

ABSTRACT
MicroRNAs (miRNAs) are small, non-coding RNA molecules that regulate gene expression post-transcriptionally. As a consequence of their function towards mRNA, miRNAs are widely associated with the pathogenesis of several human diseases, making miRNAs a target for new therapeutic strategies based on the control of their expression. Indeed, numerous works were published in the past decades showing the potential use of antisense oligonucleotides to target aberrant miRNAs (AMOs) involved in several human pathologies. New classes of chemical-modified-AMOs, including locked nucleic acid oligonucleotides, have recently proved their worth in silencing miRNAs. A correct design of a specific AMOs can help to improve their performance and potency towards the target miRNA by increasing for instance nuclease resistance and target affinity. This review outlines the technologies involved to suppress aberrant miRNAs. From the design strategies used in AMOs to its application in novel miRNA-based therapeutics and detection methodologies.
KEYWORDS: miRNA, anti-miRNA, AMO design, chemical modifications, therapeutics, miRNA detection methods
Introduction
MiRNAs are small, single-stranded and noncoding RNAs, with approximately 21 nucleotides (nt) that regulate gene expression at a posttranscriptional level. They complementarily bind to the 3′ untranslated region (3′UTR) of their target messenger RNAs (mRNAs) causing their degradation, translational repression, and/or deadenylation (Fig. 1) [1–3].
Figure 1.
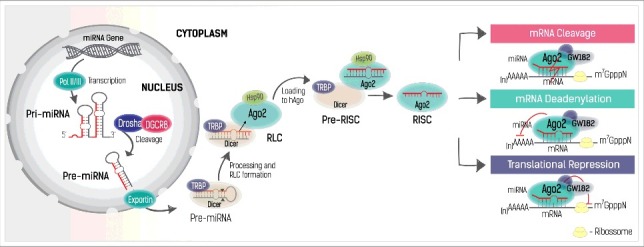
MiRNA biogenesis. Formation of a primary miRNA structure (pri-miRNA) by RNA polymerase II and III (Pol II/III), followed by Drosha's cleavage, resulting in a pre-miRNA. The pre-miRNA is then exported to the cytoplasm by Exportin-5/Ran complex (Exportin), where it is transformed in a loop-free homoduplex miRNA by the action of Dicer-TRBP. After association of the RLC complex comprised essentially by Dicer-TRBP-Ago2, the ds-miRNA is loaded into Ago2 (mediated by Hsp90), where is further processed to a mature single-stranded miRNA (guide strand). The RISC complex is then guided by the miRNA to the target complementary mRNA, leading to mRNA cleavage, translational repression or deadenylation. Upon RISC binding, other important proteins may associate to the Ago2, like GW182, to assist on the silence of the mRNA. DGCR8: Microprocessor complex subunit DGCR8 (DiGeorge syndrome critical region 8); Dicer: Double-strand-specific ribonuclease; TRBP: Trans-activation-responsive RNA-binding protein; Ago2: protein argonaute-2; Hsp90: Heat shock protein 90-KDa; RLC: RISC-loading complex; RISC: RNA-induced silencing complex; (n)AAAAA: poly-A tail of RNA; m7GpppN: 7-methyl guanosine cap structure of RNA.
In most eukaryotic cells, miRNA processing unleashes a silencing response specific to a target RNA sequence, so-called RNA interference (RNAi). The RNAi pathway is an important cellular process to control post-transcriptional gene expression, triggered endogenously by the conversion of double-stranded RNA sequences into miRNAs [4]. MiRNA transcription starts in the nucleus, typically from intronic or intergenic regions of DNA, with the formation of a primary structure called pri-miRNA as a result of the action of RNA polymerase II or RNA polymerase III [5,6] (Fig. 1). The pri-miRNA has a long stem and loop structure that is shortened to a pre-miRNA with ∼60 nt by ribonuclease-III Drosha together with the DGCR8 protein (Microprocessor complex subunit DGCR8: DiGeorge syndrome critical region 8) [7,8]. The pre-miRNA is then exported to the cytoplasm via the Exportin-5/Ran complex, [9] where it is further processed by another ribonuclease-III enzyme named Dicer (Double-strand-specific ribonuclease) [10–12] into an imperfect miRNA duplex with unpaired nucleotides (Fig. 1). In the cytoplasm, the pre-miRNA is recognized and cleaved by Dicer with the assistance of TRBP (Transactivation-responsive RNA-binding proteins), forming a smaller double-stranded miRNA (ds-miRNA) with 21–27nt. After the cleavage, the ds-miRNA dissociates from and re-associates with Dicer at a different position with the help of TRBP. Although, there are some studies reporting that TRBP is dispensable for miRNA biogenesis, recent evidences reinforce that the TRBP increases the RNA-binding affinity of Dicer and enhances cleavage accuracy, especially in a crowded environment [13]. The protein argonaute-2 (Ago2) [14] is further associated to this complex, forming a loading complex (RLC), [15] which assists in the assembling of an RNA-induced silencing complex (RISC). The ds-miRNA is then transferred to the Ago2 in a process mediated by Hsp90 (heat shock protein 90-KDa), which maintains the Ago2 in an open conformation in order to receive the miRNA, forming a pre-RISC complex. Inside the Ago2, the two miRNA strands are separated and the passenger strand is removed. The selection of the guide strand from a ds-miRNA is not arbitrary. The strand whose 5′ end is less stable serves as guide, where the other is discarded (the “asymmetry rule”). It is believed that TRBP senses the asymmetry of the ds-miRNA, and positions the miRNA in an orientation that allows a correct strand selection and loading to Ago2 proteins.
Finally, the RISC complex is then conducted by the mature miRNA to the complementary mRNA (Fig. 1) [16]. In the case in which the RISC binding is perfectly complementary to the target mRNA, the mRNA will be cleaved by Ago2. This process is frequently found in plants, where miRNAs share high homology to the 3′UTR of mRNAs. Conversely, in mammals, the RISC normally binds partially to the complementary mRNA. The seed region at positions 2–7 from the 5′-end of the miRNA sequence, is often fully paired and essential for the interaction. Due to this partial complementarity, translation repression and/or mRNA deadenylation pathways (Fig. 1)[17–19] are preferred rather than mRNA cleavage. After silencing, the miRNA remains intact and able to target other mRNA molecules with the same complementarity degree.
Translation repression and/or mRNA deadenylation processes are intimately connected and although the precise translation repression mechanism is still unclear, the deadenylation and consequent mRNA decay mediated by miRNA is better understood. The RISC complex of miRNAs can trigger mRNA deadenylation that leads to decapping and ultimately to mRNA decay. The deadenylation process starts with the association of Ago2 proteins present in the RISC complex to a GW182 protein which is responsible for the recruitment of cytoplasmic deadenylase complexes: PAN2–PAN3 (PAB-dependent poly(A)-specific ribonuclease subunit PAN2 and PAN3) and CCR4–NOT (transcription complex subunit) [20]. Deadenylated mRNAs by the deadenylase complexes are then decapped by DCP2 (m7GpppN-mRNA hydrolase) [21]. Without the protection of the cap structures mRNAs are ultimately degraded by the cytoplasmic 5′-to-3′ exoribonuclease XRN1.
To understand how miRNAs may repress translation and consequently lead to mRNA decay, it is desirable to go a step back and have a short overview of the normal mRNA translation mechanism. Translation is a multi-step process beginning with the recruitment of ribosomal subunits to the mRNA, leading to the initiation, elongation, and termination of the translation. A cap-dependent translation, which is the most frequent process for translation, requires an mRNA with a 5′-cap and a poly-A tail. The 5′-cap is recognized by the cap-binding protein, eIF4E (Eukaryotic translation initiation factor 4E), part of the eIF4F initiation complex. The eIF4F complex is responsible for recruiting the 40s ribosomal subunit to the mRNA, which is then checked for the initiation codon to start translation. The assembly of the 60s subunit is then followed by the elongation phase. The cytoplasmic poly-A-binding protein (PABPC) also play an important role in the translation. PABPC proteins recognize the poly-A tail and interact with eIF4F complex, forming a circular structure essential to protect the mRNA from degradation and thus leading to an effective translation [22].
Several studies are trying to elucidate how miRNAs interact with the mRNA translation machinery and their role in the mRNA deadenylation and decay [23–26]. A common ground of understanding is that miRNAs can inhibit cap-dependent translation at the initiation phase. More likely, miRNAs seem to interfere with the 5′-cap elF4F complex. First studies indicate that deadenylation is the cause of the translation depression. Deadenylated mRNAs fail to assume the circular conformation due to the action of GW182 proteins on PABPC [27,28] and therefore contribute to the repression and decay of mRNA. However, in 2006 it was demonstrated that although the two mechanisms are intimately connected, deadenylation and translation repression can function independently [29]. MiRNAs can repress the translation before the deadenylation step occurs, albeit the extension of the repression on deadenylated mRNAs seem to be higher in comparison to the one observed in mRNAs containing the poly-A tail [26].
Clearly, post-transcriptional mechanisms of miRNA play an important role in gene control, and in this way can influence different biological processes. For instance, they are able to trigger inflammatory responses, to control cell proliferation, apoptosis, DNA repair, and DNA methylation processes. It comes with no surprise that many miRNAs are associated with several human diseases and have already been identified in different pathologies including neurological disorders, cardiovascular diseases, inflammatory processes, and cancer. In the latter case, more than half of the miRNA genes are located in specific genomic regions frequently associated with cancer development and progression [30]. MiRNAs are able to control the expression levels of oncogenes and tumor suppressor genes, modulating apoptosis, cell migration, and angiogenesis (Fig. 2).
Figure 2.
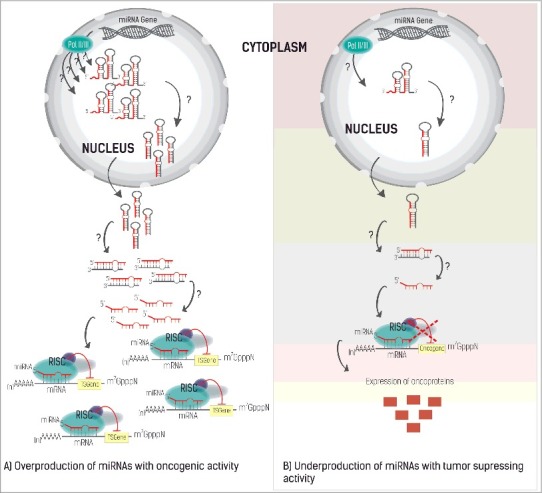
The different roles of miRNA in gene expression. A) The overexpression of miRNAs that target tumor suppressing genes, thus being responsible for the development of cancer (oncomir). B) Underexpression of miRNA with a tumor-suppressing profile leads to an increase of oncoproteins (red squares) promoting tumor formation. The question marks represent any perturbation on the miRNA level, which can occur at any stage of the miRNA biogenesis, leading either to its overproduction or degradation.
In cancer, miRNAs can act both as oncomirs (Fig. 2A) and as tumor suppressors (Fig. 2B) [31,32] depending on which genes they control. Downregulation of tumor suppressing genes or miRNAs that regulate oncogenes, promote tumor development, either by the overexpression of miRNAs with oncogenic activity (oncomirs) (Fig. 2A) or by the low expression of miRNAs with tumor suppressor activity (Fig. 2B) [33–37].
