

ABSTRACT
PAR-CLIP (photoactivatable ribonucleoside–enhanced crosslinking and immunoprecipitation) facilitates the identification and mapping of protein/RNA interactions. So far, it has been limited to select cell-lines as it requires efficient 4SU uptake. To increase transcriptome complexity and thus identify additional RNA-protein interaction sites we fused HEK 293 T-Rex cells (HEK293-Y) that express the RNA binding protein YBX1 with PC12 cells expressing eGFP (PC12-eGFP). The resulting hybrids enable PAR-CLIP on a neuronally expanded transcriptome (Fusion-CLIP) and serve as a proof of principle. The fusion cells express both parental marker genes YBX1 and eGFP and the expanded transcriptome contains human and rat transcripts. PAR-CLIP of fused cells versus the parental HEK293-Y identified 768 novel RNA targets of YBX1. We were able to trace the origin of the majority of the short PAR-CLIP reads as they differentially mapped to the human and rat genome. Furthermore, Fusion-CLIP expanded the CAUC RNA binding motif of YBX1 to UCUUUNNCAUC. The fusion of HEK293-Y and PC12-eGFP cells resulted in cells with a diverse genome expressing human and rat transcripts that enabled the identification of novel YBX1 substrates. The technique allows the expansion of the HEK 293 transcriptome and makes PAR-CLIP available to fusion cells of diverse origin.
KEYWORDS: Cell fusion, PAR-CLIP, RNAseq, RNA-binding protein, RNA processing, transcriptomics, YBX1
Introduction
The combination of immunoprecipitation and RNAseq has greatly facilitated the characterization of RNA-binding proteins and their targets. This includes high-throughput sequencing of RNA isolated by crosslinking immunoprecipitation (HITS-CLIP), photoactivatable ribonucleotide-enhanced crosslinking and immunoprecipitation (PAR-CLIP), individual-nucleotide resolution UV crosslinking and immunoprecipitation (iCLIP), enhanced UV crosslinking and immunoprecipitation (eCLIP) and the nonisotopic infrared CLIP (irCLIP).1–7 These technologies have not only expanded the protein-RNA interaction landscape but also facilitated the mapping of binding sites at nucleotide resolution. PAR-CLIP is restricted to select cell types such as HEK 293 cells due to the uptake of photoactivatable ribonucleotides such as 4-thiouridine (4SU) but provides improved resolution and signal-to-noise ratio compared to other CLIP-technologies due to the T to C transition in crosslinked RNAs – although other CLIP methods continue to be improved.2,8 To facilitate the application of PAR-CLIP to cells with a more diverse transcriptome that reflect disease relevant cell types, we used cell fusion. Cell fusion occurs during diverse natural processes such as cell differentiation, embryogenesis or morphogenesis. Induced cell-fusion is commonly used for the production of antibodies in hybridoma cells and to study cell division9 or protein shuttling.10
We used YBX1 for the proof of concept, as it binds a diverse DNA and RNA substrate spectrum related to cell proliferation and differentiation in response to stress.11 YBX1 is highly conserved among species and contains a nucleic acid-binding cold shock domain (CSD), an N-terminal arginine/proline (A/P) rich domain, and a C-terminal domain (CTD). As an oncogene it is upregulated in multiple cancers and associated with multi-drug resistance.12,13 Little is known about its functions in neuronal cells, but recent studies have suggested a role of YBX1 in the suppression of Alzheimer's disease via its interaction with β-amyloid.14
Here, we fused rat PC12 cells, which – in addition to several neuronal transcripts – express enhanced green fluorescent protein (eGFP) as a marker to human HEK 293 cells expressing YBX1 (HEK293-Y). The resulting expanded transcriptome facilitates the identification of novel protein-RNA interactions and the analysis of species-specific RNA processing. Our analysis of the HEK 293 PC12 fusion cells provide a map of YBX1 RNA-binding sites in a neuronally expanded transcriptome.
Results
Generation and characterization of fusion cells from HEK293-Y and PC12- eGFP cells
To derive neuronal transcripts that interact with YBX1, we fused the FLAG/HA-tagged YBX1 expressing human embryonic kidney cell line T-REx HEK 293 (HEK293-Y) and the eGFP expressing rat adrenal medulla cell line PC12 (PC12-eGFP; Fig. 1A). We refer to the resulting fusion cells as PC12f-Y (HEK 293 PC12 fusion cells expressing YBX1) and focus our analysis on the fusion clones PC12f-Y1 and PC12f-Y2. Their morphology is different from the parental cells and from each other as analyzed by light and fluorescence microscopy (Fig. 1B). Both fusion clones express eGFP derived from the PC12-eGFP cells and doxycycline (Dox)-inducible FLAG/HA-YBX1 derived from HEK293-Y cells (Fig. 1C). To follow the stability of the combined genome, we determined the DNA content of parental and fusion cells by DNA-staining with propidium iodide (PI) followed by flow cytometry. The DNA content of both fusion clones is increased compared to the parental cells (3-fold for PC12f-Y1 and 2-fold for PC12f-Y2 as compared to the parental HEK293-Y cell (Fig. 1D, gating details in Fig. S2).
Figure 1.
