posit that the third position initially sectors between purines and pyrimidines to add Leu, Pro, Glu and Ser. Subsequently, the third anticodon position is utilized to divide the code into 4-codon sectors adding Ile, Ser, Thr, Lys, Ter (Stop), Arg and Ser. We consider that Leu may have continued to hold two 4-codon sectors. Potentially, full sectoring of the second and third anticodon positions might correlate with the EF-TU mediated conformational tightening of the decoding center of the ribosome smaller subunit, in order to verify the accuracy of Watson-Crick base pairing of the tRNA second and third anticodon positions to mRNA [15]. At this stage, tRNASer occupies three 4-codon sectors of the codon-anticodon table, explaining how tRNASer alone of all tRNAs ends up occupying two separated and disconnected 4-codon sectors. We cannot currently explain why serine was of so much apparent importance at this stage of evolution.
Figure 5.
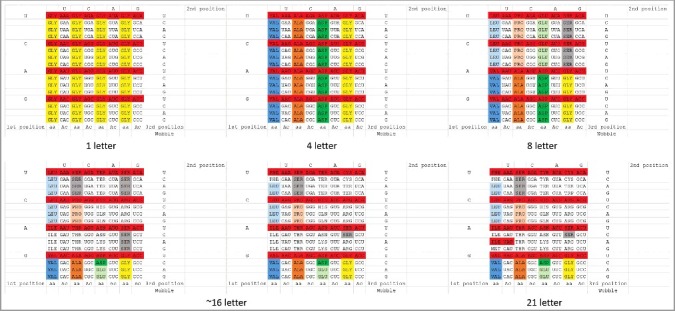
Sectoring of the genetic code. A codon-anticodon (Ac) table is shown. The code is posited to sector from a 1→4→8→∼16→21 letter code (20 aa + Ter (Stop)). Approximate intermediates are shown. Red 1-codon sectors are not utilized in archaea and are rare in bacteria because adenine in the anticodon wobble position is negatively selected. tRNAIle (UAU) is rarely utilized as the single tRNAIle in archaea and bacteria.
Next, Asn and Gln may have been added to the code. As shown in Figure 1(A–F), tRNAAsn is more diverged from tRNAAsp than tRNAGln is diverged from tRNAGlu. Perhaps, AspRS (class IIB aaRS) requires greater divergence of tRNAAsp and tRNAAsn for accurate discrimination than GluRS (class IB aaRS) requires for tRNAGlu and tRNAGln. Comparing relevant structures (PDBs 1ASY, 4WJ3, 1O0B and 1ZJW) [26–29], dimeric class IIB aaRS enzymes such as AspRS and AsnRS appear to make weaker determining acceptor stem contacts than monomeric class IB enzymes such as GluRS and GlnRS, and, therefore, may require greater divergence of tRNAs for discrimination.
Because the code initially sectors to encode a small set of amino acids (i.e. up to ∼16), but the code is forced by tRNA anticodon loop geometry to be in a register of 3 nt, wobble position ambiguity is likely to be an early advantage. If, from the onset, the code had evolved with accurate wobble specification, for instance, too many amino acids would be specified at too early a stage of evolution. Such an inflexible code puts heavy pressures on mRNA to also adopt a highly complex complementary code, slowing the pace of code evolution. So, in early stages, too few amino acids were available to justify a 3 nt code, and the barriers to code evolution were too high to allow for an inflexible 3 nt code. Also, if the wobble position were strongly specified in coding, the codon-anticodon table would likely not have broken so completely into 4-codon sectors. As the intermediate code becomes established, evolutionary pressures change to add additional chemistry to the code, and innovation at the wobble position becomes a more viable strategy. As an example, adenine in the anticodon wobble position might only pair with uridine in mRNA, supporting an inflexible 3 nt code, whereas guanine in the wobble position pairs with either cytidine or uridine. Restrictions against an overly inflexible 3 nt code may partly explain negative selection against adenine in the anticodon wobble position. Converting tRNA wobble adenine→inosine allows pairing with mRNA codon adenine, cytidine and uridine, increasing ambiguity of mRNA interpretation [12,13] Evolution of the adenine→inosine modification, therefore, provides evidence for positive selection for ambiguity at the tRNA wobble anticodon position, in order to match tRNAs with a larger set of cognate synonymous codons in mRNAs [17].