Furthermore, the same miRNA can be identified in different types of cancer, and within the same type of cancer it can have both roles, depending on its expression levels and target gene(s). For instance, miR-9 was found deregulated in leukemia, cervical, brain, breast, gastric, and colorectal cancers (Table 1) [38–46]. In the case of gastric cancer (GC), there is evidence that miR-9 is under-expressed, functioning as a tumor suppressor by targeting the oncogenes RAB34, NFKB1, CCND1, and ETS1; miR-9 was also reported to function as an oncomir by targeting the CDX2 gene (Table 1) [42,43,47,48]. In 2009, Hongchun Luo et al. [47] showed that miR-9 expression was directly linked to the RAB34 gene expression, encoding the Ras-related protein Rab-34. Rab-34 is a small GTPase protein member of the RAS oncogene family, which regulates aggregation, junction, and fusion of vesicles in exocytosis and endocytosis. The authors observed that the stimulation of the expression of miR-9 in gastric cell lines led to a decrease of Rab-34 expression. The same was observed in the work of Hai-Ying Wan et al, [48] regarding the NFKB1 gene, which is responsible for the regulation of immune responses, embryo and cell-binding development, cell-cycle progression inflammation, and oncogenesis. The authors observed that the re-expression of miR-9 in gastric cell lines led to a decrease in the expression of the NF-kB1 factor, which in its turn downregulates VEGF, resulting in the inhibition of the invasion and metastasis of tumorigenic cells.
Table 1.
Role of miR-9 and its validated targets in different cancer types.
| miR-9 role | Type of cancer | miR-9 targets | References |
|---|---|---|---|
| Onco-miRNA | Colorectal Cancer | CTNNA1 | [128] |
| Breast Cancer | CDH1 | [129-131] | |
| Gastric Cancer | CDX2 | [43] | |
| Liver Cancer | TLN1, CDH1, KLF17 | [132-134] | |
| Bladder Cancer | LASS2 | [135] | |
| Leukemia | HES1, MLL | [38,136] | |
| Osteosarcoma | GCIP | [137] | |
| Glioblastoma Multiforme (GBM) | MAPKAP/MAPK14/MAPKAP3 | [138] | |
| Squamous cell carcinoma: Esophageal Cancer and Skin Cancer, respectively | CDH1, CTNNA1 | [45,139] | |
| Tumor Suppressor | Colorectal cancer | TM4SF1, UHRF1 | [40,140] |
| Breast Cancer | NOTCH1 | [41] | |
| Gastric Cancer | NFKB1, RAB34, CCND1 and ETS1 | [47,48,139] | |
| Liver Cancer | WWTR1, IL6, AP3B1, TC10, ONECUT2, IGF2BP1, MYO1D, ANXA2 | [141,142] | |
| Cervical Cancer | CKAP2, HSPC159, IL6, and TC10 | [39] | |
| Lymphoblastic leukemia | FGFR1 and CDK6 | [143] | |
| Ovarian Cancer | NFKB1 | [144] | |
| Oral squamous cell carcinoma | CXCR4 | [145] | |
| Melanomas | NFKB1 | [146] | |
| Medulloblastoma | HES1 | [147] | |
| Neuroblastoma | MMP14 | [46] |
The CCND1 gene encodes Cyclin D1, an oncoprotein from the family of G1 cyclins that inhibits members of the retinoblastoma (RB) protein family and regulates the cell cycle during the G1/S transition. Overexpression of Cyclin D1 is often associated with carcinogenesis at early stages and is a prognostic indicator of poor survival in cancers like breast cancer [49]. Ets-1 is a transcription factor that directly controls the expression of a wide variety of genes that regulate differentiation, proliferation, and invasion [50]. Restoring the expression levels of miR-9 in GC cells, attenuated the proliferation, migration, and invasion of cancer cells, by knocking down the expression of Cyclin D1 or Ets-142.
Regarding the oncogenic activity of miR-9, Rotkrua et al. [43] suggested that the downregulation of CDX2 by miR-9 might contribute to cell proliferation in GC. The CDX2 gene encodes a homeobox protein involved in the differentiation and maintenance of the intestinal epithelial lining [51]. It is still unclear the role of CDX2 in GC, whether it functions as a tumor suppressor or an oncogene. At an initial stage, CDX2 gene expression is associated with the development of intestinal metaplasia of the stomach, a premalignant lesion that increases the risk of the patient to develop GC [52,53]. During cancer progression, the expression of CDX2 progressively decreases, suggesting it might function as a tumor suppressor gene and a prognostic marker [49].
The double role of miRNAs is an interesting example that highlights the importance of carrying out a comprehensive characterization of the role of miRNAs in cancer and their post-transcriptional mechanisms in mRNA regulation. This would allow to develop new strategies in cancer treatment and cutting-edge therapies specifically designed to control oncomirs or to promote the expression of miRNA with tumor suppressing activity.
2. Controlling miRNAs activity
It is quite straightforward that, in order to control miRNA activity, it could be desirable to restore miRNA expression with tumor suppressing activity (gain-of-function) or to block miRNAs with oncogenic activity inhibiting its function (loss-of-function) [54].
Gain-of-function strategies mainly rely on either the incorporation of double-stranded chemically synthesized miRNA mimics or on viral vectors to mediate miRNA overexpression. Chemically synthesized miRNA mimics are generally not the exact natural miRNA duplex. The sense strand is altered to be perfectly complementary to the mature miRNA (to increase stability), and should be chemically modified to inactive this now non-natural sense-strand. MiRNA mimics can be designed to target a single mRNA [55] or to incorporate multiple miRNA units for targeting different mRNAs and silence multiple target genes [54,56,57]. Viral vector-mediated miRNA overexpression consists on the use of an integrating vector system, containing short hairpin RNAs (shRNAs) driven by Pol III promoters [57]. Upon delivery, these systems are able to sturdily express the miRNA of interest.
The loss-of-function modulating system of miRNAs is mainly achieved by applying miRNA sponges; miRNA-Masking Antisense Oligonucleotides; or by using antisense oligonucleotides targeting miRNAs (AMOs) [54].
MiRNA-sponge technology consists of the expression of mRNA molecules with multiple binding sites for the target miRNA that will function as a decoy or a “sponge” to trap the desired miRNAs. That way the endogenous target mRNA will be preserved and able to function normally [58]. This approach is intensively used to inhibit functional classes of miRNAs in vitro, but so far its use in vivo has been limited [54]. MiRNA sponges are specific to a miRNA target and not to a gene of interest. Using a miRNA sponge to knockdown a miRNA or a miRNA seed family could affect all target genes of its targeted miRNAs, leading to undesirable effects.
MiRNA-Masking Antisense Oligonucleotides technology (miR-Mask, also known as BlockmiR, target protectors or as target site blockers) relies on an inverted approach: instead of blocking the target miRNA, these molecules shield the mRNA which function is desirable to preserve. In this way, the miR-Mask are able to block the access of the miRNA to its target mRNA, impeding its action to the target gene. Moreover, miR-mask technology targets the miRNA in a gene-specific manner, i.e. the oligonucleotides are designed to protect the mRNA, and consequently, the expression of the protein of interest, leaving intact the miRNA responsible for the repression, which might have other important roles in the system [59]. MiR-Mask is an alternative and valuable supplement to AMOs technology [54].
Also, this approach can be further combined with miRNA sponges (sponge-miR-mask technology) to block the access of multiple miRNA members to their binding sites on mRNA, leading to the re-expression of proteins. A sponge miR-mask is designed to bind by partial complementarity to the 3′UTR of all target mRNAs of a miRNA seed family site of 8 nt. However, sponge-miR-Mask technology has poor gene-specificity, since these molecules might block the expression of all genes associated with the same seed binding site of the miRNA family [54].
The most popular approach to correct aberrant miRNA expression is based on the synthesis of antisense oligonucleotides with a complementary sequence [60–62]. Antisense oligonucleotides (ASOs) are being used to suppress dysfunctional mRNAs for more than four decades, [63] and are currently being used to target miRNAs. In antisense oligonucleotides targeting mRNAs, ASOs mostly stimulate an RNase-dependent degradation of the target RNA. They are designed to harbor continuous DNA monomers along the sequence in order to promote RNase H activity. When applying antisense technology to miRNAs (AMOs), one may consider that the mechanisms through which AMOs act may follow the same pathway of ASOs – the binding of antisense oligonucleotides to miRNA should promote degradation via RNase, in the same way ASOs cleave mRNA. However, it is now believed that the anti-miRNAs cause miRNA silencing mostly by steric blocking the target miRNA. These findings were supported by the work of Davis et al. [64] where they tested a designed ASO sequence known to trigger the RNase H-dependent degradation to target the miR-21. The antisense oligonucleotide, despite being effective promoting mRNA degradation, failed to inhibit miR-21 activity, suggesting that the miRNA-ASO duplex may not be accessible to the RNase H.
Targeting aberrant miRNAs with AMOs is a promising approach for post-transcriptional gene control in future cancer therapies. Specific and strong AMOs' recognition and binding to the miRNA target is accomplished through Watson-Crick base pairing, which may be optimized by modifying AMOs at the chemical level.
The design of Anti-miRNAs
Anti-miR oligonucleotides-based mechanism relies on the complementary base pairing of the oligonucleotide sequence to its target miRNA. AMOs are typically designed to be a perfect match to the miRNA target, [65] especially to the seed region of the miRNA, which has been shown to influence anti-miRNA specificity and activity.
In therapeutics involving antisense oligonucleotides, AMOs must display some desirable characteristics in order to be applied successfully in a biological system. It is important to design and select AMOs with enhanced potency to silence the target miRNA and with increased binding affinity to the target. The potency of the AMOs is intrinsically associated with its capacity to efficiently reduce the expression of the target miRNA. Whereas, the binding affinity is related to the stability of the duplex upon AMO binding to its target, and thus related to the melting temperature (Tm) of the oligonucleotide. A higher Tm value represents an improved binding affinity and a stronger duplex, because more energy is required to destabilize the connection between both molecules. Overall, it is also desirable that AMOs exhibit improved extracellular and intracellular stability, and thus be resistant to exo- and endo-nucleases. They should also exhibit improved cellular uptake rates to be efficiently delivered to cells and tissues, and have no toxicity or other side effects, such as the knockdown of non-target molecules important to maintain biological function or the stimulation of undesired auto-immune responses [66].
Unmodified DNA oligonucleotides present several disadvantages that preclude their applicability as antisense technologies. As such, several sets of modifications were introduced to improve structural properties of the AMOs.
3.1. Chemical modifications – AMOs evolution
The first study reporting the use of antisense oligonucleotides applied to miRNAs used unmodified DNA sequences complementary to several miRNAs injected to Drosophila embryos [67]. Antisense oligonucleotides composed only of DNA bases are rapidly degraded in a biological system by endo- and exo-nucleases and fail to connect with the majority of the target miRNA in mammalian cells. The necessity to overcome DNA-AMOs limitations emerged rapidly and a vast array of chemical modifications have been studied applied to antisense technologies.
Chemical modifications are mainly introduced in the sugar ring, especially at the C2′ position, and/or in the backbone of the oligonucleotide structure. Backbone substitutions include either internucleotide linkage modifications or the substitution of the entire backbone for structures analogous to the DNA. Internucleotide modifications are often combined with sugar modifications to improve potency and resistance to the oligonucleotide sequence. These modifications will be further discussed in the following sections.
3.1.1. Sugar modifications
Chemically-modified sugars are a remarkable way to enhance binding affinity and nuclease stability. The C2′ position defines the conformation of the sugar ring and modifications at this position that are able to shift the conformation of the sugar moiety from a C2′-endo (southern conformation, typical of DNA duplexes) to a C3′-endo sugar pucker (northern conformation, typical of RNA duplexes), improves the binding affinity of AMOs for RNA complements [68]. Furthermore, in this conformation the 2′-modification is closer to the 3′-phosphate group, conferring higher nuclease resistance to the oligonucleotide [69].
The most popular alterations applied to miRNAs studies involve modification at the 2′ carbon of the ribose. It is the case of 2′-OMe, 2′-MOE and 2′-F alterations, that are all reported to confer AMOs increased binding affinity (ordered by increased potency: 2′-OMe ≅ 2′-MOE < 2′-F) [64] and nuclease resistance compared to DNA (Fig. 3). The use of locked nucleic acids (LNA) is very popular in AMO designs, due to their improved performance on the stabilization of the duplex by its locked configuration [64,65]. All these substitutions can be combined to improve AMOs potency. For instance, MOE/LNA, 2′-OMe/LNA or 2′-F/MOE are successful examples of oligonucleotides mixmers with improved binding affinity when compared to oligonucleotides harboring only one type of substitution [64,65,70].