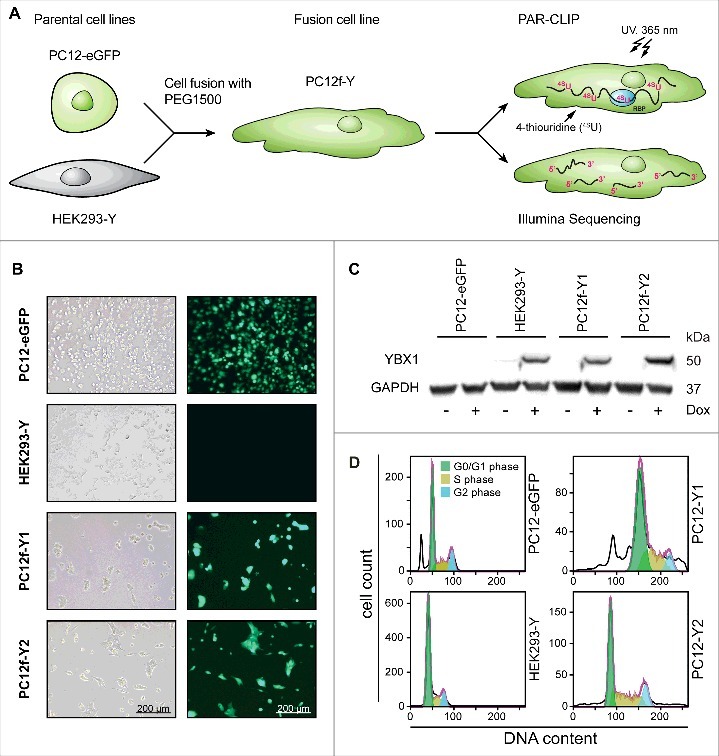
Characterization of the fusion cell model. (A) Experimental overview. (B) Characterization via light and fluorescence microscopy. Both analyzed fusion clones PC12f-Y1 and PC12f-Y2 express the marker gene eGFP derived from the parental PC12-eGFP cell line. Scale bar 200 µm. (C) Western blot of the HA-tagged YBX1. Both fusion clones express the marker gene YBX1 derived from the parental cell line HEK293-Y T-REx. The expression of YBX1 is inducible by doxycycline (Dox). (D) Propidium iodide staining of total DNA analyzed via flow cytometry. Both fusion cell lines exhibit an increased DNA content in G0/G1-phase of the cell cycle (green).
To compare the transcriptome of the fusion and parental cells we used RNAseq. Sequencing reads were mapped to the human (Ensembl GRCh38.80) and rat (Ensembl Rnor_5.0) genome and gene expression was quantified using HTSeq.15 Reads obtained from PC12-eGFP cells map almost completely to the rat (99.98%) and the parental HEK293-Y cells to the human (99.98%) genome, while the fusion cells express rat and human genes in different proportions (Fig. 2A, B). The clone PC12f-Y1 predominantly reflects the PC12 cell character as transcripts predominantly map to the rat (75.3% rat and 24.7% human). The transcripts of PC12f-Y2 predominantly map to the human genome, reflecting the HEK 293 origin (80.5% human and 19.5% rat). Their transcriptomes overlap to a higher degree as compared to the parental cell lines (84.6%; Fig. 2B), which do not correlate with each other (0.6% overlap).
Figure 2.
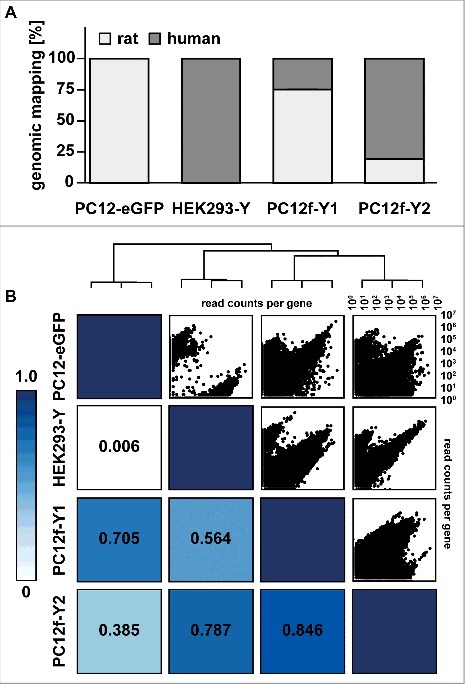
Transcriptomic analysis of the parental and fusion cells. The transcriptome of the parental and fusion cells were paired end sequenced (2 × 101bp). (A) Mapping of the sequencing reads to rat and human genome. A higher fraction of reads originating from PC12f-Y1 cells map to the rat genome (derived from the PC12-eGFP) where reads originating from PC12f-Y2 cells mainly map to the human genome (derived from HEK293-Y). (B) Scatter and heatmap correlation plot of read counts per gene comparing the four cell lines. PC12f-Y1 and PC12f-Y2 have the highest correlation. Beyond that PC12f-Y1 clusters with PC12-eGFP and PC12f-Y2 with HEK293-Y cells.
YBX1 efficiently binds rat and human RNAs in fusion cells
To evaluate the RNA binding capacity of YBX1, we performed PAR-CLIP of the parental HEK293-Y cell line and the two fusion clones PC12f-Y1 and PC12f-Y2. Differences in 4-thiouridine (4SU) uptake could affect the efficiency of the assay, but systematic data comparing cell lines is scarce. Therefore, we determined the 4SU incorporation in PC12-EGFP, PC12f-Y1 and PC12f-Y2 compared to the HEK293-Y cells (Fig. 3A, quantified from Fig. S4A). HEK293-Y cells incorporate 4SU most efficiently followed by both fusion clones (20 – 50% of the parental cells) and the PC12-eGFP cells (<15%). This is reflected in the amount of RNA crosslinked to YBX1 (Fig. 3B): the identical amount of starting material leads to a higher enrichment of YBX1-RNA-complexes in HEK293-Y cells compared to PC12f-Y1 und PC12f-Y2 cells. Gene expression of the nucleoside transporters SLC29A1 and SLC29A2, which account for the majority of the 4SU uptake16, is significantly reduced in fusion clones as determined by RNAseq analysis (read counts normalized to library size; Fig. 3C). SLC29A4 is expressed at highest levels in PC12 cells, but does function as 4SU transporter. Together, the regulation of SLCs is consistent with the differential 4SU incorporation between HEK293-Y and PC12-eGFP cells. To account for these differences, we doubled the amount of starting material for fusion cell lines compared to HEK293-Y cells.
Figure 3.