We posit that the last ∼5–6 amino acids to be added to the code may include Met, His, Cys, Phe, Tyr and Trp. The upper right 4-codon sector no longer encodes Ser but rather Cys, Ter (stop) and Trp. Arg invades another Ser 4-codon sector. Met appears to invade the Ile sector, which, judging from the unutilized tRNAIle anticodons in archaea and bacteria, may never have been fully occupied by Ile. In the archaea that are most similar to LUCA, tRNAMet appears to be derived from tRNAIle (Figure 1(A–C)), as might be expected from its position in the table [30]. In archaea and bacteria, tRNAIle generally utilizes only a single GAU, UAU or CAU anticodon from its 4-codon sector (generally GAU).
A bacterial and eukaryotic tRNA anticodon modification
Bacteria modified the genetic code in tRNA by utilizing tRNAArg (ACG), which, for the most part, is the only anticodon in bacteria with adenine encoded in the wobble position [12,13]. Bacteria utilize a tRNA adenosine deaminase to convert adenine to inosine at the wobble position. The advantage to bacteria of the ACG→ICG modification, which is missing in archaea, may be to protect the Arg (ACG, GCG, UCG, CCG) 4-codon sector from dividing into two 2-codon sectors, adding a new amino acid not encoded in archaea to the bacterial code. Inosine can interact with mRNA codons ending in wobble A, C and U. Because of this ambiguity in reading mRNA codons, it becomes difficult to subdivide this Arg 4-codon sector. Eukaryotes (and a few bacteria) have altered the genetic code further to include other tRNAs with adenine→inosine in the anticodon wobble position. In eukaryotes, tRNALeu (AAG), tRNAIle (AAU), tRNAVal (AAC), tRNASer (AGA), tRNAPro (AGG), tRNAThr (AGU), tRNAAla (AGC) and tRNAArg (ACG) with adenine converted to inosine in the wobble position are utilized [13].
Discussion
A model for tRNA evolution
A model for evolution of the tRNA cloverleaf has been proposed and strongly supported using statistical tests [3]. Essentially, all predictions of the model have been verified for archaeal and bacterial tRNAs. The model is based on ligation of three 31 nt minihelices followed by two internal, symmetrical 9 nt deletions to yield a 75 nt cloverleaf core (1–75), with the attached discriminator base (76) and 3’-CCA (77–79). By contrast, historical tRNA numbering utilizes a 72 nt core, which is based on eukaryotic tRNAs with 3 nt deleted in the D loop relative to tRNAPri. In cloverleaf evolution, one of the three ligated minihelices became the D loop, one the anticodon loop and one the T loop. 9 nt deletions are within ligated acceptor stem sequences, leaving two 5 nt relics of what were initially complementary acceptor stems surrounding the anticodon stem. The anticodon stem and loop and the T stem and loop are homologous, and obviously so, particularly for archaeal tRNAs, and homology is starkly evident from inspection of typical tRNA diagrams (i.e. of Pyrococcus tRNAs; Figure S9) [3].
Two minihelix tRNA evolution models
In a competing two minihelix model for tRNA evolution, proposed by others [31–33], the cloverleaf sequence is essentially divided through the anticodon loop, and the halves are expected to be homologous, even though, in the cloverleaf, the halves are expected to be complementary. In the two minihelix model, because, for the comparison, the anticodon stem and loop were bisected, the anticodon loop and the T loop cannot be homologs, although they clearly are, both from inspection of archaeal tRNAs (Figure S9) and using statistical tests [3]. In the two minihelix model, the D loop and the T loop ought to be homologs, although they clearly are not (in any alignment register). By contrast, the tRNA evolution model utilized here is predictive and apparently accurate, and competing models are falsified. Identification of tRNAPri based on the tRNA evolution model is highly predictive for the evolution of the genetic code (Figures 1–3; Figures S1–S8).
tRNA and rugged evolution
A tightly folded RNA such as the tRNA cloverleaf is subject to rugged evolution in which many or most substitutions are catastrophic for folding [34,35]. For instance, most substitutions in a tRNA stem are expected to require rescue by a complementary mutation (except for many C→U substitutions in stems, which allow G∼U pairing). In our model for tRNA evolution from tRNAPri, very few substitutions (if any) are required to obtain a folded cloverleaf. By contrast, in a two minihelix model for tRNA evolution, many substitutions are necessary to obtain a cloverleaf. Because of rugged RNA evolution and the required number of compensating substitutions, a two minihelix model is untenable. Furthermore, a two minihelix model requires unimaginable convergent evolution of the T stem and loop and the anticodon stem and loop to apparent structural and sequence homology. Because cloverleaf tRNA is subject to rugged evolution [3,34,35] many disqualifying criticisms are generated for a two minihelix model. Other tRNA evolution models also appear to be inconsistent with rugged evolution of RNA [36,37].
