

Abstract
Renal cell carcinoma (RCC) is the third most frequent malignancy within urological oncology. However, the mechanisms responsible for RCC metastasis are still needed further illustration. Our present study revealed that a seven-transmembrane receptor G-protein coupled estrogen receptor (GPER) was highly detected in various RCC cell lines such as ACHN, OS-RC-2 and SW839. The activation of GPER by its specific agonist G-1 significantly promoted the in vitro migration and invasion of ACHN and OS-RC-2 cells. G-1 also up regulated the expression of matrix metalloproteinase-2 (MMP-2) and MMP-9. The inhibitor of MMP-9 (Cat-444278), but not MMP-2 (Sc-204092), abolished G-1 induced cell migration, which suggested that MMP-9 is the key molecule mediating G-1 induced RCC progression. Further, G-1 treatment resulted in phosphorylation of AKT and ERK in RCC cells. PI3K/AKT inhibitor (LY294002), while not ERK inhibitor (PD98059), significantly abolished G-1 induced up regulation of MMP-9 in both AHCN and OS-RC-2 cells. Generally, our data revealed that activation of GPER by its specific agonist G-1 promoted the metastasis of RCC cells through PI3K/AKT/MMP-9 signals, which might be a promising new target for drug discovery of RCC patients.
KEYWORDS: G-1, GEPR, MMP-9, PI3K/AKT, renal cell carcinoma
Introduction
Renal cell carcinoma (RCC), which accounted for 90 ∼ 95% of neoplasms arising from the kidney and ∼3.8% of adult malignancy,1 is the third most frequent malignancy within urological oncology.2 Approximately 25 ∼ 30% of RCC patients have undiagnosed with metastatic diseases, and >95% of them have multiple metastases.3 It was reported that 20∼40% of RCC patients will relapse after resection.4 The patients with metastatic RCC have a 5-year survival of 26%.5 Therefore, the investigation about molecular pathways responsible for RCC metastasis are important for drug development and clinical treatment.
The results of statistics suggested that incidence of RCC in males is higher than that in females, with a ratio of 2:1,6 which suggested that estrogenic signal may be one of the causes of this risk. However, there were very limited data concerning the role of estrogenic signals in the tumorigenesis and development of RCC. Only one recent study revealed that estrogen receptor β (ERβ) is highly expressed in RCC cell lines such as A498, RCC-1, 786-O, ACHN, and Caki-1, and activation or over expression of ERβ suppresses the proliferation, migration and invasion of RCC cells.7 This suggested that estrogenic signal can modulate the progression of RCC via regulation its downstream genes and signals.
Recently, a seven-transmembrane receptor G-protein coupled estrogen receptor (GPER), which is structurally unrelated to nuclear ER, is suggested to mediate the rapid nongenomic signals of estrogen.8,9 The activation of GPER can induce the expression of genes and activate pathways which modulate cell migration and proliferation of endometrial,10,11 breast,12 and ovarian13 cancers. The data suggested that GPER plays an important role in modulating estrogen responsiveness and development and/or progression of cancers, which might be a promising new target for drug discovery.
In the present study, we demonstrated that GPER is highly expressed in RCC cell lines. The activation of GPER by its specific agonist G-1 promoted the migration and invasion of RCC cells and up regulation of matrix metalloproteinase-2 (MMP-2) and MMP-9. Further, the activation of GPER rapidly activated its downstream signals involved MAPK and PI3K/AKT, however, only inhibitor of PI3K/AKT signal pathway attenuated the G-1 induced up regulation of MMP-2 and MMP-9.
Materials and Methods
Reagents
All chemicals were of reagent grade or better and purchased from Sigma Chemical Co. (St. Louis, MO, USA) unless otherwise noted. Cat-444278 (MMP-9 inhibitor) and Sc-204092 (MMP-2 inhibitor) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). PD 98059 (MAPK/ERK kinase agonist) and LY294002 (PI3K/AKT antagonist) were purchased from Selleck Chemicals (Houston, TX, USA). Antibodies against GPER, MMP-2, MMP-9, AKT, p-AKT, ERK1/2, p-ERK1/2, p38-MAPK, p-p38-MAPK, JNK, p-JNK, GAPDH, and horseradish peroxidase-conjugated secondary antibody were purchased from Cell Signaling Technology (Beverly, MA, USA). PrimeScript® RT reagent kit and SYBR® Premix Ex TaqTM were products of TaKaRa. E.Z.N.A® HP Total RNA kit was bought from Omega Bio-Tek (Doraville, USA). All compounds were solubilized in DMSO with the concentrations of DMSO in the assay were less than 0.5% (v/v). Medium containing 0.5% DMSO was used as the control.
Cell line and cell culture
The human breast cancer cell line MCF-7 and RCC cell lines ACHN, A498, OS-RC-2 and SW839, which were purchased from American Type Culture Collection (Manassas, VA) and characterized at the bank by DNA fingerprinting analysis using short tandem repeat markers, were maintained in DMEM or RPMI 1640 medium, supplemented with 10% fetal bovine serum (Gibco, NY), 1% glycine (Gibco) and 1% penicillin-streptomycin in humidified 5% CO2 incubator (Thermon, USA). Twenty-four hours before the experiments, the medium was removed and replaced with RPMI 1640 or DMEM without phenol red supplemented with 5% dextran-coated, charcoal treated FBS (5% DC-FBS) to exclude estrogenic effects caused by the medium.