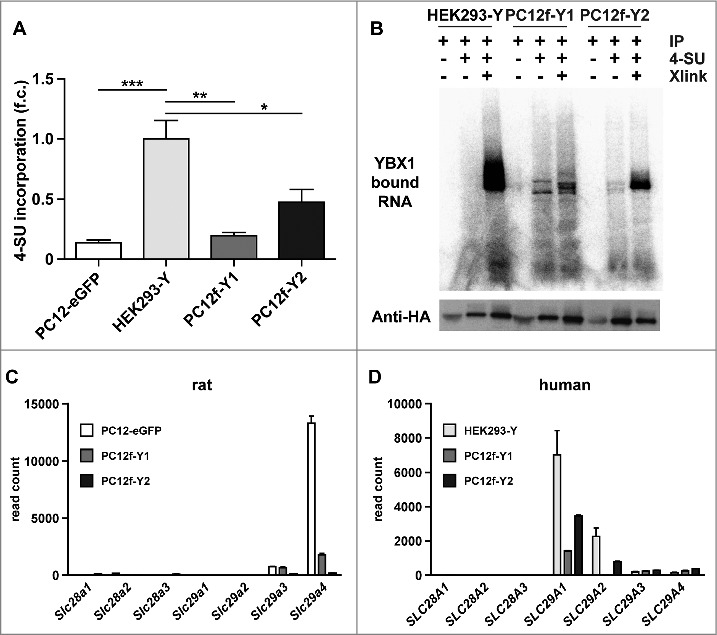
4SU incorporation and PAR-CLIP in fusion cells. (A) Quantification of the 4-Thiouridine (4SU) incorporation in total RNA extracted from the two parental cells and the two analyzed fusion clones. 4SU incorporation rate is highest in HEK293-Y cells followed by the fusion cells and PC12-eGFP. (B) Radioactive labeling of RNA bound by YBX1 after IP. HEK293-Y have the most efficient crosslinking rate. (C+D) Expression of rat and human nucleoside reporters from RNAseq data. SLC29A1 is highly expressed in HEK293-Y cells (C) compared to PC12-eGFP (D) and most important for the uptake of 4SU. *P < 0.05, **P < 0.01, ***P < 0.001, n = 3, one-way ANOVA (A).
Cell fusion expands the YBX1-RNA interactome
PAR-CLIPs of HEK293-Y, PC12f-Y1 and PC12f-Y2 cells were performed in two biological replicates. YBX1-bound RNA of each individual replicate and pooled replicate samples were mapped to the human and rat genome (Mapped genes: Table 1; Mapped reads: Tables S2+3). We observed that the mapping specificity to either human or rat genome increased with increasing read lengths (Fig. S4). Therefore PAR-CLIP reads that were shorter than 20 nt were discarded (∼50% of all PAR-CLIP reads: Table S5; Fig. S5). The genomic distribution of species specific PAR-CLIP reads and RNAseq is comparable. The PAR-CLIP reads of HEK293-Y cells map almost exclusively to the human genome (more than 90%), those derived from PC12f-Y1 mainly to the rat genome (54.1% rat vs. 45.9% human), and PC12f-Y2 reads map predominantly to the human genome (>80%; Table S3).
Table 1.
YBX1-bound RNAs mapped to rat and human genes.
| Cell line | Rat genes | Human genes | Total genes |
|---|---|---|---|
| HEK293-Y_1 | 56 | 9286 | 9342 |
| HEK293-Y_2 | 26 | 4854 | 4880 |
| HEK293-Y_union | 81 | 10420 | 10501 |
| PC12f-Y1_1 | 362 | 374 | 736 |
| PC12f-Y1_2 | 3312 | 2392 | 5704 |
| PC12f-Y1_union | 3770 | 3059 | 6829 |
| PC12f-Y2_1 | 25 | 135 | 160 |
| PC12f-Y2_2 | 68 | 937 | 1005 |
| PC12f-Y2_union | 102 | 1216 | 1318 |
Identification of YBX1 binding transcripts was highest in the parental HEK293-Y cell-line followed by PC12f-Y1 and PC12f-Y2 cells (Table 1). We merged replicates for further analyses since only few additional novel candidates were derived from the smaller replicate (Fig. 4A-C). To compare the PAR-CLIP results of the different cell lines, all rat genes were assigned into their human orthologues resulting in 9619 YBX1 targets identified in HEK293-Y cells, 5916 in PC12f-Y1 and 1119 in PC12f-Y2 (Fig. 4D). In total, 5426 genes were exclusively identified in the parental HEK293-Y cells, 648 and 88 in the fusion clones PC12f-Y1 and PC12f-Y2, respectively. 33 genes were found in both fusion lines, but not in the parental HEK293-Y cells. Taken together, additional 768 potential target transcripts of YBX1 were identified after cell fusion as compared to the parental cell line. Of those genes 515 were derived from rat transcripts, 221 from human transcripts and 32 from both. As expected, the majority of novel genes were derived from the predominantly rodent clone PC12f-Y1 (648 genes), the minority from the predominantly human clone PC12f-Y2 (88 genes; 33 from both fusion clones). The novel genes were classified according to the gene ontology (GO) term “biological processes” using the ClueGO plugin in Cytoscape.17 The GOrilla software18 was used to calculate the enriched ontology terms: The target list contained the 768 potentially new substrate RNAs of YBX1 (exclusively in PC12f-Y1 and -Y2) and the background list all genes identified by PAR-CLIP (parental and both fusion lines). The most enriched pathways are associated with cell chemotaxis, chemokines production, and DNA transposition (Fig. 4E). The newly identified genes relate to toll-like receptor 4 signaling or interferon type I response (Fig. 4F), which have both been implicated in Alzheimer's Disease.19–21
Figure 4.
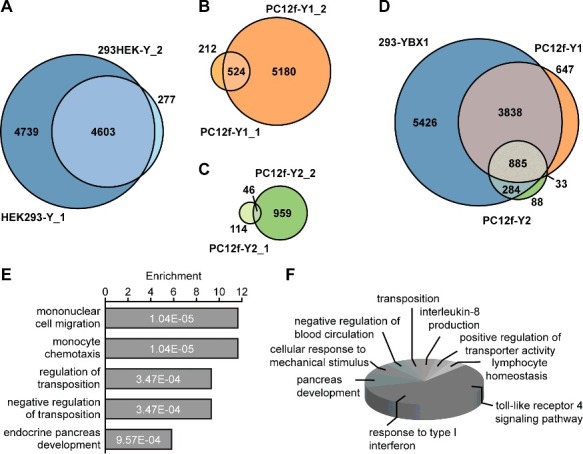
PAR-CLIP of YBX1. VENN-diagram of YBX1 bound genes in biological PAR-CLIP replicates of HEK293-Y (A), PC12f-Y1 (B) and PC12f-Y2 cells (C). (D) VENN diagram of genes bound in different cellular environments. PAR-CLIP replicates are merged and rat genes translated to human orthologues. In total 768 genes were bound by YBX1 in fusion cells but not in parental HEK293-Y cells. (E) GOrilla enrichment analysis of the cell fusion-based identified YBX1-bound genes (top five biological process ranked by p-value). (F) ClueGO classification of the YBX1-bound genes identified in the fusion cells by the gene ontology term biological process.