A root for the tRNA evolutionary tree
The model for tRNA evolution indicates a sequence for tRNAPri [3], which is most similar to archaeal tRNAGly, indicating that Gly may be the founding amino acid of the code (Figure 2) [6,7] The polyglycine hypothesis is posited, that tRNA initially evolved to synthesize short chain polyglycine to stabilize protocells. Very rapidly, every permitted anticodon was initially assigned as tRNAGly before reassignment to specify other amino acids (Figure 5). Cloverleaf tRNA and the genetic code appear to be prerequisites for cellular and DNA genome-based life, which originate at LUCA. In the RNA-protein world, genes were more independent than they subsequently became, in compact, streamlined and rapidly replicating DNA genomes encapsulated in cells. We propose, therefore, that colonies of independently replicating tRNA genes in an RNA-polymer world quickly diversified to include all permitted anticodon sequences, which, initially, encoded glycine (i.e. based on acceptor stem sequence, discriminator A (as in tRNAPri and archaeal tRNAGly (Figure 2)) and typical tRNA sequences (Figure S9)). Of course, specification of glycine attachment by tRNAPri need not have been highly accurate. It appears that errors in tRNA charging drove code evolution [2,14,25].
Degeneracy and sectoring
We favor a simple stepwise model for evolution and sectoring of the genetic code (Figure 5). The model describes why the code specifies ∼20 amino acids and is degenerate. As we argue here, the initial genetic code probably consisted of 48 and not 64 permitted anticodons, because adenine in the wobble position of the anticodon loop is destabilizing and would be expected to interact awkwardly with mRNA [12]. Furthermore, adenine in the anticodon wobble position probably supports a genetic code that is overly inflexible during initial code evolution, because adenine too strongly specifies uridine in the mRNA wobble codon position. Because of early positive selection for ambiguity in reading the anticodon wobble position, the genetic code should be considered initially to be primarily a 2 nt code encoding at most 16 amino acids (or 15 amino acids + Ter (stop)) in a register of 3 nt. Discrimination using the wobble anticodon position is only achieved with difficulty and, because of the ambiguity of tRNA anticodon-mRNA codon interactions in the ribosome decoding center [38], recognition at the wobble anticodon base is not strongly constrained by Watson-Crick base pairing. Despite early selection for ambiguity reading the mRNA wobble position, the tRNA anticodon wobble position was later innovated to add an additional ∼5–6 letters to the code (16 + 5 = 21 letters total, including stops).
Wobble pairing: the importance of being ambiguous
Negative selection against adenine in the anticodon wobble position indicates that tRNA-mRNA wobble A∼C pairing is negatively selected when A is the tRNA anticodon wobble base [17]. We note, however, that G∼U and U∼G wobble pairings are allowed. This raises the question of whether C∼A pairing might have been allowed, if C was the tRNA anticodon wobble base. Modifications of tRNA wobble C improve C∼A base pairing, including agmatidine (archaea), 2-lysidine (bacteria) and 5-formylcytidine (mitochondria, eukarya) [39]. Many tRNAs have a weak C∼A hydrogen bonding interaction between the 7 nt anticodon loop base position 1 (i.e. 2’-O-methyl-C (C = O or N)) and loop base position 7 (i.e. A (NH2)). From PDB 4TRA, it appears that the weak 1→7 C∼A interaction is modulated by Mg2+, and elevated Mg2+ is reported to induce translation errors [40,41]. During the early stages of code evolution, therefore, ambiguous wobble base pair interactions appear to have been positively selected. We posit that, for translation, a wobble tRNA base C (or modified C) may pair mRNA base A more efficiently than a wobble tRNA base A will pair mRNA base C, partly explaining the strong negative selection of A in the tRNA anticodon wobble position. It appears that tRNA anticodon wobble C is not as strongly negatively selected as wobble A. We note the possibility that tRNA anticodon wobble C modification to pair mRNA codon A may have occurred very early in evolution to compensate for an otherwise overly restrictive code. Also, there may be a selected preference for G and C over A and U during early evolution of the code. The genetic code initially evolved to be a ∼16 letter code before innovating the wobble position to expand to a 21 letter code.