Wound healing assay
For the in vitro wound healing assay, cells were seeded in 6-cm culture dishes and cultured overnight until they had reached 90% confluence. After cell adhesion, a pipette tip (300 ml) was used to draw a cell-free line (approximately 1 mm wide) on the dish. After removing the original media and washing with PBS, media containing 1 μM G-1 was added to the cells. The condition of wound healing was observed and photographed using a microscope at the indicated times.
In vitro migration and invasion assay
Cell migration and invasion assays were performed using 6-well transwell plates (Falcon cell culture inserts, 8-μm pore size, BD, NJ) according to the manufacturer's instructions. Briefly, the cells were serum-starved overnight. For the invasion assay, the transwells were coated with Matrigel Matrix (BD Biosciences) at 20 mg/ml. The top chamber of transwell was loaded with 0.2 ml of 4 × 105 cells/ml in serum-free media, and the bottom chamber was loaded with 0.6 ml of DMEM medium containing 0.2% FBS. The cells were treated with 1 μM G-1 in the transwells at 37°C in a 5% CO2 atmosphere for the indicated times. For the migration assay, the transwells were uncoated. Then the penetrated cell will be fixed with 75% ethanol, stained with 1% crystal violet solution (Fisher Scientific, PA, USA), and cells were counted under microscope. All the invasion and migration assays were performed at least 3 individual experiments.
Western blotting analysis
Subconfluent cells were washed with ice-cold PBS and lysed in cell lysis buffer, and then lysates were cleared by centrifugation and denatured by boiling in Laemmli buffer. Protein concentrations were quantified by BCA protein assay (Pierce). Aliquats of protein were separated on 10% sodium dodecyl sulfate (SDS)–polyacrylamide gels and electrophoretically transferred to nitrocellulose membranes. Following blocking with 5% non-fat milk at room temperature for 2 h, membranes were incubated with the primary antibody at 1:1000 dilution overnight at 4°C. Secondary antibody reacted with the membrane at room temperature for 2 h. The proteins were visualized using HRP-conjugated secondary antibodies and ECL chemiluminescent reagent (Pierce) for Western blotting analysis.
Quantitative real-time PCR
Cells were washed twice with ice-cold PBS. Total mRNA was extracted with TRIZOL reagent. First strand of cDNA was generated from 2 μg total RNA using oligo-dT primer and Superscript II Reverse Transcriptase (GIBCO BRL, Grand Island, NY, USA). Quantitative Real-Time PCR was run on an iCycler (Bio-rad, Hercules, USA) using validated primers and SYBR Premix Ex Taq II (Takara, Japan) for detection. The cycle number when the fluorescence first reached a preset threshold (Ct) was used to quantify the initial concentration of individual templates for expression of mRNA of genes of interest. Transcripts of the housekeeping gene GAPDH in the same incubations were used for internal normalization. Primer pairs were as follows: GAPDH, forward 5′-GCA CCG TCA AGG CTG AGA AC-3′ and reverse 5′-TGG TGA AGA CGC CAG TGG A-3′; GPER, forward 5′- CCT GGA CGA GCA GTA TTA CGA TAT C-3′ and reverse 5′- TGC TGT ACA TGT TGA TCT G-3′. CT values were reported relative to GAPDH RNA. The cycle number when the fluorescence first reached a preset threshold (Ct) was used to quantify the initial concentration of individual templates for expression of mRNA of genes of interest. All experiments were performed three times independently and the average was used for comparison.
RNA interference
Cells were seeded on a 6-well plate (2 × 105 cells/well) and left in culture until the next day. They were then transfected with 100 pmol siRNA oligomer mixed with lipofectamine 2000 reagent in serum reduced medium according to the manufacturer's instructions. Medium was changed to complete culture medium 6 h later, and the cells were incubated at 37°C in a CO2 incubator for another 24 to 48 h before harvest.
Statistical analysis
All values were reported as mean ± SD unless otherwise specified. Statistical significance among control group and various treated groups were analyzed by two-tailed unpaired Student's t-test between two groups and by One-Way ANOVA followed by Bonferroni test for multiple comparison. All statistical analyses were carried out with SPSS 17.0 (SPSS Inc., Chicago, IL). A p-value of < 0.05 was considered to be statistically significant. The data are representative of three independent experiments.
Results
GPER was highly expressed in RCC cell lines
To understand the role of GPER in RCC, its expression was detected in various RCC cell lines and compared with the positive control (MCF-7 cells). Our results showed that the protein levels of GPER in RCC cell lines were comparable with or greater than that in MCF-7 cells (Fig. 1A), suggesting that GPER is also highly expressed in human RCC cells. Further, the ACHN cells derived from metastatic RCC displayed greater GPER expression than the other two RCC cells OS-RC-2 and SW839, which were derived from the primary RCC. It was also confirmed by the results of qRT-PCR that mRNA levels of GPER in ACHN cells were greater than that in OS-RC-2 and SW839 (Fig. 1).
Figure 1.