YBX1 predominantly binds a CAUC motif in the 3′-UTR of its substrate RNAs
To characterize YBX1 binding sites we separately evaluated human and rat transcripts in PC12f-Y1 (PC12f-Y1hum and PC12f-Y1rat) and PC12f-Y2 (PC12f-Y2hum and PC12f-Y2rat) fusion cells. YBX1 mainly binds the 3′-UTR and the coding region of its target transcripts across all cell lines investigated (Fig. 5A). The spatial binding of YBX1 differs between human and rat RNAs. While all human reads map to annotated regions in the human genome approx. 25% of the rat reads map to regions in the corresponding genome that are not annotated. The binding seed sequence was determined using the motif analysis software MEME.22 Binding motifs were extracted by sequence comparison of random PAR-CLIP reads derived from the different cell lines. In all three cell lines (HEK293-Y, PC12f-Y1 and PC12f-Y2) YBX1 predominantly binds the previously described CAUC motif23 (Fig. 5B). We find the motif preceded by a yet undescribed UCUUU motif. The nucleic acid binding cold shock domain (CSD) of YBX1 is 100% identical between rat and human (the complete YBX1 amino acid sequence is 97% identical; Fig. 5C, D). Accordingly we do not expect a bias in YBX1 binding to rat or human targets, facilitating the comparison between the parental HEK293-Y cells and the fusion cells PC12f-Y1 and PC12f-Y2.
Figure 5.
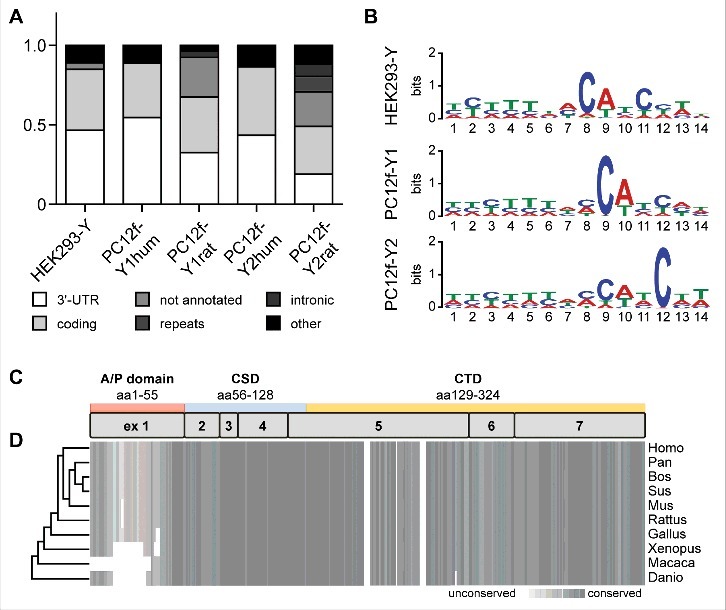
RNA binding of YBX1. (A) Spatial RNA binding of YBX1. The YBX1-bound reads of the two fusion clones were separated according to specific mapping to either the human (PC12f-Y1hum and PC12f-Y2hum) or the rat genome (PC12f-Y1rat and PC12f-Y2rat). In all analyzed cell lines YBX1 binds its targets mainly in the 3‘-UTR and the coding region. The main difference in human/rat reads bound by YBX1 in the fusion cells is due to the lower annotation of the rat genome leading to the identification of not annotated reads in the condition of PC12f-Y1rat and PC12f-Y2rat. (B) MEME binding sequence analysis. YBX1 binds to the UCUUUNNCAUC-sequence in all three cell lines. (C+D) YBX1 consists of the arginine/proline- (A/P) rich domain, the cold-shock domain (CSD) and the C-terminal domain (CTD) and is highly conserved among species. The conservation is depicted by a grey heatmap and were analyzed by the program PRALINE. Human and rat YBX1 share 97% of the protein sequence with an identical sequence of the RNA-binding CSD.
The fusion of PC12-eGFP and HEK293-Y cells generated cells with an expanded genome and transcriptome composed of human and rat. The resulting increase in transcript diversity led to the identification of 768 novel YBX1-bound RNAs in the fusion cells as compared to the parental HEK293-Y cells. YBX1 binds its target RNAs independent of the species via a UCUUUNNCAUC motif (Fig. 5C) that mainly resides in the 3′-UTR and coding regions (Fig. 5A).
Discussion
HEK293-Y and PC12-eGFP fusion cells (PC12f-Y) express both parental marker genes YBX1 and eGFP. Their morphology is not only different from the parental cells but also from each other. These differences can be expected based on the differential DNA composition of the fusion cells.
Since the DNA content of the fusion cell lines is two to three times that of the parental cells, fusion clones can represent more than two genomes allowing further diversification of the transcriptome (e.g., after fusion of more than two cell lines). We validated the contribution of each parental cell type to the merged transcriptome using RNAseq and found human and rat RNA species in fusion-cell lines with 35 to 70% rat transcripts derived from PC12 cells. Before performing PAR-CLIPs on fusion cells, we confirmed efficient uptake and incorporation of 4SU. The lower efficiency of 4SU incorporation in fusion clones compared to the parental HEK293-Y cells reflects the differential expression of nucleoside transporters which are divided in two classes: Concentrative nucleoside transporters (SLC28A1, SLC28A2 and SLC28A3) and equilibrative nucleoside transporters (SLC29A1, SLC29A2, SLC29A3 and SLC29A4). While HEK293-Y cells as well as the fusion clones PC12f-Y1 and PC12f-Y2 mainly express the uridine-transporters SLC29A1 and SLC29A224, PC12-eGFP express predominantly the nucleoside transporter SLC29A4 which a high affinity for adenine and adenosine only.24 The differential 4SU uptake and incorporation of the fusion cells leads to a lower enrichment of YBX1-RNA complexes compared to HEK293-Y cells. Accordingly, PAR-CLIP of PC12f-Y1 and PC12f-Y2 were performed with more starting material to obtain similar amounts of YBX1-RNA complexes. Taken together, the cell fusion of HEK293-Y und PC12-eGFP generated fusion cells with a heterogeneous genome consisting of rat and human which incorporate sufficient amounts of 4SU to perform PAR-CLIP experiments. While the increased uptake of 4SU could also be achieved by stable overexpression of the transporters SLC29A1 and SLC29A2 and would be suited for cell-types that do not efficiently fuse. We find the fusion process very efficient for PC12 and 293 cells, which offer the benefit of increasing the complexity of the transcriptome and facilitates interspecies comparisons that overexpression of the transporters could not achieve. This artificial environment can change expression levels of target transcripts, and indeed we find novel transcripts that relate to the process of cell fusion and maintaining a stable genome. Thus, we can evaluate transcripts that are not present in sufficient amounts in each parental cell. As the CLIP reads are normalized to the transcript levels as determined by RNAseq of the fusion cells, potential changes in expression levels can be accounted for. Still, the fusion cells could lead to the identification of protein-RNA interactions that do not occur in a natural setting, which would be similar to the results of in vitro studies using EMSA. The functional relevance would always have to be validated in the original cell system – e.g. through a loss or gain of function approach followed by quantification of isoform expression.