Covalent modifications of tRNAs are common. In Figure S10, archaeal tRNA modifications determined for Haloferax volcanii tRNAs from the Modomics database [39] are displayed on a Pyrococcus typical tRNA. In concept, tRNA modifications could be used as determinants for aaRS enzymes to discriminate different tRNAs (i.e. tRNAPhe in bacteria, which requires tRNAPhe modifications for accurate charging by PheRS) [42], although, to our knowledge, such a mechanism has not yet been clearly demonstrated for any archaeal tRNA. In archaea, many covalent modifications are found in the anticodon loop particularly at loop positions 1 and 3 (wobble). Modifications in the anticodon loop may: 1) help stabilize the tight U turn structure; 2) affect anticodon readout; and/or 3) modify weak anticodon loop positions 1→7 interactions. Contacts between loop positions 1 and 7 affect loop dynamics and modify wobble position readout [22,23]. Modifications of the D loop, T loop and V loop may stabilize loop and stem conformations, D loop-T loop interactions and/or stability of the overall cloverleaf fold. Of course, for bacteria and eukaryotes, tRNA modifications allow expansions of the anticodon repertoire, as seen for the enzymatic conversion of wobble position adenine→inosine [12,13].
Cloverleaf tRNA as an evolutionary archetype
In ancient evolution from about 3.8 to 4 billion years ago, cloverleaf tRNA was the defining innovation that made possible the RNA-protein world and then cellular life [3]. Essentially, without cloverleaf tRNA, the genetic code was impossible, and the RNA-protein world and cellular life were, therefore, impossible. 17 nt microhelices and 31 nt minihelices (17 nt microhelices with 2 × 7 nt acceptor stems) may have supported polyglycine synthesis, but there is little evidence that much more complex products were possible based on minihelix adapters [3]. For one thing, from the cloverleaf tRNAPri sequence, the 31 nt minihelix posited to have given rise to the D loop appears to have had glycine-specifying acceptor stems, indicating that, because at least two distinct minihelices (D loop and anticodon loop/T loop) appeared to have specified glycine, few products, if any, other than polyglycine were made.
In a minihelix world, the D loop minihelix could not have supported a 3 nt genetic code register, because the D loop minihelix cannot form a 7 nt U turn. By contrast, the minihelices that gave rise to the anticodon loop and the T loop form the tight 7 nt U turn loop. The anticodon loop and the T loop are homologous to each other and distinct in sequence from the D loop minihelix, except in the acceptor stems, which appear initially to be identical (GCG and CGC repeats) [3]. We posit, therefore, that polypeptide synthesis based on primitive minihelix adapters was chaotic, limited and inefficient.
Based on cloverleaf tRNA sequence, structure and evolution, we posit a strange polymer world that included acceptor stems (GCG and CGC repeats), D loop minihelices (UAGCC repeats with acceptor stems) and anticodon and T loop minihelices (∼GGCCCUUCAAAAGGGCC with acceptor stems) [3]. Replication of minihelices is expected to involve ligation and an unknown mechanism of complementary replication (i.e. ribozyme-based replication) producing complementary sequences. Ligation of 3 minihelices and symmetrical RNA processing is sufficient to generate the tRNA cloverleaf, indicating that the minihelix-polymer world quickly gave way to a world dominated by cloverleaf tRNA. We posit that cloverleaf tRNA was quickly adopted as an improved mechanism to synthesize polyglycine, which stabilized protocells to support an unknown mechanism of energy generation (i.e. ribozyme-based). As described in this paper, cloverleaf tRNA rapidly diversified to encode 20 amino acids and stop codons, sufficient to support the RNA-protein world and leading subsequently to DNA genome-based cellular life at LUCA with a relatively modern translation system and genetic code. Cloverleaf tRNA, therefore, is proposed to be the most essential and central molecular archetype that made the RNA-protein world and cellular life possible. Remarkably, particularly in archaea (i.e. Pyrococcus and Staphylothermus), cloverleaf tRNA is little changed since LUCA [3].