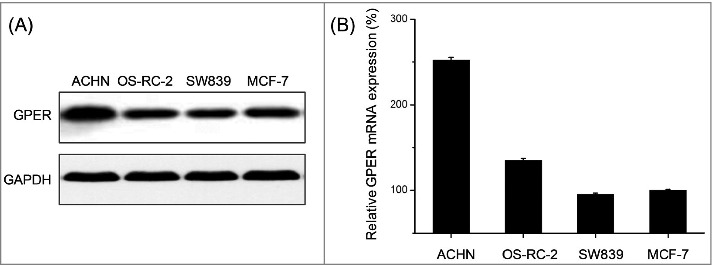
The expression of GPER in RCC cell lines. (A) The protein levels of GPER in ACHN, OS-RC-2, SW839 and MCF-7 cells were detected by Western blot. (B) The mRNA levels of GPER in ACHN, OS-RC-2, SW839 and MCF-7 cells were detected by qRT-PCR. Data are presented as means ± SD of three independent experiments.
Activation of GPER promoted the in vitro migration and invasion of RCC cells
Previous studies indicated that over expression of GPER were positively correlated with the metastasis and a poorer survival of various cancers.14 Therefore the effects of GPER on the motility of RCC cells lines were evaluated by use of wound healing and trans-well migration/invasion assays. We found that treatment with 1 μM G-1 for 48 h significantly increased wound closure as compared to the control group for both ACHN (Fig. 2A) and OS-RC-2 (Fig. 2B) cells. It was confirmed by the results of transwell migration and invasion assays, which showed that activation of GPER by 1 μM G-1 significantly promoted the migration and invasion of RCC cells such as ACHN and OS-RC-2 in vitro, as evidenced by a significant increase in the number of cells that migrated (Fig. 2C) and invaded (Fig. 2D) through the polycarbonate filter (P < 0.05). Collectively, our data revealed that activation of GPER by its specific agonist G-1 promoted the in vitro migration and invasion of RCC cells.
Figure 2.
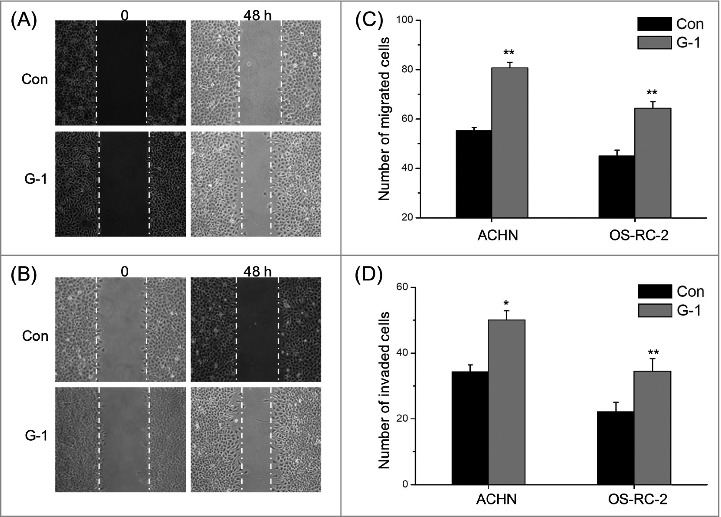
Activation of GPER promoted the in vitro migration and invasion of RCC cells. (A) Confluent monolayers of ACHN cells were scraped by a pipette tip to generate wounds and then were cultured. Representative images of wounds at 0 and 48 h in the absence or presence of 1 μM G-1; (B) Representative images of wounds of OS-RC-2 cells at 0 and 48 h in the absence or presence of 1 μM G-1; ACHN or OS-RC-2 cells were allowed to migrate or invade transwell chambers into the under-side of the filter for the indicated times in the presence or absence of 1 μM G-1. The migrated cells were fixed, stained, and photographed. The migrated cells were fixed, stained, and photographed. The number of migrated (C) or invaded (D) cells were compared with the control. Data are presented as means ± SD of three independent experiments. *P < 0.05 compared with control; **P < 0.01 compared with control.
G-1 up regulated the expression of MMP-2 and MMP-9 in RCC cells via GPER
MMPs are zinc-dependent endopeptidases which mediated the migration and invasion of various types of cancer cells.15 Whether the promotion effects of G-1 on motility of RCC cells were mediated by MMPs were investigated. We found that protein levels of MMP-2 and MMP-9 were up regulated by G-1 via a time dependent manner in both ACHN (Fig. 3A) and OS-RC-2 (Fig. 3B) cells. Further, the silence of GPER by its specific siRNA significantly attenuated G-1 induced up regulation of MMP-2 and MMP-9 in ACHN cells (Fig. 3C), which also abolished G-1 induced migration of ACHN cells (Fig. 3D). Collectively, our data revealed that activation of GPER by G-1 significantly up regulated the expression of MMP-2 and MMP-9 and then triggered the migration of RCC cells.
Figure 3.