Fusion-CLIP provides the increased specificity and low signal to noise ratio of PAR-CLIP in an increased spectrum of cell types.24 It offers the possibility to examine RNA interactions of proteins that poorly crosslink with high energy UV of 254 nm, but are amenable to 4SU PAR-CLIP.2 In addition, Fusion-CLIP allows to study protein-binding to newly synthesized transcripts by metabolic labeling of RNA using photoreactive nucleosides.25,26 Taken together, Fusion-CLIP facilitates the analysis of increased transcriptome complexity and interspecies comparisons as compared to other CLIP methods as well as to overexpression of 4SU transporters in the target cells.
Although YBX1 is highly conserved between human and rat (97% sequence identity; 100% identity of the RNA binding domain; Fig. 5C, D), we found an increased percentage of human PAR-CLIP reads as compared to RNAseq reads – especially in PC12f-Y1 (45.9% human reads in PAR-CLIP and only 24.7% in RNAseq). The shorter PAR-CLIP reads together with the incomplete rat annotation might contribute to this bias, as sequences mapping to regions that are not annotated cannot be assigned to a specific gene and were therefore discarded. This is a main reason for the for the lower identification of YBX1-bound transcripts in the fusion cells compared to the parental HEK293-Y cells since about 25% of the rat PAR-CLIP reads derived from PC12f-Y1 and PC12f-Y2 map to regions that have not previously been annotated.
Since RNA-binding sites are often highly conserved among species, long PAR-CLIP reads (> 20 nt) are needed to discriminate orthologue sequences between human and rat in the fusion cells (Fig. S4). Reads that did not map to a specific genome were discarded and led to a lower read count in the fusion cells. The number of YBX1-bound RNAs identified in fusion cells was lower than those identified in the parental HEK293-Y cells (Fig. 4A). Despite the short read length and the species homology, cell fusion led to the identification of 768 additional interactions of YBX1, which not only relate to the PC12 phenotype but also to changes resulting from the increased cell-size and DNA content associated with cell fusion.
The binding motif analysis using MEME22 validated the CAUC binding motif of YBX1 in HEK293-Y as well as in PC12f-Y1 and PC12f-Y2 cells.27 After binding this motif YBX1 activates 3′-splice sites.28 The inclusion of additional YBX1 target RNAs allowed us to identify a UCUUUNN motif upstream of the CAUC motif. The UCUUUNN motif could either promote the binding of a cofactor or also be recognized by YBX1 and influence the strength of its binding to target RNAs. YBX1's binding sites are predominantly located in the coding region or the 3′-UTR of its substrates (Fig. 5A). At the 3′-UTR YBX1 regulates polyadenylation, translation efficiency, localization, and stability of mRNAs.29,30
The expanded transcriptome of PC12f-Y fusion cells with an increased transcript complexity enables the identification of novel YBX1-bound RNAs species, which were predominantly derived from PC12 cells and partly relate to the transcriptional response to the fusion process such as DNA transposition (Fig. 4A, D). Importantly, the PC12-Y fusion transcriptome enabled the identification of genes related to toll-like receptor 4 and interferon type I signaling, which have both implications in Alzheimer's Disease.19–21 Thus, the improved transcript diversity can provide novel links to human disease, which would not have been discovered in the HEK 293 transcriptome.
Conclusions
Using cell fusion we expanded the HEK293 fibroblast transcriptome with neuronal transcripts of PC12 cells. The process combines existing technology and is therefore easy to employ. It generates suitable amounts of cells for the analysis of RNA binding proteins using PAR-CLIP in a short amount of time as compared to other types of cell engineering. In this study rat and human cells were used to differentiate the transcripts and monitor the contribution of each parental cell-line to the merged transcriptome. Future Fusion-CLIP of mouse and human cells with better genome annotation can be expected to further increase the identification of novel substrates, which are not annotated in the current version of the rat genome. This could also be achieved by revisiting our dataset when rat genome annotation has been improved. In addition to the ease of use and the possibility to compare species differences in RNA-protein interactions, a main benefit of the method is the access to transcripts for disease relevant tissue, facilitating the analysis of e.g. a cardiac or neuronal specific RNAs. Thus, cell fusion opens up the application of the PAR-CLIP to cells with a specialized transcriptome that cannot easily be engineered to take up 4SU and facilitates the analysis of tissue-specific expressed RNA binding proteins.
Materials and methods
Handling of cells
PC12-EGFP and PC12f-Y fusion cells were cultured on collagen coated plates with DMEM (Lonza) supplemented with 10% horse serum (Sigma), 5% fetal bovine serum (Sigma) and 1% PenStrep. The medium of PC12-EGFP cells were additionally supplemented with 600 µg/ml G418 (PAA), the medium of PC12-Y fusion cells with 600 µg/ml G418 and 2 µg/ml blasticidin (InvivoGen). 293 T-REx YBX1 cells were cultured in DMEM supplemented with 10% fetal bovine serum and 1% PenStrep.
Generation of stable cell lines
PC12 cells were transfected with pEGFP-C1 and for 2 weeks selected with 600 µg/ml G418. Single colonies were picked and expanded under antibiotic pressure.