Explosive evolution
Francis Crick referred to the evolution of tRNA and the genetic code as a “frozen accident”, in which, very rapidly, tRNA, the code and aaRS enzymes coevolved into existence [2,8,14,36]. Logically, tRNA must initially evolve and diversify, indicating that the first aminoacyl transferase functions were ribozymes that later were replaced by aaRS enzymes. We consider that a separate and robust (i.e. ribozyme-based) genetic code is unlikely to have existed prior to cloverleaf tRNA. Indeed, evolution to the mature and nearly universal genetic code must have been rapid. The mechanisms that brought closure to code evolution, which we begin to address here, now particularly need to be explained. We posit that the code evolved mostly in two stages. In the initial stage up to ∼16 letters, ambiguity of the wobble anticodon position was of positive value so that more mRNA wobble sequences could be tolerated and so an initial code could be established to encode proteins using a limited set of available amino acids. As we show here, rejection of inflexible code evolution can partly explain the negative selection of adenine at the tRNA anticodon wobble position. In the later stage of evolution, innovation at the wobble position was utilized to complete sectoring of the code. According to this view, closure occurs to balance initial positive selection for wobble position ambiguity and the growing requirement to accurately encode robust proteins with sufficient chemistry.
Computational approaches
We simplify and shrink the problem of evolution of the genetic code. We show that adenine is generally not utilized in the tRNA anticodon wobble position, unless adenine can be converted to inosine by a tRNA adenosine deaminase, which is missing in archaea and was probably, therefore, missing at LUCA [12]. Because adenine at the wobble position is destabilizing for the anticodon loop, and because the tRNA wobble position is selected to be ambiguous, the primordial genetic code reduces to 44 unique tRNA anticodons + 3 stop codons. The archaeal species analyzed here have 46 tRNAs, including a missing tRNAIle (UAU) and a total of 3 tRNAMet (CAU), matching the expectation of 44 unique tRNA anticodons. We propose orderly mechanisms by which the code might have sectored to produce the current genetic code and suggest mechanisms by which the code progressed to universality and closure at ∼20 amino acids + stops. These observations render the evolution of the genetic code more reasonably accessible for computational approaches, such as machine learning and artificial intelligence. Because each organism solves the problem of tRNA evolution somewhat differently, we advocate for computational methods that relate tRNAomes, compared organism to organism, as we begin here. The collection of tRNAs for each organism is a set with bound limits (i.e. because of cloverleaf structure and rugged evolution) in which tRNAs are powerfully coevolved. Clearly, machine learning can be applied to the comprehensive comparison of tRNAome evolutionary trees (i.e. used as “fingerprints” to discriminate species). We note that advances in tRNA modifications found in bacteria and eukaryotes but missing in archaea expand the possibilities for the tRNA anticodon repertoire in ways that can be predicted and/or identified, for instance, as observed for adenine→inosine conversion at the anticodon wobble position.
Methods
Cloverleaf tRNA evolution
The model for tRNA evolution [3,4] was developed by inspection of typical tRNA sequences [18], sequence logos [43] and sequences obtained from tRNA databases [18,19]. It is clear (i.e. from inspection of typical tRNA diagrams and sequence logos) that archaeal tRNAs are more similar to LUCA tRNAs than bacterial tRNAs [3], so archaeal tRNAs are mostly used in the analyses shown here. Because the tRNA database often gives genomic DNA sequences for archaeal tRNAs, DNA sequences and RNA sequences are presented here according to convenience. We use a numbering system for tRNAs based on a 75 nt core sequence [3]. Traditional tRNA numbering is based on a 72 nt core sequence, which is determined from eukaryotic tRNAs with 3 nt deleted from the D loop [3,4]. The traditional numbering system is confusing.
Sequence logos
Sequence logos were prepared using Weblogo 3.6 (http://weblogo.threeplusone.com/create.cgi) [43].
NCBI Blast searches
The tRNAPri sequence (GCGGCGGTAGCCTAGCCTGGCCTAGGCGGCCGGGTTNNNAACCCGGCCGCCCCGGGTTCAAATCCCGGCCGCCGCACCA) [3] was searched using NCBI (National Center for Biotechnology Information; https://www.ncbi.nlm.nih.gov/) nucleotide blast versus genomic archaeal and bacterial sequences and against collections of tRNA sequences from different organisms (Figure 2). A top hit in these searches is tRNAGly. Nucleotides 19 (A→G) and 67 (A→T) were adjusted in tRNAPri because these sequences are invariant in archaea and conserved in bacteria. To allow for different anticodon sequences, the anticodon is represented by NNN. The typical Pyrococcus tRNA sequence is very similar to tRNAPri and typical archaeal tRNAGly (GCGGCGGTAGCNTAGCCTGGTNNAGNGCGCCGGNCTNNNGANCCGGNGGTCCCGGGTTCAAATCCCGGCCGCNGCACC) (Figure 2) [3].