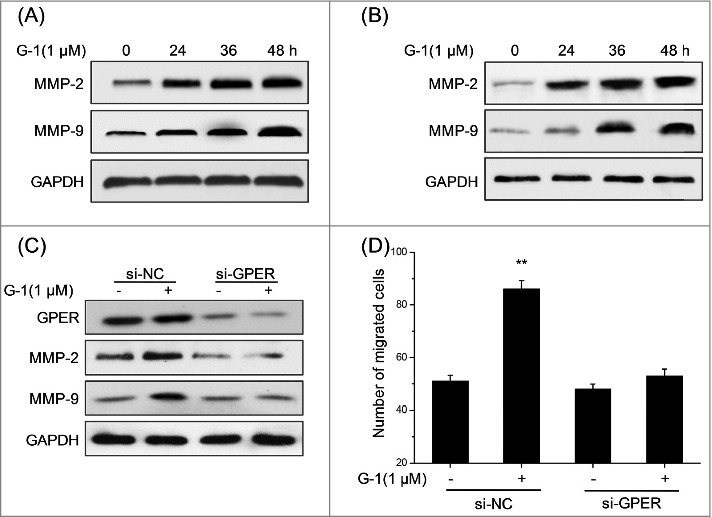
G-1 up regulated the expression of MMP2 and MMP-9 in RCC cells via GPER. (A) ACHN cells were treated with 1 μM G-1 for the indicated times, and then the protein levels of MMP-2 and MMP-9 were measured by Western blotting; (B) OS-RC-2 cells were treated with 1 μM G-1 for the indicated times, and then the protein levels of MMP-2 and MMP-9 were measured by Western blotting; ACHN cells were transfected with GPER specific si-RNA (si-GPER) or negative control si-RNA (si-NC) for 24 h before treated with or without 1 μM G-1 for 48 h, and then the proteins levels of GPER, MMP-2 and MMP-9 were measured by Western blotting (C), and the number of migrated cells were detected by use of transwell chamber (D). Data represent at least three independent experiments. **P < 0.01 compared with control.
MMP-9 was the key molecule for G-1 induced migration and invasion of RCC cells
The above results indicated that activation of GPER can increase the expression of MMP-2 and MMP-9 in RCC cells, then we further investigated which protein is responsible for G-1 induced migration and invasion of RCC cells. ACHN cells were pretreated with inhibitor of MMP-2 (Sc-204092, 10 nM) or MMP-9 (Cat-444278, 10 nM) for 60 min and then stimulated with G-1 for 48 h. Then the numbers of migrated and invaded cells were measured by transwell assays. As shown in Fig. 4, MMP-9 inhibitor Cat-444278 significantly abolished G-1 induced migration (A) and invasion (B) of ACHN cells, while MMP-2 inhibitor Sc-204092 had no obvious effect. The results suggested that MMP-9 is the key molecule for G-1 induced migration and invasion of RCC cells.
Figure 4.
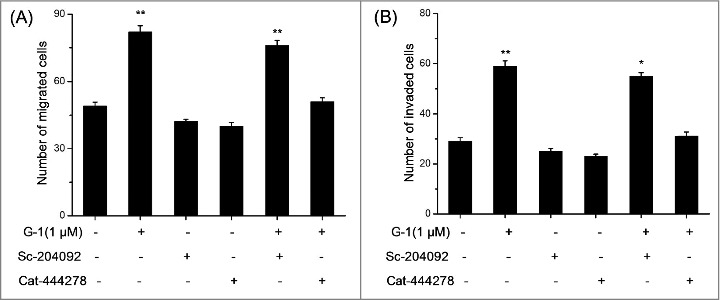
MMP-9 was the key molecule for G-1 induced migration and invasion of RCC cells. ACHN cells were pretreated with inhibitors of MMP-2 (Sc-204092, 10 nM) and MMP-9 (Cat-444278, 10 nM) for 60 min and then stimulated with 1 μM G-1 for 48 h. The number of migrated (A) or invaded (B) cells were compared with the control. Data are presented as means ± SD of three independent experiments. *P < 0.05 compared with control; **P < 0.01 compared with control.
Activation of GPER modulated the phosphorylation of MPAK and PI3K/AKT signals
Previous studies revealed that activation of GPER will lead to activation of various downstream signaling molecules such as MAPK and PI3K/AKT.16 Therefore the effects of G-1 on the phosphorylation of MAPK and PI3K/AKT were investigated. As shown in Fig. 5A, the activation of GPER increased the phosphorylation of AKT and ERK in ACHN cells treated with G-1 for 30 or 60 min, while not p38-MAPK and JNK. To further validation the effects of G-1 on phosphorylation of AKT and ERK, we checked the activation of AKT and ERK in OS-RC-2 cells . The increased phosphorylation of AKT and ERK were also observed in OS-RC-2 cells treated with G-1 for 30 min (Fig. 5B).
Figure 5.
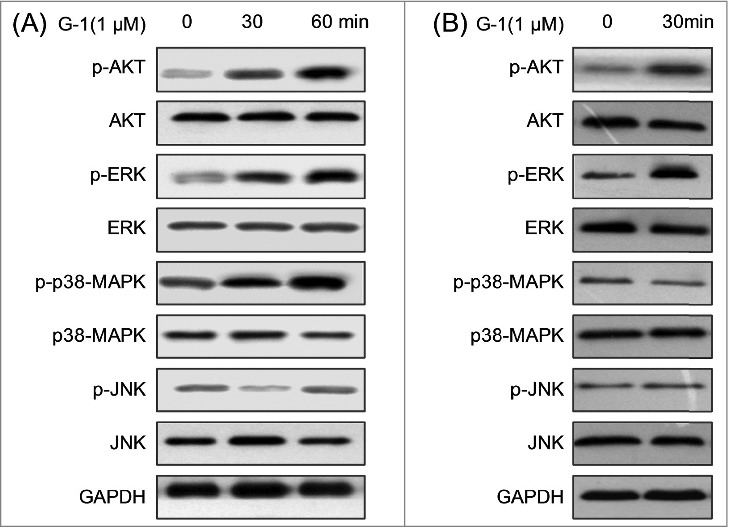
Activation of GPER modulated the activities of MPAK and PI3K/AKT signals. (A) ACHN cells were treated with 1 μM G-1 for the indicated times, and then the activation of AKT, ERK, p38-MAPK and JNK were examined by Western blot analysis; (B) OS-RC-2 cells were treated with 1 μM G-1 for the indicated times, and then the activation of AKT, ERK, p38-MAPK and JNK were examined by Western blot analysis. Data represent at least three independent experiments.