Cell fusion
6–7 × 106 PC12-EGFP and 293 T-REx YBX1 cells were mixed and spun down in serum-free medium. The whole following process was performed at 37 °C with pre-warmed solutions and serum-free medium. Under continuous stirring 100 µl of 50% PEG 1500 (Roche) were added to the cell pellet over a period of 1 min. The fusion mixture was stirred for 2 more min before 100 µl medium was added over a period of 3 min. After the addition of 1 ml medium the mixture were incubated for 5 min at 37 °C. Cells were spun down and placed in fresh medium. After 2 days the medium were replaced with medium supplemented with 600 µg/ml G418 and 2 µg/ml blasticidin (InvivoGen). Single colonies were picked and expanded.
Cell morphology
The cell morphology and expression of eGFP were analyzed using bight field and fluorescence microscopy (Leica).
Cell cycle and DNA content analysis
The cells were harvested and suspended in cold PBS, fixed in 80% ethanol and stored for at least 1 h at 4°C. The fixed cells were stained in PBS containing 40 µg/ml propidium iodide (Sigma Aldrich) and 100 µg/ml RNase A (Thermo Scientific) and incubated for 30 min at 37 °C in the dark. The cells were diluted with PBS and analyzed using a LSRFortessa (BD Bioscience). The software FlowJo was used for data analysis.
Western blot
Protein extracts of doxycycline (Clonetech) induced and not induced cells were separated by SDS-PAGE and transferred on a nitrocellulose membrane. Primary antibodies were anti-HA (Covance) and anti-GAPDH (Calbiochem).
PAR-CLIP
293 T-REx YBX1 cells and the two fusion clones PC12f-Y1 and PC12f-Y2, all expressing FLAG-tagged YBX1, were used for PAR-CLIP following the protocol in Hafner et al., 20102.
RNA sample preparation for RNA sequencing
The RNA of doxycycline induced cells was extracted using TRIzol (Invitrogen) and a clean-up was performed using RNeasy mini spin columns (QIAGEN). The concentration and integrity of the RNA were analyzed using a NanoDrop (Peqlab) and an agilent bioanalyzer 2100 (Agilent Technologies). The mRNA library preparation was performed using the TruSeq RNA Sample Preparation v2 Guide (Illumina) following the manufacturer's instructions.
Sequencing of cDNA libraries
The PAR-CLIP libraries were sequenced with 1 × 51 cycles on an Illumina HiSeq2000 (Illumina). The cDNA libraries were prepared for sequencing using the TruSeq RNA Sample Prep Kit v2 following the manufacturer's instructions and sequenced 2 × 101 paired end on a Illumina HiSeq2000 (Illumina).
Read processing
Adaptors were trimmed using Cutadapt31 with the following parameters:
-a TCGTATGCCGTCTTCTGCTTG -g AATGATACGGCGACCACCGACAGGTTCAGAGTTCTACAGTCCGACGATC -b CGTACGCGGGTTTAAACGA -b CTCATCTTGGTCGTACGCGGAATAGTTTAAACTGT -q 20 -n 1 -m 16 -e 0.1 –overlap = 4
Mapping strategy
First, we remove reads stemming from rRNA from the PAR-Clip libraries. To this end, PAR-Clip reads were first mapped against all human rRNAs from SILVA32 using STAR (version 2.3.1z1)33 with the following mapping parameters:
–chimSegmentMin 40 –chimJunctionOverhangMin 40 –outSAMattributes All –outFilterIntronMotifs RemoveNoncanonical – –alignSJoverhangMin 10 –outFilterMatchNmin 16 –alignIntronMax 10000 –outFilterMismatchNmax 2 –seedMultimapNmax 100000 –seedPerReadNmax 100000 –outQSconversion Add 33 –outFilterMultimapNmax 10000 –seedSearchStartLmax 6 –sjdbOverhang 99 –winAnchorMultimapNmax 1000
All reads that mapped to rRNAs were discarded.
Subsequently, the remaining reads were mapped with STAR to the human genome (GRCh38.80), the rat genome (Rnor_5.0) and the the union of the two genomes, using the following parameters:
–chimSegmentMin 40 –chimJunctionOverhangMin 40 –outFilterMultimapNmax 20 –outSAMattributes All –outFilterIntronMotifs RemoveNoncanonical –alignSJoverhangMin 12 –out FilterMatchNmin 19 –alignIntronMax 10000 –seedMultimapNmax 200000 –seedPerReadNmax 60000 –outQSconversionAdd 33 –seedSearchStartLmax 6 –sjdbOverhang 99 –winAnchorMultimapNmax 1000 –outFilterMismatchNmax 3
To study the effect of the read-length filtering (Fig. S4) we used the same parameters as described above, except that we allowed shorter read alignments. This was done by using the parameter –outFilterMatchNmin 14 for the alignment.
Mapping of RNA-Seq reads was performed with STAR, using the following parameters: -alignEndsType EndToEnd —chimSegmentMin 40 –chimJunctionOverhangMin 40 –outFilterMultimapNmax 20 –outSAMattributes All –outFilterIntronMotifs RemoveNoncanonical –alignSJoverhangMin 500
Read filtering
To minimize the effect of reads mapping to multiple locations on our analysis, we removed reads that did not align uniquely. Specifically, we only kept the best alignment of a read if the second best alignment had more than one mismatch more than the best alignment. Furthermore, we discarded reads that had more than two mismatches.
Quantification
Gene expressions were quantified using HTSeq15 (v. 0.6.0) with the following parameters: –minaqual = 0 –stranded = no –type = gene –mode = union
Peak calling
For peak calling, the alignments for the different samples we merged. Peaks were called with PARalyzer34 using the following parameters:
BANDWIDTH = 3
CONVERSION = T>C
MINIMUM_READ_COUNT_PER_GROUP = 5
MINIMUM_READ_COUNT_PER_CLUSTER = 2
MINIMUM_READ_COUNT_FOR_KDE = 3
MINIMUM_CLUSTER_SIZE = 11
MINIMUM_CONVERSION_LOCATIONS_FOR_CLUSTER = 2
MINIMUM_CONVERSION_COUNT_FOR_CLUSTER = 2
MINIMUM_READ_COUNT_FOR_CLUSTER_INCLUSION = 1
MINIMUM_READ_LENGTH = 20
MAXIMUM_NUMBER_OF_NON_CONVERSION_MISMATCHES = 1
EXTEND_BY_READ
Motif discovery
In order to make the motifs of different samples comparable, we down-sampled the peaks such that all samples had the same number of peaks, as the library with the smallest number of peaks (n = 1610). Subsequently, we determined motifs using MEME35 (v.4.11.1). For the motif discovery, we used the following parameters:
-mod anr -nmotifs 5 -minw 8 -maxw 14 -bfile -dna -minsites 800 -p 3 -maxsites 3000
The background nucleotide distribution was computed on all human transcripts.
