

Caspases are key enzymes responsible for inducing apoptosis, an evolutionarily conserved form of Programmed Cell Death. Upon apoptotic stimuli, initiator caspases (e.g. caspase-8, -9 and -10) are recruited to high molecular weight (HMW) protein complexes (e.g.: DISC or apoptosome), where they dimerize to be activated. Initiator caspases contain a large pro-domain, which enables their recruitment to HMW signaling complexes through adaptor proteins like Apaf-1. In contrast, executioner caspases lack large pro-domains, their activation fully depends on cleavage events conducted by initiator caspases [1]. Caspase-2 represents unique characteristics among the caspases, sharing common structural features with both initiator caspases (a large pro-domain: caspase activation and recruitment domain/CARD) and executioner caspases. Initially, caspase-2 was shown to be activated in a PIDD (p53-induced protein with a death domain) – and RAIDD (receptor-interacting protein-associated ICH-1/CED-3 homologous protein with a death domain)-dependent protein complex (PIDDosome) upon DNA damage. Intriguingly, caspase-2 was demonstrated to play different context-dependent roles in the regulation of metabolism, tumor suppression and ageing. Novel studies suggested caspase-2 activation in HMWs that do not contain PIDD or RAIDD, however, very little is known about the constituents of such complexes (reviewed in ref. [2]). Furthermore, in contrast to the well-characterized caspase-8 and -9 activation, the exact mechanism(s) driving the activation of caspase-2 as an initiator caspase is still debated.
Pore-forming toxins (PFTs) are the largest family of toxins secreted from pathogenic bacteria. α-hemolysin/alpha toxin (for example secreted by Staphylococcus aureus) is one of the most-studied PFTs. Alpha toxin forms heptameric pores in cell membranes resulting in various outcomes, including apoptotic cell death, in the host cells. In our previous study we have shown that in epithelial cells caspase-2 assembles in a PIDDosome-independent high molecular weight protein complex where it is activated as an initiator caspase upon bacterial PFTs in a potassium ion sensitive manner [3].
In our recent study [4] we set out to identify the components of the HMW casapse-2 complexes that might contribute to the regulation of caspase-2 upon PFTs. By employing an in situ caspase-trapping method and subsequent mass spectrometric analysis we have identified apoptosis inhibitor 5 (API5/AAC-11/FIF (anti-apoptosis clone 11 and fibroblast growth factor-2 interacting factor) as a novel interacting partner of caspase-2. Originally, API5 was discovered as an anti-apoptotic protein in growth factor deprivation-induced cell death [5]. Structurally, API5 comprises of several helices constituting HEAT (Huntington, Elongation Factor 3, PR65/A, TOR)-like and ARM (Armadillo)-like repeats that function as protein-protein interaction domains. A recent study demonstrated that API5 is highly expressed in solid tumors (e.g. cervical cancer) and associated with poor prognosis [6], the exact molecular mechanism of API5-mediated apoptosis inhibition, however, remains unclear.
We have observed that genetic depletion of API-5 in HeLa cells resulted in significant apoptosis sensitization upon α-toxin but not to other inducers of apoptosis. By employing in vitro protein-protein binding assays, we found that the C terminal, helix rich domain of API5 might be responsible for the interaction with caspase-2. In addition, API5 fails to bind CARD-deleted and fully processed caspase-2, suggesting that API5 specifically interacts with the CARD domain of the unprocessed initiator caspase.
Caspase-2 has been shown to be activated upon DNA damage. By employing alpha toxin and genotoxic stress inducers (camptothecin and etoposide) in parallel, we have observed that depletion of API5 sensitized cells to PFT-induced cell death, it remained entirely ineffective, however, in DNA damage-induced apoptosis. This highly specific effect of API-5 could be explained by the unique subcellular distribution of caspase-2. We anticipate caspase-2 residing in organelles (such as nucleolus) may be less available to API5 then the cytoplasmic counterparts. In accordance with this, a recent publication by Ando et al. describes the formation of a PIDD-dependent nucleolar protein complex, mainly responsible for caspase-2 activation upon genotoxic stress [7]. In contrast, our microscopic analysis by Bimolecular Fluorescence Complementation (BiFC) upon PFT treatment indicates an extra-nuclear localization of dimerized caspase-2 [3]. This suggest at least two distinct subcellular sites for caspase-2 dimerization/activation. PFTs and possibly starvation (K Rajalingam and G Imre, unpublished data) might trigger a primarily cytosolic caspase-2 activation, more vulnerable to API5 (Figure 1). Further, the stoichiometry of the API5-Caspase-2 complex has to be elucidated.