PI3K/AKT/MMP-9 mediated the G-1 induced migration of RCC cells
To explore whether both AKT and ERK are involved in G-1 induced up regulation of MMP-9, inhibitor of PI3K/AKT (LY294002) or ERK (PD98059) was used according to previous studies.17,18 Our results showed that PI3K/AKT inhibitor (LY294002), while not ERK inhibitor (PD98059), significantly abolished G-1 induced up regulation of MMP-9 in both AHCN (Fig. 6A) and OS-RC-2 (Fig. 6B) cells. It suggested that PI3K/AKT, rather than ERK, mediated G-1 induced up regulation of MMP-9 in RCC cells. Further, AHCN cells were pretreated with inhibitors of MMP-9 (Cat-444278, 10 nM) and/or LY294002 (10 μM) for 90 min respectively followed by stimulation with 1 μM G-1 for 48 h. The results showed that Cat-444278 and LY294002 alone or together significantly abolished G-1 induced migration of AHCN cells (Fig. 6C). Collectively, our results revealed that PI3K/AKT/MMP-9 mediated G-1 induced migration of RCC cells.
Figure 6.
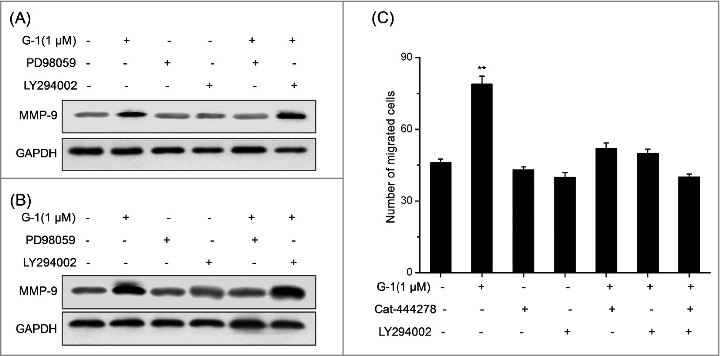
PI3K/AKT/MMP-9 mediated the G-1 induced migration of RCC cells. (A) ACHN cells were pretreated with PD98059 (10 μM) or LY294002 (10 μM) for 90 min respectively followed by stimulation with 1 μM G-1 for 48 h. The expression of MMP-9 was examined by Western blot analysis. (B) OS-RC-2 cells were pretreated with PD98059 (10 μM) or LY294002 (10 μM) for 90 min respectively followed by stimulation with 1 μM G-1 for 48 h. The expression of MMP-9 was examined by Western blot analysis. (C) ACHN cells were pretreated with inhibitors of MMP-9 (Cat-444278, 10 nM) and/or LY294002 (10 μM) for 90 min respectively followed by stimulation with 1 μM G-1 for 48 h. The number of migrated cells were compared with the control. Data are presented as means ± SD of three independent experiments. *P < 0.05 compared with control; **P < 0.01 compared with control.
Discussion
In the present study, we revealed for the first time that GPER is highly expressed in RCC cell lines. The activation of GPER by its specific agonist G-1 significantly promoted the in vitro migration and invasion of RCC cells. Further, G-1 obviously up regulated the protein expression of MMP-2 and MMP-9. Silence of MMP-9, while not MMP-2, abolished G-1 induced RCC cell migration. Further, the activation of GPER phosphorylated the downstream signals ERK1/2 and AKT. The inhibitor of PI3K/AKT, rather than ERK1/2, attenuated the G-1 induced cell migration and up regulation of MMP-9. Collectively, our results revealed that activation of GPER can trigger the motility of RCC cells via PI3K/AKT/MMP-9 signals, which might be a target for drug development against RCC.
Most studies about the roles of GPER in development of cancer focused on the effects of GPER on cell proliferation. Some studies suggested that GPER is a tumor-suppressor signal.19-22 In contrast, there were also many studies showed that activation of GPER is able to stimulate cell growth of endometrial,10 ovarian13 and breast23 cancer cells. There were limited data about the GPER and metastasis of cancer cells. The results of our study revealed that activation of GPER promotes the migration of invasion of RCC cells were consistent with the roles of GPER in endometrial cancer cell,24 breast cancer,12 and cancer-associated fibroblasts,25 but opposite of that GPER attenuated the EGF-induced cell migration in ovarian cancer cells.26 Despite these conflicting data on the roles of GPER in cancer cells, our data suggested that targeting its activity represents an important new approach for RCC therapy.