Evolutionary trees
Evolutionary trees for tRNAs were generated using PASTA (https://github.com/smirarab/pasta) [44]. All available tRNAs for an organism were collected and annotated by hand. V loop inserts were removed in alignments to eliminate detection of possible false similarity comparing tRNALeu and tRNASer, which both have V loop inserts and which test as closely related tRNAs whether or not V loop inserts are included in the alignment (the comparison is not shown). To root trees, the tRNAPri sequence was included. Evolutionary distances were determined by adding the distances of related branches. PASTA output was analyzed using FigTree (http://tree.bio.ed.ac.uk/software/figtree/). Alignments of tRNAs were checked against the tRNA databases. If PASTA is allowed to align bacterial tRNAs, gap errors are sometimes generated aligning the 5’-acceptor stem and the D loop. In some cases, apparent errors in tRNA databases (i.e. unlikely mispairing of the 5’- and 3’-acceptor stems) were detected from analysis of tRNAomes and apparent misplacing of a tRNA in the tree. Adjustment of the tRNA sequence resulted in more appropriate placement of the tRNA in the tree. Finding likely sequence errors based on tRNAome trees indicates the probable reliability of trees. Pyrococcus tRNAomes appear to have two reassigned tRNAs (tRNAPhe (from tRNATrp) and tRNATrp (from tRNAPro)) (Figure 1).
Supplementary Material
Funding Statement
This work was partly supported by National Science Foundation CAREER [Grant number DBI-0953738 to Y.S].
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgements
We thank Kevin Liu (Computer Science and Engineering, Michigan State University), Robert Root-Bernstein (Physiology, MSU) and Bruce Kowiatek (Blue Ridge Community and Technical College Martinsburg, WV) for helpful discussions and advice. This work was partly supported by National Science Foundation CAREER Grant DBI-0953738 to Y.S.
References
- [1].Koonin EV, Novozhilov AS. Origin and evolution of the universal genetic code. Annu Rev Genet. 2017;51:45–62. doi: 10.1146/annurev-genet-120116-024713. PMID:28853922 [DOI] [PubMed] [Google Scholar]
- [2].Koonin EV. Frozen accident pushing 50: stereochemistry, expansion, and chance in the evolution of the genetic code. Life (Basel). 2017;7. PMID:28545255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Pak D, Root-Bernstein R, Burton ZF. tRNA structure and evolution and standardization to the three nucleotide genetic code. Transcription. 2017;8:205–19. doi: 10.1080/21541264.2017.1318811. PMID:28632998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Root-Bernstein R, Kim Y, Sanjay A, et al. tRNA evolution from the proto-tRNA minihelix world. Transcription. 2016;7:153–63. doi: 10.1080/21541264.2016.1235527. PMID:27636862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Quigley GJ, Rich A. Structural domains of transfer RNA molecules. Science. 1976;194:796–806. doi: 10.1126/science.790568. PMID:790568 [DOI] [PubMed] [Google Scholar]
- [6].Bernhardt HS, Patrick WM. Genetic code evolution started with the incorporation of glycine, followed by other small hydrophilic amino acids. J Mol Evol. 2014;78:307–309. doi: 10.1007/s00239-014-9627-y. PMID:24916657 [DOI] [PubMed] [Google Scholar]
- [7].Bernhardt HS. Clues to tRNA evolution from the distribution of class II tRNAs and serine codons in the genetic code. Life (Basel). 2016;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Bernhardt HS, Tate WP. Evidence from glycine transfer RNA of a frozen accident at the dawn of the genetic code. Biol Direct. 2008;3:53. doi: 10.1186/1745-6150-3-53. PMID:19091122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Trifonov EN. The triplet code from first principles. J Biomol Struct Dyn. 2004;22:1–11. doi: 10.1080/07391102.2004.10506975. PMID:15214800 [DOI] [PubMed] [Google Scholar]