Figure 3.
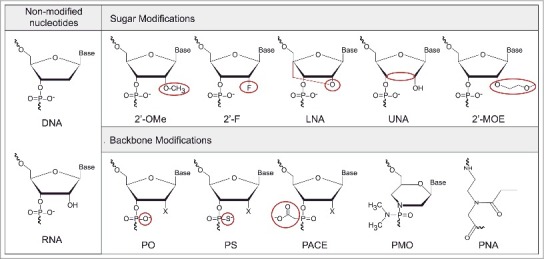
Chemical modifications applied in the AMOs design. Unmodified nucleotides – DNA: deoxyribonucleic acid, RNA: ribonucleic acid; Sugar modifications – 2′-OMe: 2′-O-methyl, 2′-F: 2′-fluoro-RNA, LNA: Locked Nucleic Acid, UNA: Unlocked Nucleic Acid, and 2′-MOE: 2′-O-methoxyethyl; Backbone modifications – PO: phosphodiester, PS: phosphorothioate, PACE: phosphonoacetate, PMO: Phosphorodiamidate Morpholino Oligomers, and PNA: Peptide Nucleic Acid. In red is highlighted the site and the type of modification.
The first generation of modified-AMOs appeared in 2004 and contained a methylated hydroxyl group at the C2 carbon of the sugar ring (2′-OMe RNAs, Fig. 3) [71,72]. Hutvagner and his co-workers [71] showed that 2′-OMe oligonucleotide are nontoxic and can efficiently suppress Caenorhabditis elegans-let-7 miRNA in vivo. In the same year, Meister et al. [72] also proved that it is possible to control miR-21 activity by blocking the RISC complex. 2′-OMe oligonucleotides offer several advantages comparing to DNA-counterparts. They increase thermal stability to the target by approximately +0.8°C/insert [73]. Also, 2′-OMe is a natural modification that occurs in mammalian RNA conferring minimal or no toxicity when used in vivo [71]. While 2′-OMe oligonucleotides are more resistant to nuclease activity when compared to DNA counterparts, they are still susceptible to exonucleases in the serum. Successful inhibition of miRNAs in vivo using 2′-OMe-AMOs was only achieved by applying the AMO directly into the cells [71]. In order to overcome this problem, additional non-oligonucleotide-modifiers can be placed near both ends of the oligonucleotide sequence to protect it from the attack of nucleases. Lennox et al. [70] described a novel nontoxic compound, N, N-diethyl-4-(4-nitronaphthalen-1-ylazo)-phenylamine (“ZEN”) placed at or near 5′- and 3′-ends of a phosphodiester (PO) 2′-OMe oligonucleotide complementary to a targeted miRNA. The 2′-OMe-ZEN, with the modifier placed in both ends of the oligonucleotide sequence (dual-ZEN AMO), completely blocks endo- and exo-nuclease attack, and shows increased potency, binding affinity, good specificity and low-toxicity in vitro when compared to the unprotected 2′-OMe-PO design. In the same work, the ZEN modifiers were also compared to other non-nucleotide blocking groups (C3 spacer) placed in the same manner in an AMO sequence. Both ZEN and C3 AMO variants showed improved potency when the modification was performed at the 5′-end of the sequence rather than at the 3′-end, and in the case of the C3-AMOs, the binding affinity of the two AMO variants was the same. The 3′-end modification was expected to give a more stable AMO, and therefore increased potency. The opposite outcome led to the speculation that the potency of an AMO depends not only of its binding affinity and nuclease resistance, but also of the role that the 5′-end plays on the ability of the AMO to invade the miRISC complex. Furthermore, the dual-ZEN AMO showed higher potency than the dual-C3 AMO, which offered limited improvement in potency regarding the unmodified 2′-OMe [70].
In a similar line of work, Vermeulen et al. [74] demonstrated that flanking a single-stranded 2′-OMe-AMO sequence with double-stranded duplexes capable of forming hairpin structures highly improves the inhibitory effect of the AMO by several fold, possibly by helping the AMO invasion to miRNA–RISC. More recently, it has been reported that cross-linked duplexes flanking the single-stranded AMO inhibited miRNA functions more efficiently than AMOs with normal single or double strands. The interstrand cross-link of DNA or RNA not only contributed to improve 2′-OMe resistance and potency properties, but also to stabilize the duplex avoiding the risk of dissociation into single strands [75].
The 2′-MOE (2′-O-methoxyethyl) oligonucleotides harbor a methoxyethyl modification at the RNA 2′-OH position (Fig. 3). This configuration increases nuclease resistance and target binding affinity by approximately +1°C/insert73 when compared with unmodified DNA oligonucleotides. Moreover, 2′-MOE oligonucleotides are effective in silencing miRNAs and display improved anti-miRNA activity when compared to 2′-OMe oligonucleotides [64,76]. Esau et al. [76] demonstrated that miR-122 function can be modulated in mice using a 2′-MOE-AMO. The inhibition of the miR-122, a miRNA overexpressed in the liver, resulted in the reduction of plasma cholesterol levels, increase of hepatic fatty acid oxidation, and a decrease of hepatic fatty acid and cholesterol synthesis rates.
The miR-122 is associated with Hepatitis C virus (HCV) infection in the liver. Currently, a company called Regulus Therapeutics is validating a 2′-MOE-AMO-122 (RG-101) conjugated with an N-Acetylgalactosamine sugar (GalNAc) for the treatment of HCV infection in humans [77,78]. The GalNAc carbohydrate structure shows high binding affinity to the asialoglycoprotein receptor (ASGPR) on hepatocytes, improving cellular uptake [79] and enhancing the potency of RG-101 by up to 10–20 times compared with unconjugated oligonucleotide. In the beginning of 2016, RG-101 entered the phase II of a clinical study, but despite the promising results in phase I, the drug is currently on a clinical hold by the American Food and Drug Administration (FDA). The same company has other promising AMOs to be applied in miRNA therapeutics. Another GalNAc-conjugated oligonucleotide, the RG-125 (AZD4076) targeting miR-103/107, is currently in phase I of clinical development. RG-125 is expected to contribute to the treatment of non-alcoholic steatohepatitis in patients with type 2 diabetes/pre-diabetes.
The 2′-Deoxy-2′-fluoro-nucleoside (2′-F) is also an RNA mimic with a fluorine 2′-hydroxyl group at the C2 carbon of the ribose (Fig. 3). Depending on the sequence, oligonucleotides with 2′-F-RNA residues show increased thermal stability of around +1.6°C/insert73, which is higher than when compared with 2′-OMe-RNA and DNA. 2′-F-AMOs with PO linkages do not have significant resistance to exonucleases, while PS linkages are highly resistant, conferring some protection to 2′-F-RNA AMOs. For in vivo applications, 2′-F-RNA can be further conjugated with other modifications, like 2′-MOE substitutions, to confer more stability [80]. In this study, a 2′-F-AMO was able to efficiently inhibit miR-122 activity, showing a mild dose-dependent immunostimulatory effect in mice. The 2′-MOE modification presents a lower binding affinity than the 2′-F, but the introduction of additional 2′-MOE modifications into the 2′-F/MOE AMO was able to minimize this immune response without affecting anti-miR-122 activity [80]. Nevertheless, the 2′-F-RNA substitution is a potent, and well-tolerated chemical modification that can be used to efficiently silence abnormal miRNAs. In addition, 2′-F-RNA residues are an effective alternative to LNA-modified oligonucleotides, since they provide a similar thermal stability and nuclease resistance, but at a lower cost [81].
LNA is a 2′-O, 4′-C-methylene-β-D-ribofuranosyl nucleotide where the ribose is locked in a 3′ endo/N-type sugar conformation with a methylene bridge between the oxygen at 2′ position and the 4′-C (Fig. 3). LNA monomers are also compatible to other RNA and DNA monomers and with phosphorothioate (PS) and PO linkages, respecting the natural base pairing binding. Due to their chemical structure, LNA-modified oligonucleotides have high resistance to nucleases and the highest binding affinity demonstrated so far of +2 to +5 °C/insert when pairing with DNA sequences and +4 to +10 °C/residue when pairing with a RNA strand [82]. The high binding affinity of LNA-AMOs makes it possible to design shorter sequences without compromising efficiency, which are useful to target the seed region of entire miRNAs families with similar biological functions. Obad et al. [83] were the first to describe tiny LNA-sequences of 8 nucleotides (so called “tiny LNAs”) targeting the seed region of both individual miRNAs and co-expressed miRNA families. Also, systemic administrations of tiny LNAs showed unassisted uptake and long-term knockdown of miR-21 in a breast cancer mouse model, emphasizing the potential use of LNA-AMOs in vivo. More recently, Zhang et al. [84] successfully applied tiny-LNAs to silence miR-155 in low-grade B-cell lymphoma cells both in vitro and in vivo. Safe in vivo applications of LNA sequences are highly dependent of sequence context, length and an overall optimized design. Shorter sequences had shown lower toxicity in vivo and in vitro, whereas long sequences had demonstrated severe cytotoxicity in vitro [70]. Also, shorter sequences are more likely to improve mismatch discrimination [85].
In terms of market, there are two drugs using LNA-AMO technology currently in clinical trials. The first drug called Miravirsen from Santaris Pharma, was developed to treat HCV infection by targeting miR-122 [86] and entered phase 2 of clinical trials. It is presently in an extension study to provide additional long-term safety and efficacy data. Another company (miRagen Therapeutics) entered phase 1 of clinical studies for MRG106-11-101 LNA-antimiR®, which was designed to control the activity of miR-155. In hematological malignancy, miR-155 has key roles in the differentiation, function, and proliferation of blood and lymph cells. In vitro tests using the developed AMO successfully inhibited miR-155 activity in lymphoma cells, restoring normal function and reducing aberrant cell proliferation.
Unlocked Nucleic Acid (UNA, Fig. 3) is another analogue of RNA, in which the C2′–C3′-bond has been cleaved, resulting in an unlocked configuration of the furanose ring. The UNA inserts additively decrease duplex stability, and depending of the configuration, the Tm can decrease −5°C to −10°C/modification [87]. However, despite the decrease of Tm, the UNA unlocked configuration makes this molecule very flexible. UNA monomers can be placed strategically within the AMO sequence to enhance mismatch discrimination, making this substitution very interesting for detection purposes (further reviewed in the “miRNA detection using AMOs” section).
3.1.2. Backbone modifications
Phosphodiester bonds (PO) are the natural internucleotide linkage present in DNA and RNA (Fig. 3). Oligonucleotides designed to harbor the natural PO bonds are in this way more tolerated in vivo, displaying less toxicity. Yet, they are more prone to be cleaved by endo- and exonucleases in mammalian cells. Chemical modifications of these bonds have been extensively studied, and several chemical strategies have been very successful on improving nuclease resistance [88–91].
The first described internucleotide modification in antisense oligonucleotides was a phosphorothioate (PS) linkage, where the non-bridging phosphate oxygen is substituted by a sulfur in the phosphodiester bond (Fig. 3) [88]. PS bonds are more resistant to the activity of exo-and endonucleases and they can be placed throughout the oligonucleotide sequence (to improve endonuclease resistance) or at strategic points within the sequence, leaving some unmodified PO linkages. Generally, the PS linkages should be added near both the 5′ and 3′ ends [65] to improve the AMO resistance to exonucleases.
The major drawback associated to PS linkages is their tendency to bind to off-target proteins. PS binding to nuclear proteins may be potentially toxic to normal cells and compromise several cell functions [92]. However, the affinity of PS oligonucleotides to serum albumin, reduces plasma clearance and increases the AMOs lifetime in serum from hours to days [93].