In the present study, the stimulatory effect of G-1 on cell migration was accompanied by up-regulation of migration-related factors MMP-2 and MMP-9. This was in line with the previous study that activation of GPER increases the production and activities of MMPs in endometrial cancer cells.24 Further, clinical data showed that GPER is more frequently expressed in malignant than benign ovarian endometriotic cysts and correlated with MMP-9 expression.27 Considering that MMPs can trigger the migration and invasion of various types of cancer cells,15 further, up regulation of MMPs is associated with poor prognosis of RCC,28 our data revealed that G-1 can stimulate the RCC cell migration by influencing the expression levels of MMP-2 and MMP-9. Furthermore, we found the up regulation of MMP-9, mediated the promotion effects of G-1 on migration of RCC cancer cells. It was also confirmed by the previous study that MMP-9, rather than MMP-2, is a candidate of predictor of disease-free survival in RCC.29
Previous studies reported that PI3K/AKT and ERK can be rapidly activated by GPER and then mediate the non genomic estrogenic responses.30-32 There were also accumulating evidences suggested that PI3K/AKT and MAPK/ERK play important roles in the regulation of cancer cell migration.33,34 Phosphorylation of AKT has been reported to be associated with a loss of cell adhesion and apico-basolateral cell polarization, decrease in cell-matrix adhesion, and induction of cell motility.35 PI3K/AKT pathway is frequently activated in various cancers and plays an important role in promoting epithelial mesenchymal transition (EMT).36 That GPER/ERK signaling stimulates cell invasion was observed in breast cancer cells such as MDA-MB-468 and MDA-MB-436 cells.37 Our results here revealed that G-1 rapidly activated both ERK1/2 and AKT, while only inhibitor of PI3K/AKT, rather than that of ERK1/2, abolished G-1 induced up regulation of MMP-9 and cell migration. It suggested that activation of GPER induced RCC cell migration via PI3K/AKT/MMP-9 signals.
In conclusion, the current study characterized that activation of GPER by its specific agonist G-1 significantly promoted the in vitro migration and invasion of human RCC cells via up regulation of MMPs by a time dependent manner. Further, GPER/PI3K/AKT/MMP-9 signals mediated G-1 induced cell migration and up regulation of MMP-9. This study provided new sight into the role of GPER in tumorigenesis and metastasis of RCC.
Funding Statement
This project was supported by the Fundamental Research Funds of the Central Universities (Grant No. 21613316).
Abbreviations
- EMT
epithelial mesenchymal transition
- ER
estrogen receptor
- GPER
G Protein Coupled Estrogen Receptor
- MMP
matrix metalloproteinase
- RCC
renal cell carcinoma
- SDS
sodium dodecyl sulfate.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2013; 62:10-29; PMID:22237781; http://dx.doi.org/ 10.3322/caac.20138 [DOI] [PubMed] [Google Scholar]
- 2. Ljungberg B, Hanbury DC, Kuczyk MA, Merseburger AS, Mulders PF, Patard JJ, Sinescu IC. Renal cell carcinoma guideline. Eur Urol 2007; 51:1502-10; PMID:17408850; http://dx.doi.org/ 10.1016/j.eururo.2007.03.035 [DOI] [PubMed] [Google Scholar]
- 3. Linehan WM, Grubb RL, Coleman JA, Zbar B, Walther MM. The genetic basis of cancer of kidney cancer: implications for gene-specific clinical management. BJU Int 2005; 95 Suppl 2:2-7; PMID:15720328; http://dx.doi.org/ 10.1111/j.1464-410X.2005.05189.x [DOI] [PubMed] [Google Scholar]
- 4. Stewart GD, O'Mahony FC, Powles T, Riddick ACP, Harrison DJ, Faratian D. What can molecular pathology contribute to the management of renal cell carcinoma? Nat Rev Urol 2011; 8:255-65; PMID:21487387; http://dx.doi.org/ 10.1038/nrurol.2011.43 [DOI] [PubMed] [Google Scholar]
- 5. Linehan WM, Bratslavsky G, Pinto PA, Schmidt LS, Neckers L, Bottaro DP, Srinivasan R. Molecular diagnosis and therapy of kidney cancer. Annu Rev Med 2010; 61:329-43; PMID:20059341; http://dx.doi.org/ 10.1146/annurev.med.042808.171650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ferlay J, Autier P, Boniol M, Heanue M, Colombet M, Boyle P. Estimates of the cancer incidence and mortality in Europe in 2006. Ann Oncol 2007; 18:581-92; PMID:17287242; http://dx.doi.org/ 10.1093/annonc/mdl498 [DOI] [PubMed] [Google Scholar]
- 7. Yu CP, Ho JY, Huang YT, Cha TL, Sun GH, Yu DS, Chang FW, Chen SP, Hsu RJ. Estrogen inhibits renal cell carcinoma cell progression through estrogen receptor-beta activation. Plos One 2013; 8:e56667; PMID:23460808; http://dx.doi.org/ 10.1371/journal.pone.0056667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Luo LJ, Liu F, Lin ZK, Xie YF, Xu JL, Tong QC, Shu R. Genistein regulates the IL-1 beta induced activation of MAPKs in human periodontal ligament cells through G protein-coupled receptor 30. Arch Biochem Biophys 2012; 522:9-16; PMID:22521737; http://dx.doi.org/ 10.1016/j.abb.2012.04.007 [DOI] [PubMed] [Google Scholar]