4SU incorporation assay
Cells were fed with 4SU for 16h and subsequently the RNA was extracted via TRIzol. 2 µg of RNA were incubated with 10 mM Tris-HCl, 10 mM EDTA and 5 µg biotin-HPDP for 2 h in the dark. The incorporated 4SU forms a disulfide bond with the reactive HPDP-biotin. In the following the biotinylated RNA was extracted via phenol/chloroform extraction. The reaction was spotted on a Hyperbond N+ membrane (Amersham). The membrane was subsequently UV-crosslinked (twice with 1200 µJ) and the biotinylated RNA was visualized using the Chemiluminescent Nucleic Acid Detection Module (Pierce).
Supplementary Material
Funding Statement
This work was supported by the Deutsche Forschungsgemeinschaft, Bonn, Germany under grant Go865/11-1 to M.G., the European Research Council under grant StG282078 to M.G., “Bundesministerium für Bildung und Forschung” under grant CaRNAtion to U.O., M.L., M.G., and the German Center for Cardiovascular Research (DZHK), Berlin to M.G.
Disclosure statement
The authors declare that they have no competing interests.
Acknowledgments
We thank Claudia Langnick and Mirjam Feldkamp (Wei Chen lab, Max Delbrück Center for Molecular Medicine) for support with NGS.
Funding
This work was supported by the Deutsche Forschungsgemeinschaft, Bonn, Germany under grant Go865/11-1 to M.G., the European Research Council under grant StG282078 to M.G., “Bundesministerium für Bildung und Forschung” under grant CaRNAtion to U.O., M.L., M.G., and the German Center for Cardiovascular Research (DZHK), Berlin to M.G.
Availability of data and material
The RNA sequencing and PAR-CLIP data have been submitted to the NCBI sequence read archive (SRP119588).
Abbreviations
Abbreviations
- 4SU
4-thiouridine
- A/P domain
arginine/proline rich domain
- CSD
cold shock domain
- CTD
C-terminal domain
- Dox
doxycycline
- eGFP
enhanced green fluorescent protein
- Fusion-CLIP
Cell fusion followed by PAR-CLIP
- GO
gene ontology
- HEK 293
Human embryonic kidney cells 293
- HEK293-Y
YBX1-expressing HEK 293 cells
- HITS-CLIP
High-throughput sequencing of RNA isolated by crosslinking immunoprecipitation
- PAR-CLIP
Photoactivatable ribonucleoside–enhanced crosslinking and immunoprecipitation
- PC12-eGFP
PC12 cells expressing eGFP
- PC12f-Y
PC12-eGFP HEK 293 fusion cells expressing YBX1
- PEG
polyethylene glycol
- PI
propidium iodide
- YBX1
Y box binding protein 1
References
- 1.Benhalevy D, Gupta SK, Danan CH, Ghosal S, Sun H-W, Kazemier HG, Paeschke K, Hafner M, Juranek SA. The Human CCHC-type Zinc Finger Nucleic Acid-Binding Protein Binds G-Rich Elements in Target mRNA Coding Sequences and Promotes Translation. Cell Rep. 2017;18:2979–2990. doi: 10.1016/j.celrep.2017.02.080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M, Jungkamp A-C, Munschauer M, Ulrich A, Wardle GS, Dewell S, Zavolan M, Tuschl T. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell. 2010;141:129–141. doi: 10.1016/j.cell.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Konig J, Zarnack K, Rot G, Curk T, Kayikci M, Zupan B, Turner DJ, Luscombe NM, Ule J. iCLIP–transcriptome-wide mapping of protein-RNA interactions with individual nucleotide resolution. J Vis Exp. 2011;50:2638. doi: 10.3791/2638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lebedeva S, Jens M, Theil K, Schwanhäusser B, Selbach M, Landthaler M, Rajewsky N. Transcriptome-wide analysis of regulatory interactions of the RNA-binding protein HuR. Mol Cell. 2011;43:340–352. doi: 10.1016/j.molcel.2011.06.008. [DOI] [PubMed] [Google Scholar]
- 5.Licatalosi DD, Mele A, Fak JJ, Ule J, Kayikci M, Chi SW, Clark TA, Schweitzer AC, Blume JE, Wang X, Darnell JC, Darnell RB. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456:464–469. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Nostrand EL, Pratt GA, Shishkin AA, Gelboin-Burkhart C, Fang MY, Sundararaman B, Blue SM, Nguyen TB, Surka C, Elkins K, Stanton R, Rigo F, Guttman M, Yeo GW. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP). Nat Methods. 2016;13:508–514. doi: 10.1038/nmeth.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zarnegar BJ, Flynn RA, Shen Y, Do BT, Chang HY, Khavari PA. irCLIP platform for efficient characterization of protein-RNA interactions. Nat Methods. 2016;13:489–492. doi: 10.1038/nmeth.3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gillen AE, Yamamoto TM, Kline E, Hesselberth JR, Kabos P. Improvements to the HITS-CLIP protocol eliminate widespread mispriming artifacts. BMC Genomics. 2016;17:338. doi: 10.1186/s12864-016-2675-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maeshima K, Funakoshi T, Imamoto N. Cell-fusion method to visualize interphase nuclear pore formation. Methods Cell Biol. 2014;49:2488 122: 239–254. [DOI] [PubMed] [Google Scholar]
- 10.Gammal R, Baker K, Heilman D. Heterokaryon technique for analysis of cell type-specific localization. J Vis Exp. 2011; doi: 10.3791/2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lyabin DN, Eliseeva IA, Ovchinnikov LP. YB-1 protein: functions and regulation. Wiley Interdiscip Rev RNA. 2014;5:95–110. doi: 10.1002/wrna.1200. [DOI] [PubMed] [Google Scholar]
- 12.Janz M, Harbeck N, Dettmar P, Berger U, Schmidt A, Jürchott K, Schmitt M, Royer H-D. Y-box factor YB-1 predicts drug resistance and patient outcome in breast cancer independent of clinically relevant tumor biologic factors HER2, uPA and PAI-1. Int J Cancer. 2002;97:278–282. doi: 10.1002/ijc.1610. [DOI] [PubMed] [Google Scholar]