Moreover, PS-DNAs present a slightly reduced binding affinity towards complementary RNA molecules in comparison to their corresponding phosphodiester oligodeoxynucleotide. The substitution to a PS-AMO decreases the melting temperature of the heteroduplex by approximately −0.5°C per insert. Davis et al. [64] compared the effect of the PS backbone substitution on anti-miRNA activity. Uniform 2′-MOE and 2′-OMe sequences, with PS or PO backbones, were tested to evaluate their potency to silence miR-21 over time. 2′-MOE-PO-AMO displayed an increase in activity compared to 2′-MOE-PS-AMO, being more active in many later time points. The 2′-OMe showed little or no improvement in both designs (PS or PO), due possibly to the poor resistance of 2′-OMe-PO-AMO in cells. The decrease in terms of thermal stability offered by PS oligonucleotides is compensated by a greater hybridization specificity, higher nuclease resistance, and an improved cellular uptake both in vitro and in vivo as compared with unmodified DNA oligonucleotides. The potency of PS-oligonucleotides can be further enhanced by combining other substitutions, especially sugar modifications.
Other internucleotide linkage modifications used to silence miRNAs include phosphonoacetate (PACE) and thio-PACE. PACE synthesis replaces the nonbridging oxygen for an acetate group in the internucleotide phosphate linkage (Fig. 3). Thio-PACE is an analog of PACE comprising a PS substitution instead of a PO bond. Both modifications are reported to be completely resistant to nuclease activity and are able to enter the cells without any assistance as neutral esters [94]. In spite of these modifications they show a slight decrease of binding affinity to complementary RNA targets when compared to PO linkages, in the order of −1.3 °C/insert for PACE duplexes and −1.8 °C/insert for thio-PACE (under high salt concentrations) [94]. Threlfall et al. [95] tested the potency of 2′-OMe oligonucleotides with PACE and thio-PACE backbones to inhibit miR-122. In most cases, both types of AMOs formed stable duplexes with their target miRNA, showing higher melting temperatures than simple DNA oligonucleotides. Furthermore, single-stranded 2′-OMe thio-PACE oligonucleotides were notably more efficient in the cellular uptake and significantly improved the potency of a 2′-OMe-based anti-miRNA targeting miRNA-122 in the absence of a transfection agent [95].
Phosphorodiamidate Morpholino oligomers (PMOs) are uncharged substitutes of the PO bonds (Fig. 3), where the morpholine ring is linked through phosphorodiamidate groups instead of phosphates [90]. Given their neutral charge, PMO oligonucleotides are more resistant to nuclease activity and exhibit a similar or a slightly improved binding affinity for target miRNAs. Morpholino oligonucleotides have proven particularly successful as anti-miRNAs in Zebrafish systems, [96,97] exhibiting specific and non-toxic inhibition of both pre-miRNA and mature miRNA activity by blocking Drosha or Dicer nucleolytic processing sites [97]. The uptake of this configuration into mammalian cells is challenging, and as such further conjugation with cell-penetrating peptides (CPPs) is required. This allows both the in vitro and in vivo uptake, enlarging the potential use of PMOs in anti-miRNA studies. It is worth noticing that the uncharged nature of these molecules facilitates their conjugation with charged cationic molecules, such as the CPPs designed to promote the delivery.
Peptide Nucleic Acid (PNA) is an uncharged synthetic DNA analog composed of N-(2-aminoethyl) glycine monomers linked by peptide bonds [91] (Fig. 3). PNAs follow the natural base-pairing rules and exhibit a significant nuclease resistance, and increased binding affinity towards DNA or RNA targets. PNA is particularly useful in anti-miRNA applications. Fabani and Gait [98] reported that PNA molecules used to silence miR-122 showed improved activity when compared with a 2′-OMe oligonucleotide. Like the PMOs, the cellular uptake of PNA-AMOs is challenging. PNA shows a more hydrophilic profile than lipophilic, which compromises the diffusion of unmodified-PNA molecules through cell membranes [99]. The PNA oligonucleotides designed by Fabani and Gait [98] had an amino-terminal cysteine to facilitate conjugation with a R6-penetratin peptide (CPP). They also tested an unconjugated AMO with four lysine residues to improve solubility. The four lysine residues were sufficient to confer charge, so that the PNA would be able to be delivered into cells using electroporation or CPPs.
4. Anti-miR oligonucleotides: Design Strategies
Apparently, the design of complementary anti-miRNAs sequences targeting miRNAs is quite straightforward. Yet, in reality, the whole process might go into a tortuous and long road before reaching a functional and non-toxic anti-miR. While highly dependent on the strategy defined for the study, some guidelines should be followed to assist in the design of AMOs. The development of a chemically-improved anti-miR oligonucleotide passes through several stages, from the search of target miRNA sequences to the design of the AMO (including the selection of the most adequate chemical structure) and its validation (Fig. 4).
Figure 4.
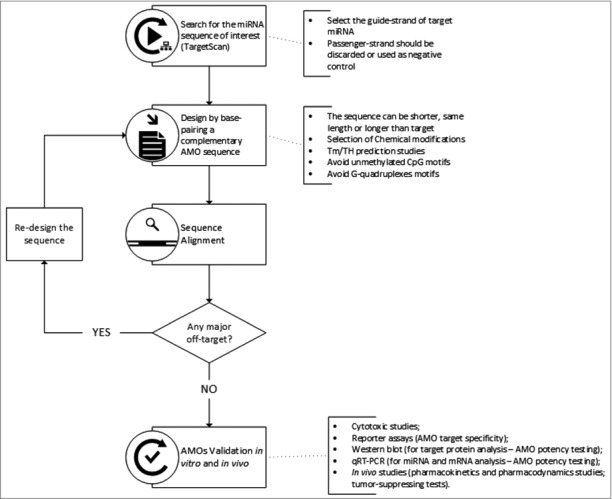
AMOs design flowchart: from the identification of target miRNA to the validation of anti-miRNA oligonucleotides.
First, the miRNA sequence of interest needs to be obtained from databases like TargetScan v7.1 [100] or miRBase [101,102]. The search should be directed to the mature sequence of the miRNA and the guide strand. The passenger strand is not an active sequence of miRNA and is generally not considered in the design of AMOs. Nonetheless, the passenger strand sequence can be used to synthesize a negative control oligonucleotide.
Next, by base-pairing, it is possible to design an anti-miR oligonucleotide sequence that is fully complementary to the 5′ end of the mature miRNA, especially matching the seed region. The designed sequence can have the same length, be shorter or even longer than the target [64,71,103]. Some examples of shorter and longer sequences were already discussed above, such as in the work of Obad et al. using tiny LNAs, [83] and in the work of Vermeulen et al. using longer 2′-OMe-AMOs with flanking nucleotides next to the complementary antisense miRNA region [74]. After selecting the desired AMO sequence, it is also advisable to perform an alignment for the human genome using for instance a Basic Local Alignment Search Tool (BLAST) [104] to check for possible off-targets of the oligonucleotide sequence.
Due to the poor stability in vivo and insufficient binding affinity of DNA sequences, chemical modifications should be employed into the design of AMOs. From all the evidence stated above, it is possible to sum-up some specific characteristics of the different chemical-modified AMOs (Table 2), especially in terms of binding affinity, nuclease resistance, cellular uptake and cytotoxicity. It is also worth mentioning that unmodified AMOs may lead to an ineffective inhibition of the miRNA activity, since they also serve as a passenger strand and thus trigger the dissociation mechanism responsible for miRNA passenger strand unwinding [64].
Table 2.
Properties of the different chemical modifications applied to the AMOs.
| Type of modification | Advantages | Disadvantages | References |
|---|---|---|---|
| PO | ▪ Natural backbone structure | ▪ Poor stability in vivo – Low resistance to exonucleases | [63] |
| ▪ Low-toxicity | |||
| ▪ Inexpensive | |||
| PS | ▪ Increased cellular uptake | ▪ Non-specific binding to proteins | [64,88] |
| ▪ High nuclease resistance | ▪ Slightly lower binding affinity compared with PO | ||
| PACE/thio-Pace | ▪ Improved and unassisted uptake▪ Combination with other modifications to improve thermal stability and binding affinity | ▪ Lower binding affinity when compared with other modifications (-1.3 °C/insert for PACE duplexes; -1.8 °C/insert for thio-PACE), but still higher than DNA-AMOs when combined with higher binding affinity modifications such as 2’-OMe | [94,95] |
| PMO | ▪ High nuclease resistance | ▪ Poor uptake/pharmacokinetic properties | [97] |
| ▪ Improved thermal stability | |||
| ▪ Uncharged – easy to conjugate with other molecules | |||
| PNA | ▪ High nuclease resistance | ▪ Poor uptake/pharmacokinetic properties | [98,99] |
| ▪ Improved thermal stability | |||
| ▪ Uncharged – easy to conjugate with other molecules | |||
| 2’-OMe | ▪ Improved nuclease resistance▪ Improved thermal stability▪ Non-toxic | ▪ Relatively poor stability in vivo: 2′-OMe-PO AMOs are less resistance to exonucleases when compared with other modifications containing a PO backbone | [64,71,73,148] |
| 2’-MOE | ▪ Improved nuclease resistance▪ Improved thermal stability▪ Non-toxic | ▪ When combined with other Tm-increasing chemical modifications (such as LNA) might form secondary structures within the AMO or present increased self-dimerization effect, lowering the potency of the AMO | [64,65,73,76] |
| 2’-F | ▪ Improved thermal stability | ▪ 2’-F-PO show no resistance to exonucleases | [73,80] |
| ▪ Non-toxic | |||
| ▪ Lower cost compared to LNA substitution | |||
| LNA | ▪ High thermal stability | ▪ Low conformational flexibility | [83,84,103,114,116,149,150] |
| ▪ Increased nuclease resistance▪ Improved potency and specificity▪ Improved and unassisted uptake▪ Mismatch discrimination▪ Efficient to target miRNA families using tiny-LNA sequences | ▪ Tm-increasing chemical modifications might form secondary structures within the AMO or present increased self-dimerization effect, lowering the potency of the AMO▪ Might present in vivo toxicity | ||
| UNA | ▪ Great conformation flexibility | ▪ Poor thermal stability | [124] |
| ▪ Mismatch discrimination within miRNA families |
Overall, some of the most widely used chemical modifications in anti-miRNA technology are LNA, 2′-F-RNA, 2′-OMe, PNA, and PMOs. Moreover, different combinations can be applied to further improve the performance of a certain AMO design. For instance, 2′-OMe oligonucleotides with a PO backbone are easily degraded in the serum by exonucleases, while introducing a PS substitution linkages at each end of a 2′-OMe-PO oligonucleotide improves nuclease resistance and is an effective AMOs substitution for a miRNA knockdown in vivo [105]. However, when applying PS substitutions in the entire sequence of a 2′-OMe-AMO, the same outcome is not achieved and the miRNA levels remain unaffected. The reduction on the activity of a PS-AMOs, comparatively to a PO counterpart, can also be explained by the higher binding affinity of PS-AMOs to many proteins. Some of those are able to relocate to the nucleus, moving the PS-AMOs along with them. As a consequence, there is a decrease of available AMOs in the cytoplasm, which may contribute to the inhibition of anti-miRNA activity [106].
Another aspect to take into consideration when designing an anti-miR oligonucleotide is the temperature at which the hybridization occurs. The hybridization temperature (TH) can be defined as the optimal temperature at which a full (or nearly full) noncovalent binding between the AMO and the target miRNA forming a duplex is observed, [107] and at which the AMO does not hybridize with sequences that contain one or more mismatches. Up to date, there are no mathematical models that accurately predict the optimal TH for a given oligonucleotide sequence. In order to overcome this and have an estimation of TH for optimization studies, the melting temperature or mid-transition temperature (Tm) is usually determined as an indicator of TH107. Thermodynamic parameters are well defined for DNA duplexes and in general, and the mathematical models to handle the thermodynamics of nucleic acid hybridization (and hence determine the Tm) are available and quite accurate. Different configurations (PNA: RNA; LNA: RNA, etc.) show different thermodynamic parameters and thus different models need to be adapted to predict the thermodynamic parameters of the hybridization [107].