- 9. Ge C, Yu M, Zhang C. G protein-coupled receptor 30 mediates estrogen-induced proliferation of primordial germ cells Via EGFR/Akt/beta-catenin signaling pathway. Endocrinology 2012; 153:3504-16; PMID:22635679; http://dx.doi.org/ 10.1210/en.2012-1200 [DOI] [PubMed] [Google Scholar]
- 10. Du GQ, Zhou L, Chen XY, Wan XP, He YY. The G protein-coupled receptor GPR30 mediates the proliferative and invasive effects induced by hydroxytamoxifen in endometrial cancer cells. Biochem Biophy Res Com 2012; 420:343-9; PMID:22425989; http://dx.doi.org/ 10.1016/j.bbrc.2012.02.161 [DOI] [PubMed] [Google Scholar]
- 11. He YY, Cai B, Yang YX, Liu XL, Wan XP. Estrogenic G protein-coupled receptor 30 signaling is involved in regulation of endometrial carcinoma by promoting proliferation, invasion potential, and interleukin-6 secretion via the MEK/ERK mitogen-activated protein kinase pathway. Cancer Sci 2009; 100:1051-61; PMID:19432902; http://dx.doi.org/ 10.1111/j.1349-7006.2009.01148.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pandey DP, Lappano R, Albanito L, Madeo A, Maggiolini M, Picard D. Estrogenic GPR30 signalling induces proliferation and migration of breast cancer cells through CTGF. EMBO J 2009; 28:523-32; PMID:19153601; http://dx.doi.org/ 10.1038/emboj.2008.304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Albanito L, Madeo A, Lappano R, Vivacqua A, Rago V, Carpino A, Oprea TI, Prossnitz ER, Musti AM, Ando S, et al. G protein-coupled receptor 30 (GPR30) mediates gene expression changes and growth response to 17beta-estradiol and selective GPR30 ligand G-1 in ovarian cancer cells. Cancer Res 2007; 67:1859-66; PMID:17308128; http://dx.doi.org/ 10.1158/0008-5472.CAN-06-2909 [DOI] [PubMed] [Google Scholar]
- 14. Wang D, Hu L, Zhang G, Zhang L, Chen C. G protein-coupled receptor 30 in tumor development. Endocrine 2010; 38:29-37; PMID:20960099; http://dx.doi.org/ 10.1007/s12020-010-9363-z [DOI] [PubMed] [Google Scholar]
- 15. Polette M, Nawrocki-Raby B, Gilles C, Clavel C, Birembaut P. Tumour invasion and matrix metalloproteinases. Crit Rev Oncol Hematol 2004; 49:179-86; PMID:15036258; http://dx.doi.org/ 10.1016/j.critrevonc.2003.10.008 [DOI] [PubMed] [Google Scholar]
- 16. Prossnitz ER, Barton M. The G-protein-coupled estrogen receptor GPER in health and disease. Nat Rev Endocrinol 2011; 7:715-26; PMID:21844907; http://dx.doi.org/ 10.1038/nrendo.2011.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang H, Wang HS, Zhou BH, Li CL, Zhang F, Wang XF, Zhang G, Bu XZ, Cai SH, Du J. Epithelial-mesenchymal transition (EMT) induced by TNF-alpha requires AKT/GSK-3beta-mediated stabilization of snail in colorectal cancer. Plos One 2013; 8:e56664; PMID:23431386; http://dx.doi.org/ 10.1371/journal.pone.0056664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Secchiero P, Gonelli A, Carnevale E, Milani D, Pandolfi A, Zella D, Zauli G. TRAIL promotes the survival and proliferation of primary human vascular endothelial cells by activating the Akt and ERK pathways. Circulation 2003; 107:2250-6; PMID:12668516; http://dx.doi.org/ 10.1161/01.CIR.0000062702.60708.C4 [DOI] [PubMed] [Google Scholar]
- 19. Ariazi EA, Brailoiu E, Yerrum S, Shupp HA, Slifker MJ, Cunliffe HE, Black MA, Donato AL, Arterburn JB, Oprea TI, et al. The G protein-coupled receptor GPR30 inhibits proliferation of estrogen receptor-positive breast cancer cells. Cancer Res 2010; 70:1184-94; PMID:20086172; http://dx.doi.org/ 10.1158/0008-5472.CAN-09-3068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Holm A, Baldetorp B, Olde B, Leeb-Lundberg LM, Nilsson BO. The GPER1 agonist G-1 attenuates endothelial cell proliferation by inhibiting DNA synthesis and accumulating cells in the S and G2 phases of the cell cycle. J Vasc Res 2011; 48:327-35; PMID:21273787; http://dx.doi.org/ 10.1159/000322578 [DOI] [PubMed] [Google Scholar]