- [10].Romaniuk JA, Cegelski L. Bacterial cell wall composition and the influence of antibiotics by cell-wall and whole-cell NMR. Philos Trans R Soc Lond B Biol Sci. 2015; 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Di Giulio M. The genetic code did not originate from an mRNA codifying polyglycine because the proto-mRNAs already codified for an amino acid number greater than one. J Theor Biol. 2014;361:204–5. doi: 10.1016/j.jtbi.2014.09.006. PMID:25218496 [DOI] [PubMed] [Google Scholar]
- [12].Saint-Leger A, Bello C, Dans PD, et al. Saturation of recognition elements blocks evolution of new tRNA identities. Sci Adv. 2016;2:e1501860. doi: 10.1126/sciadv.1501860. PMID:27386510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Rafels-Ybern A, Torres AG, Grau-Bove X, et al. Codon adaptation to tRNAs with Inosine modification at position 34 is widespread among Eukaryotes and present in two Bacterial phyla. RNA Biol. 2017;1–8. doi: 10.1080/15476286.2017.1358348. PMID:28880718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Koonin EV, Novozhilov AS. Origin and evolution of the genetic code: the universal enigma. IUBMB Life. 2009;61:99–111. doi: 10.1002/iub.146. PMID:19117371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Demeshkina N, Jenner L, Westhof E, et al. A new understanding of the decoding principle on the ribosome. Nature. 2012;484:256–259. doi: 10.1038/nature10913. PMID:22437501 [DOI] [PubMed] [Google Scholar]
- [16].Ogle JM, Murphy FV, Tarry MJ, et al. Selection of tRNA by the ribosome requires a transition from an open to a closed form. Cell. 2002;111:721–732. doi: 10.1016/S0092-8674(02)01086-3. PMID:12464183 [DOI] [PubMed] [Google Scholar]
- [17].Pak D, Burton ZF. Aminoacyl-tRNA synthetase proofreading, anticodon wobble preference and sectoring of the genetic code via tRNA charging errors. Transcription. 2018; PMID:29264963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Juhling F, Morl M, Hartmann RK, et al. tRNAdb 2009: compilation of tRNA sequences and tRNA genes. Nucleic Acids Res. 2009;37:D159–D162. doi: 10.1093/nar/gkn772. PMID:18957446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Chan PP, Lowe TM. GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016;44:D184–D189. doi: 10.1093/nar/gkv1309. PMID:26673694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Widmann J, Harris JK, Lozupone C, et al. Stable tRNA-based phylogenies using only 76 nucleotides. RNA. 2010;16:1469–1477. doi: 10.1261/rna.726010. PMID:20558546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Giege R, Eriani G. Transfer RNA recognition and aminoacylation by synthetases. John Wiley & Sons, Ltd.; 2014. doi: 10.1002/9780470015902.a0000531.pub3 [DOI] [Google Scholar]
- [22].Agris PF, Eruysal ER, Narendran A, et al. Celebrating wobble decoding: Half a century and still much is new. RNA Biol. 2017;1–17. doi: 10.1080/15476286.2017.1356562. PMID:28812932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Agris PF, Narendran A, Sarachan K, et al. The importance of being modified: the role of RNA modifications in translational fidelity. Enzymes. 2017;41:1–50. doi: 10.1016/bs.enz.2017.03.005. PMID:28601219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Sengupta S, Higgs PG. Pathways of genetic code evolution in ancient and modern organisms. J Mol Evol. 2015;80:229–243. doi: 10.1007/s00239-015-9686-8. PMID:26054480 [DOI] [PubMed] [Google Scholar]
- [25].Novozhilov AS, Koonin EV. Exceptional error minimization in putative primordial genetic codes. Biol Direct. 2009;4:44. doi: 10.1186/1745-6150-4-44. PMID:19925661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Gruic-Sovulj I, Uter N, Bullock T, et al. tRNA-dependent aminoacyl-adenylate hydrolysis by a nonediting class I aminoacyl-tRNA synthetase. J Biol Chem. 2005;280:23978–23986. doi: 10.1074/jbc.M414260200. PMID:15845536 [DOI] [PubMed] [Google Scholar]
- [27].Bullock TL, Uter N, Nissan TA, et al. Amino acid discrimination by a class I aminoacyl-tRNA synthetase specified by negative determinants. J Mol Biol. 2003;328:395–408. doi: 10.1016/S0022-2836(03)00305-X. PMID:12691748 [DOI] [PubMed] [Google Scholar]