As mentioned before, the PS linkage reduces binding affinity and a complete PS modification lowers the Tm to such extent that the AMO potency is compromised [105]. In spite of some exceptions, it is possible to establish a direct correlation between the increase of the binding affinity and the potency of the AMO to inhibit miRNA activity. However, chemical modifications presenting high Tm values, such as LNA or 2′-MOE/LNA mixmers, may result in an AMO with reduced potency, since they might show self-dimerization effects or form strong secondary structures within the oligonucleotide sequence, which can lower AMO potency. It is believed that there is a threshold in the thermal stability that directly promotes a good potency. After that point, any further affinity increases do not improve potency and higher are the chances of off-target hybridization [108].
Furthermore, other molecules can be applied in the sequence to improve uptake, delivery, or endurance of an AMO, especially when applied in vivo. Due to their polyanionic nature, these oligonucleotides do not easily diffuse across cell membranes, and most of the AMOs need to be associated with an efficient delivery system. Applying non-oligonucleotide molecules to the extremities of an AMO protects the sequence from degradation and thus increases the nuclease resistance of these designs [70,105]. Also, it has been reported that adding, for instance, a cholesterol group to the 3′-end facilitates the delivery in vivo of the 2′-OMe AMOs containing PS linkages [105,109]. Moreover, other molecules such as cell-penetrating peptides [98,110] can be applied in the extremities of an anti-miR oligonucleotide to promote cellular uptake of otherwise unattractive AMO designs for an in vivo delivery purpose. Although CPPs are an attractive way to promote the delivery of oligonucleotides, these molecules are typically conjugated with the non-charged PNA and PMOs [111].
Other technical features should be kept in mind when designing antisense oligonucleotides. Generally, oligonucleotides containing unmethylated deoxycytidine-deoxyguanosine (CpG) dinucleotides motifs can trigger immune responses in mammalian systems by activating the toll-like receptor-9 gene (TLR9) expressed in immune system cells such as the Lymphocyte B or dendritic cells. For this reason they should be avoided, especially for in vivo applications [112,113]. The CG dinucleotide is more frequently found in viral and bacterial DNA than in the human genome, suggesting that it is a marker for the immune system to recognize infection. Nevertheless, AMOs containing these motifs can be applied in situations where it is desirable to induce an immune response for treating cancer, asthma and infectious diseases [113].
The development of more stable and specific oligonucleotides, targeting miRNAs is the investigation focus of many research groups that are continually improving AMOs design and finding new miRNAs that control specific pathways in cancer epigenetics. Improved AMOs could be used not only as a therapeutic tool but also for the detection of miRNAs in tissues.
5. miRNA detection using AMOs
A field breakthrough in the miRNAs technology might pass through the use of AMOs to profile and detect in vivo miRNAs known to be involved in cancer processes. Most of these studies were validated in vitro and in vivo, yet to the best of our knowledge all the in vivo studies applying AMOs for the detection and identification of a target miRNA are performed analyzing the tissues ex-vivo. The progression of the AMOs technology allied to real-time in vivo imaging is, of course, determined by several factors, including the improved specificity and stability of the new generation of oligonucleotides, the optimization of efficient AMO delivery techniques and the advance of support instrumentation that allows the actual live detection of the molecules of interest.
Over the years, several studies have been reported to use LNA oligonucleotides to efficiently target miRNAs showing the potential of LNA-AMOs in different therapeutic and detection strategies [83,98,114-116]. In the detection field, Válóczi et al. [115] developed LNA oligonucleotides with improved sensitivity and high specificity to be applied in northern blot analysis for a highly efficient detection of miRNAs. With their work, they have opened the precedent to further explore the application of LNA oligonucleotides in other detection methods, like for instance to study the spatial expression/distribution of miRNAs in cells and tissues by in situ hybridization or to study the expression profile of miRNAs by LNA-microarrays.
In situ hybridization (ISH) analysis is a technique that applies antisense oligonucleotides complementary to a specific nucleic acid sequence to determine the precise location of miRNA within the cells. The main difference is that for detection purposes, the synthetic antisense oligonucleotides are linked either to a fluorophore, a radioactively-label or to an antigen-labeled bases so that they can be tracked respectively by fluorescence microscopy, autoradiography, or immunohistochemistry. Currently, the use of.LNA-modified oligonucleotides constitute the most promising approach to visualize miRNA expression by in situ hybridization, [117-123] but as far as we are aware only analyzed the results ex-vivo. It is desirable that the molecular probes hybridize at normal body temperature (around 37 °C) to be prone to be used in vivo and push forward the miRNA detection methods in order to determine spatial and temporal miRNA accumulation in tissues. More recently, UNA modification has proven to be an interesting substitution for mismatch discrimination within miRNAs families. Robertson et al. [124]. demonstrated the properties of UNA-AMOs for detecting specifically overexpressed miR10b in metastatic breast cancer. UNA oligonucleotides showed increased specificity towards its target and were able to discriminate miR10b from miR10a (presenting 1 mismatch) and from miR10c (presenting 2 mismatches) more efficiently than its DNA counterparts.
There is already some instrumentation in the field of fluorescence microscopy that allows the detection of fluorescence signal by using in vivo fluorescent detectors. These detectors are basically labeled-oligonucleotides attached to sophisticated and expensive instrumentation, and a lot of effort is applied in the development of smaller, disposable and low-cost detector systems [125]. Another problematic relies on the sensitivity of the detection methods. Usually, the fluorescence detection requires the high sensitivity devices that allow the measurement of very low levels of light emitted by the labeled probes. One example is a prototype developed by Stephen Bellis et al. [126] a hybrid platform consisting of a vertical cavity surface emitting laser (VCSEL) to provide the excitation and a photo-diode (APD) to detect the emitted fluorescence. The APD detectors combine the ability to count individual photons, being able to detect low levels of fluorescence, and as a plus are small in size.
Other technologies are now immersing to track and detect in real-time different molecules in vivo for biomedical research and clinical applications. It is the case of near-infrared fluorescence Imaging (NIR), a technology recently used in the field of cancer imaging. This technique uses high sensitivity molecular probes labeled with fluorophores in the near-infrared region wavelength (650 nm – 900 nm), that allows relatively deep photon penetration into tissue, minimal tissue auto-fluorescence and higher optical contrast. In vivo fluorescence imaging with near‐infrared light holds enormous potential for a wide variety of molecular diagnostic and therapeutic applications [127]. A thorough review on imaging technology for in vivo uses is beyond the scope of this review, and a detailed analysis on this matter can be found on the literature [125].
Interestingly, using the same principle of the detection techniques stated above, AMOs could be further optimized to be stable in vivo, in order to allow miRNA detection in real-time by applying a fluorescence molecule at one end of the sequence. A real-time detection of miRNAs topically in any tissue holds a great promise towards an effective, safe and immediate detection of miRNAs in humans.
6. Concluding remarks
MiRNAs are small, single-stranded and noncoding RNAs that post-transcriptionally regulate gene expression. By complementarily binding to the 3′UTR of mRNAs, the miRNAs cells mainly cause translational repression and/or mRNA deadenylation. They are involved in the control of several biological pathways, and it comes with no surprise that alteration on miRNAs levels has an extensive impact on the synthesis of different proteins associated with different human pathologies, such as cancer.
The development of new technologies to study the function and the mechanisms of regulation of miRNA in human diseases is fundamental. Numerous works show the potential use of AMOs as specific diagnostic biomarkers and/or as a therapy to control aberrant miRNA. The use of AMOs in clinical practice for cancer treatment seems a promising alternative to silence undesirable miRNA function and gene activity. A fully comprehensive study of AMOs function using different in vitro techniques is mandatory. It is also important to associate the in vitro results to the in vivo studies to validate this strategy as an efficient therapeutic route in cancer treatment.
Up to now, no therapeutic strategy has been fully developed in order to modulate miRNA function. Clinical trials are already in place for experimental AMO-based drugs targeting specifically miR-122 (Miravirsen developed by Santaris Pharma and RG-101 developed by Regulus®). Despite of the promising results, the use of anti-miRNA oligonucleotides as therapeutic agents still cares for further validation in terms of cytotoxic effects, specificity and stability of the hybridization between the oligonucleotide sequence and the target miRNA. Furthermore, allying the AMO technology to the emerging methodologies for real-time detection holds a great promise towards an effective, safe and immediate detection of miRNAs in humans. In situ detection of miRNA accumulation in tumor biopsies may provide a highly valuable approach for prognostic and diagnostic evaluation in a clinical setting.
Funding Statement
This work was financially supported by 1. The European Regional Development Fund, through COMPETE2020 — Operational Program for Competitiveness and Internationalization, and the Portuguese Foundation for Science and Technology [grant number POCI-01-0145-FEDER-006939 - UID-EQU/00511/2013, and POCI-01-0145-FEDER-007274 - PEst-C/SAU/LA0003/2013]; 2. Ipatimup integrates the i3S Research Unit, i3S and iBiMED Research Units are partially supported by the Portuguese Foundation for Science and Technology; 3. The North Portugal Regional Operational Program, under the Portugal 2020 Partnership agreement, through the European Regional Development Fund [grant number NORTE-01-0145-FEDER-000005 — LEPABE-2-ECO-INNOVATION, and NORTE-01-0145-FEDER-000029]; 4. and the Portuguese Foundation for Science and Technology, and Biomode 2, S.A. [grant number SFRH/BDE/51909/2012].