- 21. Chan QKY, Lam HM, Ng CF, Lee AYY, Chan ESY, Ng HK, Ho SM, Lau KM. Activation of GPR30 inhibits the growth of prostate cancer cells through sustained activation of Erk1/2, c-jun/c-fos-dependent upregulation of p21, and induction of G(2) cell-cycle arrest. Cell Death Differ 2010; 17:1511-23; PMID:20203690; http://dx.doi.org/ 10.1038/cdd.2010.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang C, Lv X, He C, Hua G, Tsai MY, Davis JS. The G-protein-coupled estrogen receptor agonist G-1 suppresses proliferation of ovarian cancer cells by blocking tubulin polymerization. Cell Death Dis 2013; 4:e869; PMID:24136233; http://dx.doi.org/ 10.1038/cddis.2013.397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Albanito L, Sisci D, Aquila S, Brunelli E, Vivacqua A, Madeo A, Lappano R, Pandey DP, Picard D, Mauro L, et al. Epidermal growth factor induces G protein-coupled receptor 30 expression in estrogen receptor-negative breast cancer cells. Endocrinology 2008; 149:3799-808; PMID:18467441; http://dx.doi.org/ 10.1210/en.2008-0117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. He YY, Du GQ, Cai B, Yan Q, Zhou L, Chen XY, Lu W, Yang YX, Wan XP. Estrogenic transmembrane receptor of GPR30 mediates invasion and carcinogenesis by endometrial cancer cell line RL95-2. J Cancer Res Clin Oncol 2012; 138:775-83; PMID:22270964; http://dx.doi.org/ 10.1007/s00432-011-1133-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Madeo A, Maggiolini M. Nuclear alternate estrogen receptor GPR30 mediates 17beta-estradiol-induced gene expression and migration in breast cancer-associated fibroblasts. Cancer Res 2010; 70:6036-46; PMID:20551055; http://dx.doi.org/ 10.1158/0008-5472.CAN-10-0408 [DOI] [PubMed] [Google Scholar]
- 26. Henic E, Noskova V, Hoyer-Hansen G, Hansson S, Casslen B. Estradiol attenuates EGF-induced rapid uPAR mobilization and cell migration via the G-protein-coupled receptor 30 in ovarian cancer cells. Int J Gynecol Cancer 2009; 19:214-22; PMID:19395996; http://dx.doi.org/ 10.1111/IGC.0b013e31819bcb75 [DOI] [PubMed] [Google Scholar]
- 27. Long L, Cao Y, Tang LD. Transmembrane estrogen receptor GPR30 is more frequently expressed in malignant than benign ovarian endometriotic cysts and correlates with MMP-9 expression. Int J Gynecol Cancer 2012; 22:539-45; PMID:22495744; http://dx.doi.org/ 10.1097/IGC.0b013e318247323d [DOI] [PubMed] [Google Scholar]
- 28. Kallakury BV, Karikehalli S, Haholu A, Sheehan CE, Azumi N, Ross JS. Increased expression of matrix metalloproteinases 2 and 9 and tissue inhibitors of metalloproteinases 1 and 2 correlate with poor prognostic variables in renal cell carcinoma. Clin Cancer Res 2001; 7:3113-9; PMID:11595703 [PubMed] [Google Scholar]
- 29. Cho NH, Shim HS, Rha SY, Kang SH, Hong SH, Choi YD, Hong SJ, Cho SH. Increased expression of matrix metalloproteinase 9 correlates with poor prognostic variables in renal cell carcinoma. Eur Urol 2003; 44:560-6; PMID:14572755; http://dx.doi.org/ 10.1016/S0302-2838(03)00362-2 [DOI] [PubMed] [Google Scholar]
- 30. Dong S, Terasaka S, Kiyama R. Bisphenol a induces a rapid activation of Erk1/2 through GPR30 in human breast cancer cells. Environ Poll 2011; 159:212-8; PMID:20875696; http://dx.doi.org/ 10.1016/j.envpol.2010.09.004 [DOI] [PubMed] [Google Scholar]
- 31. Ge LC, Chen ZJ, Liu HY, Zhang KS, Liu H, Huang HB, Zhang G, Wong CK, Giesy JP, Du J, et al. Involvement of activating ERK1/2 through G protein coupled receptor 30 and estrogen receptor alpha/beta in low doses of bisphenol a promoting growth of Sertoli TM4 cells. Toxicol Lett 2014; 226:81-9; PMID:24495410; http://dx.doi.org/ 10.1016/j.toxlet.2014.01.035 [DOI] [PubMed] [Google Scholar]
- 32. Ptak A, Gregoraszczuk EL. Bisphenol A induces leptin receptor expression, creating more binding sites for leptin, and activates the JAK/Stat, MAPK/ERK and PI3K/Akt signalling pathways in human ovarian cancer cell. Toxicol Lett 2012; 210:332-7; PMID:22343039; http://dx.doi.org/ 10.1016/j.toxlet.2012.02.003 [DOI] [PubMed] [Google Scholar]
- 33. Kim EK, Choi EJ. Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta 2010; 1802:396-405; PMID:20079433; http://dx.doi.org/ 10.1016/j.bbadis.2009.12.009 [DOI] [PubMed] [Google Scholar]
- 34. Reddy KB, Nabha SM, Atanaskova N. Role of MAP kinase in tumor progression and invasion. Cancer Metastasis Rev 2003; 22:395-403; PMID:12884914; http://dx.doi.org/ 10.1023/A:1023781114568 [DOI] [PubMed] [Google Scholar]
- 35. Bellacosa A, Kumar CC, Di Cristofano A, Testa JR. Activation of AKT kinases in cancer: implications for therapeutic targeting. Adv Cancer Res 2005; 94:29-86; PMID:16095999; http://dx.doi.org/ 10.1016/S0065-230X(05)94002-5 [DOI] [PubMed] [Google Scholar]
- 36. Larue L, Bellacosa A. Epithelial-mesenchymal transition in development and cancer: role of phosphatidylinositol 3' kinase/AKT pathways. Oncogene 2005; 24:7443-54; PMID:16288291; http://dx.doi.org/ 10.1038/sj.onc.1209091 [DOI] [PubMed] [Google Scholar]
- 37. Yu T, Liu M, Luo H, Wu C, Tang X, Tang S, Hu P, Yan Y, Wang Z, Tu G. GPER mediates enhanced cell viability and motility via non-genomic signaling induced by 17beta-estradiol in triple-negative breast cancer cells. J Steroid Biochem Mol Biol 2014; 143C:392-403; PMID:24874276; http://dx.doi.org/ 10.1016/j.jsbmb.2014.05.003 [DOI] [PubMed] [Google Scholar]