Figure 1.
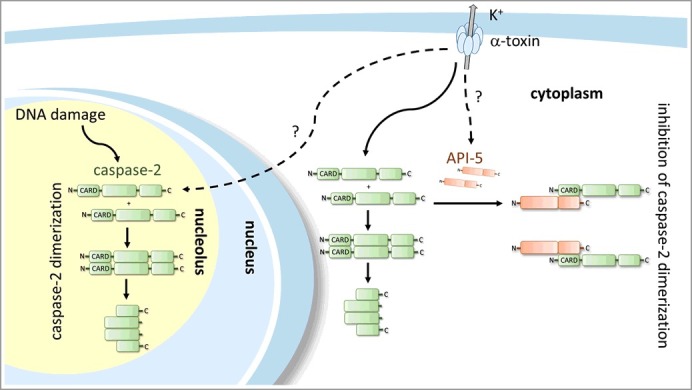
API5 directly binds to the CARD domain of caspase-2 and prevents the dimerization possibly in the cytosol. This leads to inactivation of caspase-2 and prevention of PFT-mediated apoptotic cell death.
It would be interesting to further investigate these preliminary findings by determining the subcellular distribution and the organelle-specific interaction of fluorescently labelled API5 and caspase-2 molecules. In addition, like caspase-2, API5 might undergo phosphorylation or other posttranslational modifications specific for different stimuli or organelles. These posttranslational alterations in API5 may significantly influence the binding affinity of API5 to caspase-2, further research elucidating these events may be therefore of importance.
Taken together, we have identified API5 as an endogenous inhibitor of caspase-2. Endogenous inhibitors of caspases have been described earlier: inhibitor of apoptosis proteins (IAPs) are direct caspase (casp-8/-9 and casp-3/-7) inhibitors, this is the first time, however, that an endogenously expressed direct caspase inhibitory protein is described which similarly to death effector proteins (e.g.: APAF-1) interacts via the CARD-domain. Our study unveils new segments of the so far poorly understood field of caspase-2 regulation. Future investigations of the API5 and caspase-2 interaction might provide further insights into the mechanisms of caspase-2 activation and perhaps provides the link for the poor prognosis in cancers associated with high API5 levels.
Funding Statement
This work was supported with a grant from the DFG (RA 1739/3-1).
Acknowledgments
This work was supported with a grant from the DFG (RA 1739/3-1) to KR. KR is a Heisenberg Professor of the DFG and a GFK fellow.
References
- [1].Boatright KM, Salvesen GS. Mechanisms of caspase activation. Curr Opin Cell Biol. 2003;15:725–731. doi: 10.1016/j.ceb.2003.10.009. PMID:14644197 [DOI] [PubMed] [Google Scholar]
- [2].Shalini S, Dorstyn L, Dawar S, et al. . Old, new and emerging functions of caspases. Cell Death Differ. 2015;22:526–539. doi: 10.1038/cdd.2014.216. PMID:25526085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Imre G, Heering J, Takeda AN, et al. . Caspase-2 is an initiator caspase responsible for pore-forming toxin-mediated apoptosis. EMBO J. 2012;31(11):2615–2628. doi: 10.1038/emboj.2012.93. PMID:22531785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Imre G, Berthelet J, Heering J, et al. . Apoptosis inhibitor 5 is an endogenous inhibitor of caspase-2. EMBO Rep. 2017;18(5):733–744. doi: 10.15252/embr.201643744. PMID:28336776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tewari M, Yu M, Ross B, et al. . AAC-11, a novel cDNA that inhibits apoptosis after growth factor withdrawal. Cancer Res. 1997;57:4063–4069. PMID:9307294 [PubMed] [Google Scholar]
- [6].Cho H, Chung JY, Song KH, et al. . Apoptosis inhibitor-5 overexpression is associated with tumor progression and poor prognosis in patients with cervical cancer. BMC Cancer. 2014;14:545. doi: 10.1186/1471-2407-14-545. PMID:25070070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Ando K, Parsons MJ, Shah RB, et al. . NPM1 directs PIDDosome-dependent caspase-2 activation in the nucleolus. J Cell Biol. 2017;216(6):1795–1810. doi: 10.1083/jcb.201608095. PMID:28432080 [DOI] [PMC free article] [PubMed] [Google Scholar]