- 13.Shiraiwa S, Kinugasa T, Kawahara A, Mizobe T, Ohchi T, Yuge K, Fujino S, Katagiri M, Shimomura S, Tajiri K, Sudo T, Kage M, Kuwano M, Akagi Y. Nuclear Y-Box-binding Protein-1 Expression Predicts Poor Clinical Outcome in Stage III Colorectal Cancer. Anticancer Res. 2016;36:3781–3788. [PubMed] [Google Scholar]
- 14.Bobkova NV, Lyabin DN, Medvinskaya NI, Samokhin AN, Nekrasov PV, Nesterova IV, Aleksandrova IY, Tatarnikova OG, Bobylev AG, Vikhlyantsev IM, Kukharsky MS, Ustyugov AA, Polyakov DN, Eliseeva IA, Kretov DA, Guryanov SG, Ovchinnikov LP. The Y-Box Binding Protein 1 Suppresses Alzheimer's Disease Progression in Two Animal Models. PLoS ONE. 2015;10:e0138867. doi: 10.1371/journal.pone.0138867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Young JD, Yao SYM, Baldwin JM, Cass CE, Baldwin SA. The human concentrative and equilibrative nucleoside transporter families, SLC28 and SLC29. Mol Aspects Med. 2013;34:529–547. doi: 10.1016/j.mam.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 17.Bindea G, Mlecnik B, Hackl H, Charoentong P, Tosolini M, Kirilovsky A, Fridman W-H, Pagès F, Trajanoski Z, Galon J. ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics. 2009;25:1091–1093. doi: 10.1093/bioinformatics/btp101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eden E, Navon R, Steinfeld I, Lipson D, Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tahara K, Kim H-D, Jin J-J, Maxwell JA, Li L, Fukuchi K. Role of toll-like receptor signalling in Abeta uptake and clearance. Brain. 2006;129:3006–3019. doi: 10.1093/brain/awl249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X, Long J, He T, Belshaw R, Scott J. Integrated genomic approaches identify major pathways and upstream regulators in late onset Alzheimer's disease. Sci Rep. 2015;5:12393. doi: 10.1038/srep12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taylor JM, Minter MR, Newman AG, Zhang M, Adlard PA, Crack PJ. Type-1 interferon signaling mediates neuro-inflammatory events in models of Alzheimer's disease. Neurobiol Aging. 2014;35:1012–1023. doi: 10.1016/j.neurobiolaging.2013.10.089. [DOI] [PubMed] [Google Scholar]
- 22.Machanick P, Bailey TL. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics. 2011;27:1696–1697. doi: 10.1093/bioinformatics/btr189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zasedateleva OA, Krylov AS, Prokopenko DV, Skabkin MA, Ovchinnikov LP, Kolchinsky A, Mirzabekov AD. Specificity of mammalian Y-box binding protein p50 in interaction with ss and ds DNA analyzed with generic oligonucleotide microchip. J Mol Biol. 2002;324:73–87. doi: 10.1016/S0022-2836(02)00937-3. [DOI] [PubMed] [Google Scholar]
- 24.Young JD, Yao SYM, Sun L, Cass CE, Baldwin SA. Human equilibrative nucleoside transporter (ENT) family of nucleoside and nucleobase transporter proteins. Xenobiotica. 2008;38:995–1021. doi: 10.1080/00498250801927427. [DOI] [PubMed] [Google Scholar]
- 25.Dölken L, Ruzsics Z, Rädle B, Friedel CC, Zimmer R, Mages J, Hoffmann R, Dickinson P, Forster T, Ghazal P, Koszinowski UH. High-resolution gene expression profiling for simultaneous kinetic parameter analysis of RNA synthesis and decay. RNA. 2008;14:1959–1972. doi: 10.1261/rna.1136108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rabani M, Levin JZ, Fan L, Adiconis X, Raychowdhury R, Garber M, Gnirke A, Nusbaum C, Hacohen N, Friedman N, Amit I, Regev A. Metabolic labeling of RNA uncovers principles of RNA production and degradation dynamics in mammalian cells. Nat Biotechnol. 2011;29:436–442. doi: 10.1038/nbt.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dolfini D, Mantovani R. Targeting the Y/CCAAT box in cancer: YB-1 (YBX1) or NF-Y? Cell Death Differ. 2013;20:676–685. doi: 10.1038/cdd.2013.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wei W-J, Mu S-R, Heiner M, Fu X, Cao L-J, Gong X-F, Bindereif A, Hui J. YB-1 binds to CAUC motifs and stimulates exon inclusion by enhancing the recruitment of U2AF to weak polypyrimidine tracts. Nucleic Acids Res. 2012;40:8622–8636. doi: 10.1093/nar/gks579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barrett LW, Fletcher S, Wilton SD. Regulation of eukaryotic gene expression by the untranslated gene regions and other non-coding elements. Cell Mol Life Sci. 2012;69:3613–3634. doi: 10.1007/s00018-012-0990-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pichon X, Wilson LA, Stoneley M, Bastide A, King HA, Somers J, Willis AEE. RNA binding protein/RNA element interactions and the control of translation. Curr Protein Pept Sci. 2012;13:294–304. doi: 10.2174/138920312801619475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011;17:10–12. doi: 10.14806/ej.17.1.200. [DOI] [Google Scholar]
- 32.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 2013;41:D590–596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Corcoran DL, Georgiev S, Mukherjee N, Gottwein E, Skalsky RL, Keene JD, Ohler U. PARalyzer: definition of RNA binding sites from PAR-CLIP short-read sequence data. Genome Biol. 2011;12:R79. doi: 10.1186/gb-2011-12-8-r79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bailey TL, Elkan C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc Int Conf Intell Syst Mol Biol. 1994;2:28–36. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.