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
References
- [1].Bartel DP. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–97. doi: 10.1016/S0092-8674(04)00045-5 [DOI] [PubMed] [Google Scholar]
- [2].Winter J, Jung S, Keller S, et al.. Many roads to maturity: microRNA biogenesis pathways and their regulation. Nat Cell Biol. 2009;11:228–34. doi: 10.1038/ncb0309-228 [DOI] [PubMed] [Google Scholar]
- [3].Kozlowski P, Starega-Roslan J. Structures of microRNA precursors. Curr Perspect microRNAs. 2008;1–16. Available from: http://link.springer.com/chapter/10.1007/978-1-4020-8533-8_1 [Google Scholar]
- [4].Fire A, Xu S, Montgomery MK, et al.. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature. 1998;391:806–11. doi: 10.1038/35888 [DOI] [PubMed] [Google Scholar]
- [5].Basyuk E, Suavet F, Doglio A, et al.. Human let-7 stem-loop precursors harbor features of RNase III cleavage products. Nucleic Acids Res. 2003;31:6593–7. doi: 10.1093/nar/gkg855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Borchert GM, Lanier W, Davidson BL. RNA polymerase III transcribes human microRNAs. Nat Struct Mol Biol. 2006;13:1097–101. doi: 10.1038/nsmb1167 [DOI] [PubMed] [Google Scholar]
- [7].Denli AM, Tops BBJ, Plasterk RHA, et al.. Processing of primary microRNAs by the Microprocessor complex. Nature. 2004;432:231–5. doi: 10.1038/nature03049 [DOI] [PubMed] [Google Scholar]
- [8].Gregory RI, Yan K-P, Amuthan G, et al.. The Microprocessor complex mediates the genesis of microRNAs. Nature. 2004;432:235–40. doi: 10.1038/nature03120 [DOI] [PubMed] [Google Scholar]
- [9].Kim VN. MicroRNA precursors in motion: exportin-5 mediates their nuclear export. Trends Cell Biol. 2004;14:156–9. doi: 10.1016/j.tcb.2004.02.006 [DOI] [PubMed] [Google Scholar]
- [10].Koscianska E, Starega-Roslan J, Krzyzosiak WJ. The role of Dicer protein partners in the processing of microRNA precursors. PLoS One. 2011;6:e28548. doi: 10.1371/journal.pone.0028548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zhang H, Kolb FA, Brondani V, et al.. Human Dicer preferentially cleaves dsRNAs at their termini without a requirement for ATP. Eur Mol Biol Organ J. 2002;21:5875–85. doi: 10.1093/emboj/cdf582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Provost P, Dishart D, Doucet J, et al.. Ribonuclease activity and RNA binding of recombinant human Dicer. Eur Mol Biol Organ J. 2002;21:5864–74. doi: 10.1093/emboj/cdf578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Fareh M, Yeom K-H, Haagsma AC, et al.. TRBP ensures efficient Dicer processing of precursor microRNA in RNA-crowded environments. 2016;7:13694. doi: 10.1038/ncomms13694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Chendrimada TP, Gregory RI, Kumaraswamy E, et al.. TRBP recruits the Dicer complex to Ago2 for microRNA processing and gene silencing. Nature. 2005;436:740–4. doi: 10.1038/nature03868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Gregory RI, Chendrimada TP, Cooch N, et al.. Human RISC couples microRNA biogenesis and posttranscriptional gene silencing. Cell. 2005;123:631–40. doi: 10.1016/j.cell.2005.10.022 [DOI] [PubMed] [Google Scholar]
- [16].Chekulaeva M, Filipowicz W. Mechanisms of miRNA-mediated post-transcriptional regulation in animal cells. Curr Opin Cell Biol. 2009;21:452–60. doi: 10.1016/j.ceb.2009.04.009 [DOI] [PubMed] [Google Scholar]
- [17].Zeng Y, Wagner EJ, Cullen BR. Both natural and designed micro RNAs can inhibit the expression of cognate mRNAs when expressed in human cells. Mol Cell. 2002;9:1327–33. doi: 10.1016/S1097-2765(02)00541-5 [DOI] [PubMed] [Google Scholar]
- [18].Doench JG, Petersen CP, Sharp PA. siRNAs can function as miRNAs Genes Dev. 2003;17:438–42. doi: 10.1101/gad.1064703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hutvágner G, Zamore PD. A microRNA in a Multiple-Turnover RNAi Enzyme Complex. Science. 2002;297:2056. LP-2060. doi: 10.1126/science.1073827 [DOI] [PubMed] [Google Scholar]
- [20].Wahle E, Winkler GS. RNA decay machines: Deadenylation by the Ccr4–Not and Pan2–Pan3 complexes. Biochim Biophys Acta – Gene Regul Mech. 2013;1829:561–70. doi: 10.1016/j.bbagrm.2013.01.003 [DOI] [PubMed] [Google Scholar]
- [21].Jonas S, Izaurralde E. The role of disordered protein regions in the assembly of decapping complexes and RNP granules. Genes Dev. 2013;27:2628–41. doi: 10.1101/gad.227843.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Poulin F, Sonenberg N. Mechanism of Translation Initiation in Eukaryotes. Madame Curie Biosci Database. 2000:1–33. Available from: http://www.ncbi.nlm.nih.gov/books/NBK6597/ [Google Scholar]
- [23].Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet. 2015;16:421–33. doi: 10.1038/nrg3965 [DOI] [PubMed] [Google Scholar]
- [24].Djuranovic S, Nahvi A, Green R. miRNA-Mediated Gene Silencing by Translational Repression Followed by mRNA Deadenylation and Decay. Science. 2012;336:237–40. doi: 10.1126/science.1215691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Huntzinger E, Izaurralde E. Gene silencing by microRNAs: Contributions of translational repression and mRNA decay. Nat Rev Genet. 2011;12:99–110. doi: 10.1038/nrg2936 [DOI] [PubMed] [Google Scholar]
- [26].Beilharz TH, Humphreys DT, Clancy JL, et al.. MicroRNA-mediated messenger RNA deadenylation contributes to translational repression in mammalian cells. PLoS One. 2009;4:e6783. doi: 10.1371/journal.pone.0006783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Zekri L, Huntzinger E, Heimstädt S, et al.. The silencing domain of GW182 interacts with PABPC1 to promote translational repression and degradation of microRNA targets and is required for target release. Mol Cell Biol. 2009;29:6220–31. doi: 10.1128/MCB.01081-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Huntzinger E, Kuzuoğlu-Öztürk D, Braun JE, et al.. The interactions of GW182 proteins with PABP and deadenylases are required for both translational repression and degradation of miRNA targets. Nucleic Acids Res. 2013;41:978–94. doi: 10.1093/nar/gks1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wu L, Fan J, Belasco JG. MicroRNAs direct rapid deadenylation of mRNA. Proc Natl Acad Sci U S A. 2006;103:4034–9. doi: 10.1073/pnas.0510928103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Calin GA, Sevignani C, Dumitru CD, et al.. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc Natl Acad Sci U S A. 2004;101:2999–3004. doi: 10.1073/pnas.0307323101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lu J, Getz G, a Miska E, et al.. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–8. doi: 10.1038/nature03702 [DOI] [PubMed] [Google Scholar]
- [32].Lee YS, Dutta A. MicroRNAs in Cancer. Annu Rev Pathol Mech Dis. 2009;4:199–227. doi: 10.1146/annurev.pathol.4.110807.092222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Tili E, Michaille J-J, Calin GA. Expression and function of micro RNAs in immune cells during normal or disease state. Int J Med Sci. 2008;5:73–9. doi: 10.7150/ijms.5.73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Volinia S, Calin GA, Liu C-G, et al.. A microRNA expression signature of human solid tumors defines cancer gene targets. Proc Natl Acad Sci U S A. 2006;103:2257–61. doi: 10.1073/pnas.0510565103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Esquela-Kerscher A, Slack FJ. Oncomirs – microRNAs with a role in cancer. Nat Rev Cancer. 2006;6:259–69. doi: 10.1038/nrc1840 [DOI] [PubMed] [Google Scholar]
- [36].Bandyopadhyay S, Mitra R, Maulik U, et al.. Development of the human cancer microRNA network. Silence. 2010;1:6. doi: 10.1186/1758-907X-1-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Lynam-Lennon N, Maher SG, Reynolds J V. The roles of microRNA in cancer and apoptosis. Biol Rev Camb Philos Soc. 2009;84:55–71. doi: 10.1111/j.1469-185X.2008.00061.x [DOI] [PubMed] [Google Scholar]
- [38].Tian C, You MJ, Yu Y, et al.. MicroRNA-9 promotes proliferation of leukemia cells in adult CD34-positive acute myeloid leukemia with normal karyotype by downregulation of Hes1. Tumor Biol. 2016;37:7461–71. doi: 10.1007/s13277-015-4581-x [DOI] [PubMed] [Google Scholar]
- [39].Zhang J, Jia J, Zhao L, et al.. Down-regulation of microRNA-9 leads to activation of IL-6/Jak/STAT3 pathway through directly targeting IL-6 in HeLa cell. Mol Carcinog. 2016;55:732–42. doi: 10.1002/mc.22317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Park YR, Lee ST, Kim SL, et al.. MicroRNA-9 suppresses cell migration and invasion through downregulation of TM4SF1 in colorectal cancer. Int J Oncol. 2016;48:2135–43. doi: 10.3892/ijo.2016.3430 [DOI] [PubMed] [Google Scholar]
- [41].Mohammadi-Yeganeh S, Mansouri A, Paryan M. Targeting of miR9/NOTCH1 Interaction Reduces Metastatic Behavior in Triple-negative Breast Cancer. Chem Biol Drug Des. 2015;86:1185–91. doi: 10.1111/cbdd.12584 [DOI] [PubMed] [Google Scholar]
- [42].Zheng L, Qi T, Yang D, et al.. microRNA-9 suppresses the proliferation, invasion and metastasis of gastric cancer cells through targeting cyclin D1 and Ets1. PLoS One. 2013;8:e55719. doi: 10.1371/journal.pone.0055719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rotkrua P, Akiyama Y, Hashimoto Y, et al.. MiR-9 downregulates CDX2 expression in gastric cancer cells. Int J Cancer. 2011;129:2611–20. doi: 10.1002/ijc.25923 [DOI] [PubMed] [Google Scholar]
- [44].Li Y, Xu Z, Li B, et al.. Epigenetic silencing of miRNA-9 is correlated with promoter-proximal CpG island hypermethylation in gastric cancer in vitro and in vivo. Int J Oncol. 2014;45:2576–86. doi: 10.3892/ijo.2014.2667 [DOI] [PubMed] [Google Scholar]
- [45].White RA, Neiman JM, Reddi A, et al.. Epithelial stem cell mutations that promote squamous cell carcinoma metastasis. J Clin Invest. 2013;123:4390–404. doi: 10.1172/JCI65856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Zhang H, Qi M, Li S, et al.. microRNA-9 Targets Matrix Metalloproteinase 14 to Inhibit Invasion, Metastasis, and Angiogenesis of Neuroblastoma Cells. Mol Cancer Ther. 2012;11:1454–66. doi: 10.1158/1535-7163.MCT-12-0001 [DOI] [PubMed] [Google Scholar]
- [47].Luo H, Zhang H, Zhang Z, et al.. Down-regulated miR-9 and miR-433 in human gastric carcinoma. J Exp Clin Cancer Res. 2009;28:82. doi: 10.1186/1756-9966-28-82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Wan H-Y, Guo L-M, Liu T, et al.. Regulation of the transcription factor NF-kappaB1 by microRNA-9 in human gastric adenocarcinoma. Mol Cancer. 2010;9:16. doi: 10.1186/1476-4598-9-16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Liu Q, Teh M, Ito K, et al.. CDX2 expression is progressively decreased in human gastric intestinal metaplasia, dysplasia and cancer. Mod Pathol. 2007;20:1286–97. doi: 10.1038/modpathol.3800968 [DOI] [PubMed] [Google Scholar]
- [50].Sahin A, Velten M, Pietsch T, et al.. No TitleInactivation of Ets 1 transcription factor by a specific decoy strategy reduces rat C6 glioma cell proliferation and mmp-9 expression. Int J Mol Med. 2005;15:771–6 [PubMed] [Google Scholar]
- [51].Duluc I, Lorentz O, Fritsch C, et al.. Changing intestinal connective tissue interactions alters homeobox gene expression in epithelial cells. J Cell Sci. 1997;110:1317 LP–1324. Available from: http://jcs.biologists.org/content/110/11/1317.abstract [DOI] [PubMed] [Google Scholar]
- [52].Busuttil RA, Boussioutas A. Intestinal metaplasia: A premalignant lesion involved in gastric carcinogenesis. J Gastroenterol Hepatol. 2009;24:193–201. doi: 10.1111/j.1440-1746.2008.05774.x [DOI] [PubMed] [Google Scholar]