- [28].Ruff M, Krishnaswamy S, Boeglin M, et al. Class II aminoacyl transfer RNA synthetases: crystal structure of yeast aspartyl-tRNA synthetase complexed with tRNA(Asp). Science. 1991;252:1682–1689. doi: 10.1126/science.2047877. PMID:2047877 [DOI] [PubMed] [Google Scholar]
- [29].Suzuki T, Nakamura A, Kato K, et al. Structure of the Pseudomonas aeruginosa transamidosome reveals unique aspects of bacterial tRNA-dependent asparagine biosynthesis. Proc Natl Acad Sci USA. 2015;112:382–387. doi: 10.1073/pnas.1423314112. PMID:25548166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Bhattacharyya S, Varshney U. Evolution of initiator tRNAs and selection of methionine as the initiating amino acid. RNA Biol. 2016;13:810–819. doi: 10.1080/15476286.2016.1195943. PMID:27322343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Di Giulio M. The origin of the tRNA molecule: Independent data favor a specific model of its evolution. Biochimie. 2012;94:1464–1466. doi: 10.1016/j.biochi.2012.01.014. PMID:22305822 [DOI] [PubMed] [Google Scholar]
- [32].Di Giulio M. A comparison among the models proposed to explain the origin of the tRNA molecule: a synthesis. J Mol Evol. 2009;69:1–9. doi: 10.1007/s00239-009-9248-z. PMID:19488799 [DOI] [PubMed] [Google Scholar]
- [33].Widmann J, Di Giulio M, Yarus M, et al. tRNA creation by hairpin duplication. J Mol Evol. 2005;61:524–530. doi: 10.1007/s00239-004-0315-1. PMID:16155749 [DOI] [PubMed] [Google Scholar]
- [34].Curtis EA, Bartel DP. Synthetic shuffling and in vitro selection reveal the rugged adaptive fitness landscape of a kinase ribozyme. RNA. 2013;19:1116–1128. doi: 10.1261/rna.037572.112. PMID:23798664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Novozhilov AS, Wolf YI, Koonin EV. Evolution of the genetic code: partial optimization of a random code for robustness to translation error in a rugged fitness landscape. Biol Direct. 2007;2:24. doi: 10.1186/1745-6150-2-24. PMID:17956616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Rodin AS, Szathmary E, Rodin SN. On origin of genetic code and tRNA before translation. Biol Direct. 2011;6:14. doi: 10.1186/1745-6150-6-14. PMID:21342520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Caetano-Anolles G, Wang M, Caetano-Anolles D. Structural phylogenomics retrodicts the origin of the genetic code and uncovers the evolutionary impact of protein flexibility. PLoS One. 2013;8:e72225. doi: 10.1371/journal.pone.0072225. PMID:23991065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Rozov A, Demeshkina N, Westhof E, et al. New structural insights into translational miscoding. Trends Biochem Sci. 2016;41:798–814. doi: 10.1016/j.tibs.2016.06.001. PMID:27372401 [DOI] [PubMed] [Google Scholar]
- [39].Machnicka MA, Milanowska K, Osman Oglou O, et al. MODOMICS: a database of RNA modification pathways–2013 update. Nucleic Acids Res. 2013;41:D262–D267. doi: 10.1093/nar/gks1007. PMID:23118484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Scarlat IV, Rutkovskaya NO, Ginevskaya VA, et al. Magnesium-induced errors of translation in a cell-free system from krebs-II ascites carcinoma cells. FEBS Lett. 1969;5:231–232. doi: 10.1016/0014-5793(69)80340-6. PMID:11947285 [DOI] [PubMed] [Google Scholar]
- [41].Agafonov DE, Spirin AS. The ribosome-associated inhibitor A reduces translation errors. Biochem Biophys Res Commun. 2004;320:354–358. doi: 10.1016/j.bbrc.2004.05.171. PMID:15219834 [DOI] [PubMed] [Google Scholar]
- [42].Perona JJ, Gruic-Sovulj I. Synthetic and editing mechanisms of aminoacyl-tRNA synthetases. Top Curr Chem. 2014;344:1–41. PMID:23852030 [DOI] [PubMed] [Google Scholar]
- [43].Schneider TD, Stephens RM. Sequence logos: a new way to display consensus sequences. Nucleic Acids Res. 1990;18:6097–6100. doi: 10.1093/nar/18.20.6097. PMID:2172928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Mirarab S, Nguyen N, Guo S, et al. PASTA: ultra-large multiple sequence alignment for nucleotide and amino-acid sequences. J Comput Biol. 2015;22:377–386. doi: 10.1089/cmb.2014.0156. PMID:25549288 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.