- [53].Silberg DG, Sullivan J, Kang E, et al.. Cdx2 ectopic expression induces gastric intestinal metaplasia in transgenic mice. Gastroenterology. 2017;122:689–96. doi: 10.1053/gast.2002.31902 [DOI] [PubMed] [Google Scholar]
- [54].Wang Z. MicroRNA interference technologies. Verlag Berlin Heidelberg: Springer; 2009. [Google Scholar]
- [55].Wang Z. The Guideline of the Design and Validation of MiRNA Mimics BT – MicroRNA and Cancer: Methods and Protocols In: Wu W, editor. Totowa, NJ: Humana Press; 2011:211–23. doi: 10.1007/978-1-60761-863-8_15 [DOI] [PubMed] [Google Scholar]
- [56].Wang Z. Multi-miRNA Hairpins and Multi-miRNA Mimics Technologies BT – MicroRNA Interference Technologies In: Wang Z, editor Berlin, Heidelberg: Springer Berlin Heidelberg; 2009:101–10. doi: 10.1007/978-3-642-00489-6_5 [DOI] [Google Scholar]
- [57].Xia XG, Zhou H, Xu Z. Multiple shRNAs expressed by an inducible pol II promoter can knock down the expression of multiple target genes. Biotechniques. 2006;41:64–8. doi: 10.2144/000112198 [DOI] [PubMed] [Google Scholar]
- [58].Ebert MS, Neilson JR, Sharp PA. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods. 2007;4: 10.1038/nmeth1079 Available from: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3857099/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Wang Z. The Principles of MiRNA-Masking Antisense Oligonucleotides Technology BT – MicroRNA and Cancer: Methods and Protocols In: Wu W, editor Totowa, NJ: Humana Press; 2011:43–9. doi: 10.1007/978-1-60761-863-8_3 [DOI] [PubMed] [Google Scholar]
- [60].Esau CC. Inhibition of microRNA with antisense oligonucleotides. Methods. 2008;44:55–60. doi: 10.1016/j.ymeth.2007.11.001 [DOI] [PubMed] [Google Scholar]
- [61].Stenvang J, Petri A, Lindow M, et al.. Inhibition of microRNA function by antimiR oligonucleotides. Silence. 2012;3:1. doi: 10.1186/1758-907X-3-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].He L, Xie M, Huang J, et al.. Efficient and specific inhibition of plant microRNA function by anti-microRNA oligonucleotides (AMOs) in vitro and in vivo. Plant Cell Rep. 2016;35:933–45. doi: 10.1007/s00299-016-1933-y [DOI] [PubMed] [Google Scholar]
- [63].Zamecnik PC, Stephenson ML. Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc Natl Acad Sci U S A. 1978;75:280–4. doi: 10.1073/pnas.75.1.280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Davis S, Lollo B, Freier S, et al.. Improved targeting of miRNA with antisense oligonucleotides. Nucleic Acids Res. 2006;34:2294–304. doi: 10.1093/nar/gkl183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Lennox K a, Behlke M a. A direct comparison of anti-microRNA oligonucleotide potency. Pharm Res. 2010;27:1788–99. doi: 10.1007/s11095-010-0156-0 [DOI] [PubMed] [Google Scholar]
- [66].Neudecker V, Brodsky KS, Kreth S, et al.. Emerging Roles for MicroRNAs in Perioperative Medicine. Anesthesiology. 2016;124:489–506. doi: 10.1097/ALN.0000000000000969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Boutla A, Delidakis C, Tabler M. Developmental defects by antisense-mediated inactivation of micro-RNAs 2 and 13 in Drosophila and the identification of putative target genes. Nucleic Acids Res. 2003;31:4973–80. doi: 10.1093/nar/gkg707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Freier S. The ups and downs of nucleic acid duplex stability: structure-stability studies on chemically-modified DNA:RNA duplexes. Nucleic Acids Res. 1997;25:4429–43. doi: 10.1093/nar/25.22.4429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Egli M, Minasov G, Tereshko V, et al.. Probing the Influence of Stereoelectronic Effects on the Biophysical Properties of Oligonucleotides: Comprehensive Analysis of the RNA Affinity, Nuclease Resistance, and Crystal Structure of Ten 2′-O-Ribonucleic Acid Modifications. Biochemistry. 2005;44:9045–57. doi: 10.1021/bi050574m [DOI] [PubMed] [Google Scholar]
- [70].a Lennox K, R Owczarzy, Thomas DM, et al.. Improved Performance of Anti-miRNA Oligonucleotides Using a Novel Non-Nucleotide Modifier. Mol Ther Nucleic Acids. 2013;2:e117. doi: 10.1038/mtna.2013.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Hutvágner G, Simard MJ, Mello CC, et al.. Sequence-specific inhibition of small RNA function. PLoS Biol. 2004;2:E98. doi: 10.1371/journal.pbio.0020098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].MEISTER G, LANDTHALER M, DORSETT Y, et al.. Sequence-specific inhibition of microRNA- and siRNA-induced RNA silencing. RNA. 2004;10:544–50. doi: 10.1261/rna.5235104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Freier SM, Altmann KH. The ups and downs of nucleic acid duplex stability: Structure-stability studies on chemically-modified DNA:RNA duplexes. Nucleic Acids Res. 1997;25:4429–43. doi: 10.1093/nar/25.22.4429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Vermeulen A, Robertson B, Dalby AB, et al.. Double-stranded regions are essential design components of potent inhibitors of RISC function. RNA. 2007;13:723–30. doi: 10.1261/rna.448107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Mie Y, Hirano Y, Kowata K, et al.. Function Control of Anti-microRNA Oligonucleotides Using Interstrand Cross-Linked Duplexes. Mol Ther – Nucleic Acids. 2018;10:64–74. doi: 10.1016/j.omtn.2017.11.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Esau C, Davis S, Murray SF, et al.. miR-122 regulation of lipid metabolism revealed by in vivo antisense targeting. Cell Metab. 2006;3:87–98. doi: 10.1016/j.cmet.2006.01.005 [DOI] [PubMed] [Google Scholar]
- [77].Bhat B, Hogan D. Microrna compounds and methods for modulating mir-122. 2015; Available from: https://www.google.com/patents/WO2014179446A3?cl=ko [Google Scholar]
- [78].Bhat B, Neben S, Tay J, et al.. RG-101, a GalNAC-conjugated anti-miR employing a unique mechanism of action by targeting host factor microRNA-122 (miR-122), demonstrates potent activity and reduction of HCV in preclinical studies. Hepatology. 2013;58:1393A [Google Scholar]
- [79].BIESSEN EAL, VIETSCH H, RUMP ET, et al.. Targeted delivery of oligodeoxynucleotides to parenchymal liver cells in vivo. Biochem J. 1999;340:783. doi: 10.1042/bj3400783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Davis S, Propp S, Freier SM, et al.. Potent inhibition of microRNA in vivo without degradation. Nucleic Acids Res. 2009;37:70–7. doi: 10.1093/nar/gkn904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Gene Link 2′-Fluoro deoxyadenosine (2′-F-A) [Internet]. [cited 2018January10].
- [82].Campbell MA, Wengel J, Locked vs. Unlocked nucleic acids (LNAvs.UNA): contrasting structures work towards common therapeutic goals. Chem Soc Rev. 2011;40:5680–9. doi: 10.1039/c1cs15048k [DOI] [PubMed] [Google Scholar]
- [83].Obad S, dos Santos CO, Petri A, et al.. Silencing of microRNA families by seed-targeting tiny LNAs. Nat Genet. 2011;43:371–8. doi: 10.1038/ng.786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Zhang Y, Roccaro AM, Rombaoa C, et al.. LNA-mediated anti–miR-155 silencing in low-grade B-cell lymphomas. Blood. 2012;120:1678 LP–1686. [DOI] [PubMed] [Google Scholar]
- [85].You Y, Moreira BG, Behlke MA, et al.. Design of LNA probes that improve mismatch discrimination. Nucleic Acids Res. 2006;34:e60–e60. doi: 10.1093/nar/gkl175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].van der Ree MH, van der Meer AJ, de Bruijne J, et al.. Long-term safety and efficacy of microRNA-targeted therapy in chronic hepatitis C patients. Antiviral Res. 2014;111:53–9. doi: 10.1016/j.antiviral.2014.08.015 [DOI] [PubMed] [Google Scholar]
- [87].Pasternak A, Wengel J. Unlocked nucleic acid – an RNA modification with broad potential. Org Biomol Chem. 2011;9:3591–7. doi: 10.1039/c0ob01085e [DOI] [PubMed] [Google Scholar]
- [88].Stein CA, Cheng YC. Antisense oligonucleotides as therapeutic agents–is the bullet really magical? Science. 1993;261:1004 LP–1012. Available from: http://science.sciencemag.org/content/261/5124/1004.abstract [DOI] [PubMed] [Google Scholar]
- [89].Georgakoudi I, Müller MG, Boone CW, et al.. NAD(P)H and collagen as in vivo quantitative fluorescent biomarkers of epithelial precancerous changes. Cancer Res. 2002;62:682–7 [PubMed] [Google Scholar]
- [90].SUMMERTON J, WELLER D. Morpholino Antisense Oligomers: Design, Preparation, and Properties. Antisense Nucleic Acid Drug Dev. 1997;7:187–95. doi: 10.1089/oli.1.1997.7.187 [DOI] [PubMed] [Google Scholar]
- [91].Nielsen PE, Egholm M, Berg RH, et al.. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science. 1991;254:1497 LP–1500. Available from: http://science.sciencemag.org/content/254/5037/1497.abstract [DOI] [PubMed] [Google Scholar]
- [92].Weidner DA, Valdez BC, Henning D, et al.. Phosphorothioate oligonucleotides bind in a non sequence-specific manner to the nucleolar protein C23/nucleolin. FEBS Lett. 1995;366:146–50. doi: 10.1016/0014-5793(95)00517-D [DOI] [PubMed] [Google Scholar]
- [93].Geary RS, Watanabe TA, Truong L, et al.. Pharmacokinetic Properties of 2′-O-(2-Methoxyethyl)-Modified Oligonucleotide Analogs in Rats. J Pharmacol Exp Ther. 2001;296:890 LP–897. Available from: http://jpet.aspetjournals.org/content/296/3/890.abstract [PubMed] [Google Scholar]
- [94].Sheehan D, Lunstad B, Yamada CM, et al.. Biochemical properties of phosphonoacetate and thiophosphonoacetate oligodeoxyribonucleotides. Nucleic Acids Res. 2003;31:4109–18. doi: 10.1093/nar/gkg439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Threlfall RN, Torres AG, Krivenko A, et al.. Synthesis and biological activity of phosphonoacetate- and thiophosphonoacetate-modified 2[prime or minute]-O-methyl oligoribonucleotides. Org Biomol Chem. 2012;10:746–54. doi: 10.1039/C1OB06614E [DOI] [PubMed] [Google Scholar]
- [96].Flynt AS, Li N, Thatcher EJ, et al.. Zebrafish miR-214 modulates Hedgehog signaling to specify muscle cell fate. Nat Genet. 2007;39:259–63. doi: 10.1038/ng1953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Kloosterman WP, Lagendijk AK, Ketting RF, et al.. Targeted Inhibition of miRNA Maturation with Morpholinos Reveals a Role for miR-375 in Pancreatic Islet Development. PLOS Biol. 2007;5:e203. doi: 10.1371/journal.pbio.0050203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Fabani M, Gait M. miR-122 targeting with LNA/2′-O-methyl oligonucleotide mixmers, peptide nucleic acids (PNA), and PNA–peptide conjugates. RNA. 2008;14(2):336–46. Available from: http://rnajournal.cshlp.org/content/14/2/336.short [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Wittung P, Kajanus J, Edwards K, et al.. Phospholipid membrane permeability of peptide nucleic acid. FEBS Lett. 1995;375:27–9. doi: 10.1016/0014-5793(95)00409-3 [DOI] [PubMed] [Google Scholar]
- [100].Research WI for B TargetScan. v7.1. Available from: www.targetscan.org [Google Scholar]
- [101].Griffiths-Jones S, Grocock RJ, Van Dongen S, Bateman A, et al.. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140–4. doi: 10.1093/nar/gkj112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Griffiths-Jones lab, Faculty of Life Sciences U of M miRBase. Available from: http://www.mirbase.org/
- [103].Elmén J, Lindow M, Silahtaroglu A, et al.. Antagonism of microRNA-122 in mice by systemically administered LNA-antimiR leads to up-regulation of a large set of predicted target mRNAs in the liver. Nucleic Acids Res. 2008;36:1153–62. doi: 10.1093/nar/gkm1113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].U.S National Library of Medicine. BLAST: Basic Local Alignment Search Tool. V2.6.0 January 09, 2017. Available from: https://blast.ncbi.nlm.nih.gov/Blast.cgi [Google Scholar]
- [105].Krützfeldt J, Rajewsky N, Braich R, et al.. Silencing of microRNAs in vivo with “antagomirs”. Nature. 2005;438:685–9. doi: 10.1038/nature04303 [DOI] [PubMed] [Google Scholar]
- [106].Crooke ST, Wang S, Vickers TA, et al.. Cellular uptake and trafficking of antisense oligonucleotides. Nat Biotechnol. 2017;35:230–7. doi: 10.1038/nbt.3779 [DOI] [PubMed] [Google Scholar]
- [107].Fontenete S, Guimarães N, Wengel J, et al.. Prediction of melting temperatures in fluorescence in situ hybridization (FISH) procedures using thermodynamic models. Crit Rev Biotechnol. 2015;8551:1–12. Available from: http://www.tandfonline.com/doi/full/10.3109/07388551.2014.993589%5Cnhttp://informahealthcare.com/doi/abs/10.3109/07388551.2014.993589 [DOI] [PubMed] [Google Scholar]
- [108].Lennox K a, Behlke M a. Chemical modification and design of anti-miRNA oligonucleotides. Gene Ther. 2011;18:1111–20. doi: 10.1038/gt.2011.100 [DOI] [PubMed] [Google Scholar]
- [109].Krützfeldt J, Kuwajima S, Braich R, et al.. Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res. 2007;35:2885–92. doi: 10.1093/nar/gkm024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Oh SY, Ju Y, Park H. A highly effective and long-lasting inhibition of miRNAs with PNA-based antisense oligonucleotides. Mol Cells. 2009;28:341–5. doi: 10.1007/s10059-009-0134-8 [DOI] [PubMed] [Google Scholar]
- [111].Jiang T, Olson ES, Nguyen QT, et al.. Tumor imaging by means of proteolytic activation of cell-penetrating peptides. Proc Natl Acad Sci U S A. 2004;101:17867–72. doi: 10.1073/pnas.0408191101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Vollmer J, Weeratna R, Payette P, et al.. Characterization of three CpG oligodeoxynucleotide classes with distinct immunostimulatory activities. Eur J Immunol. 2004;34:251–62. doi: 10.1002/eji.200324032 [DOI] [PubMed] [Google Scholar]
- [113].Vollmer J, Weeratna RD, Jurk M, et al.. Oligodeoxynucleotides lacking CpG dinucleotides mediate Toll-like receptor 9 dependent T helper type 2 biased immune stimulation. Immunology. 2004;113:212–23. doi: 10.1111/j.1365-2567.2004.01962.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Elmén J, Lindow M, Schütz S, et al.. LNA-mediated microRNA silencing in non-human primates. Nature. 2008;452:896–9. doi: 10.1038/nature06783 [DOI] [PubMed] [Google Scholar]
- [115].Válóczi A, Hornyik C, Varga N, et al.. Sensitive and specific detection of microRNAs by northern blot analysis using LNA-modified oligonucleotide probes. Nucleic Acids Res. 2004;32:e175. doi: 10.1093/nar/gnh171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Ørom UA, Kauppinen S, Lund AH. LNA-modified oligonucleotides mediate specific inhibition of microRNA function. Gene. 2006;372:137–41. doi: 10.1016/j.gene.2005.12.031 [DOI] [PubMed] [Google Scholar]
- [117].Chaudhuri AD, Yelamanchili S V, Fox HS. Combined fluorescent in situ hybridization for detection of microRNAs and immunofluorescent labeling for cell-type markers. Front Cell Neurosci. 2013;7:160 Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3779857&tool=pmcentrez&rendertype=abstract [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Lagendijk AK, Moulton JD, Bakkers J. Revealing details: whole mount microRNA in situ hybridization protocol for zebrafish embryos and adult tissues. Biol Open. 2012;1:566–9. doi: 10.1242/bio.2012810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Søe MJ, Møller T, Dufva M, et al.. A sensitive alternative for microRNA in situ hybridizations using probes of 2′-O-methyl RNA + LNA. J Histochem Cytochem. 2011;59:661–72. doi: 10.1369/0022155411409411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Jørgensen S, Baker A, Møller S, et al.. Robust one-day in situ hybridization protocol for detection of microRNAs in paraffin samples using LNA probes. Methods. 2010;52:375–81. doi: 10.1016/j.ymeth.2010.07.002 [DOI] [PubMed] [Google Scholar]
- [121].Nuovo GJ. In situ detection of microRNAs in paraffin embedded, formalin fixed tissues and the co-localization of their putative targets. Methods. 2010;52:307–15. doi: 10.1016/j.ymeth.2010.08.009 [DOI] [PubMed] [Google Scholar]
- [122].Obernosterer G, Martinez J, Alenius M. Locked nucleic acid-based in situ detection of microRNAs in mouse tissue sections. Nat Protoc. 2007;2:1508–14. doi: 10.1038/nprot.2007.153 [DOI] [PubMed] [Google Scholar]
- [123].Robertson KL, Thach DC. LNA flow-FISH: a flow cytometry-fluorescence in situ hybridization method to detect messenger RNA using locked nucleic acid probes. Anal Biochem. 2009;390:109–14. doi: 10.1016/j.ab.2009.04.026 [DOI] [PubMed] [Google Scholar]
- [124].Robertson NM, Toscano AE, LaMantia VE, et al.. Unlocked Nucleic Acids for miRNA detection using two dimensional nano-graphene oxide. Biosens Bioelectron. 2017;89:551–7. doi: 10.1016/j.bios.2016.02.058 [DOI] [PubMed] [Google Scholar]
- [125].Moreira L, Guimarães NM, Azevedo NF. 8 – Imaging strategies for bioinspired materials A2 – Rodrigues, Lígia. In: Mota MBT-BM for MA , editor. Woodhead Publishing; 2017;215–39. Available from: http://www.sciencedirect.com/science/article/pii/B9780081007419000085 [Google Scholar]
- [126].Bellis S, Jackson JC, Mathewson A. Towards a disposable in vivo miniature implantable fluorescence detector. 2006;60830N-60830N–8. doi: 10.1117/12.646268 [DOI]
- [127].Frangioni J V. In vivo near-infrared fluorescence imaging. Curr Opin Chem Biol. 2003;7:626–34. doi: 10.1016/j.cbpa.2003.08.007 [DOI] [PubMed] [Google Scholar]
- [128].Zhu L, Chen H, Zhou D, et al.. MicroRNA-9 up-regulation is involved in colorectal cancer metastasis via promoting cell motility. Med Oncol. 2012;29:1037–43. doi: 10.1007/s12032-011-9975-z [DOI] [PubMed] [Google Scholar]
- [129].Wang J, Zhao H, Tang D, et al.. Overexpressions of microRNA-9 and microRNA-200c in human breast cancers are associated with lymph node metastasis. Cancer Biother Radiopharm. 2013;28:283–8. doi: 10.1089/cbr.2012.1293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Gwak JM, Kim HJ, Kim EJ, et al.. MicroRNA-9 is associated with epithelial-mesenchymal transition, breast cancer stem cell phenotype, and tumor progression in breast cancer. Breast Cancer Res Treat. 2014;147:39–49. doi: 10.1007/s10549-014-3069-5 [DOI] [PubMed] [Google Scholar]
- [131].Ma L, Young J, Prabhala H, Pan E, Mestdagh P, Teruya-feldstein J, Reinhardt F, Onder TT, Valastyan S, Westermann F, et al.. miR-9, a MYC/MYCN-activated microRNA, regulates E-cadherin and cancer metastasis. 2010;12:247–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Sun J, Fang K, Shen H, et al.. MicroRNA-9 is a ponderable index for the prognosis of human hepatocellular carcinoma. Int J Clin Exp Med. 2015;8:17748–56 [PMC free article] [PubMed] [Google Scholar]
- [133].Hao-Xiang T, Qian W, Lian-Zhou C, et al.. MicroRNA-9 reduces cell invasion and E-cadherin secretion in SK-Hep-1 cell. Med Oncol. 2010;27:654–60. doi: 10.1007/s12032-009-9264-2 [DOI] [PubMed] [Google Scholar]
- [134].Sun Z, Han Q, Zhou N, et al.. MicroRNA-9 enhances migration and invasion through KLF17 in hepatocellular carcinoma. Mol Oncol. 2013;7:884–94. doi: 10.1016/j.molonc.2013.04.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Wang H, Zhang W, Zuo Y, et al.. miR-9 promotes cell proliferation and inhibits apoptosis by targeting LASS2 in bladder cancer. Tumor Biol. 2015;36:9631–40. doi: 10.1007/s13277-015-3713-7 [DOI] [PubMed] [Google Scholar]
- [136].Chen P, Price C, Li Z, et al.. miR-9 is an essential oncogenic microRNA specifically overexpressed in mixed lineage leukemia-rearranged leukemia. Proc Natl Acad Sci U S A. 2013;110:11511–6. doi: 10.1073/pnas.1310144110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Zhu SW, Li JP, Ma XL, et al.. miR-9 modulates osteosarcoma cell growth by targeting the GCIP tumor suppressor. Asian Pacific J Cancer Prev. 2015;16:4509–13. doi: 10.7314/APJCP.2015.16.11.4509 [DOI] [PubMed] [Google Scholar]
- [138].Ben-Hamo R, Zilberberg A, Cohen H, et al.. hsa-miR-9 controls the mobility behavior of Glioblastoma cells via regulation of MAPK14 signaling elements. Oncotarget. 2015;7: Available from: http://www.ncbi.nlm.nih.gov/pubmed/27036038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Song Y, Li JJ, Zhu Y, et al.. MicroRNA-9 promotes tumor metastasis via repressing E-cadherin in esophageal squamous cell carcinoma. Oncotarget. 2014;5: Available from: www.impactjournals.com/oncotarget%5Cnwww.impactjournals.com/oncotarget/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Zhu M, Xu Y, Ge M, et al.. Regulation of UHRF1 by microRNA-9 modulates colorectal cancer cell proliferation and apoptosis. Cancer Sci. 2015;106:833–9. doi: 10.1111/cas.12689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Higashi T, Hayashi H, Ishimoto T, et al.. miR-9-3p plays a tumour-suppressor role by targeting TAZ (WWTR1) in hepatocellular carcinoma cells. Br J Cancer. 2015;113:252–8. doi: 10.1038/bjc.2015.170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Zhang J, Cheng J, Zeng Z, et al.. Comprehensive profiling of novel microRNA-9 targets and a tumor suppressor role of microRNA-9 via targeting IGF2BP1 in hepatocellular carcinoma. Oncotarget. 2015;6 Available from: http://www.ncbi.nlm.nih.gov/pubmed/26547929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Rodriguez-Otero P, Román-Gómez J, Vilas-Zornoza A, et al.. Deregulation of FGFR1 and CDK6 oncogenic pathways in acute lymphoblastic leukaemia harbouring epigenetic modifications of the MIR9 family. Br J Haematol. 2011;155:73–83. doi: 10.1111/j.1365-2141.2011.08812.x [DOI] [PubMed] [Google Scholar]
- [144].Guo L-M, Pu Y, Han Z, et al.. MicroRNA-9 inhibits ovarian cancer cell growth through regulation of NF-kappaB1. FEBS J. 2009;276:5537–46. doi: 10.1111/j.1742-4658.2009.07237.x [DOI] [PubMed] [Google Scholar]
- [145].Yu T, Liu K, Wu Y, et al.. MicroRNA-9 inhibits the proliferation of oral squamous cell carcinoma cells by suppressing expression of CXCR4 via the Wnt/[beta]-catenin signaling pathway. Oncogene. 2014;33:5017–27. doi: 10.1038/onc.2013.448 [DOI] [PubMed] [Google Scholar]
- [146].Liu S, Kumar SM, Lu H, et al.. MicroRNA-9 up-regulates E-cadherin through inhibition of NF-??B1-Snail1 pathway in melanoma. J Pathol. 2012;226:61–72. doi: 10.1002/path.2964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Fiaschetti G, Abela L, Nonoguchi N, et al.. Epigenetic silencing of miRNA-9 is associated with HES1 oncogenic activity and poor prognosis of medulloblastoma. Br J Cancer. 2014;110:636–47. doi: 10.1038/bjc.2013.764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].MEISTER G. Sequence-specific inhibition of microRNA- and siRNA-induced RNA silencing. RNA. 2004;10:544–50. doi: 10.1261/rna.5235104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Fabani MM, Gait MJ. miR-122 targeting with LNA/2′-O-methyl oligonucleotide mixmers, peptide nucleic acids (PNA), and PNA-peptide conjugates. RNA. 2007;14:336–46. doi: 10.1261/rna.844108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Yamamichi N, Shimomura R, Inada K, et al.. Locked nucleic acid in situ hybridization analysis of miR-21 expression during colorectal cancer development. Clin Cancer Res. 2009;15:4009–16. doi: 10.1158/1078-0432.CCR-08-3257 [DOI] [PubMed] [Google Scholar]